

Abstract
Fe-based catalytic sites for the reduction of oxygen in acidic medium have been identified by 57Fe Mössbauer spectroscopy of Fe/N/C catalysts containing 0.03 to 1.55 wt% Fe, which were prepared by impregnation of iron acetate on carbon black followed by heat-treatment in NH3 at 950°C. Four different Fe-species were detected at all iron concentrations: three doublets assigned to molecular FeN4-like sites with their ferrous ion in low (D1), medium (D2) or high spin state (D3), and two other doublets assigned to a single Fe-species (D4 and D5) consisting of surface oxidized nitride nanoparticles (FexN, with x≤2.1). A fifth Fe-species appears only in those catalysts with Fe-contents ≥ 0.27 wt%. It is characterized by a very broad singlet, which has been assigned to incomplete FeN4-like sites that quickly dissolve in contact with an acid. Among the five Fe-species identified in these catalysts, only D1 and D3 display catalytic activity for the oxygen reduction reaction (ORR) in the acid medium, with D3 featuring a composite structure with a protonated neighbour basic nitrogen and being by far the most active species, with an estimated turn over frequency for the ORR of 11.4 e− site−1 s−1 at 0.8V vs RHE. Moreover, all D1 sites and between 1/2 to 2/3 of the D3 sites are acid-resistant. A scheme for the mechanism of site formation upon heat-treatment is also proposed. This identification of the ORR-active sites in these catalysts is of crucial importance to design strategies to improve the catalytic activity and stability of these materials.
Introduction
In recent years, the growing scarcity of easily accessible oil and its disproportionate geographical distribution have spurred interest in alternatives to the gasoline-fueled internal combustion engine currently used in most transportation applications. Proton-exchange membrane fuel cells (PEMFCs) fed with hydrogen from renewable sources constitute an efficient and environmentally responsible alternative to this gasoline-dependent technology, but have yet to overcome a number of challenges in order to reach their full commercialization potential. One of these major hurdles is the excessive cost of the platinum-based catalysts currently used to accelerate the kinetics of the two reactions that take place inside the cell,1,2 i.e. the electrooxidation of hydrogen at its anode and the complementary electroreduction of oxygen at its cathode. In their 2010 analysis of mass production cost estimation for PEMFCs, James et al. estimate that Pt electrocatalysts would account for nearly half of the fuel cell stack cost.3 The rate of the oxygen reduction reaction (ORR) is slow, even on Pt, compared to that of the hydrogen oxidation on the same metal, and thus the Pt content at the cathode is usually 5–10 times larger than that at the anode.1,2 Therefore the substitution of the Pt-catalyst at the fuel cell's cathode with an inexpensive material would significantly reduce its manufacturing cost, which could then meet the 9 $/kW Membrane-Electrode-Assembly target set by the DOE for transportation in 2017.4
Interestingly, it has been known since the 1960's that N4-macrocycles of abundant non-noble metals like cobalt and especially iron can also catalyze the ORR.5,6 Moreover, the heat-treatment of such macrocycles supported on carbon led to an enhancement of their oxygen reduction activity.6,7 Inspired by the elemental composition of these coordination compounds and a seminal paper from Yeager's group,8 most current Fe- and Co-based catalysts are prepared using separate metal and nitrogen precursors that are brought together with a carbonaceous support in a heat-treatment step at 600–1000°C; multiple examples of such preparation procedures of Fe/N/C or Co/N/C-catalysts for ORR in acid media can be found in several reviews.9–12 Moreover, recent efforts have shown that the careful choice of nitrogen and metal precursors, combined with an appropriate carbon support or host for the catalytic sites and heat-treatment conditions, can yield very active Me/N/C-catalysts that perform nearly as well as commercial Pt/C-materials.13–15 However, the processes that lead to the rapid degradation of some of the most promising catalysts still need to be clarified. To accomplish this, a thorough understanding of the particular structure for each of the active sites and their specific role in the ORR is required.
It is important to note that the high performance of the ORR catalysts that have recently been reported by our group13,15 is the logical outcome of our extensive studies previously carried out on less performing catalysts. The latter were prepared by an adaptation of the precursor-based approach proposed by Yeager and collaborators, in our case using a carbon support impregnated with iron acetate (the iron precursor) that was heat-treated at 950°C in NH3 (the nitrogen precursor). By studying these catalysts in detail, we found that their ORR-activity is governed by (i) their iron content, (ii) the concentration of nitrogen functionalities on their surface and (iii) their microporous surface area.16–18 However, despite the fundamental character of the question and after five decades of research, no consensus has yet been reached regarding the exact identity of the catalytic species at work in these catalysts. Currently, the two preponderant lines of thought in the research community are: (i) that nitrogen functionalities on the surface of the carbon-based catalysts are directly responsible for their ORR-activity,19–22 and (ii) that N-groups act as the coordinating environment for Fe- or Co-ions where the ORR takes place. The latter are believed to be structurally similar to the FeN4-moieties that are proposed to be the catalytic sites in the heat-treated FeN4-macrocycles initially used in the 60's and 70's,7,23–28 and also to a simplified version of the catalytic site of cytochrome c oxidase, the metalloenzyme that reduces oxygen in many bacteria and in mitochondria, which are billion-year-old biomimetic fuel cell analogs.29,30
Based on Time of Flight Secondary Ion Mass Spectrometry (ToF SIMS) studies of Fe/N/C-catalysts conducted previously by our group, we subscribe to the second hypothesis that the metal is at the heart of the most ORR-active catalytic sites. Those works revealed the simultaneous presence of two families of FeN4-like species, of which one was more active in acid media.31,32 Extended X-ray Absorption Fine Structure (EXAFS) and Mössbauer spectroscopies are two other analytical techniques that have proven extremely useful for the characterization of Fe-(or Co-)-based ORR-catalysts.24–26,33–44 These spectroscopies are indeed very sensitive to the immediate structural and chemical neighborhood of the probed Fe atom in the materials. Moreover, 57Fe Mössbauer spectroscopy is particularly well suited to analyze Fe-based catalysts, because it can distinguish between various Fe-sites of similar structure but in different oxidation and/or spin states. Both EXAFS and Mössbauer spectroscopies are used in this work to acquire information about all Fe-species found in our most extensively studied catalysts, prepared by impregnating a non microporous carbon support with iron acetate and heat-treating the resulting materials in NH3 at high temperature.18,28,45 Additional techniques like transmission electron microscopy (TEM), X-ray diffractometry (XRD), neutron activation analysis (NAA) and combustion analysis are also used for additional characterization.
In this work, it will be shown that the Fe/N/C catalysts with equal micropore and nitrogen contents, but different Fe-concentrations, contain up to five iron-based species, but only two of them are acid-resistant active sites for the ORR in the acid medium. This intensive characterization also enabled us to propose a scheme that describes the steps in the formation of the different iron-sites during the pyrolysis of the catalyst precursors. Besides reinforcing our belief that iron is at the heart of the catalytic activity of these materials, these results also serve two purposes: first, they can be used as a novel and unique comparison basis for the future analysis of already existing, yet much more ORR-active and performing Fe/N/C-catalysts13–15 in order to identify which site(s) should be privileged to further improve the catalytic activity; and second, they constitute a starting point in the understanding of the degradation/deactivation mechanisms affecting most of these Fe/N/C-catalysts.
Experimental section
Catalyst synthesis
1. Preparation of the original catalysts (O-catalysts)
All catalysts were synthesized following a procedure consisting of two steps. First (i), a determined loading of iron precursor was wet-impregnated onto a pristine, pore-less carbon black provided by the Sid Richardson Carbon Corporation. In the second step (ii), this Fe-loaded powder was heat-treated in ammonia at 950°C until a weight loss of 30–35 % was attained; this weight loss is known to correspond to the maximum microporous content and catalytic activity of similarly prepared catalysts made with this carbon support.18,28,45 In the following the overall procedure is described in more detail.
For the wet-impregnation, a defined mass of Fe-57 enriched iron (II) acetate (FeAc, Triportech, 95% Fe-57), mFeAc, was weighed and poured into 100–150 mL of deionized water. Next, the mass of carbon mcarbon required for the desired loading of iron on carbon, LFe (in wt% Fe), was estimated using Equation 1:
| [1] |
This mass of carbon was then added to the FeAc-solution, along with 5–10 mL of ethanol to aid its dispersion. The resulting suspension was magnetically stirred for three hours and subsequently left to dry overnight in an oven at 85°C.
The heat-treatment in ammonia started with the weighing of a defined mass of the ground, Fe-impregnated and dry powder (mi, typically 500–550 mg), that was subsequently placed into a quartz boat. The boat was introduced into a tube, along with a glass rod with a magnet embedded in the end further from the boat. The tube was then closed and the air was purged by flowing 1000 sccm of Ar for 30 minutes. The tube was subsequently placed in a hinged split-tube oven (Thermcraft), with the tube section housing the powder-containing boat and glass rod remaining outside of the heating zone. The oven was then turned on and left to stabilize at 950°C for two hours. For the first hundred minutes of this stabilization period, 100 sccm of H2 was added to the Ar; during the last 20 min, this Ar/H2-flow was substituted with 2000 sccm of NH3. After these two hours, a magnet was used to push the powder-containing boat to the centre of the oven with the glass rod. A countdown for the required duration of heat-treatment was then started and, when this time was over, the oven was opened and the tube directly removed from its interior. Once cooled down for 20 min, the tube was opened, the powder removed and the remaining mass (mf) was weighed. The weight loss W (%) caused by the heat-treatment was then estimated using Equation 2:
| [2] |
2. Preparation of Acid-washed and re-heat-treated catalysts
The conditions for acid-washing and re-heat-treatment were similar to the standard conditions used in Herranz et al.28 Acid-washed catalysts were prepared by immersing 100 mg of powder in 50 mL of pH 1, H2SO4 solution, along with 0.5 mL of ethanol to aid the powder's dispersion. The resulting suspension was magnetically stirred for 24 hours. The resulting acid-washed powder was then recovered by vacuum-filtration, rinsed with abundant deionized water (≥ 1L) and left to dry overnight in an oven at 85°C. Re-heat-treated catalysts were derivatives of the above acid-washed materials that were submitted to a subsequent heat-treatment step at 950°C similar to the catalyst preparation procedure, except using Ar instead of NH3. The weight loss caused by this re-heat-treatment was typically ≤ 5%
Characterization methods
1. Rotating-disk electrode voltammetry (RDE) in acid electrolyte
An ink of each sample was prepared by ultrasonically and mechanically mixing 10 mg of catalyst with 95 μL of 5 wt% recast Nafion solution (Aldrich) and 350 μL of ethanol. Seven microliters of this ink was then pipetted over three glassy-carbon disk electrodes of 0.196 cm2. This corresponds to a mass of catalyst (mcat) of 0.157 mg, and a catalyst loading of 0.8 mg cm−2.
An AFCBP1 Pine potentiostat controlled with Pine 2.8.0 software was used for the measurements, that were carried out using a solution of O2-saturated sulphuric acid of pH 1 as the electrolyte. A platinum wire was used as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. All voltammograms were recorded sweeping the potential between −0.25 and 0.75 V vs. SCE at a rate of 10 mV s−1. For each catalyst-loaded electrode, the measurement started by mounting it onto a Pine rotator and immersing its tip into the O2-saturated electrolyte. First, four potential cycles were recorded keeping the electrode idle. Then, a fifth cycle was recorded while rotating the electrode at 1500 rpm. Finally, O2 was purged by bubbling nitrogen into the solution for a minimum of fifteen minutes, and two more potential cycles were recorded. The current obtained in this O2-free electrolyte corresponds to the capacitive current (Icap, in mA) of the sample throughout the potential range. Using this Icap and the I current recorded in the decreasing potential scan of the previous measurement with the electrode rotating at 1500 rpm, one can estimate the Faradic current (If) throughout the potential range using Equation 3:
| [3] |
Plotting If against the potential results in a sigmoid voltammogram whose Ilim current plateau at low potentials can be used to estimate the ORR-related kinetic current ikin (in A g−1) throughout the potential range, according to Equation 4:
| [4] |
where mcat is the catalyst loading (mg cm−2). In this work, the catalysts' ORR-activity is systematically assessed as the mean value of this ikin at 0.8 V vs. RHE for the three electrodes tested for each catalyst ink. In order to transform the recorded, SCE-referred potential to the reversible hydrogen electrode (RHE) scale, every set of experiments was ended by bubbling pure H2 into the N2-saturated electrolyte for a minimum of 10 min and measuring the potential of the SCE electrode vs. the platinum wire, which acted as a RHE. This potential difference was typically of 300–305 mV.
2. 57Fe Mössbauer spectroscopy
In order to characterize the different chemical states of iron inside the catalysts, 57Fe Mössbauer spectroscopy was performed. The measurements were made at room temperature in the transmission mode using a 50 mCi 57Co/Rh-source, and the calibration was made with respect to α-Fe. For the original catalysts containing 0.27 wt% and 0.84 wt% Fe, spectra were recorded in a larger velocity range (± 10 mm s−1, measurements not shown) in order to clarify whether they contained any magnetic iron species; no sextets were found in these original catalysts. Therefore, it was decided to measure all remaining samples in a velocity range of only ± 5 mm s−1. The smaller range enables a much higher resolution of 0.02 mm s−1. This is an advantage for the detection and discrimination of different FeN4-like species. All Mössbauer spectra were plotted as relative absorption against the Doppler velocity; the relative absorption was calculated from the original transmission data (in counts) using equation 5:
| [5] |
In this equation Transmax is the maximum background transmission appearing at Doppler velocities where no absorption is observed, Trans(vDoppler) is the transmission at a specific Doppler velocity and Abs(vDoppler) is the relative absorption calculated for each Doppler velocity, and expressed as a percentage. Data were analyzed with the Recoil software, assuming Lorentzian shapes for all iron sites. The assignment of iron species was usually made by comparison to data from the literature.
3. Transmission electron microscopy
Transmission electron microscopy (TEM) was used to analyze the structure and composition of the iron-based nanoparticles contained in the catalysts. The samples were prepared by ultrasonically dispersing ~ 5 mg of catalyst into ~ 2 ml of methanol for ½ to 1 minute and dipping a copper grid (partially covered with an amorphous carbon film) into the resulting catalyst-containing suspension. For the TEM measurements, a JEOL 2100-F microscope with an acceleration voltage of 200 kV and a typical current density of 15 pA/cm2 was used. Images were obtained in bright field. Parallel electron energy loss spectroscopy (PEELS) was performed for elemental analysis.
4. Extended X-ray Absorption Fine Structure Analysis (EXAFS)
X-ray absorption spectroscopy of the iron edge was performed at beamline X3-B of the National Synchrotron Light Source, Brookhaven, NY. The measurements were performed in a setup which allows the simultaneous measurement in transmission and fluorescence mode. In transmission geometry the intensity of the incoming beam I0 and of the beam transmitted through the sample ITrans were recorded with gas ionization chambers. A Fe foil, placed after the sample, was used to establish a reference signal IRef. The fluorescence signal was measured rectangular towards the incoming beam usinga 13 element Ge detector. Measurements were made in an energy range of −200 to 800 eV with respect to the Fe K-edge (7112 eV), whereas the overall energy range was split into three parts, each with different stepwidth (SW) and accumulation time (At): Range 1: −200 to −50 eV (SW: 5 eV, At: 5 s); Range 2: −50 to 50 eV (SW: 0.5 eV, At: 2 s); Range 3: 50 to 800 eV (SW: 5 eV, At: 3 s).
The pseudo-radial distribution function χ(R) was calculated by Fourier Transformation of the original data using the following relation:
| [6] |
In this equation kMax and kMin correspond to the FT-window (approx. 2 to 12 Å−1) and χ(k) is the normalized EXAFS spectrum.
5. Other characterization methods
The final iron content of each impregnated sample was determined by Neutron Activation Analysis (NAA) that was performed at the reactors BER II in Berlin and/or SLOWPOKE-2 in Montreal. Due to the sample enrichment with 57Fe, the 57FeAc precursor, with a known amount of 57Fe, was used as standard.
Results and discussion
Identification of the iron species and their contribution to the Mössbauer spectra of Fe/N/C-catalysts
In order to get a better understanding of the catalytic site formation process and of the role of different iron species in the catalysis of the ORR, seven samples with iron contents between 0.03 and 1.55 wt% Fe were prepared and their iron content was determined by NAA. These seven catalysts were subsequently characterized by Mössbauer spectroscopy. Figure 1 shows their Mössbauer spectra, which are deconvoluted in five doublets and, for the catalysts with ≥ 0.27 wt% Fe, an additional broad singlet. Each one of these components is characterized by an isomer shift (δiso) and a quadrupole splitting (ΔEQ) that are sensitive to the electron density at the absorbing iron nucleus and the changes in its coordination environment. The isomer shift refers to the difference in Doppler velocity between the barycenter of the Mössbauer doublet or singlet and the spectrum's zero velocity. The latter corresponds to the barycenter of the Mössbauer spectral slines of a reference material (α-Fe, in this work). This δiso is related to the density of s-electrons at the absorbing Fe nucleus. For iron, a positive isomer shift is indicative of a decrease in the 4s electron density, which in turn can be due to an increased screening effect by 3d electrons (larger 3d electrons density).42 As for the quadrupole splitting, it represents the distance between the barycenters of the two absorption lines constituting each doublet. Its value depends on the chemical environment of the absorbing iron nucleus (the so-called “lattice term”) and the occupation of its 3d orbitals (or “valence electron term”). It is when one or both of these terms are different from zero that the singlet-line splits-up into two sub-lines. The values of the parameters of each component in the Mössbauer spectra are summarized in Table 1, where each component is also assigned to an iron species, when possible, in agreement with the literature.
Figure 1.

Deconvoluted Mössbauer spectra of the Fe/N/C-catalysts. The iron content of the original catalyst is given as an insert in each spectrum.
Table 1.
Mössbauer parameters and assignment of the six components featured in the deconvoluted Mössbauer spectra of Figure 1 and Figure 5. The second column features the color code of each Fespecies in these spectra.
| Component | δiso / mm●s−1 | ΔEQ / mm●s−1 | fwhm / mm● s−1 | Assignment* | References | |
|---|---|---|---|---|---|---|
| Doublet Dl | 0.39 (± 0.04) | 1.16 (± 0.06) | 0.63 (± 0.07) | FeIIN4/C, low spin | 36, 37, 41–43, 46 | |
| Doublet D2 | 0.36 (± 0.13) | 2.40 (± 0.28) | 0.61 (± 0.04) | FeIIN2+2/C, intermediate spin | 36, 37, 46, 48 | |
| Doublet D3 | 1.01 (± 0.13) | 2.32 (± 0.17) | 0.63 (± 0.07) | N-FeIIN2+2…Nprot/C, high spin** | 49–52, this work | |
| Doublet D4 | 0.43 (± 0.03) | 0.30 (± 0.03) | 0.28 (fixed) | FexN with x≤ 2.1 | 53,55 | |
| Doublet D5 | 0.29 (± 0.04) | 0.31 (± 0.02) | 0.28 (fixed) | |||
| Singlet | 0.36 (fixed) | - | 3.3 (± 0.2) | disordered FexNy | this work | |
For ferrous iron (FeII, 3d6) the low, intermediate and high spin states correspond to zero, two and four unpaired spins, as schematized in Figures 2a–2c.
According to Ref.28, this site is characterized by two different ORR-activities: (i) a low-activity one in which the basic N-group ('…Nprot') remains unprotonated (N-FeN2+2…Nprot/C), or has uptaken a proton and has already been neutralized by an anion, [N-FeN2+2…NH+/C] HSO4−; or (ii) a highly active, composite site in which the N-group is protonated and not neutralized yet, N-FeN2+2…NH+/C.
The first row in Table 1 reports the δiso and ΔEQ values of doublet D1. They are similar to those reported by Melendres for the FeN4 centers in iron phthalocyanine in interaction with the carbon support.46 In two recent works on catalysts prepared by heat-treatment of iron porphyrin Kramm (née Koslowski) et al. related D1 to the ORR-activity and assigned it to a low-spin state (S=0) FeN4/C center.41,42 The latter structure was also proposed by Bouwkamp-Wijnoltz et al.24 as the ORR-active site in heat-treated porphyrins. This D1-related center consists of a Fe-ion coordinated by four pyrrolic N-groups connected to the surface of the carbon support, therefore labelled as FeN4/C. The distribution of the six electrons in a low-spin state, S=0,47 in the 3d orbitals of its FeII ion is illustrated in Figure 2, where one possible side view is also given. In this side view we have completely integrated the FeN4-site related to D1 in the top graphene layer of a carbon crystallite.
Figure 2.

Side views and top views of the proposed structures of: (A) the FeN4/C catalytic site in heat-treated, macrocycle-based catalysts assigned to Mössbauer doublet D1, (B) the FeN2+2-like micropore-hosted site found in the catalyst prepared with iron acetate and heat-treated in ammonia assigned to doublet D2, and (C) the N-FeN2+2-like composite site, where N-FeN2+2 is assigned to doublet D3. In all side views, graphene planes are drawn as lines. In (B), the distance between the two nitrogen atoms belonging to the graphene planes above and below the FeN2+2-plane is similar to that in crystaline iron phthalocyanine (see Figure S1), thus similarly influencing the lattice contribution to the quadrupole splitting. In (C), the ironII ion in N-FeN2+2 is coordinated by five pyridinic nitrogen atoms, one of them belonging to a plane located at the vertical below the N4-plane. This axial nitrogen coordination moves the ironII ion out of the N4-plane towards the fifth coordinating nitrogen atom. Doublet 3 may exist as N-FeN2+2/C or as a composite site N-FeN2+2…Nprot/C, where Nprot is a protonable pyridinic nitrogen atom appearing at the edge of the upper plane in the side view of D3.28 Nprot is too far from NFeN2+2 to have an influence on the Mössbauer spectrum of D3, but it is close enough to drastically improve the turn-over frequency of the composite site proposed in (C).
Figure 2 also shows the energy schemes of D1, D2, and D3, as well as the filling of the molecular orbitals for the ironII ion in the structure proposed for each site according to its proposed spin state: low in D1, intermediate in D2, and high in D3.47 Part (D) of Figure 2 schematizes the changing ORR-activity of the composite N-FeN2+2…prot/C site, where N-FeN2+2/C has the D3 signature; the activity of the composite site is low when the basic N-group neighbouring the N-FeN2+2/C moiety is unprotonated (left) or protonated and anion-neutralized (right). The activity is high when this N-functionality is protonated but not neutralized (center).28
The δiso and ΔEQ values of doublet D2 are reported in the second row of Table 1. They are typical of the intermediate spin (S=1), ferrous ion in crystalline iron-phthalocyanine,46,48 whose two crystalline structures are illustrated in Figure S1. Phthalocyanines (Pcs) exist in several polymorphic forms of which the α- and β-variants are well characterized and identified by Mössbauer spectroscopy.48 The difference between these α-and β-polymorphs lies essentially in the orientation of the molecules with respect to the crystallographic axes, while the perpendicular distance between the planes of the stacked molecules is very similar (~ 3.4 nm). Additionally the same approximate distance is also found between the basal planes of graphite or carbon blacks.27
Figure 2b shows the structure of the site assigned to D2. In crystalline FePc, there are always nitrogen atoms in the plane above and below the FeN4 moiety of each FePc molecule, which is why we propose that there is also nitrogen atoms above and below the FeN4-like plane of our D2. As far as the FeN4-like moiety of D2 is concerned, this moiety has indeed the FeN2+2 configuration already proposed by our group and illustrated in Figure S2. The FeN2+2 moiety features an ironII ion coordinated by four pyridinic nitrogen functionalities attached to the edges of opposed graphene planes delimiting a micropore in the carbon.27
As for doublet D3, its isomer shift is much larger than that of D2 or D1, suggesting a larger density of 3d electrons. Its Mössbauer parameters are in fact similar to those of ferrous, high-spin FeN4-centers found in different porphyrins.49–52 To the best of our knowledge, though, this doublet is unprecedented in the literature of Fe/N/C catalysts. Considering our previous works on this type of catalyst18,27,28,32 the structure shown in Figure 2c is assigned to D3. The corresponding distribution of the six electrons in the 3d orbitals of the FeII ion that leads to a high spin state (S=2) for D3 is also illustrated in Figure 2, and is adapted from that published for FeN4-macrocycles in ref. 47. Further details about the proposed structure of D1, D2, and D3 will be given in the next sections.
Next in Table 1, doublets D4 and D5 are both related to surface-oxidized, nitrogen-rich nanoparticles of iron-nitride whose bulk has a molecular formula of FexN, with x ≤ 2.1.53 This nitride phase is found in all catalysts by transmission electron microscopy and also confirmed by X-ray diffraction for the catalysts with ≥ 0.84 wt% Fe (see Figure S3). Figure 3 displays TEM micrographs of quasi-spherical iron nitride particles having diameters between ~ 5nm and ≥ 200nm, strongly resembling those featured in other Fe/N/C catalysts that were also prepared by heat-treatment in NH3.54 Localized EELS spectra of these particles (Figure S4) show that their surface is oxidized. The fact that one crystalline structure gives rise to two doublets in the Mössbauer spectra is caused by the differences in the chemical environment and corresponding Mössbauer parameters of each absorbing iron nucleus in the nitride (i.e. number of neighboring iron and nitrogen atoms in the bulk of the nanoparticle as well as oxygen atoms at its surface).
Figure 3.

TEM images of the quasi-spherical particles of iron nitride observed in all the catalysts prepared in this study, with each catalyst's iron content specified in the label.
Lastly, the singlet at the bottom of Table 1 features an unusual broadness (large full width at half maximum, fwhm) indicative of a species in a highly disordered state. The singlet is assigned to an iron nitride with a stoichiometry in-between those of the above FexN and the FeN4-like moieties, i.e. a mixture of FexNy with various `x' and `y' values that cause slight differences in the environment of each absorbing 57Fe-nucleus and its corresponding Mössbauer parameters. As we were not able to identify these species using TEM, these FexNy species are probably subnanometric particles or of molecular character.
In the following, we will quantify the relative iron content assigned to each of the species in all seven catalysts. It is experimentally observed that the total absorption area of the Mössbauer spectra displays a linear correlation with the total iron content of these samples (see Figure S5), indicating that all iron nuclei have roughly the same probability to absorb the resonant γ-rays and contribute equally to the intensity of the Mössbauer spectra. One can therefore assume that all Fe-species have similar Debye-Waller factors, and estimate their composition in each catalyst using Equation 7:41–43
| [7] |
where FeX, expressed in wt% Fe, is the concentration of an Fe-species `X' in the catalyst; AbsX, expressed in percentage, is the relative absorption area of each Fe-species `X' in its Mössbauer spectrum; and Fecatalyst, also expressed in wt% Fe, is the total iron content in the catalyst. The results obtained for each Fe-species are plotted in Figures 4b–f.
Figure 4.

Compared evolution of the catalysts' ORR-activity and the relative composition of each Fe-species with the bulk iron content.
Assignment of the ORR-activity to the iron species in the Fe/N/C catalysts
1. Comparing Mössbauer and EXAFS experiments
Following the characterization of all seven Fe/N/C-catalysts, we proceeded to measure their ORR-activity by RDE-voltammetry. The results are presented in Figure 4a, showing a Fe-content versus ORR-activity trend similar to that reported by Jaouen and Dodelet for equivalent catalysts prepared with natural iron acetate instead of the isotopic 57FeAc used here.56 In Figure 4a, the ORR-activity increases linearly from the lowest metal concentration up to 0.27 wt% Fe, then reaches an activity maximum at 0.84 wt% Fe (but still can increase slightly when concentrations up to ≈ 1.1 wt% Fe are probed56), then drops sharply for the catalyst having the highest metal concentration (1.55 wt% Fe). When enough iron is present, the ORR-activity of the Fe/N/C-catalysts is known to be governed by nitrogen functionalities and micropores17,18,57 that are the result of the etching of the disordered carbon in the initial non-porous pristine support by the NH3 used in the heat-treatment.45,58 Figures S6a and S6b show that both micropore surface area and nitrogen content start decreasing for catalysts with iron concentrations > 0.6 wt% Fe, resulting in a drop of one order of magnitude in ORR activity for the catalyst containing 1.55 wt% Fe (Fig. 4a). This drop of activity could be related to the increasingly close packing of the first monolayer of ironII acetate on the surface of the pristine carbon during the impregnation of larger amounts of this Fe-precursor, as discussed in the text following Figure S6 in the Supplementary Information.
By comparing the ORR activity in Figure 4a with the relative composition of each Fe species shown in Figures 4b to 4f, the iron content related to both D2 and D3 strongly correlates with the ORR-activity (compare Figs. 4c and 4d with Fig. 4a). Furthermore, the contents of both sites, D2 and D3, are similar for any given catalyst, suggesting that these two families of FeN4-like species are structurally related. This is why it was concluded that D2 and D3 are both hosted in micropores, and also why both of their FeN4-like structures are derived from FeN2+2/C, as illustrated in Figure S2. The position of the FeN4-like structure in D3 in the micropore will depend on that of the fifth nitrogen atom coordinating the FeII ion; here this fifth nitrogen atom is drawn at the edge of a vertical graphene plane, as seen on the side view in Figure 2. The presence of this fifth coordinating atom will be discussed later on. Furthermore, on the basis of the spin of the FeII ion in these proposed D2 and D3 sites, we believe that D2 is not active for the ORR and that only D3 is linked to a major part of the catalytic properties of these Fe/N/C-materials. The lack of ORR-activity of D2 is related to the intermediate spin state (S=1) of its ironII ion, that translates in a completely filled 3dz2 orbital (see Fig 2b) and prevents the end-on adsorption of molecular oxygen (and its subsequent reduction) on these sites.59 On the contrary, both D3, with S=2 and a 3dz2 orbital occupied by a single electron (see Fig. 2c), and D1, with S=0 and an empty 3dz2 orbital (see Fig. 2a), are able to bind oxygen to their FeII ion in the end-on adsorption mode required to initiate the reduction of oxygen. Two other findings corroborate the lesser importance of D2 vs D3 as an active site for the ORR in these Fe/N/C catalysts. First, Fe-species with Mössbauer parameters similar to those of D2 that were detected in catalysts prepared by heat-treatment of Fe-porphyrin in an inert atmosphere (up to 1000 °C) or NH3 (at 800 °C) were not found to contribute to their ORR activity.41–43 Secondly, in a preliminary Mössbauer experiment carried out on a Fe/N/C catalyst much more ORR-active than those studied in this work (namely the one described in Ref. 13) the doublet D2 was absent from the spectrum recorded for this catalyst, while D3 was present. Thus, D3 is important for achieving a high ORR activity in Fe/N/C catalysts, whereas D2 is not.
As for doublet D1, while its relative content in catalysts prepared by heat-treatment of Fe-porphyrin in an inert atmosphere has been correlated to their ORR-activity,41,42 here it does not appear to contribute significantly to the catalytic activity (compare Figs. 4a with 4b). This minor contribution of D1 to the overall activity cannot be attributed to a low D1 site density in the catalysts, since the content of D1 sites is larger than that of D3 for all catalysts shown in Fig. 4. Therefore, this minor contribution of D1 has to be related to a lower turn-over frequency of the corresponding D1 sites vs. the turn-over frequency of D3. This assertion is supported by the turn-over frequency values already estimated in similar catalysts prepared from iron acetate and ammonia precursors vs. porphyrin-based ones.41,43,56,60
While the FeN4-like moieties assigned to D1 and D3 seem to contribute to different extents to the overall ORR-activity, the possible activity of the other Fe-species (i.e. the FexNy assigned to the singlet and the Fex≤2.1N nitride nanoparticles associated with D4 and D5) remains unclear. In order to find out more about their possible catalytic properties, we attempted to remove them by washing the catalyst originally containing 0.27 wt% Fe in a solution of sulfuric acid of pH 1 for 24 hours (see Experimental section). The Mössbauer spectrum of the resulting acid-washed catalyst (AW) is compared to that of the original-catalyst (Ocat) in Figure 5a, and the relative contents of each Fe-species in both Mössbauer spectra are shown in Figure 5b. On the one hand, acid-washing resulted in the total removal of the disordered FexNy iron species assigned to the Mössbauer singlet and a sharp decrease by 50% in the intensity of doublets D4 and D5, assigned to the nanoparticles of iron nitride illustrated in Figure 3. On the other hand, when the standard deviation of the content of each Fex species presented in Fig. 5b is taken into consideration, it seems that acid-washing had little effect on the content of the two FeN4-like catalytic sites assigned to doublets D1 and D3, while Fe(D2) was halved.
Figure 5.

Effects of acid-washing on the Fe-species in the catalyst containing 0.27 wt% Fe. (A) displays the Mossbauer spectra of the original catalyst (Ocat) and its acid-washed derivative (AW); the full bars in (B) correspond to the iron concentrations of each Fe-species in the Mössbauer spectra of the Ocat, while the patterned bars represent the concentrations in the AW sample; (C) shows the corresponding EXAFS spectra of the original catalyst Ocat and its acid-washed derivative AW. The red lines in both EXAFS spectra are that of an iron foil. The Fe content of the AW catalyst is 0.09 wt%.
The same original and acid washed catalysts were also characterized by X-ray absorption spectroscopy, and their EXAFS-spectra are shown in Figure 5c. To help the comparison, the EXAFS spectrum of a Fe foil (red curve) has been added to both frames of the Figure. There are essentially three main peaks in the EXAFS spectra of Ocat and AW. These three non-corrected peaks at ≈ 1.5, 2.4, and 4.4 Å can be assigned to the bonding distances of Fe-N, Fe-Fe (1st shell) and Fe-Fe (2nd shell), respectively, taking into account a phase correction of 0.5–0.6 Å typically found for iron nitride and FeN4-species.61–63 The FeN4-like catalytic sites assigned to D1, D2, and D3 contribute exclusively to the first EXAFS peak, while the FexNy (singlet) and FexN (D4 and D5) iron nitride particles contribute to all three peaks. After acid-washing, the two peaks corresponding to Fe-Fe distances in the 1st and 2nd shells sharply decreased, therefore revealing a total or partial dissolution of iron nitride species richer in iron than the FeN4 sites. This assertion is based on the fact that in the EXAFS spectrum of Fe3N, for instance, the contribution of the first Fe-Fe shell to the bonding distance is about three times more important than that of Fe-N.63 The conclusions drawn from the changes observed in the EXAFS spectra of the catalysts upon acid-washing are therefore in agreement with those drawn from Mössbauer spectroscopy.
2. Non-recoverable and recoverable activity decays
When the catalyst initially containing 0.27 wt% Fe is acid-washed and analyzed by NAA, its Fe-content is found to drop to 0.09 wt% Fe. This represents a loss of 67% of the original Fe content upon acid-washing. On the activity side, acid-washing of the same catalyst results in a drop of mass activity, from 3.3 to 0.33 Ag−1. This represents a decay of 90% of the original activity. However, we have demonstrated8 with a catalyst prepared identically and having a similar initial Fe content (0.20 wt% Fe), that this apparent activity decay upon acid-washing is in fact composed of two parts: (i) a non-recoverable activity decay associated with catalytic species that were proposed to simply dissolve in the acid solution, and (ii) a recoverable activity decay caused by the deactivation of acid-resistant catalytic sites that can be thermally reactivated. This is illustrated in Figure 6a, where the normalized ORR activity of the sample containing initially 0.20 wt% Fe also drops to less than 10% of its original activity after a first washing (AW1) with H2SO4 of pH1 for 24h. Upon re-heat-treatment at a temperature of 400°C (RHT1), this acid washed catalyst recovers 60% of its original activity. When a similar re-heat-treatment is applied to the acid washed catalyst initially containing 0.27 wt% Fe in this work, it recovers 39% of its original ORR activity. This is illustrated by the black squares in Figure S7. Although the general phenomenon is the same for both catalysts, the exact percentage of ORR activity recovered after re-heat treatment of the acid washed catalyst varies somewhat from one catalyst to the other. Furthermore, it is to note that the phenomenon described above is not particular to the catalysts made by impregnation, but applies also to the much more active catalyst described in reference 13 (see the half-filled circles in Figure S7). The same behavior is also displayed by an iron-free catalyst (an NH3 pyrolyzed carbon with a microporosity of about 400 m2 g−1, similar to that of the catalysts in this work). This is illustrated by the empty squares in Figure S7. It is also important to note that this iron-free catalyst is at least 50 times less active than the catalyst in this work prepared by impregnation and initially containing 0.27 wt% Fe.
Figure 6.

Activity recovered by reheat-treatment in argon of the acid-washed catalyst of reference 28, having a similar Fe content (0.20 wt% Fe) than the catalyst used in Figure 4a (0.27 wt% Fe). (a) Normalized activity, based on that of the original catalyst (Ocat), as measured in RDE (open circles) or PEM fuel cell (filled squares) at various steps of five successive acid washing / reheat-treatment cycles. Acid washed and reheat-treated catalysts are labeled AWx and RTHx, respectively, x denoting the cycle number. (b) Bulk Fe content at each step of the five acid washing / reheat-treatment cycles described in (a), as measured by neutron activation analysis.
In order to explain the decay of ORR activity upon acid washing of the iron-based catalysts and their subsequent partial recovery upon re-heat-treatment at a temperature of 400°C, it has been proposed28 that the most ORR-active sites in these catalysts have a composite structure, N-FeN2+2…Nprot/C, illustrated in Figure 2c. Note that, while in reference 28 the same catalytic site was labelled FeN2+2…Nprot/C, here we have introduced a fifth coordinating nitrogen atom in the structure of this D3-site to remain in agreement with its Mössbauer parameters, and therefore re-label it as N-FeN2+2…Nprot/C. The presence of this fifth coordinating nitrogen will be discussed later on. As far as Nprot is concerned, it represents a basic nitrogen functionality located in the vicinity of a N-FeN2+2/C site that is protonated upon contact of the catalyst with a strong acid. This composite site becomes deactivated when the resulting protonated NH+ moiety is neutralized by anions in the acid solution (HSO4− in Figure 2d), and can subsequently be reactivated by removing these anions either thermally (re-heat-treating the washed catalyst in Ar at ≥300 °C) or chemically (immersing it in an alkaline solution)28. The deactivation and reactivation mechanisms of this composite N-FeN2+2…Nprot/C catalytic site are illustrated in Figure 2d.
The similar trends for the overall ORR-activity and the iron contents of doublets D2 and D3 (Figure 4a vs. 4c and 4d) indicate that at least one of these doublets must be related to these acid-resistant sites. The fact that equal proportions of D2 and D3 are found in the catalyst (see Figs. 4c and 4d) is likely indicative of a common mechanism of formation for both species hosted in the micropores etched by ammonia during the catalyst synthesis. The physical difference between the presence or the absence of Nprot in the vicinity of N-FeN2+2…Nprot/C is that it was found that the protonation of N-FeN2+2…Nprot/C to yield N-FeN2+2…NH+/C drastically enhances its ORR-activity.28 Comparatively, unprotonated N-FeN2+2…Nprot/C, protonated and neutralized N-FeN2+2…NH+..HSO4−/C, or Nprot-free N-FeN2+2/C display an ORR-activity about 1/10th that of protonated N-FeN2+2…NH+/C, as seen in Figure 6A after AW1. Interestingly, the values of the Mössbauer parameters for doublet D3 (δiso and ΔEQ) are similar to those found for “picket-fence” porphyrins50–52 which have a high spin (S=2) caused by the positioning of their ferrous ions out-of the N4-plane. This positioning is caused by the interaction of this iron with a fifth axial nitrogen atom from a molecule (e.g. an imidazole) located perpendicularly under the FeN4 plane of the porphyrin. This will block access of O2 to the FeN4 plane on one side while leaving the other side of the picket-fence accessible for oxygen.50–52 Drawing similarities with the N-FeN2+2…Nprot/C sites responsible for doublet D3, the micropores in which these centers are hosted resemble the picket-fence surrounding the FeN4-centers in those porphyrins, and the ferrous iron in NFeN2+2…Nprot/C will be pulled out of its FeN2+2-coordinating plane by a fifth nitrogen located at the edge of a vertical graphene plane underneath the FeN2+2 site. Such a micropore configuration terminated with vertical graphene layers is not exceptional in carbon black structures, and the presence of nitrogen functionalities at their edge (see side view of Figure 2C) is the result of the etching of the carbon support by the NH3 used in the preparation of the catalysts.
As for D2, the presence or absence of Nprot in its vicinity does not make any difference in terms of its ORR-activity because, with its ironII ion in medium spin state (S=1) and its fully occupied 3dz2,47,58 this site does not to contribute to the overall ORR-activity, as discussed above and in recent works.41–43 By comparison with D3, this intermediate spin state of D2 is caused by the ferrous ion in the FeN2+2/C sites remaining co-planar with its N-coordinating environment.
3. To which Fe species is the non-recoverable activity decay attributable?
After having explained that the highly active, composite N-FeN2+2…NH+/C sites may be deactivated for ORR upon neutralization of their positive charges, while being able to remain structurally stable in acid media, we shall now discuss the Fe-species which are irreversibly lost in the catalyst during a first acid washing with H2SO4. Coming back to the catalyst containing initially 0.27 wt% Fe, the activity of the original catalyst is 3.3 Ag−1. Its activity after a first acid-washing and re-heat-treatment at 400°C is 1.3 Ag−1, which corresponds to 39% of the initial activity. This means that 61% of this initial activity was irreversibly lost upon the first acid-washing, as illustrated in Figure S7. As the Fe content of the same catalyst drops from 0.27 to 0.09 wt% Fe after acid washing and remains there after being thermally reactivated, this represents an iron loss of 67% during the first acid washing. At this stage, the remaining question is: to which Fe species is this non-recoverable activity decay attributable? In order to answer this question, we are going to combine the results presented in Figure 5b for the catalyst with an initial Fe content of 0.27 wt% (now labeled Ocat5, in reference to Figure 5) with those presented in Figures 6a and b for the catalyst containing initially 0.20 wt% Fe (now labeled Ocat6, in reference to Figure 6). In order to estimate the content of each Fe species in Ocat6, we will assume that their relative content is the same as that of Ocat5, due to the similar total Fe contents of these two samples. Experimental points of Figures 6a and b are the same as those of Figure 3 in reference 28. However, these results will now be rediscussed in the context of this work. During this discussion, we will also introduce a Figure 7 that presents the Mössbauer spectrum of the catalyst initially containing 0.20 wt% Fe and which has been submitted to four acid washing steps, each followed by a re-heat-treatment. It is the Mössbauer spectrum of the sample identified as RHT4 in Figures 6a and 6b.
Figure 7.
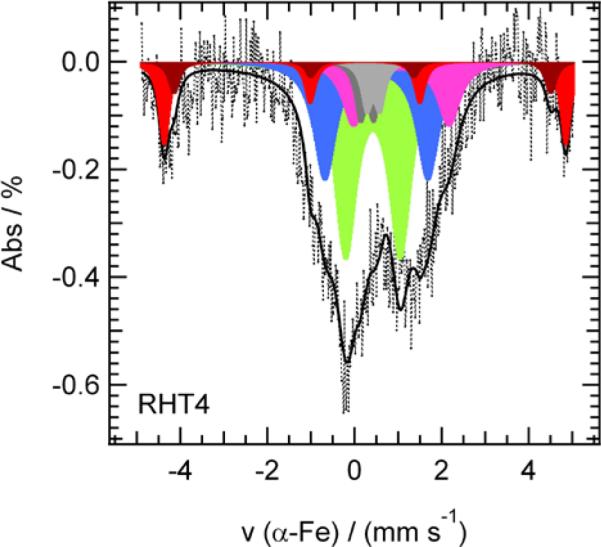
Deconvoluted Mössbauer spectrum of the sample identified as RHT4 in Figures 6a and 6b, i.e. the catalyst initially containing 0.20 wt% Fe of Figure 6 and which has been submitted to four acid washing steps, each followed by a re-heat-treatment.
In order to combine the results shown in Figures 5b and 6b, it is necessary to express the Fe contents in the same units; at% Fe was chosen here. In that case, the initial Fe content of Ocat5 with 0.27 wt% Fe is 0.058 at% Fe and that of Ocat6 with 0.20 wt% Fe is 0.042 at% Fe. The information inferred from Figure 5b (and Table 2) is that Ocat5 is composed of the following Fe-Mössbauer species : (i) 0.0278 ± 0.0015 at% Fe as a Singlet; the latter disappears completely after a first acid washing, AW1; (ii) 0.0174 ± 0.0022 at% Fe as iron nitride (D4 and D5) that half-disappears after AW1; (iii) 0.0128 ± 0.0024 at% Fe as three FeN4-like doublets (D1, D2, and D3) whose concentrations decrease somewhat after AW1, especially for D2 that loses about half its Fe content after AW1. The information inferred from Figure 6b is that changes in the total Fe content of Ocat6 may be divided in three parts, which are brought to the fore during the successive acid washing and re- heat-treatment steps. These three parts are: (i) segment 1, from 0.042 to ~ 0.022 at% Fe, which represents a normalized ORR-activity drop from 1.0 to ~ 0.68 in Figure 6a (i.e. a drop of ~ 32%, of the initially normalized activity of Ocat6 disappears after AW1 and RHT1); (ii) segment 2, from ~ 0.022 to ~ 0.012 at% Fe, which represents a drop from ~ 0.68 to ~ 0.44 in Figure 6a, or a further loss of ~ 24% of the initially normalized activity of Ocat6 that progressively disappears between AW1 and RHT3; (iii) segment 3, once ~ 0.012 at% Fe is reached, the Fe-content remains constant, and so does the normalized ORR-activity, that stabilizes at ~ 44% of the initial value of Ocat6 in Figure 6a after RHT3. Only the mean experimental values (the symbols) of Figures 6a and 6b were used to define segments 1 to 3. All numerical values introduced here will be used later on (in Figure 8) to obtain estimates for the catalytic activity that will be calculated depending on whether the nitride nanoparticles and singlet Fe-species detected by Mössbauer are ORR-active or not in Ocat5 and in Ocat6 after 4-time acid washing and re-heat-treating the catalyst (RHT4).
Table 2.
Mössbauer parameters and assignment of the six components featured in the deconvoluted Mössbauer spectrum of RHT4 (Figure 7).
| RHT4 | Calculated Ocat6 b | Ocat5c | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Component | δiso / mm●s−1 | ΔEQ / mn●s−1 | H0/G | Abs / % | 0.012 at% a | 0.042 at% | 0.058 at% | Assignment* | References | |
| Doublet Dl | 0.42 ± 0.02 | 1.26 ± 0.05 | - | 32.7 ± 2.2 | 0.0039 ± 0.0003 | 0 . 0 0 3 0 ± 0.0009 | 0 . 0 0 4 1 ± 0.0013 | FenN4/C low spin | 36, 37, 41–43, 46 | |
| Doublet D2 | 0.50 ± 0.03 | 2.37 ± 0.07 | - | 20.3 ± 2.7 | 0.0024 ± 0.0003 | 0 . 0 0 3 3 ± 0.0005 | 0 . 0 0 4 6 ± 0.0007 | FenN2+2/C intermediate spin | 36, 37, 46 | |
| Doublet D3 | 1.07 ± 0.08 | 2.19 ± 0.08 | - | 11.1 ± 2.5 | 0.0013 ± 0.0003 | 0 . 0 0 3 0 ± 0.0003 | 0 . 0 0 4 1 ± 0.0004 | FenN2+2…Nprot/C high spin** | 49–52 this work | |
| Doublet D4 | 0.43 | 0.30 | - | 3.6 ± 2.4 | 0.0009 ± 0.0006 | 0 . 0 1 2 6 ± 0.0016 | 0 . 0 1 7 4 ± 0.0022 | FexN with x < 2.1 | 53 | |
| Doublet D5 | 0.29 | 0.31 | 4.2 ± 2.3 | |||||||
| Singlet | - | - | - | - | - | 0 . 0 2 0 1 ± 0.0011 | 0 . 0 2 7 8 ± 0.0015 | disordered FexNy | this work | |
| Sextet 1 | 0.41 | 0.17 | 494 ± 4 | 20.3 ± 5.0 | 0 . 0 0 3 4 ± 0.0011 | - | - | Iron oxide | 64 | |
| Sextet 2 | 0.27 | 0.09 | 463 ± 10 | 7.9 ± 4.0 | ||||||
See footnotes of Table 1
See footnotes of Table 1
: The iron content of Ocat6 after RHT4 is 0.012 at%. The iron content of each component in this column is obtained using Eq 7: Fex = Absx. Fecatalyst/ 100, where Absx is listed in column 4 for each component and Fecatalyst is 0.012 at%.
: The total iron content in Ocat6 is 0.042 at% (0.20 wt% Fe). The iron content for each component in Ocat6 is obtained by multiplying the iron content of the same component in Ocat5 by (0.042 / 0.058), which is the ratio of the total iron content in at% for Ocat6 / Ocat5).
: Experimental iron content in at% in Ocat5, from Figure 5b. The total iron content of Ocat5 is 0.58 at% Fe (0.27 wt% Fe).
Figure 8.
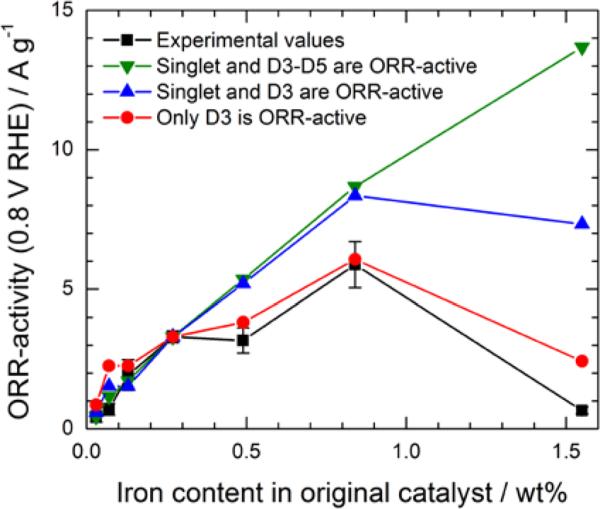
Comparison between experimental ORR activities reproduced from Figure 4a (black squares) and calculated ORR activities according to three hypotheses: (1) iron nitride nanoparticles (D4 and D5), the singlet and D3 (FeN2+2…Nprot/C composite sites) are all ORR active (green triangles); (2) the singlet and D3 are both active (blue triangles); (3) only D3-sites are ORR active (red circles).
Concerning the deconvoluted Mössbauer spectrum of Ocat6 after RHT4 in Figure 7, the first observation that springs to the eyes is that it also contains sextet lines in the higher velocity values, in contrast to the spectra of all other catalysts in this work (see Figures 1 or 5). The parameters for the deconvoluted components of Figure 7 are reported in Table 2. Besides the already known D1 to D5 Fe-species, the two supplementary sextets in Figure 7 can be assigned to iron oxides64 and arise from iron ions released by dissolution of the singlet and/or one of the D1 to D5 Fe-species during the successive acid washing steps. The presence of these iron oxides indicates that some of the released ions were first adsorbed on the carbon substrate, and then transformed into hydroxides upon rinsing with de-ionized water of the acid-washed catalyst, as also observed by Mössbauer spectroscopy of catalysts repeatedly acid-washed (e.g. AW3, AW4, not shown). Re-heat-treatment of this acid-washed and rinsed catalyst transformed the adsorbed iron hydroxides into oxides. We have already shown that iron oxides/hydroxides only have a negligible activity for ORR in acid medium.17
The fifth column after the color code in Table 2 gives the experimental iron content (in at%) of each component of the Mössbauer spectrum of Ocat6 after RHT4 (Figure 7), while the sixth column in Table 2 gives the iron content, calculated on the basis of Figures 5b and 6b, for each component of Ocat6 (Figure 6b), whose total iron content is 0.042 at%. The seventh column in Table 2 gives the experimental iron content for each components of Ocat5 (Figure 5b) whose total iron content is 0.058 at%. By comparing the sixth and fifth columns of Table 2, one sees that, after RHT4 of Ocat6, it is the nitrides (D4 and D5) that were mostly lost by progressive dissolution upon acid-washing (from 0.0126 to 0.0009 at% Fe). As for the three other doublets (D1 to D3), it seems that D3 also dissolves partially (from 0.0030 to 0.0013), while D1 remains practically constant within the experimental error (0.0030 and 0.0039). This was already suggested by Figure 5b. In this context, the fate of D2 is irrelevant since it has already been established that it does not participate to the ORR activity.
At this point in the discussion, there are two possible hypotheses concerning the decrease of activity during segment 2 of Figure 6a: either attributing it to the dissolution of nitrides as an ORR active species,43 or to the disappearance of some D3, the FeN4-like sites already known to be the site with the highest ORR-activity.
Let us consider the first hypothesis stating that the nitride FexN with x≤2.1 is ORR active and that its progressive disappearance is reflected in the activity loss occurring during segment 2 of Figure 6a. By neglecting the very minor amount of iron nitride that remains in the catalyst during segment 3, iron nitride would therefore be responsible for the loss of ~(68 - 44) ≈ 24% of the normalized activity of Ocat6. It also means that the contribution of the singlet to the activity of Ocat6 would then be about ~(100 - 68) ≈ 32%. In this first hypothesis, the catalytic activity of Ocat6 arises essentially from the singlet (~32%), the nitride (~24%) and from D3 (~44%), given that D1 is a Fe-N4-like site with a minor activity compared with that of D3. This gives us values for the activity of each of these three Fe(X) species, in [Ag−1]/[at % Fe(X)]. Applying these three site-specific activities to all other catalysts for which the at % of the same three important Fe(X) species are known (see Table 2: Calculated Ocat6), one may then estimate what would be Figure 4a if the nitrides were ORR active. The results of these calculations are illustrated by the green curve in Figure 8 and the details to build this figure are given in the supplementary information. In conclusion, this hypothesis results in an ORR activity prediction that does not fit the experimental data.
Let us now consider the second hypothesis stating that the iron nitride has a low ORR activity or that it is even ORR-inactive, and that the progressive decrease of Fe-content and ORR-activity in segment 2 would rather be the result of the loss of some of the D3 sites, responsible for ~24% of the normalized activity in Figure 6a. The latter leachable D3 sites would lack the stability of those D3 sites that remain in segment 3 and that are responsible for ~ 44% of the normalized ORR-activity in Figure 6a. Assuming that both soluble and acid-resistant D3 sites have the same ORR activity, one may conclude, in this hypothesis, that the catalytic activity of Ocat6 arises essentially from the singlet (~32%) and from D3 (~68%). By using the same procedure as the one used before, it gives us values of activity for each of these two Fe(X) species, in [Ag−1]/[at % Fe(X)]. Applying these two site-specific activities to all other catalysts for which the at % of the same two important Fe(X) species are known, another calculated curve (the blue one) for the mass activity vs iron content in original catalysts may be added in Figure 8. After a simple glance to that figure, one may conclude that this second hypothesis, although better than the first one, is not acceptable either.
In the second hypothesis used to calculate activity #2 in Figure 8, both D3 and the singlet participate in the mass activity vs iron content of the original catalyst. What happens if, in a third hypothesis, no ORR activity is attributed to the singlet in the original catalyst? In that case, D3 becomes the principal active site in the original catalyst (we are still neglecting, as previously, the small ORR activity attributed to D1). The calculated activity curve (the red one) using that third hypothesis is also given in Figure 8. In this third hypothesis, the calculated activity of the catalyst with 1.55 wt% Fe in Figure 8 becomes 2.43 A g−1. This is much better than the value calculated according to the two previous hypotheses, but 2.43 A g−1 is still about four times larger than the experimental mass activity. This discrepancy is probably attributable to the much lower microporosity and/or nitrogen content of the catalyst made with 1.55 wt% Fe compared with all the other catalysts studied in this work (see Figure S6). Therefore, it seems that most of the ORR activity of the Fe/N/C catalysts prepared for this work arises from the composite N-FeN2+2…Nprot/C sites having the D3 Mössbauer signature, while FeN4/C sites with a D1 Mössbauer signature only account for a minor contribution to the catalyst activity. It is, however, worth noting that, according to the latter hypothesis, only part (between ~ 1/2 to 2/3) of all the D3 sites, which are present in the original catalyst, are acid-resistant.
4. Turn-over frequency of D3
Given that we now know that most of the mass activity of the Fe/N/C catalysts is attributable to D3, it is therefore possible to calculate a value for the turn-over frequency (TOF) of site D3 in Ocat6 (with an initial Fe loading of 0.20 wt%) that has a mass activity, Ikin (0.8V vs RHE) = 2.8 A g−1.28 This includes the acid resistant D3 sites and also those D3 sites which are not resistant to acid, but are also present in Ocat6. The content of D3 sites in Ocat6 in wt% Fe, and labelled cFe, is estimated from Equation 8:
| [8] |
where 0.003 is the content in at%Fe calculated for D3 in Ocat6 and 0.042 at%Fe is the total iron content in Ocat6 (see Table 2)
Therefore, SD (in sites cm−3), the site density of D3 in Ocat6 becomes:
| [9] |
where NA is Avogadro's number and ρ = 0.4 is the assumed density of carbon in gram per cm3 of a porous electrode.1
Now assuming that only D3 sites are ORR active, Equation 10 can be used to calculate the TOF of D3 sites:
| [10] |
where Ikin is the kinetic current density at 0.8V [in A g−1] (i. e. the kinetic current density at 0.8V that may be attributed here to all D3 sites: ~100% of 2.8 A g−1 ≈ 2.8 A g−1), e− is the electric charge of a single electron (1.6 10−19C).
Therefore:
| [11] |
This value is only 2.2 times smaller than that of a 47 wt% Pt/C catalyst,1 and is much higher than that reported earlier by our group [0.95 e− site−1 s−1 at 0.8V vs RHE] for similar Fe/N/C catalysts in the range of 0–0.25 wt% Fe and in the same experimental conditions.56 The previously reported value was smaller because in the former publication we used the full iron loading in the catalyst as cFe to calculate SD, while what we have learned in this work is that only the concentration of the D3 sites have to be used in the calculation. Here the D3 sites were found to account for 7% only of all Fe in the Ocat6. This explains the higher TOF here.
As far as other Fe/N/C catalysts are concerned, the turn over frequencies for several porphyrin-based catalysts were also previously reported.41–43 A dependence of the turn over frequency on the heat-treatment temperature42 and on the gas atmosphere used during the heat-treatment was demonstrated for porphyrin-based catalysts.41,43 The highest TOF (0.8V vs RHE) measured for these catalysts was 0.14 e− site−1 s−1. It was reported for catalysts that were submitted to a second heat-treatment in NH3 at 800 °C after a first heat-treatment in N2 at 800 °C.43 In that case, only the number of D1 sites was considered in the TOF calculation. It is worth noting that, on the one hand, these D1 sites detected in porphyrin-based catalysts are similar to the D1 sites found in this work. On the other hand, no iron-species with a Mössbauer signature similar to that of D3 of the present work were detected in porphyrin-based catalysts. This signature, assigned to ferrous high-spin N-FeN2+2…Nprot/C composite sites, seems to be specific to the experimental conditions used to prepare Fe/N/C catalysts in this work, i. e. the impregnation of an iron acetate catalyst precursor on a carbon support, followed by a high temperature heat-treatment in ammonia. In conclusion, with targets of 2.5 e− site−1 s−1 for TOF and 3.2 × 1020 sites cm−3 for SD, which were the targets set as benchmark for non-noble metal electrocatalysts in 2005,1 it is clear that for the Fe/N/C catalyst studied in this work, the limitation is on SD. As much improved activity have been reported recently for several catalysts prepared according to other methods,13–15 it will certainly be interesting to find out if the increased activity stems from an increased TOF or SD or both.
Model for the formation of the various Fe-species
After having identified the Fe-species present in these Fe/N/C-catalysts and determined their role in the ORR-activity of these materials, we would like to finalize this work by proposing a mechanism for the formation of these species during the heat treatment step in ammonia which is necessary to obtain these catalysts. We have seen in Figure 3 that, irrespective of the original iron content, all catalysts contain iron nitride particles of formula Fex≤2.1N. The quasi-spherical shape of these particles indicates that they originate from a liquid phase that, according to the Fe-N phase diagram55 and considering the heat-treatment temperature of 950°C, is only possible for nitrogen concentrations ≥ 50 at%. Subsequently, a liquid FeNy phase with y ≥ 1 must be at the origin of these Fex≤2.1N nitride spheres. In our opinion, the same FeNy species with y ≥ 1 must also be at the origin of all other Fe-species detected in the final catalysts, as schematized in Figure 9 and explained below.
Figure 9.
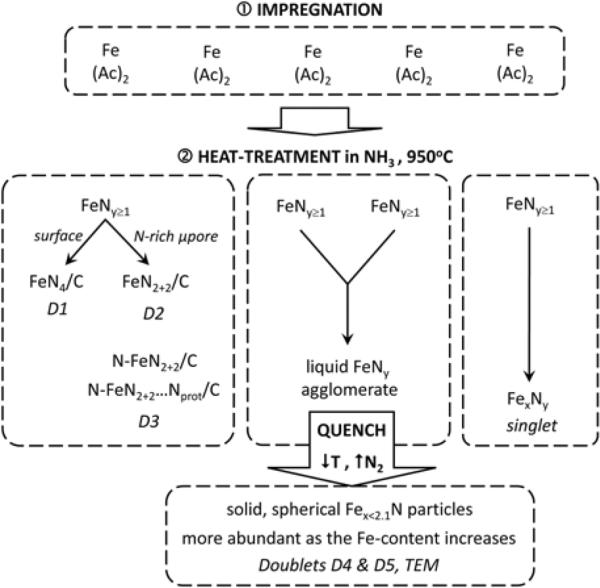
Scheme of the model proposed for the formation of the various Fe-species present in the Fe/N/C-catalysts in this study.
The first step occurring during the heat-treatment in NH3 at 950°C of the non-porous carbon black impregnated with iron acetate must be the decomposition of this ferrous salt, immediately followed by the reaction of the resulting iron atoms with NH3 to give FeNy units with y ≥ 1. All subsequent steps must greatly depend on the number of iron acetate molecules initially present on the carbon support. If the iron loading on the carbon black is low (≤ 0.13 wt%), FeNy≥1 units will only evolve into FeN4-like moieties that include FeN4/C, FeN2+2/C, N-FeN2+2/C, and N-FeN2+2…Nprot/C as well as Fex≤2.1N particles, as shown by Mössbauer spectroscopy. The latter nitride is first a liquid phase made of several FeNy≥1 units that, upon cooling to room temperature, loses nitrogen and evolves into the quasi-spherical nitride particles of formula Fex≤2.1N found in the final catalysts. The latter are responsible for the Mössbauer doublets D4 and D5. An inhomogeneous distribution of the iron acetate initially impregnated on the surface of the carbon black should certainly favor the occurrence of such nitride particles, because iron acetate molecules and FeNy≥1 will then predominantly merge into three dimensional clusters.
As for the formation of FeN4-like moieties, this is likely related to the reaction of NH3 with the carbon support at 950°C, which adds nitrogen functionalities to the carbon surface and also results in the formation of macro, meso and micropores. Instead of merging into larger FeNy≥1 agglomerates, some of the intial FeNy≥1 units will bind to these nitrogen groups or further react with ammonia to form FeN4-like moities. The latter may settle either at the surface of the carbon to end up as the low turn-over frequency FeN4/C sites (see Figure 2a) assigned to Mössbauer doublet D1 (similar to the active sites found in porphyrin-based catalysts heat-treated in inert gas24,41,42), or in micropores to end up as FeN2+2/C or N-FeN2+2/C sites (see Figs. 2b and 2c) assigned to Mössbauer doublets D2 and D3, respectively. It is probably the occurrence of some pyridinic type nitrogen atoms at the edge of one or several graphene planes in a micropore that will interact with the FeNy≥1 migrating units and result in their final presence as D2 or D3 type sites in the micropores. Among those, only the composite site N-FeN2+2…Nprot/C is highly active for ORR. The latter are characterized by the added presence of basic N functionalities in the vicinity of the N-FeN2+2/C centers, yielding composite sites with a high activity (here caused by a high TOF) in acid media (see Fig. 2c). Finally, when the iron content is increased above 0.13 wt%, a new Fe species appears in the Mössbauer spectra (see Figs. 1 and 4). The latter is characterized by a very broad singlet that certainly reflects the structural inhomogeneity of these FexNy species, and that is attributed to FexNy, metastable catalytic species that readily dissolves in contact with a H2SO4 solution of pH 1. As these species were not identified by TEM, they probably are of molecular character.
Conclusions
Combined Mössbauer and EXAFS structural studies of several Fe/N/C-catalysts prepared by impregnation of carbon black with various loadings of iron acetate and subsequent heat-treatment in NH3 have served to identify five different Fe species in these materials. The Mössbauer spectra of these Fe/N/C-catalysts feature (i) three doublets (D1, D2, and D3) assigned to FeN4-like sites (FeN4/C (D1), FeN2+2/C (D2), and N-FeN2+2/C (D3) with their ferrous ion in low, intermediate or high spin state, respectively; (ii) two more doublets (D4 and D5) assigned to surface oxidized nanoparticles of iron nitride; and (iii) a broad singlet only present in those catalysts with iron contents ≥ 0.27 wt% Fe and attributed to incomplete FeN4-like sites. Acid washing the original Fe/N/C-catalysts resulted in the total removal of this broad singlet and of a sizeable fraction of D2 and of the nanoparticles of iron nitride related to D4 and D5. D1 and D3 are much more acid-resistant.
D1 and D2 were also found in catalysts prepared by adsorbing an iron porphyrin on carbon and heat-treating the assembly in inert gas, in which D1 (FeN4/C) centers typically correspond to the majority of the active sites.24,41,42 As for D3, the N-FeN2+2 /C sites, they appear to be exclusively found in catalysts prepared by heat-treating a carbon support loaded with iron precursors in NH3 at high temperature. Based on this study and on a previous work dealing with the stability of similar catalysts,28 we propose that D3 may exist either as N-FeN2+2/ C or N-FeN2+2…Nprot/C. Among the three FeN4-like sites, only FeN4/C (D1) as well as N-FeN2+2/C (D3), especially in its composite version with a nearby basic protonable nitrogen: N-FeN2+2…Nprot/C are catalytically active for ORR, while FeN2+2/C (D2) is inactive. This conclusion is based on the electronic population of the 3d orbitals in the ironII ion, among which the 3dZ2 orbital is reputed to bind oxygen end-on. This energy level is unoccupied for D1 (S=0) or singly occupied for D3 (S=2), while it is fully occupied for D2 (S=1). Therefore, D2 is unable to accept one electron from O2 to form an adduct to the FeN2+2/C site. The surface oxidized nitride nanoparticles responsible for doublets D4 and D5 were also found to be ORR inactive. Furthermore, they slowly dissolve upon repetitive acid washing treatments. So does a part of the FeN4-like sites assigned to D3 (maybe due to an irreversible change of the carbon environment). However, between 1/2 and 2/3 of the initial population of D3 sites are acid resistant. The protonated composite version of the D3 sites (N-FeN2+2…NH+/C) are the sites which exhibit the highest activity for the ORR in these Fe/N/C catalysts with a turn over frequency of ~ 11.4 e− site−1 s−1 at 0.8V vs RHE.
EXAFS results at the iron edge corroborate the conclusions reached from the deconvolution of the Mössbauer spectra. Caution must be taken, however, when EXAFS spectra are interpreted, since they are the result of the summation of the individual contribution of each Fe-species (the Fe-N4-like sites but also the iron nitride particles) to the total spectrum and, unlike with Mössbauer spectroscopy, it is often difficult to differentiate between these components.
Possible illustrations of the three site structures responsible for doublets D1, D2, and D3 are presented in Figure 2. These illustrations respect all the constraints that we and others have accumulated over the years of works on Fe/N/C catalysts. Finally we propose that the heat-treatment step of the catalysts' synthesis starts with the reaction of iron with ammonia to form FeNy≥1units, and have presented a plausible scheme for the subsequent interactions of this initiating species with the carbon support, that yield the various Fe-species detected by Mössbauer spectroscopy.
We hope that this new knowledge will serve to develop Fe/N/C catalysts with higher ORR-activity by developing ways to make a larger fraction of the available Fe-atoms form more of the most ORR-active composite N-FeN2+2…Nprot/C sites. Future efforts should also focus on preventing the deactivation of these acid-resistant sites by the reaction of N-FeN2+2…NH+/C with anions. This remains the major challenge for the commercial application of these materials.
Supplementary Material
Broader context.
H2/O2(Air) polymer electrolyte membrane fuel cells (PEMFCs) are clean and efficient electrical power generators that have been repeatedly demonstrated as a viable alternative to combustion engines for automobile propulsion. However, the cost of automotive fuel cells remains prohibitively high because of the quantity of platinum needed for fuel cell electrodes. Since the majority of platinum resides at the cathode (the electrode where oxygen is reduced to water) the substitution of the Pt-catalyst with an inexpensive material would significantly reduce the manufacturing cost of this electrode. Recently, our group reported molecular-based Fe/N/C cathode catalysts with high activity for the oxygen reduction reaction (ORR) and enhanced mass transport properties to generate useful currents at meaningful fuel cell voltages for automotive applications. The long standing durability shortcomings of such Pt-free-based catalysts for ORR have also been addressed to some degree. However, despite five decades of intensive research to replace Pt at the cathode of PEMFCs, no consensus has yet been reached regarding the exact identity of the catalytic species at work in these catalysts. We believe that the lack of consensus in this matter stems from the large variety of species, metal-containing or not, that are produced during the pyrolysis step necessary to obtain these catalysts. In this work, it will be shown that only a few of these species exhibit catalytic activity for the ORR in Fe/N/C. Identifying these species and acquiring knowledge about their molecular structure is now essential for improving their activity and durability.
Acknowledgements
This work was supported by the MDEIE program of the “Gouvernement du Québec” and NSERC. The authors are grateful to Dr. B. Stansfield and E. Proietti for their valuable suggestions and comments. The authors are also grateful for the use of X3-B at the National Synchrotron Light Source, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract No. DE-AC02-98CH10886.
Footnotes
†Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
References
- 1.Gasteiger HA, Kocha SS, Sompalli B, Wagner FT. Applied Catalysis B: Environmental. 2005;56:9–35. [Google Scholar]
- 2.Wagner FT, Lakshmanan B, Mathias MF. J. Phys. Chem. Lett. 2010;1:2204–2219. [Google Scholar]
- 3.James BD, Kalinoski JA, Baum KN. 2010 http://www1.eere.energy.gov/hydrogenandfuelcells/pdfs/dti_80kwW _fc_system_cost_analysis_report_2010.pdf.
- 4.US Department of Energy http://www1.eere.energy.gov/hydrogenandfuelcells/mypp/pdfs/fuel_cells.pdf.
- 5.Jasinski R. Nature (London) 1964;201:1212. [Google Scholar]
- 6.Jahnke H, Schönborn M, Zimmermann G. Topics in Current Chemistry. 1976;61:133–182. doi: 10.1007/BFb0046059. [DOI] [PubMed] [Google Scholar]
- 7.van Veen JAR, van Baar JF, Kroese KJ. J. Chem. Soc., Faraday Trans. 1981;77:2827–2843. [Google Scholar]
- 8.Gupta SL, Tryk D, Bae I, Aldred W, Yeager EB. Journal of Applied Electrochemistry. 1989;19:19–27. [Google Scholar]
- 9.Dodelet JP. In: N4-Macrocyclic Metal Complexes. Zagal JH, Bedioui F, Dodelet JP, editors. Springer; New York: 2006. pp. 83–147. [Google Scholar]
- 10.Garsuch A, Bonakdarpour A, Liu G, Yang R, Dahn JR. In: Handbook of Fuel Cells – Fundamentals, Technology and Applications. Vielstich W, Lamm A, Gasteiger HA, editors. Vol. 5. Wiley; Chichester, UK: 2009. pp. 71–90. [Google Scholar]
- 11.Johnston CM, Piela P, Zelenay P. In: Handbook of Fuel Cells – Fundamentals, Technology and Applications. Vielstich W, Lamm A, Gasteiger HA, editors. Vol. 5. Wiley; Chichester, UK: 2009. pp. 48–70. [Google Scholar]
- 12.Kramm UI, Bogdanoff P, Fiechter S. In: Encyclopedia of Sustainability Science and Technology. Meyers RA, editor. Springer Science+Business Media, LLC; New York: 2012. forthcoming. [Google Scholar]
- 13.Lefèvre M, Proietti E, Jaouen F, Dodelet JP. Science. 2009;324:71–74. doi: 10.1126/science.1170051. [DOI] [PubMed] [Google Scholar]
- 14.Wu G, More KL, Johnston CM, Zelenay P. Science. 2011;332:443–447. doi: 10.1126/science.1200832. [DOI] [PubMed] [Google Scholar]
- 15.Proietti E, Jaouen F, Lefèvre M, Larouche N, Tian J, Herranz J, Dodelet JP. Nature Communications. 2011;2:416. doi: 10.1038/ncomms1427. l DOI: 10.1038/ncomms1427. [DOI] [PubMed] [Google Scholar]
- 16.Faubert G, Dodelet R. Côté, J. P., Lefèvre M, Bertrand P. Electrochimica Acta. 1999;44:2589–2603. [Google Scholar]
- 17.Jaouen F, Marcotte S, Dodelet JP, Lindbergh G. J. Phys. Chem. B. 2003;107:1376–1386. [Google Scholar]
- 18.Jaouen F, Lefèvre M, Dodelet JP, Cai M. J. Phys. Chem. B. 2006;110:5553–5558. doi: 10.1021/jp057135h. [DOI] [PubMed] [Google Scholar]
- 19.Wiesener K. Electrochimica Acta. 1986;31:1073–1078. [Google Scholar]
- 20.Franke R, Ohms D, Wiesener K. Journal of Electroanalytical Chemistry. 1989;260:63–73. [Google Scholar]
- 21.Liu G, Li X, Ganesan P, Popov BN. Applied Catalysis B: Environmental. 2009;93:156–165. [Google Scholar]
- 22.Biddinger EJ, Ozkan US. J. Phys. Chem. C. 2010;114:15306–15314. [Google Scholar]
- 23.van Veen JAR, Colijn HA, van Baar JF. Electrochimica Acta. 1988;33:801–804. [Google Scholar]
- 24.Bouwkamp-Wijnoltz AL, Visscher W, van Veen JAR, Boellaard E, Van der Kraan AM, Tang SC. J. Phys. Chem. B. 2002;106:12993–13001. [Google Scholar]
- 25.Bron M, Radnik J, Fieber-Erdmann M, Bogdanoff P, Fiechter S. Journal of Electroanalytical Chemistry. 2002;535:113–119. [Google Scholar]
- 26.Schulenburg H, Stankov S, Schünemann V, Radnik J, Dorbandt I, Fiechter S, Bogdanoff P, Tributsch H. J. Phys. Chem. B. 2003;107:9034–9041. [Google Scholar]
- 27.Charreteur F, Jaouen F, Ruggeri S, Dodelet JP. Electrochimica Acta. 2008;53:2925–2938. [Google Scholar]
- 28.Herranz J, Jaouen F, lefèvre M, Kramm UI, Proietti E, Dodelet JP, Bogdanoff P, Fiechter S, Abs-Wurmbach I, Bertrand P, Arruda T, Mukerjee S. J. Phys. Chem. C. 2011;115:16087–16097. doi: 10.1021/jp2042526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boulatov R. In: N4-Macrocyclic Metal Complexes. Zagal JH, Bedioui F, Dodelet JP, editors. Springer; New York: 2006. pp. 1–36. [Google Scholar]
- 30.Popovic DM, Leontyev IV, Beech D.Gg, Stuchebrukhov AA. Proteins. 2010;78:2691–2698. doi: 10.1002/prot.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lefèvre M, Dodelet JP, Bertrand P. J. Phys. Chem. B. 2000;104:11238–11247. doi: 10.1021/jp0529265. [DOI] [PubMed] [Google Scholar]
- 32.Lefèvre M, Dodelet JP, Bertrand P. J. Phys. Chem. B. 2002;106:8705–8713. doi: 10.1021/jp0529265. [DOI] [PubMed] [Google Scholar]
- 33.Joyner RW, van Veen JAR, Sachtler WMH. J. Chem. Soc., Farraday Trans. 1982;78:1021–1028. [Google Scholar]
- 34.Bouwkamp-Wijnoltz AL, Visscher W, van Veen JAR, Tang SC. Electrochimica Acta. 1999;45:379–386. [Google Scholar]
- 35.Yuasa M, Yamaguchi A, Itsuki H, Tanaka K, Yamamoto M, Oyaizu K. Chemical Materials. 2005;17:4278–4281. [Google Scholar]
- 36.O'Neill DG, Atanasoski R, Schmoeckel AK, Vernstrom GD, O'Brien DP, Jain M, Wood TE. Material Matters. 2006;1:17–19. [Google Scholar]
- 37.Maruyama J, Abe I. J. Electrochem. Soc. 2007;154:B297–B304. [Google Scholar]
- 38.Scherson DA, Gupta SL, Fierro C, Yeager EB, Kordesch ME, Eldridge J, Hoffman RW, Blue J. Electrochim. Acta. 1983;28:1205–1209. [Google Scholar]
- 39.Scherson DA, Yao SB, Yeager EB, Eldridge J, Kordesch ME, Hoffman RW. J. Phys. Chem. 1983;87:932–943. [Google Scholar]
- 40.Blomquist J, Lang H, Larsson R, Widelöv A. J. Chem. Soc., Faraday Trans. 1992;88:2007–2011. [Google Scholar]
- 41.Koslowski UI, Abs-Wurmbach I, Fiechter S, Bogdanoff P. J. Phys. Chem. C. 2008;112:15356–15366. [Google Scholar]
- 42.Kramm UI, Abs-Wurmbach I, Herrmann-Geppert I, Radnik J, Fiechter S, Bogdanoff P. Journal of the Electrochemical Society. 2011;158:B69–B78. [Google Scholar]
- 43.Kramm UI, Herrmann-Geppert I, Bogdanoff P, Fiechter S. Journal of Physical Chemistry C. 2011;115:23417–23427. [Google Scholar]
- 44.Hatchard TD, Harlow JE, Cullen KM, Dunlap RA, Dahn J. J. Electrochem. Soc. 2012;159:B121–B125. [Google Scholar]
- 45.Jaouen F, Serventi AM, Lefèvre M, Dodelet JP, Bertrand P. J. Phys. Chem. C. 2007;111:5971–5976. [Google Scholar]
- 46.Melendres CA. J. Phys. Chem. 1980;84:1936–1939. [Google Scholar]
- 47.Walker FA, Simonis U. In: Encyclopedia of Inorganic Chemistry: Iron Porphyrin Chemistry. Scott RA, editor. Wiley Online Library; 2006. DOI: 10.1002/0470862106.ia111. [Google Scholar]
- 48.uzmann E, Nath A, Chechersky V, Li S, ei Y, Chen X, Li J, Homonnay Z, Gal M, Garg VK, Klencsar Z, Vertes A. Hyperfine interactions. 2002;139/140:631–639. [Google Scholar]
- 49.Schulz CE, Hu C, Scheidt WR. Hyperfine Interactions. 2006;170:55–60. doi: 10.1007/s10751-006-9474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sams JR, Tsin TB. In: The Porphyrins Volume IV - Physical Chemistry, Part B. Dolphin D, editor. Academic Press; London: 1979. pp. 457–460. [Google Scholar]
- 51.Collman JP, Gagne RR, Reed CA, Halbert TR, Lang G, Robinson WT. J. Am. Chem. Soc. 1975;97:1427–1439. doi: 10.1021/ja00839a026. [DOI] [PubMed] [Google Scholar]
- 52.Collman JP, Hoard JL, Kim N, Lang G, Reed CR. J. Am. Chem. Soc. 1975;97:2676–2681. doi: 10.1021/ja00843a015. [DOI] [PubMed] [Google Scholar]
- 53.Borsa DM, Boerma DO. Hyperfine Interactions. 2003;151/152:31–48. [Google Scholar]
- 54.Meng H, Jaouen F, proietti E, Lefèvre M, Dodelet JP. Electrochimica Acta. 2010;55:6450–6461. [Google Scholar]
- 55.Schaaf P. Progress in Materials Science. 2002;47:1–161. [Google Scholar]
- 56.Jaouen F, Dodelet JP. Electrochimica Acta. 2007;52:5975–5984. [Google Scholar]
- 57.Lefèvre M, Dodelet JP. Electrochimica Acta. 2008;53:8269–8276. [Google Scholar]
- 58.Jaouen F, Dodelet JP. J. Phys. Chem. C. 2007;111:5963–5970. [Google Scholar]
- 59.Zagal JH, Paez MA, Silva JF. In: N4-Macrocyclic Metal Complexes. Zagal JH, Bedioui F, Dodelet JP, editors. Springer; New York: 2006. pp. 41–50. [Google Scholar]
- 60.Herranz J, Jaouen F, Dodelet JP. ECS Trans. 2009;25:117–128. [Google Scholar]
- 61.Hasegawa M, Yagi T. J. Alloys Comp. 2005;403:131–142. [Google Scholar]
- 62.Rechenbach D, Jacobs H. J. Alloys Comp. 1996;235:15–22. [Google Scholar]
- 63.Schmithals G. PhD thesis. Freie Universität Berlin; 2005. Structural and electrochemical characterization of the catalytic sites in non-noble metal catalysts fort he oxygen reduction (original language: German) http://www.diss.fu-berlin.de/diss/receive/FUDISS_thesis_000000001920. 212 pages. [Google Scholar]
- 64.Grennwood NN, Gibb TC. Mössbauer spectroscopy. Chapman and Hall Ltd.; London: 1971. pp. 240–258. ISBN: 0-412-10710-4. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.