

Abstract
The purpose of this work was to determine what aspect of the milled compound influences its thermal profile. For this, six different compounds with different properties were chosen and cryomilled for different times to get an amorphous solid. Differential scanning calorimetry (DSC) and X-ray powder diffraction were used to characterize the material and look at the thermal behavior. Melt-quenched samples were also prepared, and the thermal profile upon milling was determined and correlated with the thermal behavior of the cryomilled samples. Growth rates were determined by hot-stage microscopy. Ketoconazole, when cryomilled, showed only one crystallization exotherm in the DSC profile. Ursodiol, and to some extent indomethacin, initially showed a double exotherm which eventually become a single exotherm on further milling. Griseofulvin, carbamazepine, and piroxicam exhibited a double exotherm in the DSC profile upon cryomilling to the amorphous state. Surface crystal growth rates around Tg were found to be highest for compounds showing the double exotherm in the DSC. Thus, it was seen that compounds which have high surface crystallization tendency will exhibit the double exotherm during heating.
KEY WORDS: amorphous, cryomilling, crystallization, thermal analysis
INTRODUCTION
Pharmaceutical drugs and excipients are known to exist in mainly crystalline or amorphous forms. In the crystalline state, compounds can exist either as polymorphs or pseudopolymorphs (1). Transformation between these various forms is possible and quite likely during pharmaceutical unit processing, especially when it comes to operations such as drying, granulation, milling, compactions, etc. (2). Among these, milling is generally used for particle size reduction as well as for intimate mixing of powders, and polymorphic transformation of various drugs during milling has been reported for indomethacin and other drugs (3,4). Also, due to the considerable amount of energy input during the milling process, the mechanical stress involved in the process can also contribute to a reduction in crystallinity of the powder aside from reducing the particle size (5). While having a disordered material might mean having certain advantages such as higher solubility and dissolution or improved mechanical properties (6,7), it also entails the risk of product performance issues due to crystallization during further processing and storage. Disordered particles obtained via milling are also susceptible to increased water sorption which can influence its physical state as seen for albuterol sulfate (8) and also to increased chemical instability (9). However, not all compounds are influenced to the same extent upon mechanical impaction, and the propensity of a compound to form a disordered or amorphous state upon milling depends on the crystal structure of the material (10).
There are many examples of compounds which form the amorphous state upon milling. Since the milling process results in significant heating of the material, it is possible that the amorphous state is formed due to localized melting of the sample and its subsequent quenching. However, it has been shown that the main mechanism for formation of the amorphous state is due to accumulation of defects which ultimately results in a loss of long-range order (LRO) (11). In the case of cryomilling, since the sample is constantly kept in a bath of liquid nitrogen, at a temperature far below the glass transition temperature (Tg), the formation of amorphous regions is essentially due to the accumulation of defects which can be gradual or spontaneous.
It has been seen for most compounds that the starting material has a definite impact on the behavior of the final milled product. For example, when indomethacin was cryomilled to yield an X-ray amorphous form, it was seen that the milled γ form reverted back to the same solid form upon crystallization as the starting polymorph, while the α form resulted in a mixture of α and γ forms upon recrystallization at 30°C (12). This result is said to be due to the presence of small crystallites which have not completely transformed to the amorphous state thus acting as seeds or due to a “memory effect” where there is some LRO associated with the powder. This also results in faster crystallization of cryomilled crystals than ground melt-quenched amorphous samples.
The creation of disorder in the material due to a loss of long-range order can be due to thermodynamic or kinetic disordering (13). Thermodynamic disordering refers to the direct appearance of random close packing, due to complete loss of long-range-order symmetry operators (translational, orientational, and conformational), in the amorphous state without any local packing arrangement. Kinetic disordering is a continuous process where the long-range order is reduced to a shorter-range-order nanocrystalline where the local crystalline packing symmetry is preserved (giving very broad crystalline peaks in the powder X-ray pattern). Compounds that are milled can reach the glassy state via an intermediate nanocrystalline state or remain in a nanocrystalline state depending on the properties of the compound. In this case, nanocrystalline can refer to having an order of the scale of a few nanometers and can be distinguished from amorphous phase by a lack of Tg in the differential scanning calorimetry (DSC) profile.
Recently, there had been reports about cryomilling of griseofulvin wherein it was claimed that the final product after cryomilling is not amorphous but results in a defective or “mesomorphous” state, and this conclusion was drawn mostly due to an unusual (bimodal) recrystallization event in the DSC profile and the apparent lack of a Tg (14,15). Similar thermal behavior has been observed for another compound, etravirine, where cryomilling resulted in an amorphous solid exhibiting a double exotherm in the crystallization event (16). However, upon further examination of the double exotherm, it was concluded that griseofulvin actually forms an amorphous state on milling and that the double exotherms coincide with two distinct events: crystallization of nucleated surfaces and subsequent crystallization of the bulk and non-nucleated surfaces (17). The question why compounds such as griseofulvin may exhibit this interesting type of crystallization behavior and what underlying physicochemical properties are responsible for it remains unanswered to date.
In this paper, we investigated the effect of cryomilling on the thermal behavior of a diverse set of six drug compounds (provided in Fig. 1) to see if any of these compounds show an effect similar to that of griseofulvin and whether there are any similarities in their physicochemical properties which could provide a clue to explaining this unusual thermal behavior.
Fig. 1.

Chemical structures of the compounds used in this study: a griseofulvin (GSF), b piroxicam (PXM), c indomethacin (IMC), d carbamazepine (CBZ), e ketoconazole (KTZ), and f ursodeoxycholic acid (USD)
MATERIALS AND METHODS
Materials
Griseofulvin (GSF), piroxicam (PXM), and carbamazepine (CBZ) were obtained from Sigma-Aldrich Co. (St. Louis, MO); indomethacin (IMC) and ketoconazole (KTZ) were purchased from Hawkins, Inc. (Minneapolis, MN, USA); and ursodeoxycholic acid (USD) was purchased from Spectrum Chemicals (New Brunswick, NJ, USA).
Preparation of Cryomilled Form
Crystalline drugs were cryomilled using a SPEX centriprep 6750 cryogenic impact mill (Metuchen, NJ, USA). Approximately 1 g of the drug compound was filled into a cylindrical polycarbonate vial and milled for different time intervals. Cycles of 2 min of milling were alternated with a 2-min cooling phase to prevent any temperature buildup. The intensity of milling was set to ten impacts per second. Subsequent to milling, the sample was allowed to come to room temperature in a desiccator containing P2O5 (to prevent condensation of environmental moisture from influencing the physical state of the samples) prior to removing it for analysis.
Milling of Amorphous Form
Amorphous GSF and CBZ were prepared by melting pure crystalline drug on a hot plate set a few degrees above its melting temperature under nitrogen flow. After the samples were kept on the hot plate for a sufficient period of time (to ensure complete melting), the melts were quench cooled by immersing in liquid nitrogen since cooling over ice resulted in partial crystallization. These amorphous solids were immediately placed in a desiccator over P2O5 to prevent water sorption. The other drugs were also prepared in the same way, except that they were quenched over ice instead of liquid nitrogen. Subsequently, the samples were milled for 15 s in a small stainless steel vial with one steel ball bearing using a Wig-L-Bug crescent amalgamator (Dentsply International, Surrey, UK).
Differential Scanning Calorimetry
The samples were analyzed using a DSC 2920 (TA instruments, New Castle, DE, USA) in the standard mode at a rate of 10°C per minute. The instrument was calibrated for temperature and enthalpy using indium. Nitrogen was used as a purge gas at 50 ml/min. The samples were sealed in hermetic aluminum pans and run from 15°C to above their melting temperatures. The results were analyzed using Universal Analysis software version 4.7A (TA Instruments, New Castle, DE, USA).
X-Ray Powder Diffraction
The samples were analyzed using a Siemens D-5000 diffractometer using Cu Kα radiation with a wavelength of 1.54 Å. Scans were taken from 4° to 40° 2θ at a step size of 0.02° and a scan rate of 1 s per step. The X-ray generator voltage and amperage was set to 40 kV and 40 mA, respectively. The divergence and scattering slits were 1.0 mm wide, and the receiving slit was 0.1 mm wide. The samples were mounted in a silicone sample holder with a well diameter of 1.3 cm and well depth of 1 mm.
Determination of Surface Crystal Growth Rates Using Hot-Stage Microscopy
For determination of surface crystal growth rates, the compounds were dissolved in dichloromethane and spin coated on coverslips using a KW-4A spin coater (Chemat Technology Inc, Northridge, CA, USA). The polymorphs that are stable at room temperature and which were used for cryomilling were sprinkled on the sample to nucleate the respective polymorphs. The samples were then loaded on the microscope hot stage (Linkam THMS 600, Surrey, UK), allowing the sample temperature to be precisely controlled at selected values. Once crystals started growing on the surface of the amorphous matrix, it was centered in the field of the microscope (Nikon Eclipse E600, Nikon Corp, Tokyo, Japan), and pictures of the growing face of the crystals were taken at regular intervals using an image capturing software “Metavue” (Molecular Devices Inc., CA, USA). The rate of crystal growth was calculated from the slope of the length vs. time plot of the growing face, which was linear at the different temperatures studied. The radial growth rate was then plotted against temperature using a log scale since the growth changed exponentially with temperature.
RESULTS
Griseofulvin
GSF exhibits a melting temperature of around 218–220°C and has a very low water solubility of 30.56 μg/ml in water at 25°C (18). GSF became more and more disordered upon cryomilling as seen from the X-ray profile where a reduction in the peak intensity and an associated increase in the background amorphous halo occurred (Fig. 2a). The GSF crystalline peaks did not show any increase in peak width but only a decrease in intensity with milling time indicating that the transition from crystal to amorphous was a direct transformation and is best modeled as a mixture of amorphous and crystalline material (13).
Fig. 2.

a XRPD of unmilled and cryomilled GSF, b DSC profiles of cryomilled GSF. Cryomilling was done for 10, 30, and 60 min and shows a gradual development of a double exothermic peak
The DSC profiles of the cryomilled samples are shown in Fig. 2b. A broad exotherm (~15 J/g) developed after 10 min of cryomilling overlapping the Tg of the melt quench which is around 88°C (data not shown). After 30 min, it could be clearly seen that two separate exothermic thermal events were obtained leading to the formation of a double exotherm which became even more pronounced after 60 min resulting in an overall recrystallization enthalpy of around 44 J/g. As shown in a previous publication, the first exotherm is due to the crystallization of particles having nuclei on their surface (surface crystallization); the second exotherm corresponds to crystallization of particle surfaces without nuclei along with the crystallization of the interior of the particles (bulk crystallization) (17). The fact that a considerable increase in surface self-diffusion relative to bulk diffusion occurs at temperatures close to Tg results in the surface crystallization occurring close to and sometimes overlapping the glass transition event (19). It was also seen that the two exothermic peaks were due to crystallization to the same polymorph (which is to be expected since GSF has only one reported form).
Indomethacin
It is known that IMC exists in at least three polymorphic forms and many solvated forms (20). Cryomilling of the three anhydrate forms has been carried out, and it was seen that all of these forms result in the formation of the amorphous state after around 60 min of processing (21).
In our experiments, the stable form of IMC (γ-form) was taken as obtained from the vendor and cryomilled for 10, 30, and 60 min. From the XRPD (Fig. 3a), it can be seen that the X-ray diffraction peaks broadened as milling proceeded. This observation points to the possibility that amorphization of indomethacin occurs via a continuous disordering process instead of the direct crystal to amorphous transition as seen for griseofulvin (13,22). The DSC profiles for these samples are shown in Fig. 3b. It can be noted that IMC initially forms a broad exotherm of 20 J/g after milling for 10 min. After 30 min, a hint of formation of a double exotherm can be observed. However, this develops into a clear recrystallization event of 38 J/g after 60 min of milling. From the melting behavior, it could be concluded that the same form (γ) was obtained after the recrystallization. Finally, for IMC, a Tg could be identified around 40°C which is a few degrees lower than that of the undisturbed melt-quenched sample.
Fig. 3.

a XRPD of unmilled and cryomilled IMC, b DSC profiles of cryomilled IMC. Cryomilling was done for 10, 30, and 60 min, and only one exothermic peak is observed
Piroxicam
PXM (form I) was cryomilled for 10, 30, 60, and 120 min to ensure complete amorphization. XRPD confirmed the formation of an X-ray amorphous powder (Fig. 4a). DSC evaluation showed that while the unmilled piroxicam form I exhibited only an endothermic peak at 201°C corresponding to melting of the crystal form, the DSC of the milled samples showed the presence of two crystallization exotherms (Fig. 4b) before melting. After 10 min of milling, the exotherms (totaling around 24 J/g) were seen at 60°C and 83°C, respectively. The second exotherm was dominant upon short periods of milling but reduced in magnitude as the milling progressed. For the first exotherm, the opposite was observed as it increased upon cryomilling and dominated the crystallization event after 120 min. The total crystallization enthalpy was around 56 J/g after 120 min of cryomilling. The Tg of melt-quenched amorphous PXM is around 61°C, and it can be seen that the crystallization exotherm in the cryomilled samples overlapped the glass transition temperature. The melting temperature of the recrystallized samples did not change upon milling indicating that the same polymorph (form I) is obtained for all samples. XRPD (data not shown) confirmed that the same polymorphic form was obtained after both exotherms and that the second exotherm did not represent any type of polymorphic transformation.
Fig. 4.

a XRPD of unmilled and cryomilled PXM, b DSC profiles of cryomilled PXM. Cryomilling for 10, 30, 60, and 120 min shows development of a double exotherm dominated by the first exotherm
Ketoconazole
For KTZ, crystallization in different solvents and recrystallizing from amorphous melt-quenched samples have resulted in only one polymorph as determined by XRPD (23).
Cryomilling of KTZ has not been reported in literature to date. Upon cryomilling KTZ for 10, 30, and 60 min, XRPD (Fig. 5a) showed that KTZ rapidly became disordered and was completely amorphous after 60 min of milling. The width of the diffraction peaks increased only slightly and then started to reduce in intensity as the amorphous halo developed. This suggested again a direct crystal to amorphous transition. The DSC results (Fig. 5b) showed just a single exothermic event, representing crystallization prior to melting. The enthalpy of this exotherm increased with milling time (from 18 J/g after 10 min to 42 J/g after 60 min). The formation of the amorphous state in the powder was clear and discrete since crystallization (1) occurred at temperatures 20–30°C higher than the Tg which was seen around 38° for the milled sample and (2) the crystallization exotherm was very sharp, unlike that seen for PXM and GSF.
Fig. 5.

a XRPD of unmilled and cryomilled KTZ, b DSC profiles of cryomilled KTZ. Cryomilling for 10, 30, and 60 min shows only one exotherm
Ursodeoxycholic Acid
USD is known to form the amorphous state both upon grinding and melt quenching (24). However, it has been reported that the amorphous form prepared by these two methods resulted in a different crystallization behavior (25). Therefore, USD was considered to be an interesting compound for inclusion into the current study. From the XRPD data (Fig. 6a), it could be seen that USD formed the amorphous form quite easily with the diffraction peaks drastically reducing after just 10 min and an X-ray amorphous material being observed after 30 min. The DSC results (Fig. 6b) showed that after 10 min, there was a broad exotherm (25 J/g) due to crystallization of the disordered regions. Upon further milling, a double exotherm developed with the first exotherm occurring just above the Tg. The fact that two exotherms were observed after 30 min of cryomilling suggests that crystal nuclei are still present at that point, a phenomenon XRPD was unable to pick up. Increasing the milling time resulted in the first exotherm reducing in size and the second exotherm increasing and moving to higher temperatures. After 120 min of cryomilling, only one exotherm (56 J/g) was observed with a clear Tg indicating complete amorphization of the solid. Thus, while USD shows a double exotherm in the beginning as GSF does, its behavior is different from GSF since it ended up with a single exotherm (with a visible Tg) after prolonged milling.
Fig. 6.

a XRPD of unmilled and cryomilled USD, b DSC profiles of cryomilled USD. Cryomilling for 10, 30, 60, and 120 min shows a double exotherm in the beginning which develops into only one exotherm
Carbamazepine
CBZ is known to exist in various polymorphic forms and has been studied by multiple authors. It is not completely clear as to how many polymorphic forms of CBZ actually exist as different numbers of forms have been reported (26–28). For the sake of clarity, the stable form at room temperature will be called form III here; the stable form at higher temperatures will be termed form I, in line with the nomenclature of McMahon (29) and Krahn (27).
CBZ form III was cryomilled for 10, 30, and 60 min and analyzed by DSC and XRPD. The X-ray data (Fig. 7a) showed that the intensity of the diffraction peaks decreased without any significant broadening of the peaks. After 60 min of cryomilling, there were the remnants of some of the crystalline peaks indicating that the sample was not completely amorphous. DSC results (Fig. 7b) showed a broad exotherm (10 J/g) after 10 min. This exotherm occurred over the Tg of amorphous CBZ which is normally observed around 50–55°C. As milling time was increased, the exotherm increased and eventually resulted in two recrystallization exotherms after 60 min, similar to what was seen for GSF, with a total crystallization enthalpy of around 41 J/g. Interestingly, while the DSC results of the starting material showed the melting of form III and subsequent crystallization of form I (which is more stable at higher temperatures), the thermograms of the cryomilled samples showed direct enantiotropic transformation of form III to form I at an onset temperature of 150°C followed by a large melting endotherm of form I at around 189°C.
Fig. 7.

a XRPD of unmilled and cryomilled CBZ, b DSC profiles of cryomilled CBZ. Cryomilling for 10, 30, and 60 min shows a gradual development of double crystallization exotherm
Effect of Milling on Melt-Quenched Samples
Melt-quenched amorphous samples were milled for 15 s, and the powder was analyzed by DSC. The thermograms of the compounds showing the double exotherm upon cryomilling of the crystals are shown in Fig. 8. GSF showed a thermal profile showing a small exotherm immediately after the Tg and before the main recrystallization exotherm which also occurs at a lower temperature than for the undisturbed sample (data not shown). This suggests that the thermal behavior of the triturated melt quench is similar to the behavior of the milled crystalline compound. Milling of the amorphous melt-quenched CBZ sample also showed the development of a small exotherm before the main exotherm (as seen for GSF), but the main exotherm was more broadened than for GSF. PXM crystals, when analyzed by DSC, melted at around 200°C and did not crystallize when cooled at a rate of 20°C/min. Reheating the sample showed the Tg of melt-quenched piroxicam to be around 61°C, and no recrystallization was observed during the heating phase. However, when melt-quenched piroxicam was milled and analyzed by DSC, the sample showed the appearance of a double crystallization exotherm where the first exotherm overlapping the Tg was more pronounced than for the previous two compounds and the second exotherm was also broader than observed for CBZ. These results show that the double exotherm in the thermogram observed for these compounds is due to crystallization of the amorphous phase and should not be ascribed to the formation of a new “mesomorphous” phase formed from the crystal, as suggested previously (14).
Fig. 8.

DSC thermograms of ball-milled melt-quenched a CBZ, b GSF, and c PXM show the development of a small exotherm at temperatures around the T g followed by the main crystallization exotherm
When IMC, KTZ, and USD were quenched from the melt to form an amorphous solid and analyzed after milling the sample, all three samples showed a similar recrystallization behavior. Crystallization of the amorphous drug particles occurred as one exotherm at temperatures far above their Tg and relatively close to the melting temperature for IMC and KTZ (Fig. 9).
Fig. 9.

DSC thermograms of ball-milled melt-quenched a IMC, b KTZ, and c USD show that for these compounds, the recrystallization occurs at temperatures considerably higher than the T g
Surface Crystal Growth Rates
The surface crystal growth rates of the different compounds are shown in Fig. 10 where the temperatures have been normalized to the glass transition temperature. Radial growth rates were measured starting from temperatures below Tg to around 30°C above the Tg. It was seen that PXM and GSF had the fastest growth rate among all the compounds, both of which were higher by nearly two orders of magnitude when compared to IMC and KTZ. CBZ had an intermediate growth rate, close to one order of magnitude higher than IMC and KTZ. The CBZ growth rate appeared to move closer to that of GSF and PXM with increasing temperature. USD had significantly lower growth rates (more than one order of magnitude) compared to GSF and PXM, but they were slightly higher than those for IMC and KTZ. IMC showed a slight jump in growth rate close to Tg with reducing temperature. The exact reason is not known, but a similar increase in growth rate has been observed in the bulk as well, and it was suggested to be due to a change in the mechanism of growth and crystal morphology (30,31).
Fig. 10.

Surface crystal growth rates of PXM (squares), GSF (diamonds), CBZ (triangles), USD (plus signs), IMC (circles), and KTZ (multiplication signs) as a function of the temperature below and above the T g
DISCUSSION
Crystallization of the amorphous phase is a spontaneous process, the rate of which depends on a combination of the thermodynamic driving force and molecular mobility. It is made up of two subprocesses, nucleation and growth, and may be studied either at isothermal temperatures or non-isothermally as has been done in this study. In a non-isothermal analysis, a crystallization exotherm is detected at temperatures where the rate of crystallization is high enough to result in a release of heat that can be detected by the instrument. According to classical crystallization theory for amorphous compounds, the rate of crystal growth exhibits a maximum at a certain temperature between the Tg and the Tm, depending on the opposing influences of the thermodynamic driving force (increases with decrease in temperature) and molecular mobility (decreases with decrease in temperature) (32). Therefore, one would expect a recrystallization exotherm at a temperature above Tg and below the Tm upon heating an amorphous sample. However, the compounds selected clearly show different recrystallization behavior. While KTZ always showed a single crystallization exotherm above Tg, IMC and USD initially showed a double exotherm which evolved to a single exotherm upon further milling. On the other hand, GSF, PXM, and CBZ always showed a clear double exotherm with one exotherm occurring below Tg and, in the case of PXM, the first exotherm being dominant. Given the clear differences observed in crystallization behavior, it is of interest to further try to understand these in terms of crystallization tendency and physicochemical drug compound characteristics.
Recently, the crystallization tendency upon cooling drugs from the melt has been classified (Table I) (33). According to this classification, class I solids are drug compounds that crystallize during cooling of the melt at 20°C/min; class II compounds are those that turn amorphous on cooling but crystallize upon heating at 10°C/min; and class III compounds remain amorphous upon cooling and on the subsequent reheating step. Class I compounds were further sub-divided into type I-A, which crystallize even at higher rates of cooling, and type I-B, which can form the amorphous phase upon using fast rates of cooling (using liquid nitrogen) (33). A cursory glance at the classification, applied to our current set of model compounds, shows that GSF and CBZ which show double exotherms are present in class I-B, i.e., they show high crystallization tendencies. The fact that PXM is classified as a class III even though it results in a double exotherm may possibly be due to undisturbed PXM having a low nucleation rate, resulting in the absence of a recrystallization exotherm. In contrast, upon cryomilling, one can expect nuclei to be already present in the material, thereby triggering overall crystallization more easily.
Table I.
Classification of Drugs Based on Their Glass Formation Ability (Adapted from (33))
| Class | Behaviora | Examples | |
|---|---|---|---|
| Class 1 | A | Always crystallize on cooling | Atenolol, felbinac |
| B | Amorphous on quench cooling | Griseofulvin, carbamazepine | |
| Class 2 | Cooling—no crystallization; heating—crystallization | Acetaminophen, cinnarizine | |
| Class 3 | Cooling and heating—no crystallization | Piroxicam, ketoconazole, indomethacin, ursodiol | |
aCooled at 20°C/min, heated at 10°C/min in a DSC
The reduced crystallization temperature of an amorphous solid is a normalized measure of the temperature at which spontaneous crystallization occurs during heating so that different compounds can be compared on their crystallization tendency (34). It is given by the equation,
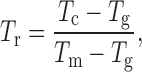 |
where Tc is the temperature of crystallization, Tg is the glass transition temperature, and Tm is the melting temperature. In the present study, onset temperatures were used for the calculation, and Tc was determined only after the triturating of the sample to ensure that all the compounds crystallized upon heating at 10°C/min. The Tr values are reported in Table II. Interestingly, it can be seen that compounds which had a low Tr were also the ones which exhibited the double exotherm in the cryomilled samples. Thus, it can be concluded that the crystallization tendency of these compounds determines the type of crystallization exotherm observed upon cryomilling.
Table II.
Summary of Physicochemical Properties of the Compounds Studied
| Compound | Mol wt. (g/mol) | T m (°C) | T g (°C) | T r a | ΔC p (J/g·K) | Rotatable bonds | ΔH f (kJ/mol) | ΔS f (J/mol·K) |
|---|---|---|---|---|---|---|---|---|
| Carbamazepine | 236 | 190 | 55 | 0.18 | 0.38 | 0 | 24.7 | 53.5 |
| Piroxicam | 331 | 201 | 61 | 0.22 | 0.46 | 2 | 34.8 | 73.5 |
| Griseofulvin | 352 | 218 | 88 | 0.24 | 0.46 | 3 | 39.6 | 80.8 |
| Ursodiol | 392 | 203 | 103 | 0.49 | 0.48 | 4 | 37.6 | 78.9 |
| Indomethacin | 357 | 160 | 45 | 0.58 | 0.47 | 5 | 38.4 | 88.9 |
| Ketoconazole | 531 | 148 | 45 | 0.68 | 0.47 | 7 | 52.2 | 123.9 |
aReduced crystallization temperature T r = (T c − T g)/(T m − T g)
In the last few years, an improved understanding of the crystallization process from amorphous solids has been obtained. Briefly, the crystallization process can be separated into two processes, one occurring at the surface and the other happening in the bulk (35,36). Surface crystallization occurs at a much faster rate compared to bulk especially at temperatures near and below Tg. This difference in crystal growth rates reduces as the temperature is increased with no differences to be seen above a certain temperature. In a previous publication of our group, surface crystallization has been suggested to be the most probable explanation for the appearance of a double exotherm observed upon heating cryomilled griseofulvin (17). Upon comparison of the surface crystal growth rates of the different compounds used in this study (Fig. 10), it can be seen that GSF indeed has a very fast surface growth rate which apparently allows sufficient crystallization to occur at Tg. It has also been observed that crystallization of GSF and IMC bulk powders analyzed by XRPD shows a two-stage crystallization with the first stage exhibiting fast surface crystallization and the second, bulk crystallization that is much slower (35,37). In a DSC experiment, crystallization occurs in the same order resulting in two distinct exotherms. In general, the compounds showing the double exotherm were found to be the ones with a much higher crystal growth rate on the surface while the samples exhibiting unimodal recrystallization exotherm had lower surface growth rates. It should be noted that the heating rate used in the experiment can have an influence on the resulting crystallization behavior. For example, when high heating rates are used, very little surface crystallization may occur at Tg, even for compounds with high surface growth rates resulting in a clear observation of the glass transition in the thermogram (38). Upon using lower heating rates, the latter can potentially be overshadowed by the surface crystallization exotherm. In addition, reducing the heating rate for compounds with low surface growth rates can also result in exhibiting a crystallization exotherm around Tg, to the extent that the heating rate used allows detectable crystallization to take place (39). The faster the surface growth rate, the harder it will be to prevent its crystallization at Tg by increasing the heating rate in the experiment.
The reason why some compounds crystallize faster on their surface as compared to others is expected to further depend on their physicochemical properties, some of which are provided in Table II where the Tg values reported are for the undisturbed melt-quenched samples heating at 10°C/min. Why cryomilled compounds show differences in terms of their crystallization behavior is currently not very well understood. In what follows, we will attempt to relate the latter to a number of physicochemical attributes of the model compounds that have been shown before to have an influence on crystallization behavior when prepared by cooling from the melt (molecular weight, heat of fusion, entropy of melting, number of rotatable bonds, etc.) (33). With respect to molecular weight, it has been observed that compounds with smaller molecular weights tend to crystallize faster in general which is most likely due to the fact that these molecules will have higher diffusivity due to their low mass which will allow them to move to the growing crystal face more quickly. It will also help the molecules to attain the appropriate conformation more easily since less energy will be required to move lighter side groups. For our compounds, it can indeed be observed that the highest molecular weight of KTZ coincides with the fact that this compound showed the lowest crystallization tendency. CBZ, on the other hand, had the lowest molecular weight but crystallized slower than PXM and GSF. Also, GSF and IMC which had very similar molecular weights showed very different crystallization behavior. Therefore, molecular weight alone does not suffice to fully explain the observed rank order. As the number of rotatable bonds has previously been found to influence crystallization behavior from amorphous products prepared by cooling from the melt, it is of interest to further evaluate this property with respect to the current study (33). In the amorphous state, the number of possible conformations can be thought to be smaller for molecules having a lower number of rotatable bonds, i.e., more rigid molecules, resulting in the lowering of the configurational entropy and thus making it easier to attain a conformation matching that in the crystalline form (40). Therefore, compounds that have a fewer number of rotatable bonds are expected to crystallize out faster. The entropy of fusion is related to the number of rotatable bonds, since an increase in the latter would allow a large number of configurations to be achieved by the molecules in the melt and thus increase the entropic difference between the crystal and the melt at the Tm. In this study, we observe that the compounds with the highest crystallization rates are also those with a fewer number of rotatable bonds. CBZ is the most rigid with zero freely rotatable bonds, GSF has three, and while PXM has two rotatable bonds, one of which is hindered due to an intramolecular H bond (41). While entropic considerations are of importance, the reason why GSF and PXM had faster surface crystal growth rates than CBZ which has a fewer number of rotatable bonds and had a lower molecular weight could be due to enthalpic reasons (as seen from the higher heat of fusion for PXM and GSF). It is also possible that there is some contribution from hydrogen bonding patterns between the molecules in the amorphous phase which leads to the observed order of crystallization. Clearly, it is a combination of molecular properties which determine the rates of crystallization of the different compounds.
Other than thermodynamic considerations, kinetic factors such as relaxation time of molecular motion can also have an influence on the crystal growth rate (42). When a sample is melted and cooled, the molecular mobility is highest close to the melting temperature and decreases as the temperature is reduced. The rate at which the mobility/viscosity changes with temperature in the super-cooled region depends on whether the system is “strong” or “fragile” and may have an impact on bulk crystallization rates (43). A fragile system is one in which the mobility or viscosity follows a non-arrhenius relationship with temperature in the super-cooled region. The differences between the fragility of the compounds studied can contribute to different crystallization rates at temperatures between the Tg and the Tm. Also, fragility is a bulk property, and since the double exotherm is observed due to crystallization on the surface, it may not have much correlation to this study. Moreover, the bulk mobility for all compounds will be the same at and close to the Tg since by definition, the glass transition temperature occurs when the viscosity is around 1012 Pa·s or relaxation time constant is around 100 s (at a heating rate of 10°C/min) (44). In this study, since we are concerned about the crystallization that occurs at or close to the Tg, molecular mobility in the bulk will be very similar for all the compounds studied and is therefore not expected to contribute to the difference in the crystallization behavior near the Tg. It has also been shown that the mobility or self-diffusion of molecules on the surface of an amorphous solid is significantly higher than that in the bulk, and this can result in crystallization at temperatures around the Tg (36). Experimental determination of this surface self-diffusion has recently been done using AFM imaging (19). It is possible that PXM, GSF, and CBZ have the optimum surface self-diffusion rates compared to the other compounds which may be causing an increased surface crystal growth rate.
Since crystallization is a combination of nucleation and crystal growth, mention has to be made of the importance of the nucleation behavior of the amorphous solids. Melt-quenched samples will not show any crystallization unless they have nuclei present as seen for PXM when it is quenched and heated in a DSC. Milling of these melt-quenched samples creates a large amount of surface and also creates nuclei. The larger the total number of nuclei on the surface, the greater will be the amount of solid crystallizing at Tg, while a small number of nuclei will result in incomplete surface crystallization around Tg during heating leading to a small first exotherm. In compounds with slow surface crystal growth rates, the presence of a small number of nuclei may not be enough to cause any noticeable crystallization during heating while a large number of nuclei will allow detection of surface crystallization in a DSC. For example, in the milled amorphous IMC sample, we observed a unimodal exotherm, while it is seen in literature that when melt-quenched IMC was milled for 2 h and heated at 5°C/min, it developed a bimodal crystallization exotherm (45). When the sample was milled at higher milling rates, however, the exotherm favored a unimodal shape. Since the crystal growth rates do not change, the only reason why this could be happening is a large number of crystalline nuclei being formed on the surface at low milling rates which result in a larger amount of sample crystallizing and being detected. At higher milling rates, it is likely that most of the nuclei are forced back into the amorphous phase resulting in a fewer number of nuclei.
Clearly, milling will result in the creation of a large amount of surface on the amorphous solid which can increase the extent of crystallization occurring around the glass transition temperature. As shown in this work, not all compounds subjected to milling result in detectable surface crystallization at a temperature close to Tg at the same heating rate in a DSC study. Whether a milled amorphous compound exhibits a single or a double recrystallization exotherm will depend on the number of nuclei formed and the crystal growth rate at the surface as well as the heating rate employed to analyze the sample. Therefore, it is not just the creation of surface but also the physicochemical properties of the compound that influences nucleation behavior and crystal growth rates and thus the extent of crystallization around Tg.
CONCLUSIONS
GSF, PXM, and CBZ showed a double exotherm upon cryomilling of the crystals to the X-ray amorphous state while only one crystallization exotherm was observed for IMC, USD, and KTZ. The compounds exhibiting the double exotherm showed higher surface crystal growth rates as measured by light microscopy. High crystal nucleation and growth rates which are influenced by multiple physicochemical compound properties as well as the creation of a large amount of surface from cryomilling, where crystal growth rates are higher than the bulk (especially around the Tg), appear to be the cause for this unusual thermal behavior.
ACKNOWLEDGMENTS
The authors acknowledge financial support from the McKeehan Graduate Assistantship and would like to thank Dr. Katsuhiro Kobayashi for his help with the cryomilling of the drugs.
REFERENCES
- 1.Haleblian J, McCrone W. Pharmaceutical applications of polymorphism. J Pharm Sci. 1969;58(8):911–929. doi: 10.1002/jps.2600580802. [DOI] [PubMed] [Google Scholar]
- 2.Morris KR, Griesser UJ, Eckhardt CJ, Stowell JG. Theoretical approaches to physical transformations of active pharmaceutical ingredients during manufacturing processes. Adv Drug Deliv Rev. 2001;48(1):91–114. doi: 10.1016/S0169-409X(01)00100-4. [DOI] [PubMed] [Google Scholar]
- 3.Lin SY, Hsu CH, Ke WT. Solid-state transformation of different gabapentin polymorphs upon milling and co-milling. Int J Pharm. 2010;396(1–2):83–90. doi: 10.1016/j.ijpharm.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 4.Otsuka M, Matsumoto T, Kaneniwa N. Effect of environmental temperature on polymorphic solid-state transformation of indomethacin during grinding. Chem Pharm Bull. 1986;34(4):1784–1793. doi: 10.1248/cpb.34.1784. [DOI] [PubMed] [Google Scholar]
- 5.Willart JF, De Gusseme A, Hemon S, Odou G, Danede F, Descamps M. Direct crystal to glass transformation of trehalose induced by ball milling. Solid State Commun. 2001;119(8–9):501–505. doi: 10.1016/S0038-1098(01)00283-6. [DOI] [Google Scholar]
- 6.Longuemard P, Jbilou M, Guyot-Hermann AM, Guyot JC. Ground and native crystals: comparison of compression capacity and dissolution rate. Int J Pharm. 1998;170(1):51–61. doi: 10.1016/S0378-5173(98)00120-3. [DOI] [Google Scholar]
- 7.Duddu SP, Grant DJW. The use of thermal-analysis in the assessment of crystal disruption. Thermochim Acta. 1995;248:131–145. doi: 10.1016/0040-6031(94)01951-C. [DOI] [Google Scholar]
- 8.Ward GH, Schultz RK. Process-induced crystallinity changes in albuterol sulfate and its effect on powder physical stability. Pharm Res. 1995;12(5):773–779. doi: 10.1023/A:1016232230638. [DOI] [PubMed] [Google Scholar]
- 9.Shalaev E, Shalaeva M, Zografi G. The effect of disorder on the chemical reactivity of an organic solid, tetraglycine methyl ester: change of the reaction mechanism. J Pharm Sci. 2002;91(2):584–593. doi: 10.1002/jps.10066. [DOI] [PubMed] [Google Scholar]
- 10.Wildfong PLD, Hancock BC, Moore MD, Morris KR. Towards an understanding of the structurally based potential for mechanically activated disordering of small molecule organic crystals. J Pharm Sci. 2006;95(12):2645–2656. doi: 10.1002/jps.20672. [DOI] [PubMed] [Google Scholar]
- 11.Willart JF, Descamps M. Solid state amorphization of pharmaceuticals. Mol Pharm. 2008;5(6):905–920. doi: 10.1021/mp800092t. [DOI] [PubMed] [Google Scholar]
- 12.Otsuka M, Kaneniwa N. A kinetic study of the crystallization process of noncrystalline indomethacin under isothermal conditions. Chem Pharm Bull. 1988;36(10):4026–4032. doi: 10.1248/cpb.36.4026. [DOI] [PubMed] [Google Scholar]
- 13.Bates S, Zografi G, Engers D, Morris K, Crowley K, Newman A. Analysis of amorphous and nanocrystalline solids from their X-ray diffraction patterns. Pharm Res. 2006;23(10):2333–2349. doi: 10.1007/s11095-006-9086-2. [DOI] [PubMed] [Google Scholar]
- 14.Feng T, Bates S, Carvajal MT. Toward understanding the evolution of griseofulvin crystal structure to a mesophase after cryogenic milling. Int J Pharm. 2009;367(1–2):16–19. doi: 10.1016/j.ijpharm.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Feng T, Pinal R, Carvajal MT. Process induced disorder in crystalline materials: differentiating defective crystals from the amorphous form of griseofulvin. J Pharm Sci. 2008;97(8):3207–3221. doi: 10.1002/jps.21219. [DOI] [PubMed] [Google Scholar]
- 16.Qi S, Weuts I, De Cort S, Stokbroekx S, Leemans R, Reading M, et al. An investigation into the crystallisation behaviour of an amorphous cryomilled pharmaceutical material above and below the glass transition temperature. J Pharm Sci. 2010;99(1):196–208. doi: 10.1002/jps.21811. [DOI] [PubMed] [Google Scholar]
- 17.Trasi NS, Boerrigter SXM, Byrn SR. Investigation of the milling-induced thermal behavior of crystalline and amorphous griseofulvin. Pharm Res. 2010;27(7):1377–1389. doi: 10.1007/s11095-010-0129-3. [DOI] [PubMed] [Google Scholar]
- 18.Zili Z, Sfar S, Fessi H. Preparation and characterization of poly-e-caprolactone nanoparticles containing griseofulvin. Int J Pharm. 2005;294(1–2):261–267. doi: 10.1016/j.ijpharm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 19.Zhu L, Brian CW, Swallen SF, Straus PT, Ediger MD, Yu L. Surface Self-Diffusion of an Organic Glass. Phys Rev Lett. 2011;106:256103-1–256103-4. doi: 10.1103/PhysRevLett.106.256103. [DOI] [PubMed] [Google Scholar]
- 20.Nicolai B, Ceolin R, Rietveld IB. Polymorphism and solvation of indomethacin. J Therm Anal Calorim. 2010;102(1):211–216. doi: 10.1007/s10973-009-0412-x. [DOI] [Google Scholar]
- 21.Crowley KJ, Zografi G. Cryogenic grinding of indomethacin polymorphs and solvates: assessment of amorphous phase formation and amorphous phase physical stability. J Pharm Sci. 2002;91(2):492–507. doi: 10.1002/jps.10028. [DOI] [PubMed] [Google Scholar]
- 22.Rani M, Govindarajan R, Surana R, Suryanarayanan R. Structure in dehydrated trehalose dihydrate—evaluation of the concept of partial crystallinity. Pharm Res. 2006;23(10):2356–2367. doi: 10.1007/s11095-006-9058-6. [DOI] [PubMed] [Google Scholar]
- 23.Viseras C, Salem II, Galan ICR, Galan AC, Galindo AL. The effect of recrystallization on the crystal growth, melting point and solubility of ketoconazole. Thermochim Acta. 1995;268:143–151. doi: 10.1016/0040-6031(95)02319-4. [DOI] [Google Scholar]
- 24.Yonemochi E, Ueno Y, Ohmae T, Oguchi T, Nakajima S, Yamamoto K. Evaluation of amorphous ursodeoxycholic acid by thermal methods. Pharm Res. 1997;14(6):798–803. doi: 10.1023/A:1012114825513. [DOI] [PubMed] [Google Scholar]
- 25.Yonemochi E, Inoue Y, Buckton G, Moffat A, Oguchi T, Yamamoto K. Differences in crystallization behavior between quenched and ground amorphous ursodeoxycholic acid. Pharm Res. 1999;16(6):835–840. doi: 10.1023/A:1018817801444. [DOI] [PubMed] [Google Scholar]
- 26.Kaneniwa N, Yamaguchi T, Watari N, Otsuka M. Hygroscopicity of carbamazepine crystalline powders. Yakugaku Zasshi-J Pharm Soc Jpn. 1984;104(2):184–190. doi: 10.1248/yakushi1947.104.2_184. [DOI] [PubMed] [Google Scholar]
- 27.Krahn FU, Mielck JB. Relations between several polymorphic forms and the dihydrate of carbamazepine. Pharmaceutica Acta Helvetiae. 1987;62(9):247–254. [PubMed] [Google Scholar]
- 28.Umeda T, Ohnishi N, Yokoyama T, Kuroda K, Kuroda T, Tatsumi E, et al. Kinetics of the thermal transition of carbamzepine polymorphic forms in the solid state. Yakugaku Zasshi-J Pharm Soc Jpn. 1984;104(7):786–792. doi: 10.1248/yakushi1947.104.7_786. [DOI] [PubMed] [Google Scholar]
- 29.McMahon LE, Timmins P, Williams AC, York P. Characterization of dihydrates prepared from carbamazepine polymorphs. J Pharm Sci. 1996;85(10):1064–1069. doi: 10.1021/js960117e. [DOI] [PubMed] [Google Scholar]
- 30.Ishida H, Wu TA, Yu LA. Sudden rise of crystal growth rate of nifedipine near Tg without and with polyvinylpyrrolidone. J Pharm Sci. 2007;96(5):1131–1138. doi: 10.1002/jps.20925. [DOI] [PubMed] [Google Scholar]
- 31.Wu T, Yu L. Origin of enhanced crystal growth kinetics near Tg probed with indomethacin polymorphs. J Phys Chem B. 2006;110(32):15694–15699. doi: 10.1021/jp062771g. [DOI] [PubMed] [Google Scholar]
- 32.Okui N. Relationship between crystallization temperature and melting temperature in crystalline materials. J Mater Sci. 1990;25(3):1623–1631. doi: 10.1007/BF01045361. [DOI] [Google Scholar]
- 33.Baird JA, Van Eerdenbrugh B, Taylor LS. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J Pharm Sci. 2010;99(9):3787–3806. doi: 10.1002/jps.22197. [DOI] [PubMed] [Google Scholar]
- 34.Zhou DL, Zhang GGZ, Law D, Grant DJW, Schmitt EA. Physical stability of amorphous pharmaceuticals: importance of configurational thermodynamic quantities and molecular mobility. J Pharm Sci. 2002;91(8):1863–1872. doi: 10.1002/jps.10169. [DOI] [PubMed] [Google Scholar]
- 35.Wu T, Yu L. Surface crystallization of indomethacin below Tg. Pharm Res. 2006;23(10):2350–2355. doi: 10.1007/s11095-006-9023-4. [DOI] [PubMed] [Google Scholar]
- 36.Zhu L, Wong L, Yu L. Surface-enhanced crystallization of amorphous nifedipine. Mol Pharm. 2008;5(6):921–926. doi: 10.1021/mp8000638. [DOI] [PubMed] [Google Scholar]
- 37.Zhu L, Jona J, Nagapudi K, Wu TA. Fast surface crystallization of amorphous griseofulvin below T (g) Pharm Res. 2010;27(8):1558–1567. doi: 10.1007/s11095-010-0140-8. [DOI] [PubMed] [Google Scholar]
- 38.Willart JF, Carpentier L, Dannede F, Descamps M. Solid-state vitrification of crystalline griseofulvin by mechanical milling. J Pharm Sci. 2012;101(4):1570–1577. doi: 10.1002/jps.23041. [DOI] [PubMed] [Google Scholar]
- 39.Chattoraj S, Bhugra C, Telang C, Zhong L, Wang ZR, Sun CC. Origin of two modes of non-isothermal crystallization of glasses produced by milling. Pharm Res. 2012;29(4):1020–1032. doi: 10.1007/s11095-011-0644-x. [DOI] [PubMed] [Google Scholar]
- 40.Zhou D, Zhang GGZ, Law D, Grant DJW, Schmitt EA. Thermodynamics, molecular mobility and crystallization kinetics of amorphous griseofulvin. Mol Pharm. 2008;5(6):927–936. doi: 10.1021/mp800169g. [DOI] [PubMed] [Google Scholar]
- 41.Vrecer F, Vrbinc M, Meden A. Characterization of piroxicam crystal modifications. Int J Pharm. 2003;256(1–2):3–15. doi: 10.1016/S0378-5173(03)00057-7. [DOI] [PubMed] [Google Scholar]
- 42.Korhonen O, Bhura C, Pikal MJ. Correlation between molecular mobility and crystal growth of amorphous phenobarbital and phenobarbital with polyvinylpyrrolidone and L-proline. J Pharm Sci. 2008;97(9):3830–3841. doi: 10.1002/jps.21273. [DOI] [PubMed] [Google Scholar]
- 43.Ediger MD, Angell CA, Nagel SR. Supercooled liquids and glasses. J Phys Chem. 1996;100(31):13200–13212. doi: 10.1021/jp953538d. [DOI] [Google Scholar]
- 44.Crowley KJ, Zografi G. The use of thermal methods for predicting glass-former fragility. Thermochim Acta. 2001;380(2):79–93. doi: 10.1016/S0040-6031(01)00662-1. [DOI] [Google Scholar]
- 45.Desprez S, Descamps M. Transformations of glassy indomethacin induced by ball-milling. J Non-Cryst Solids. 2006;352(42–49):4480–4485. doi: 10.1016/j.jnoncrysol.2006.02.130. [DOI] [Google Scholar]