

Abstract
The highly conserved HIV-1 gp41 “pocket” region is a promising target for inhibiting viral entry. PIE12-trimer is a protease-resistant trimeric D-peptide inhibitor that binds to this pocket and potently blocks HIV entry. PIE12-trimer also possesses a reserve of binding energy that provides it with a strong genetic barrier to resistance (“resistance capacitor”).
Here we report the design of a modular scaffold employing PEGs of discrete lengths for the efficient optimization and synthesis of PIE12-trimer. This scaffold also allows us to conjugate PIE12-trimer to several membrane-localizing cargoes, resulting in dramatically improved potency and retention of PIE12-trimer’s ability to absorb the impact of resistance mutations. This scaffold design strategy should be of broad utility for the rapid prototyping of multimeric peptide inhibitors attached to potency- or pharmacokinetic-enhancing groups.
Introduction
HIV entry is mediated by the trimeric viral envelope glycoprotein (Env), which is cleaved into surface (gp120) and transmembrane (gp41) subunits1, 2. Viral entry is triggered by binding of gp120 to a primary receptor (CD4) and subsequently a coreceptor (typically CXCR4 or CCR5), which induces large conformation changes in gp120 that activate gp41 for fusion3. gp41 then adopts an extended prehairpin conformation, embedding its N-terminal hydrophobic fusion peptide into the host cell membrane, bridging the virus and host cell (Fig. 1). In this state, the gp41 N-peptide region forms a trimeric coiled-coil (N-trimer) while the C-peptide region is in a structurally undefined state. This prehairpin intermediate then slowly collapses into a hairpin structure, with the C-peptide folding back upon the N-trimer to pack in an antiparallel orientation into the grooves of the N-trimer. The formation of this trimer-of-hairpins structure brings the viral and host membranes into close proximity and drives membrane fusion.4, 5
Figure 1. HIV entry pathway.
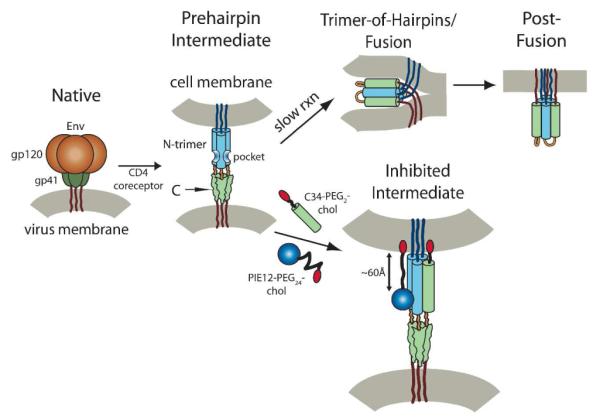
Upon engagement with cellular receptor and coreceptor, gp120 and gp41 undergo a conformational change resulting in extension of gp41 into the prehairpin intermediate, exposing the hydrophobic pocket region of the N-trimer. gp41 collapses into the trimer-of-hairpins structure, juxtaposing the viral and host membranes and causing membrane fusion. The hydrophobic pocket targeted by PIE12 is an estimated 60 Å from the cell membrane, which can be bridged by a relaxed PEG24 linker. In contrast, the C-peptide C-terminus is directly adjacent to the membrane. Cholesterol (red) conjugated with PEG spacers (black lines) are shown.
In the prehairpin intermediate, gp41 is vulnerable to inhibitors that bind to either the N-trimer or C-peptide2, 6 and prevent hairpin formation7-9. This vulnerability has been exploited by the C-peptide-derived therapeutic Fuzeon (enfuvirtide). Fuzeon binds to a portion of the N-trimer groove, preventing fusion with nM potency. Though effective, Fuzeon is currently utilized only as “salvage therapy” for patients with multi-drug resistance because of its high cost (~$30,000/year/patient), dosing requirements (90 mg twice daily), injection site reactions, and the rapid emergence of resistant strains10, 11.
The gp41 N-trimer contains a functionally critical and highly conserved deep hydrophobic pocket at its C-terminus4, 12, 13. The genomic region that encodes for the pocket also forms the structured RNA region of the Rev-responsive element (RRE), which is critical for the export of viral mRNA to the cytoplasm14, further constraining evolution of this region on the nucleotide level. Fuzeon binds to the N-trimer groove region just outside the pocket, an area that is more tolerant of resistance mutations. Second/third-generation C-peptide inhibitors (e.g, T1249, T2635) bind the groove and pocket and are much less susceptible to resistance10, 15-20.
We have utilized structure-guided mirror-image phage display to generate D-peptide inhibitors that bind with high affinity to the pocket13, 21, 22. D-peptides are protease resistant (as proteases have stereochemical specificity and generally only cleave L-substrates)23, giving them the potential for a much longer lifetime in the body.PIE12, our most potent monomeric D-peptide, is a pocket-specific inhibitor of HIV-1 with high-nM potency against the difficult-to-inhibit primary HIV isolate JRFL. Since the N-trimer contains three symmetric pockets, we designed a trimeric version of PIE12 that uses PEG to link three monomers and greatly improves affinity and potency via avidity. PIE12-trimer inhibits all major HIV clades with high-pM to low-nM potency21 and is a promising preclinical candidate for the treatment and prevention of HIV-1. Here we describe a novel modular PEG scaffold used to optimize the production and the potency of PIE12-trimer.
While designing D-peptide inhibitors with progressively greater potency, we encountered a potency limit that could not be overcome by affinity optimization because the target is only available in the short-lived prehairpin intermediate. Due to the finite target exposure and the limits of diffusion, the potency of inhibitors with very high affinities (and on-rates) is limited by the diffusion-limited on-rate rather than binding affinity. For such diffusion-limited inhibitors, a potency plateau is reached beyond which further improvements in affinity do not improve potency. Similar potency plateaus have been observed for several inhibitors that target the transient prehairpin intermediate21, 22, 24-26. “Over-engineering” our inhibitors with improved affinity, but no corresponding improvement in potency, provides a reserve of binding energy and slows the evolution of resistance mutations. This “resistance capacitor” eliminates the selective advantage conferred by affinity-disrupting resistance mutations, since viruses bearing mutations that reduce affinity are still inhibited with equal potency, depriving HIV of an efficient evolutionary pathway to resistance. A profoundly disruptive mutation could escape the resistance capacitor, but such severe pocket mutations are discouraged due to the high potential cost to viral fitness. With high pM to low nM potency but sub-fM binding affinity, PIE12-trimer has a very strong resistance capacitor21.
We hypothesize that potency could be improved beyond the plateau by pre-positioning inhibitor on the cell surface, the site of viral entry, thus increasing the association rate beyond the diffusion limit. Using our novel modular PEG scaffold we conjugate PIE12-trimer to membrane-localizing groups (cholesterol and alkyl chains) that improve potency up to ~160-fold. This approach greatly simplifies trimer synthesis and improves yield. Importantly, our data show that this gain in potency does not disrupt the resistance capacitor, leaving intact PIE12-trimer’s strong barrier to resistance mutations. Using a discrete PEG scaffold with orthogonal reactive groups and defined geometry allows for rapid optimization of multimeric inhibitors and scouting of various potency-enhancing cargoes and should be of broad utility for the design of other multimeric peptide inhibitors.
Experimental Procedures
Peptide synthesis
Peptides were synthesized using a PTI PS3 peptide synthesizer or by RS Synthesis as previously described21, 22 to generate either PIE12-GK or ΔHP-PIE12-GK (lacks two N-terminal residues, D-His and D-Pro). PIE12-dPEG4/5-NH2 (the precursor to PIE12-trimer synthesis) was synthesized as follows: PIE12-GK (10 mM in dimethylacetamide, DMAC) was reacted with 250 mM Fmoc-N-amido-dPEG4/5-NHS ester (Quanta BioDesign 10994 and 10053) in dry DMAC (Acros Organics, septa sealed with molecular sieves) at a 1:1 molar ratio buffered by triethylamine (200 mM, pH 7.5) for 60 minutes at RT. This reaction was quenched by addition of acetic acid to 5% and purified by reverse-phase HPLC (water/acetonitrile gradient in 0.1% TFA) on a Waters BEH X-Bridge 10 μm, 300 Å C18 column (RP-HPLC). Purified product was lyophilized, then resuspended in 20% piperidine in DMAC for 20 minutes to remove Fmoc and produce PIE12-PEG4/5-NH2, which was then purified by RP-HPLC.
Trimer synthesis
PIE12-PEG4/5-NH2 (10 mM) was reacted with 250 mM trimethylolethane-triNHS ester (Fig 2A, Quanta BioDesign 10674) in DMAC at a 3.3:1 (peptide/scaffold) ratio in DMAC buffered by triethylamine (200 mM, pH 7.5) for 60 minutes at RT. Product was purified by RP-HPLC. All masses were confirmed by ESI-MS (AB Sciex API-3000).
Figure 2. Trimeric and heterotetrameric PEG scaffolds and cargoes.

A. Trimeric trimethoylester triNHS. B. Heterotetrameric PEG scaffold. The fourth maleimide arm is available for reaction with thiol-containing cargoes, such as 1-octadecanethiol (C18-SH) and thiocholesterol.
Cholesterol-PIE12-trimer and alkyl-PIE12-trimer were synthesized as follows: PIE12-PEG4-NH2 (10 mM) was reacted with Maleimide-PEG12-triNHS ester (Quanta BioDesign 10676, 250 mM in DMAC) or Maleimide-PEG24-triNHS ester (Fig. 2B, Quanta BioDesign 10680, 250 mM in DMAC) at a 3.3:1 (peptide/scaffold) ratio in DMAC buffered by triethylamine (200 mM, pH 7.5) for 45 minutes at RT. Thiocholesterol (Sigma Aldrich, 136115, 250 mM in chloroform), 1-octanethiol (Sigma-Aldrich 471836), 1-Hexadecanethiol (Sigma-Aldrich 52270) or 1-octadecanethiol (Sigma Aldrich 01858) were then added to a final concentration of 4.5 mM and reacted for an additional 60 minutes. For PEG16, PIE12-PEG4-NH2 was first reacted with Mal-PEG12-triNHS ester, followed by reaction with D-Cysteine (5 mM) to yield (PIE12-PEG4)3-PEG12-Cys. This product was then purified by RP-HPLC before sequential reaction with Maleimide-PEG4-NHS and thiocholesterol under conditions identical to those used to generate chol-PEG24-PIE12-trimer. PEG36, PEG57, and PEG132-trimer were produced through conjugation of PIE12-PEG4-NH2 to Maleimide-PEG24-triNHS ester, followed by addition of D-Cysteine. This intermediate was then conjugated to Mal-PEG12-NHS ester (Quanta Biodesign, 10284), Mal-PEG2K-NHS ester (Creative PEGWorks, PHB-950, ~45 PEG units), or Mal-PEG5K-NHS ester (Creative PEGWorks, PHB-952, ~120 PEG units) to yield Chol-PEG36-PIE12-Trimer, Chol-PEG57-PIE12-trimer, and Chol-PEG132-PIE12-trimer, respectively. The reaction was quenched by addition of acetic acid to 5% before purification by RP-HPLC.
Viral infectivity assays
Pseudovirion infectivity assays were carried out as previously described21, 22 using HXB2 and JRFL luciferase reporter pseudovirions (NL4-3 strain) and HOS-CD4-CXCR4 (for HXB2) or HOS-CD4-CCR5 (for JRFL) target cells. Inhibitors curves were generated using six concentration points measured in quadruplicate, and luciferase counts were normalized to an uninhibited control. Inhibition curves were fit using a standard IC50 equation (1-c/(IC50+c)) weighting each concentration point by its standard error in KaleidaGraph™ (Synergy Software). Reported IC50 values are the average of at least 2 independent assays.
Results
Our first goal was to simplify the synthesis of PIE12-trimer while also optimizing the linkages between PIE12 monomers. In our previous work, we synthesized PIE12-trimer by attaching bis-NHS ester PEG5 spacers to PIE12-GK. After purification, two of these PEGylated monomers were reacted with a central PIE12-GKK monomer (two primary amines) to produce PIE12-trimer21. This method is cumbersome for large-scale production because it requires the synthesis of two distinct D-peptides and a series of HPLC purifications to assemble the trimer, resulting in low yields. In addition, our PIE12 crystal structure suggested that shorter PEG linkers might adequately bridge the neighboring pockets and improve avidity. To address these goals, we redesigned PIE12-trimer using a scaffold strategy. We designed a homotrimeric scaffold containing three NHS ester arms for conjugation to PIE12-GK (Fig. 2a) in a single-pot reaction. PEG linkers of various lengths can be appended to the PIE12-GK peptide, allowing for the simple production of PIE12-trimers with varying PEG lengths.
PIE12-trimer’s estimated sub-fM affinity for the N-trimer makes direct comparative KD measurements (e.g., by surface plasmon resonance) very challenging. Although antiviral potency can be used as a surrogate for affinity, PIE12-trimer’s potency plateau can mask even large changes in affinity. To overcome this problem, we designed a PIE12 variant with weakened affinity to allow comparative evaluation of different trimer geometries by measuring inhibitor potency. We previously observed that PIE12’s two N-terminal residues make important contacts with the N-trimer and reasoned that deletion of these residues (D-His and D-Pro) would significantly reduce binding affinity without disrupting the overall orientation of PIE12 binding to the gp41 pocket or the local structure at the C-terminal PEG linkage site. ΔHP-PIE12 is 84-fold less potent than PIE12 (Table 1). In the context of the homotrimeric scaffold, ΔHP-PIE12 connected via our standard PEG5 linkers has an IC50 of 380 nM against HXB2 (a standard lab-adapted strain) and is therefore well outside of the potency plateau (~500 pM for HXB2). Using ΔHP-PIE12-trimer, we can now detect changes in potency due to linker changes that subtly alter affinity.
Table 1. D-peptide Inhibition data.
Antiviral potency against HXB2 and JRFL HIV-1 strains (*from21).
| Inhibitor | IC50 (nM) |
|
|---|---|---|
| HXB2 | JRFL | |
| PIE12* | 37 ± 2.3 | 580 ± 21.4 |
| ΔHP-PIE12 | 3100 ± 783 | nd |
| Chol-PEG2-PIE12 | 69 ± 11 | nd |
| Chol-PEG12-PIE12 | 12 ± 3.6 | nd |
| Chol-PEG24-PIE12 | 0.64 ± 0.25 | nd |
| C34 | 1.4 ± 0.3 | 13.4 ± 0.1 |
| C34-PEG2-Chol | 0.044 ± 0.0004 | 0.05 ± 0.01 |
| C34-PEG11-Chol | 0.021 ± 0.0014 | 0.024 ± 0.005 |
| C34-PEG80-Chol | 0.022 ± 0.0004 | 0.1 ± 0.045 |
| PEG4-ΔHP-PIE12-trimer | 300 ± 7.2 | nd |
| PEG5-ΔHP-PIE12-trimer | 380 ± 13 | nd |
| PEG4-PIE12-trimer | 0.72 ± 0.04 | 2.1 ± 0.28 |
| Chol-PEG12-PIE12-trimer | 0.052 ± 0.02 | 0.06 ± 0.004 |
| Chol-PEG16-PIE12-trimer | 0.02 ± 0.002 | 0.017 ± 0.0002 |
| Chol-PEG24-PIE12-trimer | 0.013 ± 0.0013 | 0.019 ± 0.003 |
| Chol-PEG36-PIE12-trimer | 0.011 ± 0.0015 | 0.015 ± 0.005 |
| Chol-PEG57-PIE12-trimer | 0.007 ± 0.0013 | 0.013 ± 0.003 |
| Chol-PEG132-PIE12-trimer | 0.012 ± 0.0015 | 0.025 ± 0.002 |
| C8-PIE12-trimer | 0.42 ± 0.01 | nd |
| C16-PIE12-trimer | 0.09 ± 0.014 | 0.11 ± 0.012 |
| C18-PIE12-trimer | 0.054 ± 0.018 | 0.087 ± 0.012 |
Our initial exploration of PEG linker lengths in PIE12-trimer showed that PEG2 and PEG3 were slightly less potent than the original PEG5. To determine whether PEG4 or PEG5 was the optimal arm length, both PEG5 and PEG4 ΔHP-PIE12 conjugates were attached to the homotrimeric scaffold, and we observed that a PEG4 linker was slightly more optimal (Table 1). Therefore, PEG4 was selected as the new standard linker for conjugating PIE12 to the scaffold. The scaffold synthesis strategy is dramatically simpler than our previous method for generating trimer since it requires only one peptide and a single purification. Additionally, the yields are considerably higher due to the reduced number of purification and lyophilization steps that led to loss of active NHS esters in the previous strategy. Finally, the high activity of the scaffold and single-pot reaction allow for near stoichiometric concentrations of peptide and scaffold, further improving yield.
Heterotetrameric scaffold
With the optimal PEG linker length in place, we next turned our attention to improving PIE12-trimer’s potency via localization to sites of viral entry (the cell surface). To enable the conjugation of membrane-localizing groups to PIE12-trimer, we designed a heterotetrameric scaffold containing three short arms with NHS ester groups (for addition of PIE12-PEG4-NH2) and a fourth PEG arm of variable length functionalized with maleimide (an orthogonal reactive group for the addition of thiol-containing cargoes) (Fig. 2b).
Our first cargo for the heterotetrameric scaffold was cholesterol. Several recent studies have shown that cholesterol conjugation improves both the potency and circulating half-life of C-peptide inhibitors of HIV27 and paramyxoviruses28, 29. Cholesterol conjugation has also been shown to specifically localize dyes to the membrane surface30, 31. A challenge of applying this approach to PIE12 is that while the N-terminus of the C-peptide lies immediately adjacent to the membrane, PIE12 targets a pocket that we estimate is ~60 Å from the membrane (Fig. 1). We used flexible PEG linkers of varying lengths to span this distance. PEG12 is sufficiently long if stretched taut, but PEG typically assumes an average length approximately half of its fully stretched distance32.
To study the potency effects of cholesterol (chol) conjugation to PIE12 and the length of the linker between chol and PIE12, we used monomeric PIE12, which is not in a potency plateau and therefore should be a sensitive reporter for optimal linker length. We began by generating chol-PIE12 conjugates using heterobifunctional PEG2, PEG12, and PEG24 NHS ester/maleimide crosslinkers to conjugate thiocholesterol (cholesterol with a thiol replacing its hydroxyl group) to PIE12’s C-terminal Lys (its only primary amine). We observed that the PEG2 conjugate, much too short to bridge the membrane to pocket distance, causes a two-fold loss of potency (HXB2 strain) compared to unconjugated PIE12. In contrast, chol-PEG12-PIE12 shows 3-fold improved potency, while PEG24 provides an even greater 58-fold increase in potency compared to PIE12. For comparison, we also synthesized C-peptide (C34) cholesterol conjugates of varying lengths (Table 1). We reproduce Ingalinella’s finding of ~40-fold improved potency27 using a short PEG2 linker, but surprisingly, a longer linker (PEG11) provides an additional 2-fold improvement in potency, and a much longer linker (PEG80) maintains the same potency (HXB2 strain). A similar pattern is seen with the JRFL strain, but with significant attenuation at very long PEG linker lengths (4-fold worse than the optimal PEG length).
Based on these dramatic potency gains, we next conjugated cholesterol to PIE12-trimer using the heterotetrameric scaffold. Using the optimal PEG4 linker determined earlier for the three NHS ester (PIE12) arms, we synthesized chol-PIE12-trimers with a variety of fourth arm (maleimide) lengths to confirm the relationship between PEG length and potency observed with the monomer. In the context of chol-PIE12-trimer, we did not need to utilize ΔHP-PIE12, as the cholesterol-mediated improvement in potency was discernable using PIE12. This sensitivity was expected because membrane localization affects the association rate rather than changing affinity (masked by the resistance capacitor). We varied the fourth arm from 12 to 132 PEG units, covering a distance range of ~60 to 480 Å (fully-extended).
Cholesterol conjugation dramatically improves PEG4-PIE12-trimer potency against both HXB2 and JRFL entry (up to 160-fold, Table 1 and Fig. 3). Comparison of varying 4th arm lengths in chol-PIE12-trimer shows that inhibitor potency varies modestly in an optimal range between PEG24 and PEG57. A shorter PEG12 linker is suboptimal, though it performs better than seen in the monomer series, likely due to the additional length provided by the PEG4 arms. Only a slight decrease in potency is observed with the longest (PEG132) linker. Despite being slightly less potent than Chol-PEG57-PIE12-trimer, we have chosen Chol-PEG24-PIE12-trimer as our lead candidate due to its ease of synthesis and the availability of monodisperse PEG24. A monodisperse PEG scaffold will ease future preclinical studies of chol-PIE12-trimer purity, metabolism, pharmacokinetics, and stability. Importantly, cholesterol conjugates retain high (mM) aqueous solubility.
Figure 3. JRFL pseudovirion infectivity assay.
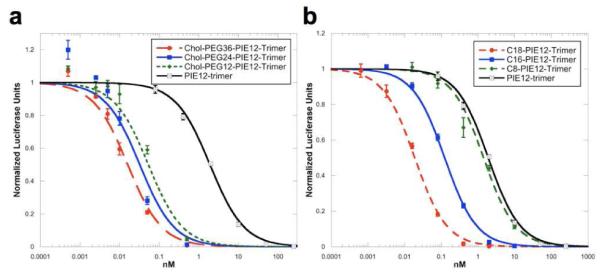
A. Dependence of linker length on chol-PIE12-trimer potency. B. Thio-alkane-conjugated PIE12-trimer series with differing alkane lengths.
Another established strategy for localizing inhibitors to membranes is fatty acid conjugation33-37. Using the same heterotetramer scaffold strategy described above with cholesterol, we synthesized PIE12-trimers conjugated to aliphatic chains of 8, 16, and 18 carbons (C8/C16/C18-PIE12-trimer). While C8 conjugation has little effect on PIE12-trimer potency, C16 and C18 both provide substantial gains in potency, though to a lesser degree than seen with cholesterol (Table 1). C18-PIE12-trimer was slightly more potent than C16-PIE12-trimer.
Effect of membrane localization on the resistance capacitor
Drug resistance is a constant threat to the effectiveness of HIV inhibitors. PIE12-trimer is an attractive drug candidate in part because of its strong resistance capacitor, which provides a high genetic barrier to resistance21. The resistance capacitor depends on the diffusion-limited association rate for PIE12-trimer binding to gp41. The cholesterol and C16/18 conjugation strategies described here break through this kinetic barrier via inhibitor localization to viral entry sites (i.e., increasing effective inhibitor concentration and overcoming the diffusion rate limitation). In theory, this improvement in potency could come at the cost of weakening the resistance capacitor. To test for this possibility, we measured the potency of chol- and C16/C18-conjugated PIE12-trimer against resistance mutations we have previously identified21.
Previous selection for resistance to PIE7-dimer (an earlier-generation D-peptide inhibitor)22 generated E560K/V570I, which minimally affects the potency of PIE12-trimer, but dramatically reduces PIE7-dimer potency21. Selection of resistance to PIE12-trimer required more than a year of viral passaging, but ultimately resulted in the Q577R mutation, which decreases PIE12-trimer potency by >1000-fold21. The effect of these resistance mutations on chol- and C16/18-PIE12-trimer potency is shown in Table 2. The relative effects of both resistance mutations are similar for PIE12-trimer and the cholesterol/alkane-conjugated PIE12-trimers. However, because of the greatly improved potency of the conjugated PIE12-trimers, these inhibitors maintain nM potency even against the severe Q577R resistance mutation. The impact of the less severe E560K/V570I resistance mutation is absorbed by all of the conjugated PIE12-trimers, as well as plain PIE12-trimer. These data suggest that the improvement in potency through C16/C18 and cholesterol conjugation retains enough excess binding energy to maintain an effective resistance capacitor.
Table 2. Antiviral potency against resistant strains.
Antiviral potency against identified resistant strains (HXB2 background). The IC50 standard error of the mean values are <35% for all samples.
| Inhibitor | IC50 (nM) |
||
|---|---|---|---|
| WT HXB2 | E560K/V570I | Q577R | |
| PEG4-PIE12-trimer | 0.72 | 0.89 | >3 μM |
| Chol-PEG24-PIE12-trimer | 0.013 | 0.01 | 10.1 |
| C8-PIE12-trimer | 0.42 | 0.86 | 452 |
| C16-PIE12-trimer | 0.09 | 0.045 | 39 |
| C18-PIE12-trimer | 0.054 | 0.035 | 32.5 |
Discussion
PIE12-trimer, our previously described D-peptide inhibitor, is a promising preclinical candidate for the treatment and prevention of HIV-1 due to its strong potency, wide breadth, and highly charged resistance capacitor that slows the emergence of resistance mutations. However, the transient nature of PIE12-trimer’s target means that its potency is restricted by its diffusion-limited association rate with the gp41 pocket. In an attempt to break through this potency barrier, we designed a heterotetrameric scaffold to allow us to conjugate various membrane-localizing cargoes to PIE12-trimer. This scaffold also allows us to produce PIE12-trimer variants much more efficiently than previously reported. As hoped, conjugation of PIE12-trimer to cholesterol or C16/C18 reduces the kinetic limitation and greatly improves potency up to 160-fold.
We hypothesize that this increased potency is due to local concentration of inhibitor at membrane sites of viral entry. Cholesterol is specifically enriched at sites of viral entry (lipid rafts, where CD4 and co-receptor are localized)38, 39. The mechanism by which cholesterol improves potency is the focus of ongoing work. Preliminary evidence suggests that the interaction between cholesterol and the membrane is readily reversible, which may explain why there is a broad range of compatible linker lengths. It may also be the case that cholesterol-conjugated inhibitors interact directly with Env, as a cholesterol recognition/interaction amino acid consensus sequence (CRAC) has been identified in the membrane proximal region of gp4140.
By comparison, C16 and C18 conjugates are less potent than the cholesterol conjugate. Saturated fatty acids C16:0 (palmitate) and C18:0 (stearate) are also enriched in lipid rafts41, but are abundant in the general plasma membrane as well42. The reduced potency of alkylated PIE12-trimer compared to cholesterol may therefore be explained by a relatively lower affinity of alkyl chains for lipid rafts. Another possible explanation is fatty-acid sequestration by albumin, which is known to bind fatty acids with high affinity (compared to cholesterol)43, though it is not known how loss of the acid group (leaving an alkane chain) affects this binding.
GPI anchors in lipid rafts contain C16 and C18 alkyl chains as well as acylated C16 and C18 fatty acids44. Originally we synthesized alkyl conjugates, and noted that they improved potency through membrane association (overcoming the potency plateau). For completeness we also synthesized an acylated C16 (fatty acid) conjugate. Surprisingly, the C16 acyl conjugate was much less potent than the C16 alkyl conjugate (data not shown), presumably because it does not associate as effectively with plasma membranes. This finding may explain why a recent study did not observe a potency enhancement with C16 acylation of C3427.
Importantly, we show that membrane localization does not impair the resistance capacitor. Both chol- and C16/C18-conjugated PIE12-trimer are able to absorb the affinity-disrupting impact of PIE7-dimer resistance mutations (E560K/V570I). For the more severe PIE12-trimer resistance mutation Q577R, the relative loss of potency for both conjugates is comparable to that seen with PIE12-trimer. The full resistance profile of these conjugates will be determined by ongoing viral passaging studies starting from both wild-type and PIE12-trimer resistant virus.
Although PIE12-trimer has ideal antiviral properties, its relatively small size (~8 kD) will likely lead to a short serum half-life due to renal filtration. In addition to their potency-boosting effects, we hypothesize that both cholesterol and alkyl conjugation will also lead to improvements in the pharmacokinetic (PK) properties of these inhibitors via interaction with cell membranes and albumin that slow renal clearance. Albumin serves as a carrier for both cholesterol45 and fatty acids46, reducing the rate of renal filtration. Adherence to membrane surfaces may also slow the absorption of inhibitor from the subcutaneous space, enabling prolonged dosing via a slow-release depot effect. This type of depot would be especially attractive for non-degradable D-peptides.
This work demonstrates the successful application of modular PEG scaffold-based design to peptide drug optimization (both peptide geometry and localization to the site of action via conjugated localizing cargoes). This approach allows for alterations in the scaffold to accommodate a variety of cargoes and chemistries (e.g., “click” chemistry), as well as rapid optimization of PEG arm lengths. For viruses that undergo membrane fusion within the endosome, such as Ebola, this strategy could be employed to attach an endosome-targeting moiety to localize inhibitor to the site of entry and increase potency. Additionally, the scaffold allows for conjugation to a variety of cargoes to modulate PK properties (e.g., large branched PEGs, albumin, or albumin-binding peptides)47, 48. The scaffold itself is inexpensive to produce and can be used directly for cost-effective large-scale production.
PK and animal toxicity studies for chol- and C16/C18-PIE12-trimer are underway to determine how conjugation alters serum half-life and to determine if any specific toxicity arises as a result of conjugation. Fatty acid conjugation has been used to prolong serum half-life of a GLP-1 peptide (liraglutide, C16) and insulin (detemir, C14). Alkane toxicity in the context of peptide conjugates has not been studied.
The in vivo efficacy of these conjugates will be determined in future studies of systemic treatment via subcutaneous injection or as a vaginally/rectally applied preventative (microbicide) in human tissue and animal models. Our D-peptide scaffold is especially advantageous for application as a microbicide due to its protease resistance, which should enable it to persist for extended periods in the vaginal/rectal mucosa’s harsh protease-rich environment. The addition of membrane-binding groups may also improve microbicide tissue penetration and retention.
Acknowledgements
We thank Paul Davis, James Guyo, and Robert Woodman of Quanta BioDesign for custom synthesis of the PEG scaffolds. This research was funded by an NIH grant AI076168 to M.S.K.
J.N.F. is supported by an NIH Microbial Pathogenesis Predoctoral Training Grant (AI055434). Special thanks to Michael Root for providing cloned resistant strains and Brett Welch for critical review of the manuscript.
References
- (1).Freed EO, Martin MA. The role of human immunodeficiency virus type 1 envelope glycoproteins in virus infection. J Biol Chem. 1995;270:23883–6. doi: 10.1074/jbc.270.41.23883. [DOI] [PubMed] [Google Scholar]
- (2).Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- (3).Jones PL, Korte T, Blumenthal R. Conformational changes in cell surface HIV-1 envelope glycoproteins are triggered by cooperation between cell surface CD4 and co-receptors. J Biol Chem. 1998;273:404–9. doi: 10.1074/jbc.273.1.404. [DOI] [PubMed] [Google Scholar]
- (4).Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–73. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- (5).Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–30. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- (6).Root MJ, Kay MS, Kim PS. Protein design of an HIV-1 entry inhibitor. Science. 2001;291:884–8. doi: 10.1126/science.1057453. [DOI] [PubMed] [Google Scholar]
- (7).Jiang S, Lin K, Strick N, Neurath AR. HIV-1 inhibition by a peptide. Nature. 1993;365:113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- (8).Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc Natl Acad Sci U S A. 1992;89:10537–41. doi: 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci U S A. 1994;91:9770–4. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rimsky LT, Shugars DC, Matthews TJ. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J Virol. 1998;72:986–93. doi: 10.1128/jvi.72.2.986-993.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Golding H, Zaitseva M, de Rosny E, King LR, Manischewitz J, Sidorov I, Gorny MK, Zolla-Pazner S, Dimitrov DS, Weiss CD. Dissection of human immunodeficiency virus type 1 entry with neutralizing antibodies to gp41 fusion intermediates. J Virol. 2002;76:6780–90. doi: 10.1128/JVI.76.13.6780-6790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chan DC, Chutkowski CT, Kim PS. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci U S A. 1998;95:15613–7. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99:103–15. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- (14).Pollard VW, Malim MH. The HIV-1 Rev protein. Annual review of microbiology. 1998;52:491–532. doi: 10.1146/annurev.micro.52.1.491. [DOI] [PubMed] [Google Scholar]
- (15).Derdeyn CA, Decker JM, Sfakianos JN, Zhang Z, O’Brien WA, Ratner L, Shaw GM, Hunter E. Sensitivity of human immunodeficiency virus type 1 to fusion inhibitors targeted to the gp41 first heptad repeat involves distinct regions of gp41 and is consistently modulated by gp120 interactions with the coreceptor. J Virol. 2001;75:8605–14. doi: 10.1128/JVI.75.18.8605-8614.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Eggink D, Baldwin CE, Deng Y, Langedijk JP, Lu M, Sanders RW, Berkhout B. Selection of T1249-resistant human immunodeficiency virus type 1 variants. J Virol. 2008;82:6678–88. doi: 10.1128/JVI.00352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Eggink D, Bontjer I, Langedijk JP, Berkhout B, Sanders RW. Resistance of human immunodeficiency virus type 1 to a third-generation fusion inhibitor requires multiple mutations in gp41 and is accompanied by a dramatic loss of gp41 function. J Virol. 2011;85:10785–97. doi: 10.1128/JVI.05331-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ray N, Harrison JE, Blackburn LA, Martin JN, Deeks SG, Doms RW. Clinical resistance to enfuvirtide does not affect susceptibility of human immunodeficiency virus type 1 to other classes of entry inhibitors. J Virol. 2007;81:3240–50. doi: 10.1128/JVI.02413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Reeves JD, Lee FH, Miamidian JL, Jabara CB, Juntilla MM, Doms RW. Enfuvirtide resistance mutations: impact on human immunodeficiency virus envelope function, entry inhibitor sensitivity, and virus neutralization. J Virol. 2005;79:4991–9. doi: 10.1128/JVI.79.8.4991-4999.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lalezari JP, Bellos NC, Sathasivam K, Richmond GJ, Cohen CJ, Myers RA, Jr., Henry DH, Raskino C, Melby T, Murchison H, Zhang Y, Spence R, Greenberg ML, Demasi RA, Miralles GD. T-1249 retains potent antiretroviral activity in patients who had experienced virological failure while on an enfuvirtide-containing treatment regimen. J Infect Dis. 2005;191:1155–63. doi: 10.1086/427993. [DOI] [PubMed] [Google Scholar]
- (21).Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP, Root MJ, Kay MS. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J Virol. 2010;84:11235–44. doi: 10.1128/JVI.01339-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D-peptide inhibitors of HIV-1 entry. Proc Natl Acad Sci U S A. 2007;104:16828–33. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Milton RC, Milton SC, Kent SB. Total chemical synthesis of a D-enzyme: the enantiomers of HIV-1 protease show reciprocal chiral substrate specificity. Science. 1992;256:1445–8. doi: 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- (24).Kahle KM, Steger HK, Root MJ. Asymmetric deactivation of HIV-1 gp41 following fusion inhibitor binding. PLoS Pathog. 2009;5:e1000674. doi: 10.1371/journal.ppat.1000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Platt EJ, Durnin JP, Kabat D. Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus. J Virol. 2005;79:4347–56. doi: 10.1128/JVI.79.7.4347-4356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Steger HK, Root MJ. Kinetic dependence to HIV-1 entry inhibition. J Biol Chem. 2006;281:25813–21. doi: 10.1074/jbc.M601457200. [DOI] [PubMed] [Google Scholar]
- (27).Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci U S A. 2009;106:5801–6. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Porotto M, Rockx B, Yokoyama CC, Talekar A, Devito I, Palermo LM, Liu J, Cortese R, Lu M, Feldmann H, Pessi A, Moscona A. Inhibition of Nipah virus infection in vivo: targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog. 2010;6:e1001168. doi: 10.1371/journal.ppat.1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Porotto M, Yokoyama CC, Palermo LM, Mungall B, Aljofan M, Cortese R, Pessi A, Moscona A. Viral entry inhibitors targeted to the membrane site of action. J Virol. 2010;84:6760–8. doi: 10.1128/JVI.00135-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knolker HJ, Simons K. Efficient inhibition of the Alzheimer’s disease beta-secretase by membrane targeting. Science. 2008;320:520–3. doi: 10.1126/science.1156609. [DOI] [PubMed] [Google Scholar]
- (31).Teruya K, Nishizawa K, Doh-ura K. Semisynthesis of a protein with cholesterol at the C-terminal, targeted to the cell membrane of live cells. Protein J. 2010;29:493–500. doi: 10.1007/s10930-010-9278-9. [DOI] [PubMed] [Google Scholar]
- (32).Lee H, Venable RM, Mackerell AD, Jr., Pastor RW. Molecular dynamics studies of polyethylene oxide and polyethylene glycol: hydrodynamic radius and shape anisotropy. Biophys J. 2008;95:1590–9. doi: 10.1529/biophysj.108.133025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wexler-Cohen Y, Ashkenazi A, Viard M, Blumenthal R, Shai Y. Virus-cell and cell-cell fusion mediated by the HIV-1 envelope glycoprotein is inhibited by short gp41 N-terminal membrane-anchored peptides lacking the critical pocket domain. FASEB J. 2010;24:4196–202. doi: 10.1096/fj.09-151704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wexler-Cohen Y, Shai Y. Demonstrating the C-terminal boundary of the HIV 1 fusion conformation in a dynamic ongoing fusion process and implication for fusion inhibition. FASEB J. 2007;21:3677–84. doi: 10.1096/fj.07-8582com. [DOI] [PubMed] [Google Scholar]
- (35).Wexler-Cohen Y, Shai Y. Membrane-anchored HIV-1 N-heptad repeat peptides are highly potent cell fusion inhibitors via an altered mode of action. PLoS Pathog. 2009;5:e1000509. doi: 10.1371/journal.ppat.1000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bader B, Kuhn K, Owen DJ, Waldmann H, Wittinghofer A, Kuhlmann J. Bioorganic synthesis of lipid-modified proteins for the study of signal transduction. Nature. 2000;403:223–6. doi: 10.1038/35003249. [DOI] [PubMed] [Google Scholar]
- (37).Peisajovich SG, Gallo SA, Blumenthal R, Shai Y. C-terminal octylation rescues an inactive T20 mutant: implications for the mechanism of HIV/SIMIAN immunodeficiency virus-induced membrane fusion. J Biol Chem. 2003;278:21012–7. doi: 10.1074/jbc.M212773200. [DOI] [PubMed] [Google Scholar]
- (38).Luo C, Wang K, Liu de Q, Li Y, Zhao QS. The functional roles of lipid rafts in T cell activation, immune diseases and HIV infection and prevention. Cell Mol Immunol. 2008;5:1–7. doi: 10.1038/cmi.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Waheed AA, Freed EO. The Role of Lipids in Retrovirus Replication. Viruses. 2010;2:1146–1180. doi: 10.3390/v2051146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Vincent N, Genin C, Malvoisin E. Identification of a conserved domain of the HIV-1 transmembrane protein gp41 which interacts with cholesteryl groups. Biochim Biophys Acta. 2002;1567:157–64. doi: 10.1016/s0005-2736(02)00611-9. [DOI] [PubMed] [Google Scholar]
- (41).Van Laethem F, Liang X, Andris F, Urbain J, Vandenbranden M, Ruysschaert JM, Resh MD, Stulnig TM, Leo O. Glucocorticoids alter the lipid and protein composition of membrane rafts of a murine T cell hybridoma. J Immunol. 2003;170:2932–9. doi: 10.4049/jimmunol.170.6.2932. [DOI] [PubMed] [Google Scholar]
- (42).Schumann J, Leichtle A, Thiery J, Fuhrmann H. Fatty acid and peptide profiles in plasma membrane and membrane rafts of PUFA supplemented RAW264.7 macrophages. PLoS One. 2011;6:e24066. doi: 10.1371/journal.pone.0024066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Spector AA. Fatty acid binding to plasma albumin. J Lipid Res. 1975;16:165–79. [PubMed] [Google Scholar]
- (44).Benting J, Rietveld A, Ansorge I, Simons K. Acyl and alkyl chain length of GPI-anchors is critical for raft association in vitro. FEBS Lett. 1999;462:47–50. doi: 10.1016/s0014-5793(99)01501-x. [DOI] [PubMed] [Google Scholar]
- (45).Peng L, Minbo H, Fang C, Xi L, Chaocan Z. The interaction between cholesterol and human serum albumin. Protein Pept Lett. 2008;15:360–4. doi: 10.2174/092986608784246542. [DOI] [PubMed] [Google Scholar]
- (46).Charbonneau DM, Tajmir-Riahi HA. Study on the interaction of cationic lipids with bovine serum albumin. J Phys Chem B. 2010;114:1148–55. doi: 10.1021/jp910077h. [DOI] [PubMed] [Google Scholar]
- (47).Fishburn CS. The pharmacology of PEGylation: balancing PD with PK to generate novel therapeutics. J Pharm Sci. 2008;97:4167–83. doi: 10.1002/jps.21278. [DOI] [PubMed] [Google Scholar]
- (48).Dennis MS, Zhang M, Meng YG, Kadkhodayan M, Kirchhofer D, Combs D, Damico LA. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J Biol Chem. 2002;277:35035–43. doi: 10.1074/jbc.M205854200. [DOI] [PubMed] [Google Scholar]