

Abstract
Deep intronic mutations are often ignored as possible causes of human diseases. A deep intronic mutation in the MTRR gene, c.903+469T>C, is the most frequent mutation causing the cblE type of homocystinuria. It is well known to be associated with pre-mRNA missplicing, resulting in pseudoexon inclusion; however, the pathological mechanism remains unknown. We used minigenes to demonstrate that this mutation is the direct cause of MTRR pseudoexon inclusion, and that the pseudoexon is normally not recognized due to a suboptimal 5′ splice site. Within the pseudoexon we identified an exonic splicing enhancer (ESE), which is activated by the mutation. Cotransfection and siRNA experiments showed that pseudoexon inclusion depends on the cellular amounts of SF2/ASF and in vitro RNA-binding assays showed dramatically increased SF2/ASF binding to the mutant MTRR ESE. The mutant MTRR ESE sequence is identical to an ESE of the alternatively spliced MST1R proto-oncogene, which suggests that this ESE could be frequently involved in splicing regulation. Our study conclusively demonstrates that an intronic single nucleotide change is sufficient to cause pseudoexon activation via creation of a functional ESE, which binds a specific splicing factor. We suggest that this mechanism may cause genetic disease much more frequently than previously reported. Hum Mutat 31:437–444, 2010. © 2010 Wiley-Liss, Inc.
Keywords: MTRR, ESE, SF2/ASF, Pre-mRNA splicing, cblE, homocystinuria
Introduction
In the eukaryotic protein expression pathway pre-mRNA splicing is a fundamental process, in which splice site sequences have to be recognized, so that exons can be correctly joined to form an mRNA that encodes the desired protein. Due to the degenerate nature of the consensus splice site sequences [Shapiro and Senapathy, 1987], these relatively short sequence elements are not always sufficient to ensure precise discrimination between exonic and intronic sequences. This is illustrated by the fact that typical mammalian pre-mRNAs harbor many pseudosplice sites that fit the consensus splice site sequences, but which are normally not used [Sun and Chasin, 2000], and conversely, many functional splice sites do not conform well to the consensus splice site sequences. Thus, in addition to the splice site sequences other cis-regulatory elements, which act as splicing enhancers or silencers, direct the splicing machinery to use the correct splice sites, and to avoid the use of pseudosplice sites. Exonic splicing enhancers (ESE) stimulate splicing and serve as binding sites for various splicing factors of which the best characterized are the serine/arginine-rich proteins (SR proteins) [Cartegni et al., 2002]. Exonic splicing silencers (ESS) and intronic splicing silencers (ISS) repress splicing, and often function by binding of proteins from the heterogenous nuclear ribonucleoprotein (hnRNP) family [Cartegni et al., 2002]. Most reported splicing mutations directly abolish an authentic splice site or create a novel splice site, but an increasing number of disease-associated mutations that alter splicing enhancers or silencers have been reported in recent years [Buratti et al., 2006; Cartegni et al., 2002; Wang and Cooper, 2007]. The majority of reported mutations in this type of splicing regulatory sequences are located in exons and believed to disrupt ESEs, thereby causing aberrant splicing [Lorson et al., 1999; McVety et al., 2006; Nielsen et al., 2007; Pagani et al., 2003]. There has been only few reports of disease-causing mutations in introns that cause missplicing by inducing inclusion of intronic sequences as exons (pseudoexon inclusion) [Buratti et al., 2006], and the vast majority of this type of mutation functions by strengthening of preexisting weak pseudosplice sites or by creating new splice sites. Only very few examples have been reported where disease-causing mutations cause pseudoexon inclusion without changing the splice sites directly [Davis et al., 2009]. Consequently, little is known about the mechanisms by which intronic sequence variations can activate pseudoexons and cause disease. It is likely that this is an important disease mechanism, which is heavily underreported, because the causative mutations are typically located in intronic sequences, which are not routinely investigated in diagnostic laboratories and because it requires analysis of patient RNA to observe the effect directly.
The complementation group cobalamin E (cblE) type of homocystinuria (cblE; MIM# 236270) [Wilson et al., 1999; Zavadakova et al., 2005] is a rare autosomal recessive disorder, which presents early in life and is characterized by failure to thrive, megaloblastic anemia, and neurological complications such as developmental delay and brain atrophy. Patients also exhibit homocystinuria, hyperhomocysteinaemia, and often hypomethioninaemia [Rosenblatt and Fenton, 2001; Zavadakova et al., 2005]. The cblE type of homocystinuria is caused by mutations in the MTRR gene (MIM# 602568) that lead to a deficiency of methionine synthase reductase (MTRR, EC 1.16.1.8), an enzyme that participates in the remethylation pathway of the methionine cycle [Leclerc et al., 1998; Wilson et al., 1999].
The most frequent mutation in the MTRR gene is a T>C transition located deep in intron 6 (c.903+469T>C, p.S301fsX315) [Wilson et al., 1999; Zavadakova et al., 2005]. The mutant allele causes severe missplicing of the MTRR mRNA and only very small amounts of normal mRNA. The vast majority of the mRNA produced from the mutant allele contains 140 bp of intronic sequence, a pseudoexon, inserted between exons 6 and 7 (r.903_904ins140). It is not clear if and how the T>C substitution, which is located 23 bp downstream from the 5′ end of the pseudoexon, may result in pseudoexon inclusion, because it does not abolish or create any splice site. Wilson and coworkers [1999] suggested that it creates a potential ESE, but did not address this possibility experimentally. The pseudoexon insertion causes a frameshift, and creates a downstream premature termination codon, which results in dramatically decreased amounts of MTRR mRNA possibly via nonsense-mediated decay and prevents production of a functional protein.
To date, the consequences of the c.903+469T>C mutation in the MTRR gene have not been characterized functionally. Therefore, we have in the present study investigated the underlying mechanism experimentally using splicing reporter minigenes and RNA affinity purification. We show that the mutation exerts its pathogenicity by creating a general ESE motif, which is also important in another gene and functions by binding of the splicing regulatory protein SF2/ASF (splicing factor 2/alternative splicing factor).
Materials and Methods
Splicing Minigene Constructs
HIV-tat minigene
MTRR-EcoRI-S (5′-ttatgagcttgaattctctcagcacagtaatattgtca-3′) and MTRR-PstI-AS (5′-ttatatattctgcagctgggttcaagtgaccg-3′) primers were used to amplify the region of the MTRR intron 6 spanning the MTRR pseudoexon. PCR products were cloned between the EcoRI and PstI site of the pSPL3 exon-trapping vector (Invitrogen, Carlsbad, CA) to produce HIV-tat minigenes (pSPL3/wt and pSPL3/mut).
β-Globin minigene
The region of exon 1–intron 1–exon 2 of the β-globin gene was amplified by β-NotI-S (5′-ttattagcggccgcattgcttacatttgcttctga-3′) and β-HindIII-AS (5′-tgatcggttaagcttgcccttgaggttgtcca-3′) primers and ligated into pCMV-Tag-1 (Stratagene, La Jolla, CA) to produce the pCMV/β vector. The wild-type and mutant MTRR pseudoexon and their flanking intronic sequences omitting upstream VNTR region, which can cause difficulties in cloning, were amplified from the genomic DNA from a subject who is heterozygous for the c.903+469T>C mutation using primers MTRR-BpiI-S (5′-tgagacgaagactcttgggtcatcttgccattttattcc-3′) and MTRR-BpiI-AS (5′-tgatttacccaagagtcttcctgggttcaagtgaccg-3′) and cloned into BpiI site of the pCMV/b vector to produce β-globin minigenes (β/wt and β/mut).
5′ splice site optimization
Using PCR-based mutagenesis and the mutagenic primer MTRR OPT5-S (5′-gctgtccactcaggtragtcaggaagttaga-3′), we prepared four different variants of the pCMV/β minigene: pCMV/β-MTRR WT+3A and pCMV/β-MTRR MUT1+A, which has an optimal splice site with A at position +3 of the pseudoexon 5′ splice site. pCMV/β-MTRR WT1+3G and pCMV/β-MTRR MUT+3G, which has an optimal splice site with G at position +3 of the pseudoexon 5′ splice site, making it slightly weaker than the +3A variants. The integrity of all final constructs was confirmed by sequencing.
All nucleotide numbering regarding the MTRR gene (NM_002454.2) reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to journal guidelines (http://www.hgvs.org/mutnomen). The initiation codon is codon 1.
Functional Splicing Assays
Eukaryotic COS-7 cells were transiently transfected with 5 μg plasmid DNA using Lipofectamine 2000 (Invitrogen). After 24 hr incubation total RNA was extracted by ToRNAzol (GeneAge Technologies, Czech Republic), treated with DNAseI, and reversely transcribed with ImProm-II™ Reverse Transcriptase (Promega, Madison, WI) using oligo(dT)12–18 primers. Splicing products were analysed by PCR with vector specific primers T3 (5′-aattaaccctcactaaaggg-3′) and T7 (5′-taatacgactcactataggg-3′) used for the β-globin minigenes and SD6 (5′-tctgagtcacctggacaacc-3′) and SA2 (5′-atctcagtggtatttgtgagc-3′) for the HIV-tat minigenes, respectively. The identity of the PCR products was confirmed by sequencing.
In Silico Analysis of the Splice Site Strength
The strength of pseudoexon- and authentic-splice sites was assessed by Web-based tools: GeneSplicer (http://www.tigr.org/tdb/GeneSplicer/) [Pertea et al., 2001]; NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) [Brunak et al., 1991]; NNSplice (http://www.fruitfly.org/seq_tools/splice.html) [Reese et al., 1997]; SpliceView (http://zeus2.itb.cnr.it/∼webgene/wwwspliceview_ex.html) [Rogozin and Milanesi, 1997], MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) [Yeo and Burge, 2004], and SpliceSiteFinder (http://www.genet.sickkids.on.ca/∼ali/splicesitefinder.html) [Shapiro and Senapathy, 1987], which predicts the presence of donor or acceptor sites. The programs were used to scan the entire sequence of the human MTRR gene (GenBank accession no. NC_0000005.9), authentic and pseudoexon splice sites were selected from the results and their scores were compared.
In Silico Analyses of the ESE and ESS Motifs
Predictive programs RESCUE-ESE (http://genes.mit.edu/burgelab/rescue-ese/) [Fairbrother et al., 2004], ESE finder 3.0 (http://rulai.cshl.edu/tools/ESE/) [Cartegni et al., 2003] were used for ESE motifs prediction and program FASS-ESS Web server (http://genes.mit.edu/fas-ess/) [Wang et al., 2004] using both set of hexamers (FAS-hex2 and FAS-hex3) for prediction of ESS motifs in sequence of wild-type or mutant pseudoexon with default settings for human sequences. Alternative Splicing Database–Splicing Rainbow (http://www.ebi.ac.uk/asd-srv/wb.cgi) [Stamm et al., 2006] was utilized for searching of both ESE and ESS motifs.
Cotransfection Experiments
HEK-293 cells were transiently cotransfected with 0.8 μg of wild-type or mutant HIV-tat minigene (pSPL3/wt or pSPL3/mut) and 0.8 μg of the relevant vector expressing SF2/ASF, SRp40, and SRp55 [Hanamura et al., 1998], U1snRNA [Pagani et al., 2002], or empty vector pcDNA3.1+ using FuGENE 6 Transfection Reagent (Roche Molecular Biochemicals, Indianapolis, IN). Total RNA was isolated after 48 hr incubation and splicing products were analyzed as described above.
RNA Interference
For RNA interference SF2/ASF SMARTpool siRNA oligonucleotides were purchased (Dharmacon, Boulder, CO). Approximately 200,000 HEK-293 cells cultured in 24-well plates were transfected with 100 pmol of the double-stranded oligo-nucleotides or the negative control (5′-UAGCGACUAAACACAUCAA-3′). RNA using Dharmafect1 transfection reagent (Dharmacon). Twenty-fours hours later the cells were transfected with 0.09 μg of either the β-globin minigenes or the HIV-tat minigenes using FuGene 6 transfection reagent (Roche Molecular Biochemicals). Cells were harvested 72 hr after the initial siRNA transfection. SF2/ASF down regulation was monitored by Western blotting using an SF2/ASF monoclonal antibody (AK96, Zymed Laboratories, San Fransicso, CA; Invitrogen).
In Vitro Transcription of MTRR Pseudoexon
Primers MTRR-KpnI-S (5′-cctattttggtaccacttcagggaaggact-3′) and MTRR-HindIII-AS (5′-ctctatttcaagcttcctggctgaccttag-3′) were used to amplify the MTRR pseudoexon region from heterozygous gDNA. The PCR product was cloned between KpnI and HindIII sites in the pTRAP V5 vector (CytoStore, Canada). The resulting plasmids were sequenced to exclude PCR-derived errors and to identify clones containing either wild-type or mutant MTRR pseudoexon (pTRAP/MTRRwt or pTRAP/MTRRmut, respectively). Vectors were subsequently linearized by XhoI digestion and used as templates (1 μg DNA) for in vitro transcription with MEGAshortscript™ High Yield Transcription Kit (Ambion, Austin, TX).
Pull-Down Assay
Short biotinylated RNA oligonucleotides (MTRR-wt: 5′-gaatggctggaggagattcagcctgax-3′ and MTRR-mut: 5′-gaatggccggaggagattcatcctgax-3′; x = biotin) spanning the ESE motif or in vitro transcribed RNA comprising the S1 aptamer [Srisawat and Engelke, 2001] fused to the 140-bp MTRR pseudoexon (wild type or mutant) were used for affinity purification of interacting nuclear proteins. Pull-down assay and immunodetection of purified proteins were carried out as previously described [Nielsen et al., 2007].
Databases
MTRR; MIM#s 236270, 602568; GenBank: NC_000005.9 (g.DNA), NM_002454.2 (mRNA).
Results
The Deep Intronic Mutation c.903+469T>C Is by Itself Sufficient to Cause Aberrant Splicing of the MTRR Transcript
To investigate if the c.903+469T>C mutation directly causes inclusion of the pseudoexon into MTRR mRNA or alternatively is just a marker linked to another causative mutation, we cloned a segment of intron 6 containing the pseudoexon sequence with its flanking sequences into the context of two different splicing reporter minigenes: a β-globin minigene and a HIV-tat minigene (Fig. 1A). In the β-globin minigene the MTRR intronic sequence harboring the pseudoexon sequence is inserted in β-globin intron 1 and flanked by β-globin exon 1 and exon 2, whereas in the HIV-tat minigene the MTRR intronic sequence harboring the pseudoexon sequence is inserted in an intron of the HIV-tat gene and flanked by two exons. Eukaryotic COS-7 cells were transiently transfected with each minigene and splicing products were analyzed by RT-PCR (Fig. 1A).
Figure 1.
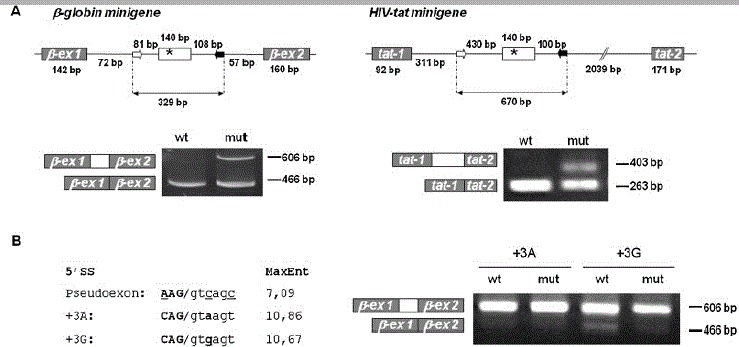
Recognition of the MTRR pseudoexon with weak 5′ splice site is dependent on the presence of the c.903+469T>C mutation. A: Splicing minigene assay. Upper panel depicts the β-globin and HIV-tat minigenes harboring the wild-type or mutant MTRR pseudoexon. Minigenes were used to transiently transfect COS-7 cells. After RNA isolation the splicing products were analyzed by RT-PCR using minigene-specific primers. The lower bands represent correctly spliced minigene exons, the upper bands represent MTRR pseudoexon inserted between minigene exons. B: 5′ splice site optimization. The suboptimal pseudoexon 5′ splice site (AAG/gtcagc) was converted to an optimal 5′ splice site (variant +3A: CAG/gtaagt) or to the nearly optimal 5′ splice site (variant 13G: CAG/gtgagt), and wild-type and mutant minigenes were analyzed by transfection/RT-PCR. The scores of the different 5′ splice sites assessed by the MaxEntScan program (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) [Yeo and Burge, 2004] are shown in the left panel. Right panel shows results from RT-PCR analysis of splicing products.
The results obtained with both minigenes show that inclusion of the MTRR pseudoexon is caused by the c.903+469T>C mutation per se, and thus that this mutation is by itself responsible for inducing aberrant splicing of the MTRR transcript.
The MTRR Pseudoexon Is Flanked by a Strong 3′ Splice Site and a Weak 5′ Splice Site
We next focused our attention on the possible mechanism underlying the observed aberrant splicing. The entire sequence of the MTRR gene was scanned by six different programs for splice site prediction (for details see Materials and Methods) and the scores of the splice sites of all the MTRR constitutive exons and the pseudoexon were compared (Table 1 and Supp. Table S1). Although each program calculates the score according to a different algorithm [Brunak et al., 1991; Pertea et al., 2001; Reese et al., 1997; Rogozin and Milanesi, 1997; Shapiro and Senapathy, 1987; Yeo and Burge, 2004], all analyses gave similar results. In comparison with the authentic MTRR 3′ splice sites, the pseudoexon 3′ splice site is among the strongest 3′ splice sites because it was identified by all programs and its score exceeds the score of the majority of the authentic MTRR 3′ splice sites. Moreover, three programs assessed the pseudoexon 3′ splice site as being stronger than all the constitutive MTRR 3′ splice sites (Table 1 or Supp. Table S1). On the other hand, the 5′ splice site of the pseudoexon is suboptimal. It was recognized as a splice site only by three of five predictive programs, although the scores oscillate around the median value found for the authentic MTRR 5′ splice sites (for details, see Supp. Table S1). However, the intrinsic strength of the MTRR pseudoexon 5′ splice site is low due to mismatches to U1snRNA at the −3, +3 and 16 positions [Roca et al., 2008]. We hypothesized that it is because of this weak 5′ splice site that the pseudoexon is normally not recognized and included in the MTRR mRNA, and that strengthening of this weak splice site by changing positions −3, +3, and +6 to the consensus nucleotides would improve splicing, so that the pseudoexon is always included irrespective of the presence of the c.903+469T>C mutation. To test this hypothesis we generated the wild-type and mutant MTRR- β-globin minigenes in which the 5′ splice site was converted to (1) a strong splice site with C at −3, A at +3, and T at the +6 position, and (2) other constructs with a slightly weaker splice site also with C at −3 and T at +6, but with a G at the +3 position. All the constructs with the optimized 5′ splice sites, except the wild-type construct with the suboptimal +3G, showed complete exon inclusion (Fig. 1B). The wild-type construct with the +3G still showed a small amount of pseudoexon exclusion, consistent with +3G being a slightly weaker splice site [Madsen et al., 2006; Ohno et al., 1999; Roca et al., 2008]. This showed that it is this weak 5′ splice site, which ensures that there is no pseudoexon inclusion into wild-type MTRR mRNA.
Table 1.
In Silico Analyses of MTRR Authentic and Pseudoexon Splice Sites
| GeneSplicer | NetGene2 | NNSplice | SpliceView | SpliceSiteFinder | MaxEnt | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Program | 5′SS | 3′SS | 5′SS | 3′SS | 5′SS | 3′SS | 5′SS | 3′SS | 5′SS | 3′SS | 5′SS | 3′SS |
| Constitutive exons | ||||||||||||
| Na | 3 | 10 | 13 | 12 | 12 | 11 | 13 | 12 | 13 | 13 | 13 | 13 |
| Min. score | 4.91 | 1.50 | 0.30 | 0.07 | 0.71 | 0.62 | 74 | 77 | 68.4 | 77.6 | 5.68 | 5.39 |
| Max. score | 9.37 | 7.20 | 0.95 | 0.96 | 1.00 | 0.99 | 90 | 89 | 95.4 | 92.7 | 11.11 | 10.41 |
| Median | n.d. | 5.46 | 0.67 | 0.33 | 0.97 | 0.90 | 84 | 83 | 84.3 | 86.4 | 8.54 | 7.89 |
| Pseudoexon | n.f. | 12.58 | n.f. | 0.94 | 0.90 | 0.98 | 83 | 92 | 84.1 | 96.9 | 7.09 | 10.41 |
Number of authentic MTRR splicing donors or acceptors identified by a particular predictive program from the total number of 13 authentic MTRR splicing donors and acceptors, respectively.
SS, splice site; n.d., not determined; n.f., not found. Reference sequence of the cytosolic isoform of the MTRR gene, GenBank Accession No. NC_000005.9 (g.DNA), was used for in silico analyses. All nucleotide numbering regarding the MTRR gene (NM_002454.2) reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1.
The Intronic Mutation c.903+469T>C Creates a New ESE
Because recognition of weak splice sites can either be assisted by splicing enhancers or suppressed by splicing silencers, we hypothesized that the c.903+469T>C mutation could function by creating a splicing enhancer or by disrupting a splicing silencer. Therefore, we scanned the wild-type and c.903+469T>C mutant pseudoexon sequence for ESEs and ESSs using different web based programs (for details see Materials and Methods). The results generated by ESE finder [Cartegni et al., 2003] indicated that the c.903+469C nucleotide dramatically increases the score of a SF2/ASF binding motif (TGGAGGA > CGGAGGA) from being close to the lower threshold (2.341) to having a very high score (5.262). In addition, the c.903+469T>C mutation creates a new high score SF2/ASF motif (GGCCGGA) with a score of 2.967 (Table 2). An overlapping SC35 binding motif (GGCTGGAG > GGCCGGAG) is not affected by the mutation. Analysis using the ASD program [Stamm et al., 2006] gave similar results, but the RESCUE-ESE program [Fairbrother et al., 2004] did not show any difference between the distribution of ESE motifs in the wild-type and the mutant pseudoexon sequence. Moreover, neither the FASS-ESS program [Wang et al., 2004] nor the ASD program indicated that c.903+469>T disrupts an ESS. Altogether, these data suggested that the c.903+469T>C mutation activates the pseudoexon by strengthening and/or creating an ESE, which potentially could function by binding of the splicing factor SF2/ASF.
Table 2.
ESEfinder 3.0 Analyses of the Wild-Type and Mutant Pseudoexon
| SF2/ASF threshold = 1.956 | SC35 threshold = 2.383 | |||||
|---|---|---|---|---|---|---|
| Splicing factor | Pseudoexon Positiona (nt) | Motif | Score | Positiona (nt) | Motif | Score |
| Wild type mutant | 23 | TGGAGGA | 2.341 | 20 | GGCTGGAG | 3.385 |
| 20 | GGCCGGA | 2.967 | 20 | GGCCGGAG | 3.616 | |
| 23 | CGGAGGA | 5.262 | ||||
Position of the first nucleotide of an ESE motif counted from the 5′ boundary of the pseudoexon.
SF2/ASF Regulates MTRR Pseudoexon Inclusion
To investigate whether inclusion of the MTRR pseudoexon can be regulated by SF2/ASF, HEK-293 cells were cotransfected with the MTRR-β-globin minigenes and vectors expressing SF2/ASF, or either of two other SR-proteins, namely, SRp40 and SRp55. Whereas SRp40 and SRp55 overexpression had no effect on pseudoexon inclusion, overexpression of SF2/ASF had a strong positive effect on the mutant construct (Fig. 2A), but only a very weak effect on the wild type. Together these results suggested that the c.903+469T>C mutation strengthens and/or creates an SF2/ASF binding ESE, and that the cellular amounts of SF2/ASF determines if the pseudoexon is included by binding to this ESE.
Figure 2.
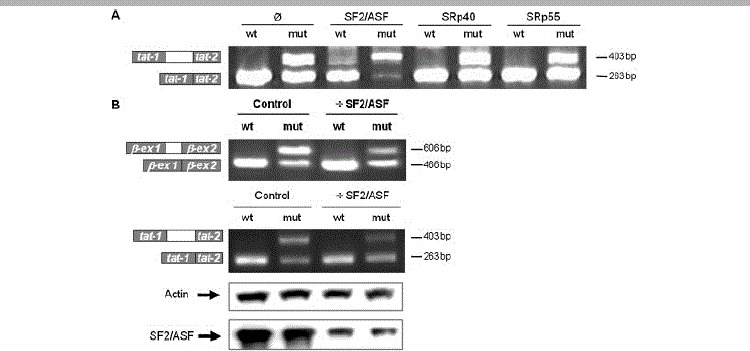
SF2/ASF regulates MTRR pseudoexon inclusion by binding to an MTRR ESE. A: SF2/ASF overexpression. HEK293 cells were cotransfected by the wild-type or c.903+469T>C mutant MTRR β-globin, and vectors expressing SF2/ASF, SRp40 or SRp55, respectively. After RNA isolation the splicing products were analyzed by RT-PCR using minigene specific primers. The lower band represents correctly spliced minigene exons, the upper band represents the MTRR pseudoexon inserted between the minigene exons. B: RNA interference. HEK-293 cells were transfected with double-stranded RNA oligonucleotides directed toward the SF2/ASF mRNA or a negative control, followed by transfection with the MTRR β-globin or HIV-tat minigenes (left panels). The degree of SF2/ASF downregulation was tested by Western blotting (right panel). The results of the Western blot is shown below the corresponding lanes.
Consequently, we speculated that a decrease in cellular SF2/ASF amounts would inhibit pseudoexon inclusion from the mutant minigene. Therefore, we performed siRNA mediated downregulation of SF2/ASF in HEK-293 cells and transfected them with the wild-type and mutant MTRR-β-globin minigenes (Fig. 2B). This showed a clear decrease in pseudoexon inclusion from the mutant constructs despite the fact that downregulation of SF2/ASF was not complete. Identical results were observed when we performed this analysis using the HIV-tat minigene with wild-type and mutant MTRR pseudoexon (Fig. 2B). This further corroborates the notion that MTRR pseudoexon inclusion is caused by SF2/ASF binding to an ESE, which is created and/or strengthened by the c.903+469T>C mutation.
SF2/ASF Binds to the Mutant MTRR ESE
Because both in silico analysis and the overexpression/knock-down experiments indicated that the main effect of the c.903+469T>C mutation is to create and/or dramatically improve a binding site for SF2/ASF, we analyzed the binding of nuclear proteins to in vitro transcribed wild-type and mutant MTRR pseudoexon RNAs by a pull-down assay and Western blotting (Fig. 3). This showed very clearly that the c.903+469T>C mutation results in the binding of SF2/ASF to the pseudoexon, whereas there was no indication of a change in hnRNP A1 binding. To more specifically investigate binding of SF2/ASF to the predicted MTRR ESE motifs we performed RNA affinity chromatography with RNA oligonucleotides that harbor the wild-type and mutant ESE motifs as part of a much shorter sequence. This analysis confirmed that the c.903+469T>C mutation dramatically increases the ability of the ESE motifs to bind SF2/ASF, whereas there is no change in the ability to bind hnRNPA1 (Fig. 3). Thus, the results confirmed that the c.903+469T>C mutation creates a high-affinity ESE, which in turn, binds SF2/ASF.
Figure 3.
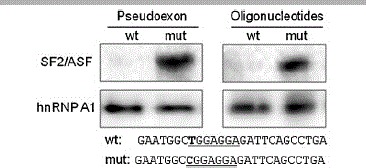
SF2/ASF exclusively binds to the mutant MTRR ESE. Pull-down assay. In vitro transcribed wild-type or mutant MTRR pseudoexon RNAs coupled to biotinylated magnetic beads, or oligonucleotides harboring either the wild-type or mutant MTRR ESE sequence were incubated with HeLa nuclear extract, and the interacting proteins were identified by Western blotting with the use of SF2/ASF or hnRNPA1 antibodies. Sequences of the used oligonucleotides are listed below the pictures.
Discussion
In recent years our view of aberrant splicing as a disease mechanism has changed from a situation in which this possibility was considered to be relatively rare and only relevant when one out of a few critical nucleotides in the splice site consensus sequences at the exon/intron borders are changed, to a situation where all sequence variants in a gene may potentially disrupt splicing. Despite the fact that introns typically make up more than 90% of the sequence of a gene, the vast majority of reported mutations that cause aberrant splicing without directly affecting the splice sites are located in exons [Buratti et al., 2006; Cartegni et al., 2002; Wang and Cooper, 2007]. This overrepresentation does of course reflect the importance of exonic sequences in splicing regulation, but may also in part be caused by the current routine mutation–detection techniques, where intronic sequences are not analyzed. With the rapidly evolving improvements in sequence analysis techniques, an enormous amount of intronic sequence variation, like intronic single-nucleotide polymorphisms (SNPs), are detected. These sequence variations may potentially affect splicing and thus be involved in human disease. It is therefore crucial that we learn more about the mechanisms by which intronic sequence variations may cause aberrant splicing.
In the present study we have investigated in detail how the most frequent disease-causing mutation in the MTRR gene, the deep intronic mutation c.903+469T>C, leads to pseudoexon inclusion. We first used two different splicing reporter minigenes to demonstrate that the pseudoexon inclusion, which is observed in patient cells [Wilson et al., 1999; Zavadakova et al., 2005], is caused by the c.903+469T>C mutation per se and not by another closely linked sequence variation. Although the degree of pseudoexon inclusion was not as pronounced as previously observed in patient cells [Zavadakova et al., 2005], the fact that we observed a similar splicing pattern in two different splicing reporter minigenes, each representing a different heterologous context for the pseudoexon, shows that the c.903+469T>C mutation is decisive for the MTRR pseudoexon inclusion, but that the nature of the genomic context is an important determinant for the efficiency of pseudoexon inclusion. We next wanted to explore the molecular mechanism underlying pseudoexon inclusion. From our in silico analysis it is obvious that the MTRR pseudoexon possesses a strong 3′ splice site, whereas the 5′ splice site is suboptimal. When we substituted the weak 5′ splice site with an optimal 5′ splice site having −3C, +3A, and +6T, the wild-type pseudoexon was completely included, whereas substitution with a slightly weaker optimal 5′ splice site having −3C, +3G, and +6T slightly resulted in a marginally lower lever of inclusion of the wild-type pseudoexon. These results confirmed that it is the weak 5′ splice site that ensures that the wild-type pseudoexon is normally not activated, and that activation depends on splice site strength. Our results demonstrate that a single suboptimal splice site is sufficient to distinguish a pseudoexon from a functional exon, and it is in line with the many reported examples of pseudoexon activation caused by single nucleotide substitutions that strengthen a weak splice site [Buratti et al., 2006]. There are, however, numerous examples of functional 5′ splice sites, which are functional despite being weaker than the natural MTRR pseudosplice. Moreover, a search in the SpliceRack database (http://katahdin.cshl.edu:9331/splice/index.cgi?database=spliceNew) for human 5′ splice sites shows that 10 different genes have functional exons with a 5′ splice site with the same consensus (AAG|GTCAGCC) as the nonfunctional site in the MTRR pseudoexon [Sheth et al., 2006]. This nicely illustrates that this splice site sequence is, in fact, functional in other exons, and points to the fact that the nature of sequences outside of the splice sites is fundamental for splicing regulation, and that exon recognition is determined by a balance between splice site strength and the local distribution of splicing silencers and splicing enhancers.
A search for potential splicing regulatory elements within the MTRR pseudoexon showed that the c.903+469T>C mutation created a new one and strengthened another binding motif for the splicing factor SF2/ASF. The hypothesis that the activation of the mutant pseudoexon is, in fact, caused by creation and/or strengthening of an SF2/ASF binding ESE was confirmed by our knock-down and overexpression experiments, which showed that inclusion of the mutant pseudoexon is regulated by the cellular amounts of SF2/ASF. We confirmed that this effect was directly mediated by the ESE sequence created by the c.903+469T>C mutation, because our in vitro binding studies very clearly showed that SF2/ASF binding was dependent on the c.903+469T>C mutant sequence. Interestingly, an identical motif (GGCGGAGGAAG) has previously been identified as a functional SF2/ASF binding ESE in the alternatively spliced RON (MST1R) proto-oncogene [Ghigna et al., 2005]. In this gene, the ESE sequence in exon 12 was also proven to bind SF2/ASF in vitro and SF2/ASF overexpression was shown to promote recognition of the weak 3′ splice site of exon 12 in model minigenes. The data from RON and MTRR splicing demonstrates that the shared core sequence CGGAGGA is a strong ESE motif, which is recognized by SF2/ASF. Identification of an identical regulatory sequence in two different and functionally diverse genes suggests that this could be a frequent motif that can function in SF2/ASF mediated recognition of weak 5′ splice sites as well as weak 3′ splice sites, and suggests that this motif could also function as a fundamental splicing regulatory element in other genes.
To our knowledge, only four other examples of intronic mutations that activate pseudoexons by altering splicing regulatory elements within the pseudoexon have been reported so far [Davis et al., 2009; Ishii et al., 2002; King et al., 2002; Pagani et al., 2002]. A G>A substitution in intron 4 of the α-GalA gene [Ishii et al., 2002], a G>A substitution in intron 6 of the COL4A5 gene [King et al., 2002] and a A>G substitution in intron 1 of the FGB gene [Davis et al., 2009], are all believed to activate pseudoexons either by creating ESE motifs or abolishing ESS motifs, but in none of these examples the functional binding motif or the involved splicing regulatory protein have been identified experimentally. However, introduction of secondary mutations in a minigene indirectly suggests that the mutation in intron 1 of the FGB gene, similar to the MTRR mutation, activates the pseudoexon by creating a SF2/ASF-binding motif [Davis et al., 2009], and thus also suggests ESE creation as the mechanism for pseudoexon activation. Pagani and coworkers [Lewandowska et al., 2005; Pagani et al., 2002] reported pseudoexon activation due to a deep intronic 4 bp deletion in the ATM gene. They demonstrated experimentally that the deletion abolishes a splicing silencer, which functions by binding of the U1snRNP. Also, studies from the GHR gene [Akker et al., 2007] have suggested that U1 snRNP can act as a splicing suppressor of pseudoexons, and this was supported by testing of U1snRNP binding to other pseudoexons that had sequences complementary to U1snRNA [Akker et al., 2007]. Whereas both the ATM and GHR pseudoexons harbor a suitable binding site for U1snRNA within the pseudoexon sequence, there are no similar binding sites in the MTRR pseudoexon sequence. Consistent with this, we could not observe any suppressing effect on mutant MTRR pseudoexon inclusion from overexpression of U1snRNA (Supp. Fig. S1), and there is therefore no evidence that U1snRNP binding outside of the 5′ splice site contributes to MTRR pseudoexon silencing. Examination of the pseudoexon sequences from the α-GalA gene, the COL4A5 gene, and the FGB gene also showed that there were no U1snRNP binding sites, and consequently, that it is only the wild-type ATM pseudoexon that harbors a motif that is complementary to U1snRNA (Supp. Fig. S1). This indicates that although suppression of pseudoexon splicing by U1snRNP binding outside of the splice site can suppress some pseudoexons, it is not a general feature of all pseudoexons.
When Sun and Chasin [2000] systematically studied the importance of various splicing regulatory elements (5′- and 3′-splice site strength in combination with splicing enhancer and silencer sequences) for their ability to cause inclusion of a pseudoexon from the human HPRT gene, they concluded that more than one element had to be changed to activate the pseudoexon. This would suggest that pseudoexon activation is a rare event. However, an increasing number of examples of pseudoexons, which have been activated by a single nucleotide substitution and resulted in disease, has been reported in recent years [Buratti et al., 2006]. The majority of these mutations simply improve splice site strength, which is consistent with splice site strength being the major determinant for exon recognition. Whereas such intronic nucleotide substitutions that create a new splice site or strengthen an existing pseudo splice site are relatively easy to discover, substitutions that either abolish a splicing silencer or creates a splicing enhancer are more difficult to assess due to the degeneracy of the binding motifs. The present study shows, however, that this mechanism does occur, and may lead to dramatic changes in the splicing pattern and ultimately result in human disease.
In conclusion, we present for the first time functional evidence that conclusively demonstrates that an intronic single nucleotide sequence mutation is sufficient to induce pseudoexon activation by creating a functional ESE, which binds the splicing factor SF2/ASF. We suggest that this molecular mechanism may cause genetic disease much more frequently than reported so far.
Acknowledgments
This work was supported partially by the Grant Agency of Charles University, Czech Republic (30/2004 to K.H. and P.Z.); by the Research Project of Charles University, Ministry of Education of the Czech Republic (MSM0021620806), and by the Danish Research Council for Health and Disease (22-04-395 and 271-07-342 to B.S.A.). We thank Miroslav Janosik and Henriette S. Andersen for helpful discussion and comments. We thank Adrian Krainer for the generous gift of the SF2/ASF, SRp40, and SRp55 expression vectors and Franco Pagani for the U1snRNA expression vector.
Supplementaary material
References
- Akker SA, Misra S, Aslam S, Morgan EL, Smith PJ, Khoo B, Chew SL. Pre-spliceosomal binding of U1 small nuclear ribonucleoprotein (RNP) and heterogenous nuclear RNP E1 is associated with suppression of a growth hormone receptor pseudoexon. Mol Endocrinol. 2007;21:2529–2540. doi: 10.1210/me.2007-0038. [DOI] [PubMed] [Google Scholar]
- Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle M, Baralle FE. Defective splicing, disease and therapy: searching for master checkpoints in exon definition. Nucleic Acids Res. 2006;34:3494–3510. doi: 10.1093/nar/gkl498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Homer VM, George PM, Brennan SO. A deep intronic mutation in FGB creates a consensus exonic splicing enhancer motif that results in afibrinogenemia caused by aberrant mRNA splicing, which can be corrected in vitro with antisense oligonucleotide treatment. Hum Mutat. 2009;30:221–227. doi: 10.1002/humu.20839. [DOI] [PubMed] [Google Scholar]
- Fairbrother WG, Yeo GW, Yeh R, Goldstein P, Mawson M, Sharp PA, Burge CB. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004;32:W187–W190. doi: 10.1093/nar/gkh393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, Green MR, Riva S, Biamonti G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell. 2005;20:881–890. doi: 10.1016/j.molcel.2005.10.026. [DOI] [PubMed] [Google Scholar]
- Hanamura A, Cáceres JF, Mayeda A, Franza BR, Jr, Krainer AR. Regulated tissue-specific expression of antagonistic pre-mRNA splicing factors. RNA. 1998;4:430–444. [PMC free article] [PubMed] [Google Scholar]
- Ishii S, Nakao S, Minamikawa-Tachino R, Desnick RJ, Fan JQ. Alternative splicing in the alpha-galactosidase A gene: increased exon inclusion results in the Fabry cardiac phenotype. Am J Hum Genet. 2002;70:994–1002. doi: 10.1086/339431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King K, Flinter FA, Nihalani V, Green PM. Unusual deep intronic mutations in the COL4A5 gene cause X linked Alport syndrome. Hum Genet. 2002;111:548–554. doi: 10.1007/s00439-002-0830-3. [DOI] [PubMed] [Google Scholar]
- Leclerc D, Wilson A, Dumas R, Gafuik C, Song D, Watkins D, Heng HH, Rommens JM, Scherer SW, Rosenblatt DS, Gravel RA. Cloning and mapping of a cDNA for methionine synthase reductase, a flavoprotein defective in patients with homocystinuria. Proc Natl Acad Sci USA. 1998;95:3059–3064. doi: 10.1073/pnas.95.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowska MA, Stuani C, Parvizpur A, Baralle FE, Pagani F. Functional studies on the ATM intronic splicing processing element. Nucleic Acids Res. 2005;33:4007–4015. doi: 10.1093/nar/gki710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen PP, Kibaek M, Roca X, Sachidanandam R, Krainer AR, Christensen E, Steiner RD, Gibson KM, Corydon TJ, Knudsen I, Wanders RJ, Ruiter JP, Gregersen N, Andresen BS. Short/branched-chain acyl-CoA dehydrogenase deficiency due to an IVS3+3A>G mutation that causes exon skipping. Hum Genet. 2006;118:680–690. doi: 10.1007/s00439-005-0070-4. [DOI] [PubMed] [Google Scholar]
- McVety S, Li L, Gordon PH, Chong G, Foulkes WD. Disruption of an exon splicing enhancer in exon 3 of MLH1 is the cause of HNPCC in a Quebec family. J Med Genet. 2006;43:153–156. doi: 10.1136/jmg.2005.031997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen KB, Sorensen S, Cartegni L, Corydon TJ, Doktor TK, Schroeder LD, Reinert LS, Elpeleg O, Krainer AR, Gregersen N, Kjems J, Andresen BS. Seemingly neutral polymorphic variants may confer immunity to splicing-inactivating mutations: a synonymous SNP in exon 5 of MCAD protects from deleterious mutations in a flanking exonic splicing enhancer. Am J Hum Genet. 2007;80:416–432. doi: 10.1086/511992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Brengman JM, Felice KJ, Cornblath DR, Engel AG. Congenital end-plate acetylcholinesterase deficiency caused by a nonsense mutation and an A→G splice-donor-site mutation at position +3 of the collagenlike-tail-subunit gene (COLQ): how does G at position +3 result in aberrant splicing? Am J Hum Genet. 1999;65:635–644. doi: 10.1086/302551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani F, Buratti E, Stuani C, Bendix R, Dork T, Baralle FE. A new type of mutation causes a splicing defect in ATM. Nat Genet. 2002;30:426–429. doi: 10.1038/ng858. [DOI] [PubMed] [Google Scholar]
- Pagani F, Stuani C, Tzetis M, Kanavakis E, Efthymiadou A, Doudounakis S, Casals T, Baralle FE. New type of disease causing mutations: the example of the composite exonic regulatory elements of splicing in CFTR exon 12. Hum Mol Genet. 2003;12:1111–1120. doi: 10.1093/hmg/ddg131. [DOI] [PubMed] [Google Scholar]
- Pertea M, Lin X, Salzberg SL. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 2001;29:1185–1190. doi: 10.1093/nar/29.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- Roca X, Olson AJ, Rao AR, Enerly E, Kristensen VN, Borresen-Dale AL, Andresen BS, Krainer AR, Sachidanandam R. Features of 5′-splice-site efficiency derived from disease-causing mutations and comparative genomics. Genome Res. 2008;18:77–87. doi: 10.1101/gr.6859308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozin IB, Milanesi L. Analysis of donor splice sites in different eukaryotic organisms. J Mol Evol. 1997;45:50–59. doi: 10.1007/pl00006200. [DOI] [PubMed] [Google Scholar]
- Rosenblatt DS, Fenton WA. Inherited disorders of folate and cobalamin transport and metabolism. In: Scriver CR, Beaudet AL, Valle D, Sly WS, editors. The metabolic & molecular bases of inherited disease. New York: McGraw-Hill, Inc; 2001. pp. 3897–3933. [Google Scholar]
- Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987;15:7155–7174. doi: 10.1093/nar/15.17.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth N, Roca X, Hastings ML, Roeder T, Krainer AR, Sachidanandam R. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. 2006;34:3955–3967. doi: 10.1093/nar/gkl556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srisawat C, Engelke DR. Streptavidin aptamers: affinity tags for the study of RNAs and ribonucleoproteins. Rna. 2001;7:632–641. doi: 10.1017/s135583820100245x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm S, Riethoven JJ, Le Texier V, Gopalakrishnan C, Kumanduri V, Tang Y, Barbosa-Morais NL, Thanaraj TA. ASD: a bioinformatics resource on alternative splicing. Nucleic Acids Res. 2006;34:D46–D55. doi: 10.1093/nar/gkj031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Chasin LA. Multiple splicing defects in an intronic false exon. Mol Cell Biol. 2000;20:6414–6425. doi: 10.1128/mcb.20.17.6414-6425.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Wilson A, Leclerc D, Rosenblatt DS, Gravel RA. Molecular basis for methionine synthase reductase deficiency in patients belonging to the cblE complementation group of disorders in folate/cobalamin metabolism. Hum Mol Genet. 1999;8:2009–2016. doi: 10.1093/hmg/8.11.2009. [DOI] [PubMed] [Google Scholar]
- Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- Zavadakova P, Fowler B, Suormala T, Novotna Z, Mueller P, Hennermann JB, Zeman J, Vilaseca MA, Vilarinho L, Gutsche S, Wilichowski E, Horneff G, Kozich V. cblE Type of homocystinuria due to methionine synthase reductase deficiency: functional correction by minigene expression. Hum Mutat. 2005;25:239–247. doi: 10.1002/humu.20131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.