

Abstract
RAD51 is important for restarting stalled replication forks and for repairing DNA double-strand breaks (DSBs) through a pathway called homology-directed repair (HDR). However, analysis of the consequences of specific RAD51 mutants has been difficult since they are toxic. Here we report on the dominant effects of two human RAD51 mutants defective for ATP binding (K133A) or ATP hydrolysis (K133R) expressed in mouse embryonic stem (ES) cells that also expressed normal mouse RAD51 from the other chromosome. These cells were defective for restarting stalled replication forks and repairing breaks. They were also hypersensitive to camptothecin, a genotoxin that generates breaks specifically at the replication fork. In addition, these cells exhibited a wide range of structural chromosomal changes that included multiple breakpoints within the same chromosome. Thus, ATP binding and hydrolysis are essential for chromosomal maintenance. Fusion of RAD51 to a fluorescent tag (enhanced green fluorescent protein [eGFP]) allowed visualization of these proteins at sites of replication and repair. We found very low levels of mutant protein present at these sites compared to normal protein, suggesting that low levels of mutant protein were sufficient for disruption of RAD51 activity and generation of chromosomal rearrangements.
INTRODUCTION
Replication fork maintenance is essential for maintaining the structural integrity of chromosomes, and replication defects were proposed to cause copy number variation (CNV) and complex genomic rearrangements (CGRs) (8, 35). CNV is a natural change in the number of copies of one or more sections of DNA in the genome of a population ranging from one to thousands of kilobases and occurs between repeat segments (22, 23). CNV accounts for about 12% of the human genome and is important for murine (11) and primate (5, 46) evolution. CGRs cause genomic disorders and cancer in humans (13, 23, 74, 75). They consist of at least two rearrangements with multiple breakpoints all closely aligned in the same chromosomal region, suggesting they derived from a single event. The genesis of structural chromosomal rearrangements is not known, yet compromised replication fork progression may evoke novel error-prone mechanisms, such as fork stalling and template switching (FoSTeS) (32) or microhomology-mediated break-induced replication (MMBIR) (22), which suppress broken replication forks but at the risk of structurally rearranging the genome. FoSTeS and MMBIR are not well understood at a mechanistic level but were proposed after sequencing of CGRs found in human genomic disorders and cancers. The sequence information showed that these events involved multiple chromosome segments and small levels of homology at some of the rearrangement junctions (22, 23). Therefore, defects in replication fork maintenance may promote CGRs and CNV.
RAD51 is a RecA recombinase that is essential for replication fork maintenance. RAD51 performs two functions to suppress broken replication forks. First, RAD51 enables replication restart when a replication fork encounters DNA damage or reduced nucleotide pools (49). Without restart, the fork may collapse to form a double-strand break (DSB). Second, RAD51 repairs DNA DSBs that occur after exposure to some genotoxins or at broken replication forks through a pathway called homology-directed repair (HDR). To promote restart and HDR, RAD51 forms a filament on the 3′ end of single-stranded DNA (ssDNA). This end then invades and anneals to a homologous template most often provided by the sister chromatid during replication (55). Strand invasion is highly regulated to suppress inappropriate annealing to a nonallelic repeat that could cause a structural rearrangement. Thus, proper RAD51 function is essential for genome maintenance, while faulty RAD51 function has the potential to be mutagenic.
RAD51 contains conserved Walker A and B motifs that bind ATP. In the Walker A motif, a highly conserved lysine (K133 in mammalian RAD51) is important for binding ATP. The K133A mutant causes a defect in ATP binding, while the K133R mutant causes a defect in ATP hydrolysis. K133A, but not K133R, disables the ability of RAD51 to induce topological changes in duplex DNA in an ATP-dependent manner. Thus, the K133A mutant is biochemically more severe than the K133R mutant. These mutants do not affect RAD51-protein interactions and have little effect on the equilibrium binding affinity of RAD51 to ssDNA (19). However, ATP binding is critical for assembly and stabilization of a catalytically active nucleoprotein filament, while ATP hydrolysis promotes filament disassembly and RAD51 release from DNA (12). Therefore, K133 is essential for proper RAD51 function.
A full understanding of the RAD51 K133 mutant proteins (e.g., RAD51K133A) in a biological context is lacking because of their toxic effects; consequently, their stable expression in cells is problematic. Cells are sensitive to changes to the dose of wild-type (WT) and mutant RAD51, limiting most RAD51 studies to in vitro assays or short-time-course, tissue culture assays without the opportunity to observe the long-term effects of defective RAD51 expressed at physiological levels (29). As expected, ectopic expression of RAD51K133A and RAD51K133R proved extremely toxic to cells, making it difficult to generate euploid cells that stably express these mutant proteins at physiological levels, especially the K133A mutant (19, 31, 43, 54, 64). For the K133A mutant, only hybridoma cells that stably express a FLAG-tagged hRAD51K133A have been generated (33, 54). Ectopic expression of either RAD51 mutant inhibited HDR, intrachromosomal recombination, and gene targeting, despite the presence of endogenous wild-type RAD51. In addition, ATP binding and hydrolysis influence the repair of I-SceI-induced DSBs (31, 64, 65). Ectopic expression of RAD51 with either the K133A or K133R mutant decreased HDR but increased single-strand annealing (SSA). Thus, ATP binding and hydrolysis support HDR but suppress SSA. For cells that stably expressed RAD51K133R, the gene conversion tracts were longer than in cells stably expressing RAD51WT (65), supporting biochemical observations that the K133R mutant stabilizes the RAD51 nucleoprotein filament (60). Thus, these RAD51 mutants impart a dominant-negative phenotype since wild-type RAD51 was still expressed. However, we do not know the consequences of the long-term expression of the K133 mutants and their impact on replication fork maintenance and chromosomal stability.
To bridge this gap, we knocked in Homo sapiens RAD51 (HsRAD51) cDNAs (WT, K133A, and K133R) adjacent to the endogenous Mus musculus (MmRad51) promoter on chromosome 2 in two lines of mouse embryonic stem (ES) cells (Lex1 and AB2.2) using our previously described high-throughput knock-in methodology (27). These cells still express wild-type MmRad51 from the unaltered chromosome. We found that expression of either K133 mutant caused a similar phenotype, suggesting faulty replication. Both mutants exhibited impaired cellular proliferation, reduced replication fork restart, reduced sister chromatid exchanges (SCEs), hypersensitivity to camptothecin (CPT), enhanced ATR/CHK1 response, elevated chromosomal instability, and loss of heterozygosity (LOH), with HsRAD51K133A causing a more severe phenotype. To distinguish MmRAD51 from HsRAD51, we expressed eGFP-HsRAD51 cDNA (WT, K133A, and K133R) using the same knock-in approach and found that MmRAD51 and eGFP-HsRAD51WT were efficiently located to the nascent DNA strand during replication and formed foci after CPT exposure. However, only very low levels of the HsRAD51 K133 mutant proteins localized to these sites compared to MmRAD51 and eGFP-HsRAD51WT, even though they were present at equal levels in the chromatin fraction of cells. These differences suggest that low levels of K133 mutant protein are sufficient to disrupt replication restart and HDR and cause chromosomal abnormalities.
MATERIALS AND METHODS
Generation of MmRad51 targeting vector.
The mouse MmRad51 targeting vector (illustrated in Fig. 1A and B) was constructed by cloning the amplified left (5′) and right (3′) arms using genomic DNA extracted from AB2.2 ES cells by high-fidelity PCR using iProof DNA polymerase (Bio-Rad Laboratories). After amplification, the left arm (3.9 kb) was digested with SalI and NotI and cloned into a pKO plasmid backbone (24) cut with XhoI and NotI. The right arm (3.0kb) was cut with XhoI and NotI and cloned into the same backbone adjacent to the left arm digested with SalI and NotI. The region from exons 2 to 4 of MmRad51 was deleted in the targeting vector, and then the floxed SAβgeo-HPRT minigene (expressing hypoxanthine phosphoribosyltransferase) was cloned into unique SfiI sites as described previously (24).
Fig 1.

High-throughput knock-in at MmRad51. (A) The SAβgeo-miniHPRT selection cassette. SAβgeo is a fusion of β-galactosidase and neomycin phosphotransferase genes with a splice acceptor (SA) instead of a promoter so that cells will survive G418 selection only if a promoter/splice donor is trapped (20). miniHPRT (hypoxanthine phosphoribosyltransferase) (24, 51) is commonly used to select for transfected ES cells previously mutated for Hprt and offers an advantage over other selection cassettes in that one may select for either its presence in HAT (hypoxanthine, aminopterin, thymidine) or its absence in 6-thioguanine (6-TG). miniHPRT has a phosphoglycerate kinase (PGK) promoter (1) and an intron that separates the coding sequence from exons 1 and 2 to exons 3 to 8. An RE mutant lox (2) is in the intron. In addition, another RE mutant lox is 5′ to SAβgeo. An FRT is at the 3′ end of miniHPRT. (B) Replacement of MmRad51 exons 2 to 4 (exon 2 is the first coding exon) with the SAβgeo-miniHPRT selection cassette. PCR is used to screen G418-HAT-resistant ES cell clones for gene targeting (clone 2) using primers H13F and SR3. (C) Removal of SAβgeo, the 5′ half of miniHPRT and a RE mutant lox by Cre-mediated recombination selected by 6-TG resistance. Correct recombinant clones were identified by PCR screening using primers RCF1 and AS2. (D) Knock-in of wild-type HsRAD51 cDNA by Cre-mediated recombination. The Cre-mediated targeting vector contains the 5′ half of miniHPRT, an LE mutant lox, an FRT, and the cDNA (with a Kozak ATG [30], shown as a red box, and SV40 polyadenylation sequences, shown as a dark blue box) with endogenous splicing sequences (light blue area shown in panel B). HAT was used to select for miniHPRT restoration, with correct integration screened by PCR (top panel with black half-arrow primers; lanes 1 and 10 are negative controls, and lanes 2 to 11 are all knock-ins) and RT-PCR (bottom panel with red half-arrow primers; lanes 1 to 4 are negative controls, and lanes 5 and 6 are knock-ins). (E) Removal of bacterial backbone, miniHPRT, one FRT, and the wild-type loxP by FLP recombinase. Screen TG-resistant clones by PCR (black half-arrow primers; lanes 1 and 7 are negative controls, and lanes 2 to 6 have had the sequences removed).
PCR conditions for both left and right arms.
All reactions were performed in 25-μl reaction volumes containing 5 μl of 5× iProof HF buffer, 0.5 μl of 10 mmol/liter deoxynucleoside triphosphates (dNTPs), 0.75 μl of 4-μmol/liter forward or reverse primers (described below), 100 ng of genomic DNA, and 0.25 μl of iProof DNA polymerase. There was 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, a 64.7- to 70.2°C gradient for 1 min, and 72°C for 1 min 30s, followed by 1 cycle of 72°C for 10 min. The left (5′) arm primers were Rad51KiLA for (5′-CACACTCGAGTCCCCTCTACGCTGAGAAGCCGGAGAAAG-3′) and Rad51KiLA rev (5′-CACAGCGGCCGCAGGCCACTAAGGCCAGAACTGCAGCTGGCCCTCCCTATCCAC-3′), and the right (3′) arm primers were Rad51KiRA for (5′-CACAGCGGCCGCAGGCCTGCGTGGCCGGATTATAGGAATGTCAGCTTCTCATAGAC-3′) and Rad51KiRA rev (5′-CACAGTCGACGGTACTGGTTAGTTCATAATGTTGTTCCA-3′).
Gene targeting MmRad51 in ES cells.
The K133A and K133R mutants were introduced into mouse Lex1 and AB2.2 ES cells by our high-throughput knock-in system (Fig. 1) (24, 27). For this study, both lines were mutated at the hypoxanthine phosphoribosyltransferase (Hprt) gene (51). A random retroviral insertion mutated the Hprt gene in AB2.2 cells, while an Hprt targeting vector mutated Hprt exon 3 in the Lex1 cells by a standardized protocol (16) that includes the use of the puroΔtk double-selection cassette (10) with subsequent removal of puroΔtk. Lex1 and AB2.2 ES cells were maintained in M15 medium: high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 15% fetal bovine serum, 100 μmol/liter β-mercaptoethanol, 1 mmol/liter glutamine, 3 mg/ml penicillin, 5 mg/ml streptomycin, and 1,000 U/ml ESGRO leukemia inhibitory factor (LIF). These cells were grown on plates with 2.5 × 106 gamma-irradiated murine embryonic fibroblasts (mitotically inactive feeders) seeded on 0.1% gelatin-coated plastic (for about 1 h). The cells then were expanded in 5% CO2 in a 37°C incubator at atmospheric O2. A total of 5 × 106 cells in 800 μl Dulbecco's phosphate-buffered saline (DPBS) were electroporated with 5 μg of MmRad51 targeting vector linearized with PacI. The condition for electroporation was as follows: 230 V, 500 μF (Bio-Rad Gene Pulsar apparatus). Next, 250 μl of electroporation mixture was seeded onto a 10-cm feeder plate. The next day, final concentrations of 1× HAT (1 mM sodium hypoxanthine, 4 μM aminopterin, 160 μM thymidine) and 2× G418 (360 μg active ingredient/ml) were added. After 8 to 10 days of HAT and G418 selection, resistant colonies were picked and seeded onto a 96-well feeder plate and maintained in HAT and G418 selection. These colonies were replica plated, and then one was frozen and the other was used to isolate DNA by the genomic DNA microextraction method (50) for screening correctly targeted ES cells by genomic PCR. The PCR screen used the same reaction conditions as described for generating the targeting vector but detects a 3-kb fragment derived from amplifying a region from outside the right arm of the targeting construct in the endogenous genomic DNA.
The PCR conditions were 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 67°C for 1 min, and 72°C for 1 min 30s, followed by 1 cycle of 72°C for 10 min. The following primers (Fig. 1) were used: H13F (in miniHPRT), 5′-GTAAATGAAAAAATTCTCTTAAACCACAGCACTATTGAG-3′; and SR3 (outside the right arm), 5′-AGCCAGGTATAGTCTCAAAGGAATCTGCAATCC-3′.
Cre-mediated deletion of SAβgeo and 5′ miniHPRT.
SAβgeo and the 5′ half of miniHPRT were deleted using Cre recombinase (Fig. 1C). Targeted ES cells were expanded in 1× HAT medium to remove HPRT-negative cells that survive due to cross feeding. HAT selection was removed 2 days before transfection and cultured in 1× HT (1 mM sodium hypoxanthine and 160 μM thymidine); a total of 5 × 106 cells in 800 μl DPBS were electroporated with 10 μg of pPGKcrepA using a Bio-Rad Gene Pulsar at 230 V, 500 μF. After transfection, 200 μl of electroporated cells were seeded onto a 10-cm feeder plate without selection for 2 to 4 days to allow for miniHPRT removal and time for degradation of HPRT mRNA and protein. Next, 4 × 104 cells were seeded onto a 10-cm feeder plate in 10 μM 6-thioguanine (6-TG) selection medium. 6-TG-resistant colonies were picked about 8 to 10 days later. Cells were expanded in 10 μM TG selection medium and replica plated. One plate was frozen, and the other was used to isolate genomic DNA by the microextraction method (50). Cre-mediated deletion was confirmed by PCR using the same reaction conditions described for generating the targeting vector. A 1.4-kb fragment was detected.
The PCR conditions were 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 64°C for 1 min, and 72°C for 30 s, followed by 1 cycle of 72°C for 10 min. The following PCR primers were used: RCF1 (in RAD51 intron 1), 5′-GTGCTGAATCTCCTAGAACTG-3′; and AS2 (in exon cluster 3 to 8 of miniHPRT), 5′-TGTCCCCTGTTGACTGGTCA-3′.
Generation of CMKIPs.
The knock-in plasmids were generated using the Cre-mediated knock-in plasmid (CMKIP) backbone (27). This backbone contains the 5′ half of miniHPRT, an LE mutant lox, simian virus 40 (SV40) polyadenylation sequences, and an FLP recombination target gene (FRT) (Fig. 1D). Two different sequences were cloned into CMKIP to make the HsRAD51 knock-in plasmids. The first sequence was the mouse intron 1-exon 2 splice acceptor (light blue in Fig. 1D). The second sequence was HsRAD51 cDNA (red rectangle in Fig. 1D). Both sequences were amplified by PCR under the same reaction conditions described for generation of the targeting vector. The template was either 100 ng of AB2.2 genomic DNA for the splice acceptor or 50 ng of plasmid for the cDNA.
The PCR conditions for the splice acceptor were 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 57.6°C to 70.2°C for 1 min, and 72°C for 20 s, followed by 1 cycle of 72°C for 10 min. The following PCR primers for the splice acceptor were used: mRAD51 5′g Ki for (5′-GCGCGGCGCGCCTCAAAGGTATGTCGGGAAC-3′) and mRAD51 5′g Ki rev (5′-GCGCTAGCCATGGCTAAAAAACACAG-3′).
The PCR conditions for the cDNA were 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 67°C for 1 min, and 72°C for 30 s, followed by 1 cycle of 72°C for 10 min. The following PCR primers were used for the cDNA: Kozak hRAD51 cDNA for (5′-CGCCACCATGGCAATGCAGCT-3′) and hRAD51 cDNA rev (5′-CACACTCGAGTCAGTCTTTGGCATCGCCC-3′).
The HsRAD51WT knock-in plasmid was constructed by a three-way ligation, including (i) the splice acceptor (digested with AscI and NcoI), (ii) the cDNA (digested with NcoI and SalI), and (iii) the CMKIP backbone (digested with AscI and SalI). Next, the HsRAD51K133A and HsRAD51K133R cDNAs were PCR amplified from plasmids (gifts from Patrick Sung, Yale University), as described for the HsRAD51WT cDNA, and then cloned into the HsRAD51WT CMKIP after releasing of HsRAD51WT with NcoI and SalI. All knock-in plasmids were sequenced to ensure fidelity.
Knock-in of HsRAD51 and eGFP-HsRAD51 cDNAs.
A pool of 6-TG-resistant cells generated after Cre-mediated deletion of SAβgeo and 5′ miniHPRT were expanded without 6-TG for 4 days. Next, 5 × 106 cells in 800 μl DPBS were electroporated with 20 μg of HsRAD51 CMKIP (or control) and 10 μg of pPGKcrepA using a Bio-Rad Gene Pulsar at 230 V, 500 μF. An aliquot of these cells was seeded onto a 10-cm feeder plate with murine embryonic fibroblasts mutated for Hprt (250 μl for HsRAD51WT, KS, and eGFP-HsRAD51WT cDNAs and 400 μl for human HsRAD51K133A, HsRAD51K133R, eGFP-HsRAD51K133A, and eGFP-HsRAD51K133R cDNAs). After transfection (48 h), a final concentration of 1× HAT was added to the medium. For HsRAD51, 8 to 10 days later, 4 to 8 colonies were picked, pooled, and expanded, while for eGFP-HsRAD51, single colonies were expanded. Cells were maintained in HAT to eliminate HPRT-negative cells that survive by cross-feeding. In addition, 16 individual colonies for HsRAD51WT and 1 to 4 colonies for the eGFP-HsRAD51 cDNAs were picked and expanded in HAT medium to test the knock-in efficiency. This plate was replica plated, one plate was frozen, and the other was used to isolate genomic DNA using the microextraction method (50) for screening targeted clones by genomic PCR. PCRs were performed under the same reaction conditions described for generation of the targeting vector to amplify a 500-bp fragment.
The PCR conditions were 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 64°C for 1 min, and 72°C for 45 s, followed by 1 cycle of 72°C for 10 min. The following PCR primers were used: RCF1 (in mouse RAD51 intron 1; 5′-GTGCTGAATCTCCTAGAACTG-3′) and 285R (in human RAD51 cDNA; 5′-GCCTTTGGTGGAATTCAGTTGC-3′).
Further proof of correct knock-in was obtained by RT-PCR using primers that detected a mouse-human fusion transcript from MmRad51 exon 1 and SV40 polyadenylation sequences. This amplified a 1.2-kb band for HsRAD51 knock-in and a 200-bp band for KS knockin. As a control for RNA integrity, Brca2 exons 26 and 27 were amplified (300 bp).
The reverse transcription-PCR (RT-PCR) conditions for RAD51 were 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 67°C for 1 min, and 72°C for 45 s, followed by 1 cycle of 72°C for 10 min. The following RT-PCR primers were used; for RAD51: R51E1F (mouse exon 1; 5′-AGGCGCTCCCACAGGTGTGG-3′) and SV40R (5′-ATATCGGTCCGTGATCATAATCAGCCATAC-3′).
The RT-PCR conditions for Brca2 were 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 67°C for 1 min, and 72°C for 30 s, followed by 1 cycle of 72°C for 10 min. The following RT-PCR primers were used for Brca2: PICTFF1 (5′-AAAAGAATTCTCCACACCGAACAAAGACCC 3′) and 3226R (5′-AAAAGCGGCCGCCTAGCTCCGTGGCGGCTGAAAA-3′).
Removal of the plasmid backbone and miniHPRT by FLP recombinase.
HAT-resistant HsRAD51 wild-type cDNA clones were pooled and expanded in HAT selection medium for 1 to 2 weeks and then expanded without HAT for 2 days. Next, 5 × 106 cells in 800 μl DPBS were electroporated with 5 μg of FLP recombinase expression vector (pCAGGS-FLPe; from A. Francis Stewart, Max Planck Institute) using a Bio-Rad Gene Pulsar at 230 V, 500 μF. 6-TG-resistant colonies were processed as described for Cre-mediated deletion of SAβgeo and the 5′ half of miniHPRT. Flippase-mediated deletion of the plasmid backbone and miniHPRT was confirmed by PCR using the same reaction conditions described for generation of the targeting vector to amplify a 1.5-kb fragment.
The PCR conditions were 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 64°C for 1 min, and 72°C for 45 s, followed by 1 cycle of 72°C for 10 min. The following PCR primers were used: CMKIF (in hRAD51 cDNA; 5′-TCACGGTTAGAGCAGTGTG-3′) and FDR2 (in mouse Rad51 intron 4; 5′-GACTAATGCCTATGATCTC-3′).
Quantification of the number and surface area of HAT-resistant knock-in colonies.
HAT-resistant colonies were counted 12 days after addition of HAT to select for knock-in cells. Colony number counting was repeated for three transfections. Next, 7 to 8 colonies from each plate were photographed at a magnification of ×40 on days 3, 6, and 8 and printed onto paper. To quantify colony surface area, the colony was cut out of the paper and weighed in grams. The average of grams for colonies from each plate was considered a representation of the colony surface area.
Pools of colonies were observed. For Lex1 cells, these included HsRAD51WT (4), HsRAD51KS (4), HsRAD51K133A (12), and HsRAD51K133R (8). For AB2.2 cells, these included HsRAD51WT (8), HsRAD51KS (8), HsRAD51K133A (8), and HsRAD51K133R (8).
shRNA knockdown.
Sense and antisense primers (see below) were inserted into pSUPER.retro.puro (Origoengine) to construct a short hairpin RNA (shRNA) plasmid that knocks down MmRad51 and eGFP-HsRAD51 (4). An shRNA plasmid that specifically knocks down MmRad51, but not eGFP-HsRAD51 (TRCN0000012658), was purchased from Open Biosystems. To knock down MmRad51 and eGFP-HsRAD51, these shRNA expression plasmids (5 μg) were electroporated into cells (2.5 × 106 cells in 800 μl DPBS; Bio-Rad Gene Pulsar at 230 V, 500 μF) that express eGFP-HsRAD51WT, eGFP-HsRAD51K133A, or HsRAD51K133R. After transfection, 250 μl of the electroporated cells was seeded onto a 6-well feeder plate in M15. The next day, puromycin was added to a final concentration of 3 μg/ml and selection was maintained for 8 to 10 days. Puromycin-resistant colonies were counted, and two colonies expressing eGFP-HsRAD51WT were picked and expanded for Western blot analysis to determine the levels of MmRAD51 and eGFP-HsRAD51WT using rabbit polyclonal anti-HsRAD51 (H92, 1:1,000; Santa Cruz Biotechnology). An anti-β-actin antibody was used as a control.
The following primers were used for shRNA that targets both MmRad51 and HsRAD51: sense primer 5′-GATCCCCGGGAATTAGTGAAGCCAAATTCAAGAGATTTGGCTTCACTAATTCCCTTTTTC-3′ and antisense primer 5′-CCCGGGAATTAGTGAAGCCAAATTCAAGAGATTTGGCTTCACTAATTCCCTTTTTCTCGA-3′.
Microfiber analysis.
DNA fiber analysis was performed as described previously (49). Mouse ES cells were pulse-labeled with 25 μM chlorodeoxyuridine (CldU) for 20 min and washed three times with medium followed by 2 mM hydroxyurea (HU) for 2 h. Next, HU was removed, and the cells were washed three times with fresh medium, and pulse-labeled with 250 μM iododeoxyuridine (IdU) for 20 min. Labeled cells were harvested, and DNA fiber spreads were prepared as described previously (57). Fibers were then fixed in methanol-acetic acid (3:1) and subsequently air dried. Slides were treated with 2.5 M HCl to denature the DNA fibers for 75 to 80 min and washed with PBS two times before being blocked with 1% bovine serum albumin (BSA) plus 0.1% Tween 20 for 1 h. Slides were incubated with primary antibodies against CldU [rat anti bromodeoxyuridine (anti-BrdU); BU1/75(ICR1), 1:500; Abcam] and IdU (mouse anti-BrdU B44, 1:200) for 1.5 h at room temperature. Slides were then fixed with 4% paraformaldehyde and washed with PBS three times. Alexa Fluor 555-conjugated goat anti-rat IgG (1:500; Molecular Probes) and Alexa Fluor 488-conjugated goat anti-mouse IgG (1:400; Molecular Probes) were applied to slides and incubated for 2 h at room temperature. Slides were washed and mounted in Fluoroshield (Sigma). Fibers were examined using a Zeiss fluorescence microscope (Axioplan2).
SCE assay.
The sister chromatid exchange (SCE) assay was performed as described previously (16).
Cell survival assay in response to CPT and ICRF-193.
The fraction of cells that survive exposure to either CPT or ICRF-193 was determined as described previously (39). These cells include those that can proliferate and those that are senescent.
Colony-forming assay.
On day 0, 2,000 cells were seeded onto the wells of a 6-well plate. The next day, the medium was changed with camptothecin (CPT) at the doses shown in Fig. 4. Eight days later, colonies were stained with 0.2% methylene blue (in 70% ethanol) and counted.
Fig 4.

Dose response to topoisomerase inhibitors. In Lex1 cells, the dose response to camptothecin (CPT) (A) and ICRF-193 (B) were determined using a cell survival assay that counts both replicating and nonreplicating cells (39). Lex1 unaltered cells are the control. These graphs represent the average of three experiments. (C) Comparison of eGFP-tagged and untagged proteins using a colony-forming assay for AB2.2 cells that express WT (left), KA (middle), or KR (right). Shown is the average of three experiments. The control (+/−) is the same for all three graphs and can be used as a standard for comparison.
Phosphorylated CHK1 analysis.
Cells were incubated in medium containing 100 nM CPT for 16 h. Cells were harvested at the indicated time points after release from CPT. Western blot analysis was performed by a standard procedure. Mouse anti-Chk1 (1:1,000; Santa Cruz) and rabbit anti-phospho-CHK1 (1:1,000; Cell Signaling) were used for detection. β-Actin was used as a loading control (1:10,000; Sigma).
Three-color FISH.
The methods for three-color fluorescent in situ hybridization (FISH) have been described previously (28).
SKY.
Frozen mouse ES cells were resuspended in fresh M15 in LIF. Next, the cells were centrifuged and plated onto gelatin-coated 6-well plates. The cells were then harvested for metaphase spreads (MPSs) after 48 to 72 h of culture.
Spectral karyotyping (SKY) was performed as described earlier (56). For details, see www.riedlab.nci.nih.gov. Metaphase cell suspensions were dropped onto clean glass slides inside a humidity chamber. Slides were then hybridized with the combinatorially labeled whole-chromosome painting probes (Applied Spectral Imaging, Inc., Carlsbad, CA), and metaphase images were captured using the Applied Spectral Imaging interferometer (Applied Spectral Imaging, Inc.) on an epifluorescence microscope (Zeiss). SKY karyotypes were then analyzed with SKY view v 1.62 software (Applied Spectral Imaging, Inc.); a total of 11 metaphases were captured and analyzed using the nomenclature approved by the International Committee on Standardized Genetic Nomenclature for Mice (http://www.informatics.jax.org).
LOH.
Loss-of-heterozygosity (LOH) analysis was performed as described previously (26, 37), with minor modifications. ES cells were grown in 1× HAT for at least 4 days, followed by growth in 1× HT for 2 days and then no selection for 1 day. Cells were counted and seeded at 2 × 105 cells/10-cm feeder plate (3 plates) and selected in 10 μM 6-thioguanine (6-TG). For plating efficiency, 2,000 cells were seeded on two wells of a 6-well feeder plate. Eight days after plating, the number of 6-TG-resistant colonies was counted. Some of the 6-TG-resistant clones were expanded, and genomic DNA was extracted to test by PCR for the presence of miniHPRT and the junction between mouse genomic sequences and HsRAD51. (The primer locations are shown in Fig. 7C.) We tested 8 WT, 7 KS, 10 K133A, and 10 K133R colonies. A fragment of FancB was also amplified as a control for DNA quality.
Fig 7.

Analysis of LOH in AB2.2 cells that express HsRAD51 (WT, K133A, and K133R). (A) The percentage of colonies that survived in 6-TG. The numbers of 6-TG-resistant colonies from three replica plates are as follows: KS, 12, 14, and 13; HsRAD51WT, 16, 12, and 10; HsRAD51K133A, 2,038, 2,457, and 2,558; and HsRAD51K133R, 911, 951, and 951. The numbers of colonies that grew without selection are as follows: KS, 55,800; HsRAD51WT, 95,800; HsRAD51K133A, 78,000; and HsRAD51K133R, 57,600. Statistics (t test): 1 versus 2, 0.062; 1 versus 3, 0.0047; 1 versus 4, 0.0002; 2 versus 3, 0.0046; 2 versus 4, 0.0002; and 3 versus 4, 0.0167. (B) Location of primers for PCR analysis to detect the presence/absence of the HsRAD51 cDNA (blue half-arrows) and miniHPRT (red half-arrows). The total numbers of 6-TG resistant colonies observed are as follows: WT, 8; KS, 7; KA, 10; and KR, 10. (C) The percentage of metaphase spreads that contain the number of chromosomes shown on the x axis. The inset shows the total percentages of metaphase spreads with <40, 40, and >40 chromosomes, showing there is more chromosome loss than gain. The total numbers of metaphase spreads observed are as follows: WT, 147; KA, 144; and KR, 129.
The conditions for PCR analysis to detect miniHPRT (1.35-kb PCR product) were 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 65°C for 1 min, and 72°C for 45 s, followed by 1 cycle of 72°C for 10 min. The primers were Hf1 (5′-GATGAACCAGGTATGACCTTG-3′) and AS2 (5′-TGTCCCCTGTTGACTGGTCA-3′).
The conditions for PCR analysis to detect the junction between mouse genomic sequences and HsRAD51 cDNA (550-bp PCR product) were 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 65°C for 1 min, and 72°C for 20 s, followed by 1 cycle of 72°C for 10 min. The primers were mR51int1F (5′-GTGCTGAATCTCCTAGAACTG-3′) and HsR51-285R (5′-CACAGTCGACTCAGCCTTTGGTGGAATTCAGTTGC-3′).
The conditions for PCR analysis to detect Fancb (820-bp PCR product) were 1 cycle of 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 65°C for 1 min, and 72°C for 30 s, followed by 1 cycle of 72°C for 10 min. The primers were FBF1 (5′-TGCTGCTTTGCATATTGCAG-3′) and FBR1 (5′-TGTGTTCTCATCCAATGCATG-3′).
Detection of MmRAD51 and eGFP-HsRad51 in the chromatin fraction.
Protein fractionation was performed as described previously (44) with minor modifications. To remove the cytoplasmic fraction, 1 × 107 cells were resuspended in buffer A (10 mM HEPES [pH 7.5], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT], 0.05% NP-40, and protease inhibitor cocktail [Roche]), incubated for 10 min on ice, and centrifuged at 1,300 × g for 5 min at 4°C. To remove the nuclear soluble fraction, the pellet was resuspended in buffer B (10 mM HEPES [pH 7.0], 1.5 mM MgCl2, 10 mM KCl, 0.34 M sucrose, 10% glycerol, 150 mM NaCl, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], and protease inhibitor cocktail [Roche]), incubated for 40 min on ice, and centrifuged at 1,700 × g for 5 min at 4°C. To extract the chromatin-bound protein fraction, the pellet was resuspended in buffer C (20 mM Tris-Cl [pH 8.1], 2 mM EDTA [pH 8.0], 500 mM NaCl, 0.1% SDS, 1% Triton X-100, and protease inhibitor cocktail [Roche]), sonicated, treated with micrococcal nuclease for 10 min at 37°C, and then centrifuged at 13,000 rpm for 5 min at 4°C. After centrifugation, the supernatant containing the released chromatin-bound proteins was used for Western blotting with rabbit polyclonal anti-HsRAD51 (GTX100469, 1:1,000; GeneTex) and rabbit polyclonal anti-histone H3 (C-16, 1:1,000; Santa Cruz Biotechnology).
iPOND.
For isolation of proteins on nascent DNA (iPOND), mouse ES cells were incubated for 10 min with 10 μM 5′-ethynyl-2′-deoxyuridine (EdU; Invitrogen). EdU-labeled cells were washed once with EdU-free medium and then treated with 100 nM CPT. After labeling and CPT exposure, cells were cross-linked with 1% formaldehyde–PBS for 20 min at room temperature, quenched with 0.125 M glycine, and washed three times with PBS. Collected cell pellets were resuspended in 0.25% Triton X–PBS solution and incubated at room temperature for 30 min for permeabilization. Permeabilized cells were washed once with 0.5% BSA–PBS and once with PBS. Cells were incubated in Click reaction buffer [10 μM biotin-azide (Jena Bioscience), 10 mM Na ascorbate (Sigma), 2 μM Cu(II) sulfate] for 1 h at room temperature. Dimethyl sulfoxide (DMSO) was used instead of biotin-azide as a negative-control sample. Cells were washed once with 0.5% BSA–PBS and once with PBS. Cells were resuspended in lysis buffer (1% SDS, 50 mM Tris-HCl [pH 8.0], 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml PMSF), sonicated, and then centrifuged for 10 min at 14,000 rpm. Supernatants were collected and diluted 1:1 (vol/vol) with PBS containing protease inhibitor (1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml PMSF). Streptavidin-agarose beads (Novagen) were washed twice in lysis buffer and once in PBS containing protease inhibitor. Washed beads were incubated with samples for 16 h at 4°C. Captured proteins were eluted by boiling for 25 min at 95°C. Eluted proteins were detected by Western blotting.
Detection of eGFP-HsRAD51 foci by observing green fluorescence.
To visualize eGFP-HsRAD51, 2 × 104 cells were seeded on 4-well chamber slides (Nalge Nunc International Corp., Naperville, IL). After 48 h, cells were processed as described below in the dark chamber to prevent GFP from being bleached. Fluorescence was visualized with a Zeiss fluorescence microscope (Axioplan2).
Detection of MmRAD51 foci by anti-HsRAD51 antibody.
Immunostaining was performed as described previously (28). First, 2 × 104 cells were seeded on the chamber slide (Nalge Nunc International, Corp., Naperville, IL). After 48 h, cells were treated with 1 μM CPT for 3 h. Following CPT treatment, cells were rinsed with PBS and fixed with 2% formaldehyde (diluted in PBS) by incubation at room temperature for 10 min, washed three times with PBS, and permeabilized with 0.5% Triton in PBS for 10 min at room temperature. Following permeabilization, the cells were washed three times in PBS and then blocked with blocking buffer (4% nonfat milk in PBS) for 1 to 2 h at room temperature. After being washed, anti-HsRAD51 antibody (H92, 1:200 dilution; Santa Cruz Biotechnology) in 4% nonfat milk in PBS was applied to cells, and the slides were incubated overnight at 4°C. Upon completion of antibody incubation, the cells were washed with PBS three times and then incubated with blocking buffer containing a fluorescently labeled secondary antibody, the Alexa Fluor 594 (AB′)2 fragment of goat anti-rabbit IgG (H+L) (Molecular Probes) at a working dilution of 1:1,000 for 1 h at room temperature. After four to five rinses with PBS, 4′,6-diamidino-2-phenylindole (DAPI)-containing mounting medium (Vectashield mounting medium; Vector Laboratories, Inc.) was added to the culture slide, which was mounted for imaging under a Zeiss fluorescence microscope (Axioplan2).
RESULTS
Knock-in of HsRAD51 cDNAs adjacent to the endogenous MmRad51 promoter.
Deletion of RAD51 is lethal to proliferating vertebrate cells (62); therefore, biological data concerning MmRad51 are mostly confined to early null embryos that exhibited chromosomal loss, hypersensitivity to gamma radiation, and apoptosis (34). Complicating matters further, isolation of cells that stably express some HsRAD51 mutants like the K133A mutant has been difficult due to toxicity (19, 31, 43, 54, 64). To overcome these obstacles, we used a highly efficient four-step knock-in procedure (27) to integrate HsRAD51 cDNAs (WT, K133A, and K133R) adjacent to the endogenous MmRad51 promoter. These cDNAs were transfected into two ES cell lines, Lex1 and AB2.2, both derived from 129S5 mice (18) and mutated for the hypoxanthine phosphoribosyltransferase (Hprt) gene (described in Materials and Methods). In addition, the eGFP gene was fused to the N terminus of HsRAD51 (WT, K133A, and K133R) since previous reports showed GFP-HsRAD51 was capable of rescuing deletion of endogenous RAD51 (73). GFP-RAD51 was also shown to coimmunoprecipitate with BRCA2 (73), endogenous RAD51 (19, 48), and XRCC3 (19). GFP-RAD51 forms S-phase nuclear foci, ionizing radiation-induced nuclear foci (19), nuclear foci that colocalize with RAD52 and RAD54 (17), and nuclear foci that were disrupted by coexpression of BRCA2 polypeptide spanning the BRC repeats 3 and 4 (48). Furthermore, GFP-RAD51 restored the cells' capacity to repair DNA DSBs, as indicated by comet assay measurements (19). These eGFP-HsRAD51 fusion cDNAs were also integrated adjacent to the MmRad51 promoter in AB2.2 cells by the same four-step knock-in procedure.
Steps 1 and 2 of the knock-in procedure generated the parental clones used to knock-in all cDNAs. For step 1, MmRad51 exons 2 to 4 were replaced with a floxed selection cassette, SAβgeo-miniHPRT (Fig. 1A). SAβgeo is a fusion of β-galactosidase and neomycin phosphotransferase genes with a splice acceptor (SA) instead of a promoter so that cells will survive G418 selection only if a promoter/splice donor is trapped (20). miniHPRT is the HPRT minigene (24, 51) used for selection in HAT (hypoxanthine, aminopterin, thymidine) or in 6-TG (6-thioguanine) for the presence or absence of expression, respectively. RE mutant lox (2) was placed 5′ to SAβgeo and in the intron of miniHPRT. FRT was placed 3′ to miniHPRT (Fig. 1A, orange arrow). After transfection, G418-HAT-resistant clones were isolated and screened by PCR for targeted clones. About 20 to 30% of G418-HAT-resistant clones were verified as targeted by PCR (Fig. 1B). For step 2, SAβgeo and the 5′ half of miniHPRT were removed by transient transfection of a Cre recombinase expression vector and selection in 6-TG. About 1,000 6-TG-resistant colonies were recovered, and all 6-TG-resistant clones tested were shown by PCR to have correctly deleted the sequences in between the RE mutant lox sites (Fig. 1C). Steps 1 and 2 need to be done only once since they generated the parental clone of Lex1 and AB2.2 cells used for the remaining steps.
Step 3 integrates the HsRAD51 cDNAs adjacent to the MmRad51 promoter by cotransfecting a Cre-mediated targeting vector and a Cre recombinase expression vector into the parental clones of ES cells. The Cre-mediated targeting vector contains a fusion of MmRad51 exon 2 splice acceptor plus HsRAD51 cDNA sequences and the 5′ half of miniHPRT with an LE mutant lox (Fig. 1D, green-blue arrow). After Cre-mediated recombination, the RE mutant lox and the LE mutant lox fused to form an RE LE mutant lox (blue arrow) and an unaltered loxP (green arrow). In addition, the cDNA sequences are juxtaposed to the endogenous MmRad51 promoter and miniHPRT is restored. Cells were selected in HAT, and all HAT-resistant colonies were verified to contain the HsRAD51 cDNA sequences adjacent to the MmRAD51 promoter by PCR (Fig. 1D). This is a powerful selection step since the only efficient way to achieve HAT resistance is through Cre-mediated integration of the Cre-mediated targeting vector.
Step 4 is optional and removes the plasmid backbone, loxP, and miniHPRT by transient transfection of an FLP recombinase expression cassette, leaving behind the cDNA, RE LE mutant lox, and an FRT. Thus, other knock-in locations can be established since the cells no longer contain miniHPRT. In addition, this system was designed to remove loxP and leave the RE LE mutant lox since the double mutant lox is a poorer substrate for Cre recombinase (3). The RE LE mutant lox was left behind since it will not greatly interfere with future knock-ins at the second location. After transient FLP recombinase transfection, ∼100 TG-resistant colonies were recovered and all clones tested by PCR were correct (Fig. 1E). Thus, these genetically altered ES cells can be directly compared since there is only one cDNA copy and all cDNAs are in the same location and expressed by the endogenous MmRad51 promoter.
The K133A and K133R mutants impaired clonal expansion.
Previous experiments showed that expression of either HsRAD51K133A or HsRAD51K133R was toxic to cells, especially the K133A mutant (19, 31, 43, 54, 64). Therefore, we were unsure if HAT-resistant colonies would be recovered after expression of these mutant proteins. We found diminished levels of HAT-resistant colonies for cells transfected with HsRAD51K133A (P = 0.0138, t test) and HsRAD51K133R (P = 0.0019) compared to HsRAD51WT, with the K133A mutant being more severely affected (P = 0.0423) (Fig. 2A). Similarly, recovery of HAT-resistant colonies was diminished for cells transfected with eGFP-HsRAD51K133A (P = 0.00035) and eGFP-HsRAD51K133R (P = 0.00048) compared to eGFP-HsRAD51WT, with the K133A mutant being more severe (P = 0.019). A direct comparison of the K133 mutant proteins with and without eGFP suggests the eGFP tag did not significantly impact toxicity. Thus, expression of the HsRAD51 mutant proteins diminished the recovery of HAT-resistant colonies, suggesting they were toxic.
Fig 2.

Expression of HsRAD51 K133 mutants reduces colony number and size. (A) Ratios of HAT-resistant colonies were compared for HsRAD51WT (WT) and the negative control (KS) to HsRAD51K133A (KA) and HsRAD51K133R (KR) with and without eGFP. Shown is the total number of HAT-resistant colonies observed from four experiments for the untagged Cre-mediated knock-in plasmids. Colony numbers are listed in sequential order from the first to the fourth experiment, and some of plasmids were not examined (shown by “ND” [not done]) for some experiments: HsRAD51WT, 337, 262, 106, and 208; KS, 337, ND, ND, and 123; HsRAD51K133A, 20, 26, ND, and ND; HsRAD51K133R, 84, 100, 18, and ND. The total numbers of HAT-resistant colonies observed from three experiments for the eGFP-tagged Cre-mediated knock-in plasmids are as follows: eGFP-HsRAD51WT, 52, 227, and 261; eGFP-HsRAD51K133A, 7, 19, and 20; and eGFP-HsRAD51K133R, 15, 75, and 76. AB2.2 cells are shown. (B) Surface area of HAT-resistant colonies after knock-in. The total numbers of colonies observed are as follows: HsRAD51WT, 8; KS, 8; HsRAD51K133A, 7; and HsRAD51K133R, 8. AB2.2 cells are shown. (C) RT-PCR on cells expressing untagged HsRAD51. AB2.2 cells are the control (Con). (D) Western analysis of nuclear extracts using anti-RAD51 antibody on cells expressing eGFP-HsRAD51. AB2.2 cells are the control. (E) Nonspecific eGFP-HsRAD51 and MmRad51 shRNA knockdown. The total colony numbers are shown. (F) MmRad51-specific shRNA knockdown. Three experiments were performed. The number of colonies that grow after stable transfection of shRNA plasmid is divided by the number of colonies that grow after stable transfection of empty vector for cells that express the following: eGFP-HsRAD51WT, 98/188, 103/180, and 99/176; eGFP-HsRAD51K133A, 14/118, 17/144, and 15/106; and eGFP-HsRAD51K133R, 15/110, 15/118, and 16/114. AB2.2 cells are shown. (G) Western analysis after no transfection (NT) or transfection with empty vector (EV) or shRNA for MmRad51. Two clones were observed for cells that express eGFP-HsRAD51WT in AB2.2 cells. α-HsRAD51, anti-HsRAD51; α-β-actin, anti-β-actin.
To estimate the impact of toxicity on clonal expansion, the colony surface area was measured for the HAT-resistant clones as they were cultured in selection over 8 days. The HsRAD51 K133 mutants generated small colonies compared to controls (HsRAD51WT or KS) (Fig. 2B) (P < 0.0001). Thus, colony growth appeared to be slower for cells expressing the K133 mutant proteins compared to those expressing wild-type HsRAD51.
HAT-resistant colonies were picked and expanded. Cells were easily expanded from individual colonies for HsRAD51WT and KS controls. It was also possible to expand individual colonies that expressed HsRAD51K133R. Cells from these colonies proliferated very slowly for ∼3 passages, but then expanded the same as controls. In contrast, we were unable to expand cells derived from individual colonies that expressed HsRAD51K133A. Therefore, multiple colonies were pooled into the same well of a 24-well plate with the expectation that increasing cell density would improve recovery. Although proliferation was initially very slow, cells from the pooled colonies adapted after ∼5 passages and began to proliferate similar to controls.
It is possible that the mutant clones adapted simply by turning off the mutant cDNA, rendering them useless for further experimentation. Therefore, we tested the level of HsRAD51 expression by RT-PCR using primers specific to the human cDNA (primers shown in Fig. 1E and described in Materials and Methods). We found HsRAD51WT was expressed at similar levels to HsRAD51K133A and HsRAD51K133R, although this was not quantitative, and it was not possible to compare to endogenous MmRad51 mRNA (Fig. 2C). However, these results show that cell adaptation was not due to turning off expression of the K133 mutant cDNAs. Initially, we wished to test expression directly by Western analysis, but were unable to distinguish HsRAD51 from MmRAD51 since they share 99% amino acid identity (42, 59) and anti-RAD51 antibodies cross-react with both proteins. Therefore, we tested the eGFP fusion proteins by Western blotting and found similar levels of eGFP-HsRAD51 (WT, K133A, K133R) and endogenous MmRAD51 in the nuclear extract (Fig. 2D). Thus, the eGFP-tagged proteins were expressed in cells at physiological levels (suggesting the untagged proteins were also expressed at physiological levels since their toxicity was about the same, judging by recovery of HAT-resistant clones). Therefore, these cells express the HsRAD51 K133 mutant proteins at physiological levels and were analyzed for their phenotype.
eGFP-HsRAD51WT, but not the K133 mutants, maintained cell viability after MmRad51 depletion.
Gene knockouts showed that MmRAD51 deletion is cell lethal (34, 70). In keeping with these results, we found stable integration of an shRNA expression vector that depletes both MmRad51 and HsRAD51 (4) resulted in very few colonies for cells expressing eGFP-HsRAD51WT, eGFP-HsRAD51K133A, and eGFP-HsRAD51K133R, further demonstrating that RAD51 deletion is cell lethal (Fig. 2E).
We used shRNA knockdown that specifically depletes MmRad51 to test for the ability of eGFP-HsRAD51 (WT, K133A, and K133R) to maintain cell viability. Recovery of cells that express the HsRAD51 K133 mutants is difficult, especially HsRAD51K133A (19, 31, 43, 54, 64). Therefore, we predicted expression of eGFP-HsRAD51WT, but not the tagged K133 mutants, would efficiently rescue MmRad51 depletion. We found that stable expression of an shRNA vector that depletes only MmRad51 resulted in many more colonies for cells that expressed eGFP-HsRAD51WT compared to eGFP-HsRAD51K133A (P < 0.0001, t test) or eGFP-HsRAD51K133R (P = 0.0005). This confirmed that eGFP-HsRAD51WT, but not the K133 mutants, is able to compensate for MmRad51 depletion when expressed at physiological levels (Fig. 2F). Western blot analysis showed the MmRad51-specific shRNA effectively depleted MmRAD51, but not eGFP-HsRAD51WT (Fig. 2G). Thus, eGFP-HsRAD51WT, but not the K133 mutants, was able to rescue cell lethality in the absence of MmRAD51. In addition, the eGFP tag did not interfere with cell rescue.
Analysis of replication fork restart and SCE.
RAD51 promotes stalled replication fork restart after brief exposure to hydroxyurea (HU) (49). Therefore, we tested the K133 mutants for their impact on replication fork restart by using microfiber analysis. Cells were cultured in CldU (chlorodeoxyuridine) for 20 min to label the nascent replication strand and then exposed to 2 mM HU for 2 h to stall replication forks (Fig. 3A). Finally, cells were cultured in IdU (iododeoxyuridine) for 20 min to label post-HU replication, either from the restart or a new origin. If replication restarts after HU exposure, then the fiber will be red (CldU), followed by a short black gap (HU-induced stall), and then green (IdU). However, if replication fails to restart, then the DNA fiber will be labeled only red, while a fiber generated from a new replication origin will be labeled only green. Compared to AB2.2 control cells, we observed more stalled forks for cells expressing the K133A (P < 0.0001) and K133R (P < 0.0001) mutant proteins, with the K133A mutant proving more severe (P < 0.0001). Thus, we conclude that ATP binding and hydrolysis are important for replication fork restart.
Fig 3.
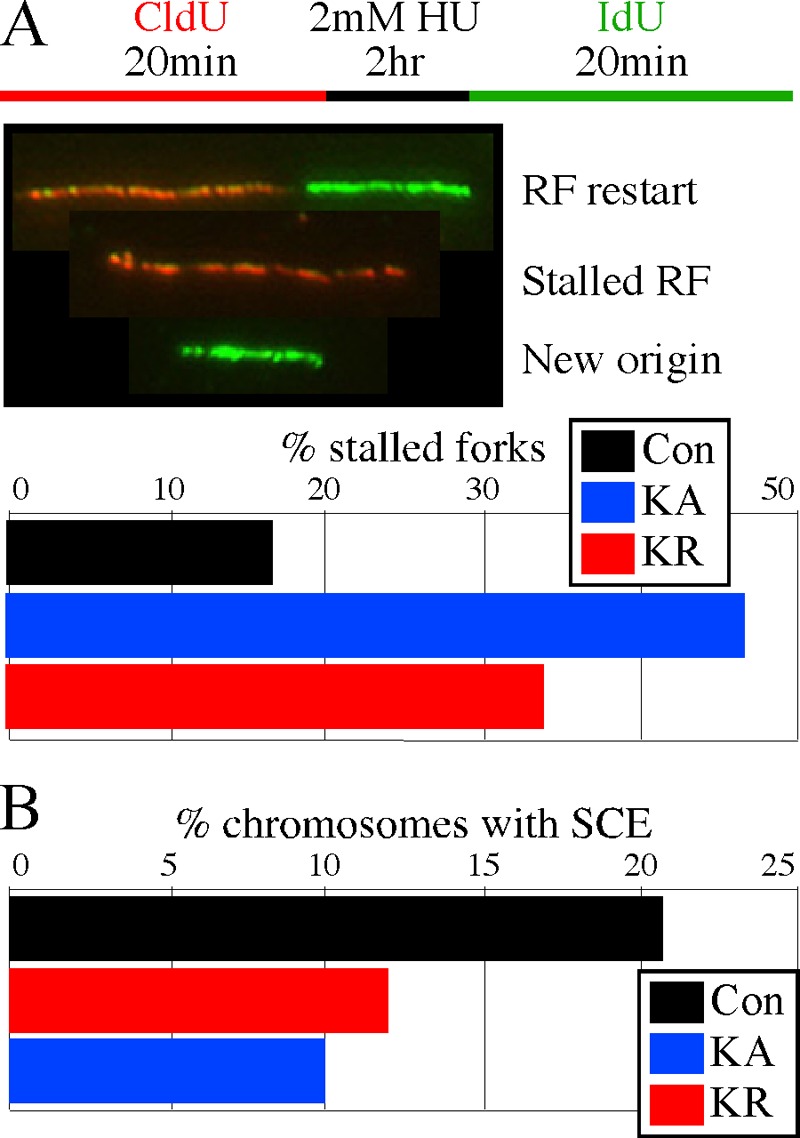
Replication fork restart and sister chromatid exchanges. Control is unaltered AB2.2 cells. (A) Microfiber analysis to observe replication fork restart. The experimental design is shown at the top, and DNA fibers shown in the middle illustrate replication forks (RFs) that have restarted (red-black-green), stalled (red), or initiated from a new origin (green). Shown is a quantification of the percentage of fibers that were stalled after exposure to 2 mM HU for 2 h (bottom). The numbers of fibers observed that were stalled, restarted, or from a new origin, respectively, are as follows: control, 69, 351, and 5; HsRAD51K133A, 267, 288, and 19; and HsRAD51K133R, 264, 515, and 9. (B) Graph depicting the percentage of chromosomes observed to undergo spontaneous SCEs. The total number of SCEs and total number of chromosomes, respectively, observed are as follows: control, 197 and 945; HsRAD51K133A, 234 and 1,927; and HsRAD51K133R, 153 and 1,572.
RAD51-mediated HDR is important for inducing SCEs (14, 15, 63, 67, 68). SCE is the result of a single strand of DNA at a break invading and recombining with the intact sister chromatid during replication such that duplicated chromosomes are reciprocally exchanged (72). A previous report showed the K133R mutant inhibited spontaneous SCEs and the repair of an induced DSB (64, 65). In agreement, we found cells that expressed the K133A (P < 0.0001, Yates corrected chi-square test) and K133R (P < 0.0001) mutant proteins exhibited about a 50% reduction in SCEs compared to AB2.2 control cells, with the K133A mutant being slightly more severe (P = 0.027) (Fig. 3B). Thus, ATP binding and hydrolysis are important for HDR.
Sensitivity to topoisomerase inhibitors.
A defect in replication fork restart and HDR-mediated SCEs indicate that cells expressing the K133 mutant proteins are defective in overcoming replication-fork-blocking lesions. Therefore, we performed a survival analysis after exposure to the topoisomerase inhibitors camptothecin (CPT) and ICRF-193 (39). CPT is a type I topoisomerase (topo I) poison that stabilizes a ternary complex between topo 1 and double-stranded DNA, thus resulting in DSBs at replication forks (52). ICRF-193 is a type II topoisomerase (topo II) catalytic inhibitor that stabilizes the enzyme to DNA, thereby suppressing DNA decatenation and the decatenation G2 checkpoint response without interfering with DNA synthesis and without causing replication-associated DSBs (although DSBs may occur at mitotic exit) (7). We compared Lex1 control cells and MmRAD51+/− cells to knock-in cells (KS, HsRAD51WT, HsRAD51K133A, and HsRAD51K133R). Cells that expressed the K133 mutants were hypersensitive to CPT compared to cells that expressed HsRAD51WT or compared to the other cells that did not express the K133 mutants (Lex1, MmRAD51+/−, and KS) (Fig. 4A), which shows that defective ATP binding and hydrolysis diminished the repair of DSBs at stalled replication forks. In contrast, we found that HsRAD51K133A, HsRAD51K133R, MmRAD51+/−, and KS cells were resistant to ICRF-193 compared to control Lex1 cells and HsRAD51WT cells (Fig. 4B). This result shows that an MmRAD51 haploinsufficiency caused ICRF-193 resistance that was corrected by HsRAD51WT but not the K133 mutants and suggests that reduction of RAD51-mediated strand annealing ameliorates ICRF-193 toxicity by reducing the formation of DNA structures typically resolved by topo II, such as hemicatenanes and DNA tangles. We previously reported a similar observation with brca2Lex1/Lex2 cells (deleted for Brca2 exon 27), which proved hypersensitive to CPT but resistant to ICRF-193 (38). These observations are consistent since BRCA2 is a RAD51-interacting protein (55) and suggest that HDR is needed to correct CPT-induced damage or topo I depletion but is toxic in the presence of ICRF-193-induced anomalies or topo II depletion.
Next we tested AB2.2 cells that express either eGFP-tagged or untagged HsRAD51WT, HsRAD51K133A, or HsRAD51K133R using a colony-forming assay to specifically look at the impact CPT has on cell proliferation. This experiment also addresses the effect the eGFP tag may have on HsRAD51 function. Cells expressing the K133 mutants were hypersensitive to CPT compared to MmRAD51+/− cells, and the eGFP tag did not diminish the phenotype (Fig. 4C). Thus, the colony-forming assay reproduced the results from the cell survival assay and the eGFP-tagged and untagged K133 mutant proteins are equally defective in correcting CPT-induced damage, indicating the tag does not influence HsRAD51 function.
Analysis of the ATR-CHK1 response.
Considering the specific sensitivity of the K133 mutants to CPT, we examined CHK1 phosphorylation, a hallmark of replication stress. CHK1 is a downstream effector kinase for the ATR response that is phosphorylated in response to impaired replication fork progression to induce a replication checkpoint (47). In Lex1 cells, we found that expression of HsRAD51K133A, but not HsRAD51K133R, increased levels of phosphorylated CHK1 16 h after CPT removal (Fig. 5). Interestingly, in AB2.2 cells, expression of either HsRAD51K133A or HsRAD51K133R increased levels of phosphorylated CHK1 16 h after CPT removal. These observations suggest that expression of K133 mutant proteins resulted in replication fork defects inducing an ATR response.
Fig 5.
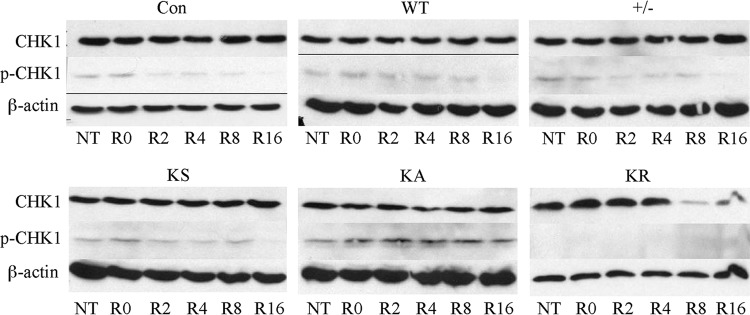
The ATR response as measured by CHK1 phosphorylation. No treatment (NT) is compared to the time in hours after release (R) from camptothecin (100 nM, 16 h). Compared are the levels of total CHK1 to phosphorylated CHK1 (p-CHK1).
Chromosomal abnormalities.
Since replication fork maintenance is important for genome integrity, we determined whether expression of the K133 mutants impacted chromosomal stability by using three-color fluorescence in situ hybridization (FISH) on Lex1 and AB2.2 cells (Table 1). Cells were treated with colcemid (1 μg/ml) for 4 h to arrest cells in metaphase and stained with a telomere probe (green), a major satellite repeat (MSR) probe in the pericentromere (red), and DAPI (blue) (21).
Table 1.
Summary of three-color FISH analysis resultsa
| Cell typeb | Strain | No. of MPSs | Treatment | No. (%) of: |
||||
|---|---|---|---|---|---|---|---|---|
| Chromatids | Isochromatid breaks and fragments | Radials | EPT-1 | EPT-2 | ||||
| Con | Lex1 | 96 | NT | 1 (1.0) | 2 (2) | 0 | 0 | 0 |
| WT | Lex1 | 112 | NT | 1 (0.9) | 1 (0.9) | 0 | 0 | 0 |
| +/− | Lex1 | 112 | NT | 2 (1.7) | 2 (1.7) | 0 | 0 | 0 |
| KS | Lex1 | 106 | NT | 3 (2.8) | 0 | 0 | 0 | 0 |
| KA | Lex1 | 101 | NT | 0 | 62 (28.5) | 0 | 0 | 0 |
| KR | Lex1 | 89 | NT | 7 (7.9) | 6 (6.7) | 0 | 0 | 0 |
| Con | AB2.2 | 168 | NT | 2 (1.2) | 4 (2.4) | 0 | 0 | 0 |
| WT | AB2.2 | 163 | NT | 3 (1.8) | 11 (6.7) | 0 | 0 | 0 |
| +/− | AB2.2 | 163 | NT | 11 (6.7) | 17 (10.4) | 0 | 0 | 0 |
| KS | AB2.2 | 179 | NT | 11 (5.8) | 15 (7.9) | 0 | 0 | 0 |
| KA | AB2.2 | 153 | NT | 27 (17.6) | 29 (19) | 7 (4.6) | 23 (15.6) | 92 (60) |
| KR | AB2.2 | 131 | NT | 17 (12.9) | 28 (21.3) | 6 (4.6) | 0 | 0 |
| Con | AB2.2 | 166 | MMC | 6 (3.6) | 20 (12) | 0 | 0 | 0 |
| KA | AB2.2 | 154 | MMC | 41 (26.6) | 59 (38.3) | 19 (12.3) | 24 (15.5) | 79 (51) |
| KR | AB2.2 | 63 | MMC | 14 (20.6) | 30 (44.1) | 6 (8.8) | 0 | 0 |
| Con | AB2.2 | 178 | CPT | 7 (3.9) | 22 (12.3) | 0 | 0 | 0 |
| KA | AB2.2 | 165 | CPT | 38 (23.1) | 43 (26.2) | 4 (2.4) | 80 (46) | 32 (21) |
| KR | AB2.2 | 130 | CPT | 24 (18.3) | 42 (32.0) | 1 (0.8) | 0 | 0 |
| WT | AB2.2 | 132 | HU | 7 (5.3) | 35 (26.5) | 2 (1.5) | 0 | 0 |
| KA | AB2.2 | 91 | HU | 33 (36.3) | 61 (67) | 7 (7.7) | 75 (82.4) | 3 (3.2) |
| KR | AB2.2 | 133 | HU | 39 (29.3) | 78 (59.4) | 13 (7.7) | 2 (1.5) | 0 |
Shown is a summary of data for MPSs derived from Lex1 and AB2.2 cells. Cells were exposed to no treatment (NT) or were treated with MMC (30 nM for 16 h), CPT (50 nM for 16 h), or HU (1 mM for 2 h). The percentage of MPSs with at least one event is shown in parentheses. Note that some of the metaphase spreads have more than one event.
Con, control; +/−, MmRAD51+/−.
Metaphase spreads derived from Lex 1 cells that expressed HsRAD51K133R showed increased levels of spontaneous chromatid breaks compared to control, HsRAD51WT, MmRAD51+/−, KS, and HsRAD51K133A (Fig. 6A and Table 1) (P < 0.006, Fisher's exact test). Metaphase spreads derived from AB2.2 cells that expressed HsRAD51K133A (P < 0.0001) or HsRAD51K133R (P < 0.0001) showed increased levels of spontaneous chromatid breaks compared to control, HsRAD51WT, MmRAD51+/− and KS (Table 1). Chromatid breaks indicate failed replication fork restart and the subsequent generation of one-ended DSBs, which is in agreement with our observation that the K133 mutants inhibited replication fork restart. In addition, both HsRAD51K133A (P < 0.0001 for both Lex1 and AB2.2 cells) and HsRAD51K133R (Lex1 cells; P = 0.016; AB2.2 cells, P < 0.0001) metaphase spreads showed increased levels of spontaneous isochromatid breaks and fragments compared to control, HsRAD51WT, MmRAD51+/−, and KS metaphase spreads. Isochromatid breaks suggest the presence of failed-strand-exchange intermediates that break both chromatids, which can result in fragments. Unlike the chromatid breaks, the isochromatid breaks and fragments were usually in the pericentromere, a highly repetitive region composed of major satellite repeats (21). This observation is interesting compared to trex2null cells that also exhibited spontaneous isochromatid breaks and fragments in the pericentromere (16). TREX2 is a 3′→5′ exonuclease that removes 3′ mismatches from single-stranded DNA (40). trex2null cells have elevated levels of HDR (16), suggesting TREX2 and HDR perform complementary functions through parallel pathways. Thus, expression of the K133 mutants increased the level of spontaneous breaks in LEX1 and AB2.2 cells.
Fig 6.

Evaluation of metaphase spreads (MPS) for spontaneous chromosomal abnormalities in Lex1 (A to C) and AB2.2 (D to G) cells. (A) Chromatid breaks in the long arm (blue). (B) Isochromatid breaks (left) in the pericentromere (red). (C) Fragments containing the pericentromere (red). (D) Radial. (E) An EPT-1. (F) An EPT-2. (G) SKY analysis. (Row 1) der(11)fusion. This is an EPT. (Row 2) Duplication 1 der(1)t(1:6). This is likely an EPT. (Row 3) der(3)t(3:5). (Row 4) der(5)t(5:3). (Row 5) der(8)t(8:14). This may be an EPT but is difficult to discern. (Row 6) Duplication 2.
For AB2.2 cells, expression of the K133 mutant proteins caused spontaneous chromosomal rearrangements (Table 1). Elevated levels of spontaneous radials were seen in HsRAD51K133A (P = 0.006, Fisher's exact test) and HsRAD51K133R (P = 0.004) metaphase spreads compared to the sum of the controls (con, WT, MmRAD51+/−, and KS) (Fig. 6D). Radials are interesting since they are commonly observed in cells derived from Fanconi anemia (FA) patients (71). FA is a rare autosomal recessive disease caused by a mutation in one of multiple genes involved in a cellular signaling and DNA-processing pathway in response to DNA interstrand cross-linking agents like mitomycin C (MMC). Cells deleted for the FA pathway are extremely sensitive to MMC. The FA pathway interacts with RAD51 since one of the FA proteins, FANCD2, complexes with RAD51 (69) and binds to DNA ends and Holliday junctions in vitro (45). In addition, mouse ES cells deleted for FancB exon 2 (fancbΔex2) showed reduced levels of MmRAD51 foci in response to MMC, suggesting a defect in HDR (28). Therefore, we exposed cells expressing the K133 mutants to MMC (30 nM for 16 h) and found that cells expressing the K133A mutant exhibited a mild increase in radials (P = 0.025; Yates-corrected chi-square test), but not to the extent typically seen in FA-defective ES cells (28). We also exposed these cells to CPT (50 nM, 16 h) and found little change in the level of radials (P > 0.1). The MMC observation suggested a replication fork defect since a DSB is formed when a replication fork collides with a cross-link (6).
We also found abnormal chromosomes with spontaneous extra pericentromeres and telomeres, simply called EPTs in AB2.2 cells expressing HsRAD51K133A but not HsRAD51K133R (Fig. 6E and F) (P < 0.0001). These abnormal chromosomes contained 1 to 5 extra pericentromeres and telomeres. EPTs are unusual and to our knowledge have not been described before. We noticed that some EPTs contained only one terminal pericentromere, while others contained two, classified as EPT-1 (Fig. 6E) and EPT-2 (Fig. 6F), respectively. We found that exposure to MMC (30 nM, 16 h) did not alter the absolute number or proportion of EPT-1s and EPT-2s. However, exposure to CPT (50 nM, 16 h) increased the proportion of EPT-1s compared to EPT-2s (P < 0.0001) without changing the absolute number, suggesting one type of EPT leads to another under the control of type I topoisomerases. A comparison between radials and EPTs showed these are fundamentally different structures formed by different mechanisms since the former is responsive to MMC, while the latter is responsive to CPT. This distinction is interesting since both MMC and CPT block replication forks to cause DSBs, yet these agents still induce different molecular responses with different biological outcomes.
It is possible that EPTs were generated from duplication of a single chromosome or after end joining of broken fragments from multiple chromosomes. Unfortunately, three-color FISH is unable to make these distinctions. Therefore, to better define spontaneous EPTs and observe other rearrangements that evade detection by three-color FISH, we performed spectral karyotyping (SKY) on HsRAD51K133A metaphase spreads (Fig. 6G and Table 2). Previously we showed by SKY that wild-type AB2.2 metaphase spreads exhibited a total of 3 spontaneous chromosomal rearrangements (all deletions) out of 597 chromosomes analyzed from 16 metaphase spreads (9). HsRAD51K133A metaphase spreads exhibited significantly more spontaneous chromosomal rearrangements (P < 0.0001), with a total of 73 out of 462 chromosomes analyzed from 11 metaphase spreads; interestingly, every metaphase spread exhibited multiple rearrangements, showing that HsRAD51K133A is extremely mutagenic (Table 2). Some of the more common spontaneous chromosomal changes are shown in Fig. 6G1 to -G6. EPTs were mostly duplications of chromosome 11 [Fig. 6G1, der(11) fusion] and not the product of fusion between different chromosomes. However, some EPTs may be a duplication of chromosome 1 in addition to a translocation with chromosome 6 [Fig. 6G2, duplication 1 der(1)t(1:6)]. Thus, EPTs contain multiple breakpoints in the same chromosome, suggesting a complicated genomic rearrangement that is likely the product of faulty replication in concert with the CPT data (23). Other rearrangements included fragments (n = 5), deletions (n = 8), duplications (n = 13), a Robertsonian translocation (n = 1), duplications plus derivative translocations (n = 6), derivative translocations (n = 34), and derivative fusions (n = 6). Derivative chromosomes were scored as structurally rearranged chromosomes generated by a rearrangement involving two or more chromosomes (derivative translocations) or as multiple aberrations within the same chromosome (duplications plus derivative translocations and derivative fusions). Additional common observations included duplications (chromosomes 1 and 2), fragments (chromosome 2), and translocations (chromosomes 1:6, 3:5, and 8:11). The SKY analysis showed a large number of complicated genomic rearrangements, indicating that the K133A mutant caused defective replication that could lead to multiple breakpoints in the same chromosome.
Table 2.
Summary of SKY data
| MPS | No. of abnormal chromosomesa: |
||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| del 1 | dup 1 | dup 1 der(1)t(1:6) | Rb(1:10) | del 2 | dup 2 | der(2)t(2:14) | der(3)t(3:5) | der(3)t(3:7) | frag 4 | der(5)t(5:3) | del 7 | der(7)t(7:3) | der(8)t(8:11) | der(8)t(8:14) | del 8 | dup 11 | del 11 | der(11)fusion | der(11)t(11:5) | der(11)t(11:6) | dup 13 | der(13)t(13:?) | der(14)t(14:3) | Marker | |
| 1 | 1 | 1 | 1 | (+1) | 1 | ||||||||||||||||||||
| 2 | 1 | 1 | 1 | 1 | 1 | (+1) | (+1) | ||||||||||||||||||
| 3 | 1 | 1 | 1 | (+1) | |||||||||||||||||||||
| 4 | 1 | (+1) | 1 | 1 | 1 | 1 | 1 | (+1) | |||||||||||||||||
| 5 | (+1) | 1 | 1 | (+1) | 1 | 1 | (+1) | ||||||||||||||||||
| 6 | 2 | (+2) | 1 | 1 | (+1) | 1 | (+1) | (+1) | (+1) | 1 | |||||||||||||||
| 7 | (+2) | 1 | (+1) | (+2) | 1 | (+2) | (+1) | (+1) | 1 | ||||||||||||||||
| 8 | |||||||||||||||||||||||||
| 9 | 1 | 1 | 1 | 1 | 1 | (+1) | 1 | ||||||||||||||||||
| 10 | 1 | 1 | 1 | (+1) | 1 | (+1) | 1 | 1 | |||||||||||||||||
| 11 | 1 | 1 | (+1) | 1 | |||||||||||||||||||||
Numbers in parentheses indicate the number of extra abnormal chromosomes in addition to two normal chromosomes. A value of 1 indicates one of the two chromosomes is abnormal, and a value of 2 indicates that both chromosomes are abnormal.
Metaphase spreads were observed for cells expressing the K133 mutants after exposure to HU to discern the influence replication fork stalling has on the genesis of at least some of the chromosomal abnormalities summarized in Table 1. Cells were exposed to 1 mM HU for 2 h (the same as for the microfiber analysis) and then treated with colcemid for 4 h. Observation of metaphase spreads shows that HU exposure increased the level of chromatid breaks for cells expressing HsRAD51WT (although not significantly for the numbers observed; P = 0.19) and the K133A (P = 0.002) and K133R (P = 0.002) mutants (Table 1). Furthermore, the K133A (P < 0.0001) and K133R (P < 0.0001) mutants increased the level of chromatid breaks compared to HsRAD51WT after HU treatment. Similarly, HU exposure increased the level of isochromatid breaks for cells expressing HsRAD51WT (P < 0.0001) and the K133A (P = 0.02) and K133R (P < 0.0001) mutants, while the K133A (P < 0.0001) and K133R (P < 0.0001) mutants increased the level of isochromatid breaks compared to HsRAD51WT after HU treatment. Once again the isochromatid breaks were predominately in the pericentromere, suggesting the stalled replication forks are especially problematic in this highly repetitive area. Thus, HU-induced stalled replication forks increased chromatid and isochromatid breaks and expression of the K133 mutants further exacerbated the level of these breaks, once again demonstrating the importance for RAD51 K133 ATP binding/hydrolysis in the suppression of breaks during replication fork maintenance.
HU exposure had a substantial impact on the levels of EPT-1s and EPT-2s. Cells expressing HsRAD51WT did not exhibit EPTs with or without HU exposure (Table 1). However, for cells expressing the K133A mutant, HU exposure dramatically increased the levels of EPT-1s (P = 0.0001) but also decreased the levels of EPT-2s (P = 0.0001). HU exposure also slightly increased the level of EPT-1s for cells expressing the K133R mutant (although insignificantly for the numbers tested; P = 0.253), but still at a much lower level than for cells expressing the K133A mutant (P < 0.0001). The impact HU has on EPT levels is similar to that seen for CPT (but HU exposure is more dramatic), suggesting replication fork anomalies are critical for their genesis.
LOH.
Chromosomal instability can lead to loss of heterozygosity (LOH), a potential cancer-causing mechanism (41). In addition, HsRAD51 is implicated in tumor suppression since it associates with a variety of tumor suppressors, including BRCA2 and RAD51C (55). However, direct evidence that ATP binding or hydrolysis suppresses LOH is lacking. Therefore, we tested the knock-in cells harboring miniHPRT (Fig. 1D) for LOH. Using these cells, LOH can be measured by loss of miniHPRT function and survival in 6-thioguanine (6-TG). Compared to HsRAD51WT cells, the KS cells exhibited a mild increase in 6-TG-resistant colonies, suggesting that a RAD51 haploinsufficiency contributed to LOH (Fig. 7A) (P = 0.06, t test). Cells expressing either HsRAD51K133A (P = 0.004) or HsRAD51K133R (P = 0.0002) exhibited elevated levels of 6-TG-resistant cells compared to KS cells, with the K133A mutant being more severe (P = 0.017). Thus, the K133 mutants facilitate LOH.
We next used PCR to examine for the presence or absence of the mutated and wild-type chromosomes. First we tested for retention of miniHPRT in the 6-TG-resistant cells. Retention of miniHPRT would indicate de novo mutations or epigenetic changes, while absence of miniHPRT would indicate mitotic recombination, chromosome loss, or an internal chromosome deletion. The PCR results show that miniHPRT was absent from all 6-TG-resistant clones generated from control, KS, K133A, and K133R clones (8, 7, 10, and 10 clones tested, respectively). To confirm these results, we then tested the same K133 mutant clones for the presence or absence of the HsRAD51 cDNA and again found it was not present. However, all clones retain the wild-type MmRAD51 locus, as seen using PCR with primers that span the knock-in site. These results implicate inappropriate mitotic recombination, chromosome loss, or an internal deletion as the mechanism that causes LOH.
Metaphase spreads were examined for ploidy to determine if mitotic recombination or chromosome loss accounted for LOH. Mitotic recombination seems an unlikely mechanism for LOH since the K133 mutants are defective for HDR (64), while chromosome deletion seems more likely since MmRAD51 deletion caused chromosomal loss likely due to unrepaired DSBs (34) and HsRAD51 depletion or expression of the K133 mutants caused defects in chromosome separation during anaphase (31). Cells that expressed HsRAD51K133A (P < 0.0001, Yates-corrected chi-square test) and HsRAD51K133R (P < 0.0001) exhibited elevated levels of aneuploidy compared to cells that expressed HsRAD51WT (Fig. 7C). Both chromosome loss and gain were observed, but chromosome loss was more common, suggesting a defect in DSB repair contributed to aneuploidy in addition to a defect in chromosomal segregation.
eGFP-HsRAD51 focus formation and chromatin localization after CPT exposure.
To determine the impact the K133 mutants have on protein localization, Western analyses were performed on the fraction of proteins bound to chromatin for cells expressing eGFP-HsRAD51 (WT, K133A, and K133R). Cells were observed without CPT exposure (left), 16 h after 100 μM CPT exposure (middle), or 16 h after CPT removal (right). For all three conditions, the eGFP-HsRAD51 proteins (WT, K133A, and K133R) were found in the chromatin fraction at about the same level relative to MmRAD51 and histone H3 (Fig. 8A). Thus, the K133 mutants did noticeably reduce the level of protein isolated in the chromatin fraction.
Fig 8.
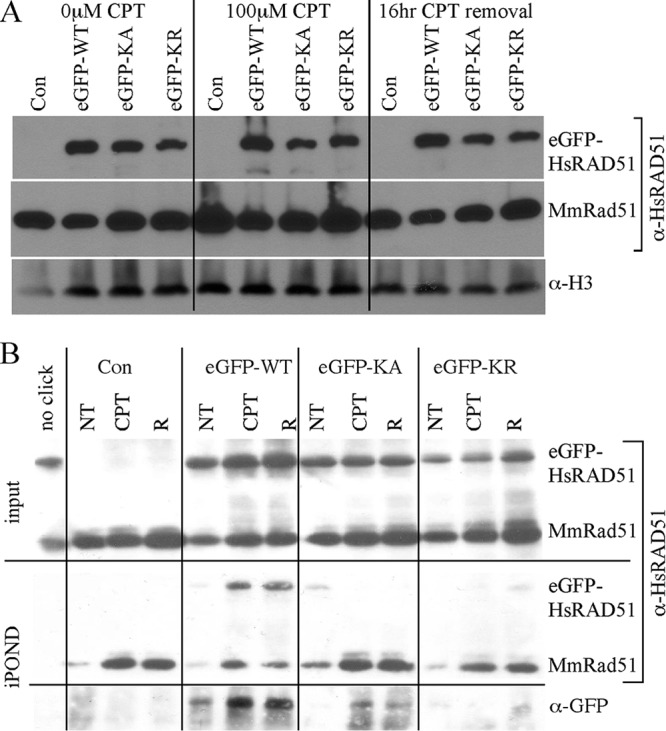
Protein localization. AB2.2 cells that express eGFP-HsRAD51WT, eGFP-HsRAD51K133A, or eGFP-HsRAD51K133R. (A) Chromatin fraction. Screened with anti-HsRAD51 (α-HsRAD51) antibody to detect eGFP-HsRAD51 (top) and MmRAD51 (bottom). Histone H3 is the loading control. α-H3, anti-histone H3. (B) Observation of proteins on or adjacent to the nascent replication strand. Input screened with anti-HsRAD51 antibody (top). Shown are purified nascent DNA-protein complexes screened with anti-HsRAD51 antibody (middle) or with anti-GFP antibody (bottom). NT, no treatment; R, release.
We performed isolation of proteins on nascent DNA (iPOND) (61) to observe proteins bound to the nascent DNA strand during replication. iPOND will also detect proteins on ssDNA immediately adjacent to the nascent strand. Cells were tested without CPT exposure (NT), 16 h after 100 μM CPT exposure, or 12 h after CPT removal (R). The input shows equal levels of MmRAD51 and eGFP-HsRAD51 (WT, K133A, and K133R), similar to the chromatin fraction analysis. The iPOND procedure purified MmRAD51 and eGFP-HsRAD51WT at about equal levels in unexposed cells (Fig. 8B). Exposure to CPT equally increased both proteins. However, iPOND did not purify the K133 mutants at the same level as for MmRAD51; by this analysis, the K133 mutant proteins were barely detectable. Therefore, the K133 mutants did not efficiently associate with newly replicated DNA even though they were detected in the chromatin fraction at normal levels, suggesting a defect specific to replication.
RAD51 forms foci at active sites of repair (49). Therefore, we used fluorescence to detect the formation of eGFP-HsRAD51 foci with and without exposure to CPT (1 μM, 16 h). We found more spontaneous green foci in cells expressing eGFP-HsRAD51WT compared to eGFP-HsRAD51K133A (P < 0.0001, Yates-corrected chi-square test) or eGFP-HsRAD51K133R (P < 0.0001) (Fig. 9A and B). Exposure to CPT (1 μM, 3 h) equally increased green foci for all genotypes, such that cells expressing eGFP-HsRAD51WT still exhibited more green foci than cells expressing eGFP-HsRAD51K133A (P < 0.0001) or eGFP-HsRAD51K133R (P < 0.0001) after exposure to CPT. Thus, the K133 mutants decreased spontaneous and CPT-induced green foci. Foci indicate sites of DSB repair (49), suggesting that the K133 mutants interfered with the repair of CPT-induced DSBs. This observation corroborates a previous study that showed the K133R mutant interfered with repair of an I-SceI-induced DSB (64, 65).
Fig 9.
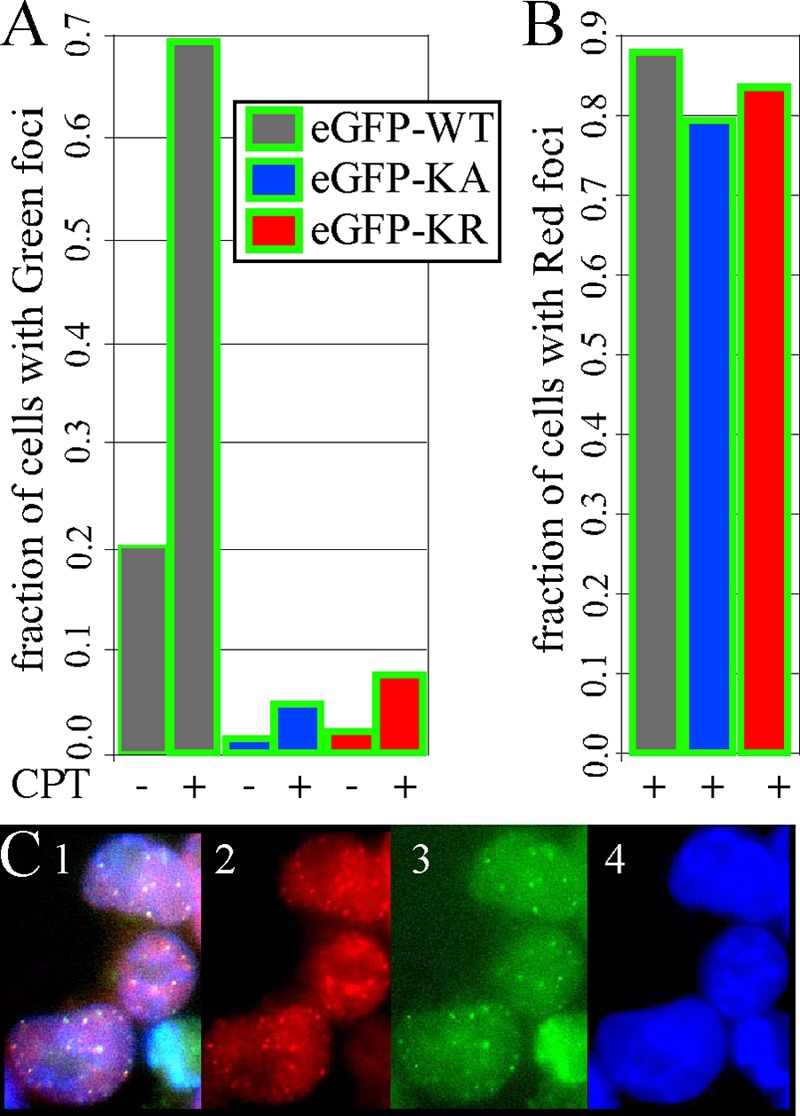
Spontaneous and CPT-induced foci in AB2.2 cells. (A) Graph showing the fraction of cells that exhibit green foci before and after 3 h of exposure to 1 μM CPT. The total numbers of cells observed before and after CPT, respectively, are as follows: eGFP-WT, 162 and 154; eGFP-KA, 112 and 87; and eGFP-KR, 199 and 253. (B) Graph showing the fraction of cells that exhibit red foci after 3 h of exposure to 1 μM CPT. The same cells were observed as for the green foci shown in panel A. (C) Cells expressing eGFP-HsRAD51WT after 3 h of exposure to 1 μM CPT. Panel 1, merge; panel 2, anti-HsRAD51 antibody; panel 3, green fluorescence; panel 4, DAPI.
Next, we measured the combination of MmRAD51 and eGFP-HsRAD51 foci after exposure to CPT by immunofluorescence using anti-RAD51 antibody that cross-reacts with both mouse and human proteins. By using a secondary antibody with a red fluorophore for immunofluorescence, it was possible to compare and contrast these foci with those emitting green fluorescence. Cells expressing eGFP-HsRAD51WT exhibited spontaneous and CPT-induced red foci in similar numbers to green foci (Fig. 9C). Furthermore, most of the red and green foci colocalized, indicating that MmRAD51 and eGFP-HsRAD51WT associated with each other to form a mixed filament in accordance with coimmunoprecipitation studies (19). Interestingly, there was the same level of red foci in cells expressing eGFP-HsRAD51WT compared to eGFP-HsRAD51K133A (P = 0.18) and eGFP-HsRAD51K133R (P = 0.41). This is surprising since the K133 mutant cells did not efficiently form green foci. Thus, our observation that cells expressing the K133 mutant proteins produce red but not green foci suggests the K133 mutants were selectively absent from foci.
These results are in contrast to a previous report that showed GFP-HsRAD51K133A and GFP-HsRAD51K133R formed wild-type levels of foci in response to ionizing radiation (19). However, differences in experimental design likely led to these contrasting results. For example, Forget et al. depleted endogenous HsRAD51 by siRNA to almost undetectable levels, so unlike our experiments, the K133 mutant proteins did not compete with wild-type protein. In addition, unlike our experiments, these mutant proteins were ectopically expressed at higher-than-physiological levels (2- to 10-fold). Finally, ionizing radiation causes replication-independent DSBs, while CPT causes replication-dependent DSBs. Considering these experimental differences, it is possible the K133 mutants cause some defects only if they are present with wild-type protein, expressed at physiological levels and in response to replication associated DSBs.
DISCUSSION
The present work describes the phenotype of mouse ES cells that stably expressed two HsRAD51 mutants defective for either ATP binding (K133A) or ATP hydrolysis (K133R). Cells that expressed these mutants showed reduced cellular proliferation, defective replication fork restart, reduced spontaneous SCEs, hypersensitivity to CPT, increased chromosomal aberrations, and elevated LOH. Both K133 mutants caused a similar phenotype, but the effects of the K133A mutation were often more severe. These data showed that the K133 mutants had an adverse effect on both replication fork restart and HDR. Expression of eGFP-HsRAD51 fusions showed that the K133 mutants were present in the chromatin fraction at normal levels but not at sites of replication and repair. However, the K133 mutants did not prohibit MmRAD51 localization to these sites. Therefore, a selective process reduced levels of the K133 mutant proteins at these active sites, indicating K133 ATP binding/hydrolysis was necessary for efficient and/or stable localization to replication and repair sites.
Models that address the role of ATP binding and hydrolysis in RAD51 localization and function.
Previously published data show that in cells depleted for endogenous HsRAD51, transiently expressed GFP-HsRAD51K133A (but not GFP-HsRAD51K133R) repaired ionizing radiation-induced DNA DSBs according to the comet assay (19). Furthermore, both GFP-HsRAD51K133A and GFP-HsRAD51K133R formed S-phase foci and ionizing radiation-induced foci. The GFP-HsRAD51 K133 mutants also coimmunoprecipitated with endogenous HsRAD51 and XRCC3. By yeast two-hybrid assay, the K133 mutants associated with HsRAD51, XRCC3, and the BRCA2 BRC3 repeat and both K133 mutants bound to ssDNA and ATP. Thus, the K133 mutant proteins are capable of performing many important physiological functions, yet they are dominant negative (19).
Analysis of cells that stably express the eGFP-HsRAD51 proteins (WT, K133A, and K133R) along with endogenous MmRAD51 allowed us to make three important observations about protein dynamics that are relevant to the dominant-negative phenotype for the K133 mutants. First, all proteins were present in the chromatin fraction at nearly equal levels. Second, eGFP-HsRAD51WT efficiently colocalized with MmRAD51 at or adjacent to newly replicated DNA and localized to spontaneous and CPT-induced foci. Thus, MmRAD51 and eGFP-HsRAD51WT likely formed functional mixed filaments, as seen in biochemical assays (19). Third, eGFP-HsRAD51K133A and eGFP-HsRAD51K133R failed to efficiently colocalize with MmRAD51 to newly replicated DNA and foci. Thus, a failure in ATP binding and hydrolysis did not impact the level of chromatin-bound protein but negatively affected the level of protein found at important areas of replication and repair.
Low levels of the K133 mutant proteins likely formed a mixed filament with wild-type protein at replication sites. The iPOND procedure detected small amounts of the K133 mutant protein, and there were low levels of eGFP-HsRad51K133A and eGFP-HsRad51K133R foci; therefore, low levels of K133 mutant protein at just a few sites may be sufficient to cause a dominant-negative phenotype. Taken to the extreme, just one mutant protein in just one filament would be toxic if this mixed filament caused an inactive or rogue intermediate. This is supported by the observation that HsRAD51K133R reduced the efficiency of HsRAD51WT to shorten dsDNA even at a 1:10 ratio (53). In addition, the K133 mutant proteins associated with wild-type RAD51 (19). Thus, a low level of K133 mutant protein likely formed a mixed filament with wild-type MmRAD51 to impair replication fork restart and HDR.
The K133 mutant proteins caused a dominant-negative phenotype since wild-type MmRAD51 was still present. However, the nature of this dominant-negative phenotype is uncertain. We present two models. The inactive RAD51 model proposes that the K133 mutant and wild-type RAD51 proteins form an inactive mixed filament that fails to restart replication forks and repair DSBs, thus, allowing more error-prone mechanisms like FoSTeS or MMBIR to take over. Support for this model comes from our observation that cells expressing the K133 mutants exhibited reduced replication fork restart and previous observations of reduced DSB repair (64, 65). These defects would lead to increased levels of breaks as seen in cells that expressed the K133 mutant proteins, in particular after HU exposure. The rogue RAD51 model proposes that the K133 mutant and wild-type RAD51 proteins form an active, but rogue, mixed filament that is error prone. Support for this possibility comes from a previous report that shows the K133A mutant is capable of repairing DSBs when expressed in cells depleted for endogenous wild-type RAD51 (19). Furthermore, the K133 mutant proteins bound to ssDNA and associated with RAD51 binding proteins (19). Therefore, disabling ATP binding/hydrolysis does not affect many RAD51 functions, suggesting K133 mutant protein may be functional but act inappropriately if mixed with wild-type RAD51. If true, the Walker A motif performs a regulatory function. Both models explain the large number of chromosomal rearrangements observed in ES cells expressing the RAD51 K133 mutants, and these models are not mutually exclusive.
A perspective of RAD51 in maintaining the structural integrity of chromosomes.
Replication pathways like FoSTeS (32) and MMBIR (22) may suppress collapsed replication forks but may also cause CNV and CGRs. CGRs cause some cancers (66) and genomic disorders (36) in humans. Sequence information derived from these patients implicates faulty replication in the generation of CGRs since there were multiple rearrangements in one chromosome, yet other explanations are possible (8). Thus, the etiology of these CGRs is not known.
Our data suggest that imperfect RAD51 activity could contribute to CNV and CGRs by the inactive or rogue models presented above since cells expressing HsRAD51K133A exhibited EPTs. The metaphase spreads suggest EPTs contain multiple breakpoints in the same chromosome, similar to CGRs. EPTs were principally der(11) fusion (Fig. 6G1) and dup 1 der(1)t(1:6) (Fig. 6G2). Even though two separate unrelated events could cause the latter alteration (a duplication and a translocation), they were reproduced in 5 of 11 metaphase spreads, suggesting a common event. Likewise, cells expressing HsRAD51K133A showed many chromosomal alterations that were reproduced in multiple metaphase spreads, suggesting a recurrent problem associated with replication and not the rejoining of randomly generated broken ends since a purely stochastic process would not be so reproducible.
The causal factors for EPTs may be at least partly different from those for CNV and CGRs in humans since the latter do not contain large tracts of duplicated pericentromeres and telomeres. However, limited segmental duplications involving the pericentromere have occurred in vivo during primate evolution (25, 58), especially as a pericentromere transitions into the chromosome arm. These segmental duplications contain novel genes that likely contributed to speciation and would be difficult to detect from metaphase spreads. Furthermore, EPTs may be able to survive only in tissue culture. Thus, we do not know if EPTs share an etiology with segmental duplications, CNVs, and CGRs.
Our data reveal some mechanistic insight into the genesis of EPTs that suggest they arise from defective replication. (i) The K133 mutants caused defects in replication fork restart and the repair of replication-associated DSBs. (ii) The K133 mutants also increased chromatid breaks, indicating one-end DSBs at broken replication forks. (iii) Exposure to HU at levels that stall replication forks increased chromatid breaks, which was further exacerbated by expression of the K133 mutants. (iv) HU increased EPT-1s at the expense of EPT-2s, suggesting they are different outcomes emanating from the same initiating event. (v) CPT exposure had a similar effect to HU on levels of EPT-1s and EPT-2s, suggesting that topo I played an important mechanistic role in their genesis. Thus, the HU and CPT observations suggest replication fork stalls and breaks induce EPT-1s but suppress EPT-2s for cells that express HsRAD51K133A. A more in-depth analysis will be required to understand the genesis of EPTs and the other rearrangements seen in cells expressing HsRAD51K133A, which would include sequencing and an epistatic analysis.
Our results implicate imperfect replication fork maintenance in the genesis of CGRs and CNV. In particular, decreased or faulty RAD51 activity may cause structural chromosomal changes found in genomic disorders and cancer and support a multiple de novo CNV model (35) that proposes imperfections in proteins involved in replication as a causal factor in the genesis of CNV and CGRs.
ACKNOWLEDGMENTS
We thank Charnae Williams for technical support and the Molecular Cytogenetic Core at Albert Einstein College of Medicine for helping with the execution of these experiments. We thank Patrick Sung for the human RAD51K133A and RAD51K133R cDNAs, Eva Petermann for a detailed protocol for the microfiber analysis, and David Cortez for a detailed protocol for iPOND.
This work was supported by the following grants from the NIH: R01 CA123203-03 to P.H. and 2P01AG017242-12 to P.H. and J.V. and 1R01 HD063791-01 to C.M. We also thank the CTRC (CA054174).
Footnotes
Published ahead of print 9 July 2012
REFERENCES
- 1. Adra CN, Boer PH, McBurney MW. 1987. Cloning and expression of the mouse pgk-1 gene and the nucleotide sequence of its promoter. Gene 60:65–74 [DOI] [PubMed] [Google Scholar]
- 2. Araki K, Araki M, Yamamura K. 1997. Targeted integration of DNA using mutant lox sites in embryonic stem cells. Nucleic Acids Res. 25:868–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Araki K, Okada Y, Araki M, Yamamura K. 2010. Comparative analysis of right element mutant lox sites on recombination efficiency in embryonic stem cells. BMC Biotechnol. 10:29 doi:10.1186/1472-6750-10-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Badie S, et al. 2010. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat. Struct. Mol. Biol. 17:1461–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bailey JA, Eichler EE. 2006. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat. Rev. Genet. 7:552–564 [DOI] [PubMed] [Google Scholar]
- 6. Bessho T. 2003. Induction of DNA replication-mediated double strand breaks by psoralen DNA interstrand cross-links. J. Biol. Chem. 278:5250–5254 [DOI] [PubMed] [Google Scholar]
- 7. Bowater R, Doherty AJ. 2006. Making ends meet: repairing breaks in bacterial DNA by non-homologous end-joining. PLoS Genet. 2:e8 doi:10.1371/journal.pgen.0020008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carr AM, Paek AL, Weinert T. 2011. DNA replication: failures and inverted fusions. Semin. Cell Dev. Biol. 22:866–874 [DOI] [PubMed] [Google Scholar]
- 9. Chen MJ, et al. 2007. Cisplatin depletes TREX2 and causes Robertsonian translocations as seen in TREX2 knockout cells. Cancer Res. 67:9077–9083 [DOI] [PubMed] [Google Scholar]
- 10. Chen YT, Bradley A. 2000. A new positive/negative selectable marker, puDeltatk, for use in embryonic stem cells. Genesis 28:31–35 [DOI] [PubMed] [Google Scholar]
- 11. Cheung J, et al. 2003. Recent segmental and gene duplications in the mouse genome. Genome Biol. 4:R47 doi:10.1186/gb-2003-4-8-r47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chi P, Van Komen S, Sehorn MG, Sigurdsson S, Sung P. 2006. Roles of ATP binding and ATP hydrolysis in human Rad51 recombinase function. DNA Repair (Amst.). 5:381–391 [DOI] [PubMed] [Google Scholar]
- 13. Colnaghi R, Carpenter G, Volker M, O'Driscoll M. 2011. The consequences of structural genomic alterations in humans: genomic disorders, genomic instability and cancer. Semin. Cell Dev. Biol. 22:875–885 [DOI] [PubMed] [Google Scholar]
- 14. Dong Z, Fasullo M. 2003. Multiple recombination pathways for sister chromatid exchange in Saccharomyces cerevisiae: role of RAD1 and the RAD52 epistasis group genes. Nucleic Acids Res. 31:2576–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dronkert ML, et al. 2000. Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol. Cell. Biol. 20:3147–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dumitrache LC, et al. 2011. Trex2 enables spontaneous sister chromatid exchanges without facilitating DNA double-strand break repair. Genetics 188:787–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Essers J, et al. 2002. Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J. 21:2030–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Festing MF, Simpson EM, Davisson MT, Mobraaten LE. 1999. Revised nomenclature for strain 129 mice. Mamm. Genome 10:836. [DOI] [PubMed] [Google Scholar]
- 19. Forget AL, Loftus MS, McGrew DA, Bennett BT, Knight KL. 2007. The human Rad51 K133A mutant is functional for DNA double-strand break repair in human cells. Biochemistry 46:3566–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Friedrich G, Soriano P. 1991. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 5:1513–1523 [DOI] [PubMed] [Google Scholar]
- 21. Guenatri M, Bailly D, Maison C, Almouzni G. 2004. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol. 166:493–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hastings PJ, Ira G, Lupski JR. 2009. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 5:e1000327 doi:10.1371/journal.pgen.1000327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hastings PJ, Lupski JR, Rosenberg SM, Ira G. 2009. Mechanisms of change in gene copy number. Nat. Rev. Genet. 10:551–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holcomb VB, et al. 2007. HPRT minigene generates chimeric transcripts as a by-product of gene targeting. Genesis 45:275–281 [DOI] [PubMed] [Google Scholar]
- 25. Horvath JE, et al. 2003. Using a pericentromeric interspersed repeat to recapitulate the phylogeny and expansion of human centromeric segmental duplications. Mol. Biol. Evol. 20:1463–1479 [DOI] [PubMed] [Google Scholar]
- 26. Hu Y, et al. 2007. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 21:3073–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim TM, Choi YJ, Ko JH, Hasty P. 2008. High-throughput knock-in coupling gene targeting with the HPRT minigene and Cre-mediated recombination. Genesis 46:732–737 [DOI] [PubMed] [Google Scholar]
- 28. Kim TM, Ko JH, Choi YJ, Hu L, Hasty P. 2011. The phenotype of FancB-mutant mouse embryonic stem cells. Mutat. Res. 712:20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klein HL. 2008. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst.) 7:686–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kozak M. 1989. The scanning model for translation: an update. J. Cell Biol. 108:229–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Laulier C, Cheng A, Stark JM. 2011. The relative efficiency of homology-directed repair has distinct effects on proper anaphase chromosome separation. Nucleic Acids Res. 39:5935–5944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee JA, Carvalho CM, Lupski JR. 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131:1235–1247 [DOI] [PubMed] [Google Scholar]
- 33. Lee SA, Roques C, Magwood AC, Masson JY, Baker MD. 2009. Recovery of deficient homologous recombination in Brca2-depleted mouse cells by wild-type Rad51 expression. DNA Repair (Amst.) 8:170–181 [DOI] [PubMed] [Google Scholar]
- 34. Lim DS, Hasty P. 1996. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol. Cell. Biol. 16:7133–7143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu P, Carvalho CM, Hastings P, Lupski JR. 2012. Mechanisms for recurrent and complex human genomic rearrangements. Curr. Opin. Genet. Dev. 22:211–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu P, et al. 2011. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 146:889–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luo G, et al. 2000. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat. Genet. 26:424–429 [DOI] [PubMed] [Google Scholar]
- 38. Marple T, Kim TM, Hasty P. 2006. Embryonic stem cells deficient for Brca2 or Blm exhibit divergent genotoxic profiles that support opposing activities during homologous recombination. Mutat. Res. 602:110–120 [DOI] [PubMed] [Google Scholar]
- 39. Marple T, Li H, Hasty P. 2004. A genotoxic screen: rapid analysis of cellular dose-response to a wide range of agents that either damage DNA or alter genome maintenance pathways. Mutat. Res. 554:253–266 [DOI] [PubMed] [Google Scholar]
- 40. Mazur DJ, Perrino FW. 2001. Excision of 3′ termini by the Trex1 and TREX2 3′→5′ exonucleases. Characterization of the recombinant proteins. J. Biol. Chem. 276:17022–17029 [DOI] [PubMed] [Google Scholar]
- 41. Michor F, Iwasa Y, Vogelstein B, Lengauer C, Nowak MA. 2005. Can chromosomal instability initiate tumorigenesis? Semin. Cancer Biol. 15:43–49 [DOI] [PubMed] [Google Scholar]
- 42. Morita T, et al. 1993. A mouse homolog of the Escherichia coli recA and Saccharomyces cerevisiae RAD51 genes. Proc. Natl. Acad. Sci. U. S. A. 90:6577–6580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morrison C, et al. 1999. The essential functions of human Rad51 are independent of ATP hydrolysis. Mol. Cell. Biol. 19:6891–6897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Motegi A, et al. 2008. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. U. S. A. 105:12411–12416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Park WH, et al. 2005. Direct DNA binding activity of the Fanconi anemia D2 protein. J. Biol. Chem. 280:23593–23598 [DOI] [PubMed] [Google Scholar]
- 46. Patterson N, Richter DJ, Gnerre S, Lander ES, Reich D. 2006. Genetic evidence for complex speciation of humans and chimpanzees. Nature 441:1103–1108 [DOI] [PubMed] [Google Scholar]
- 47. Paulsen RD, Cimprich KA. 2007. The ATR pathway: fine-tuning the fork. DNA Repair (Amst.) 6:953–966 [DOI] [PubMed] [Google Scholar]
- 48. Pellegrini L, et al. 2002. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 420:287–293 [DOI] [PubMed] [Google Scholar]
- 49. Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. 2010. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 37:492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ramirez-Solis R, et al. 1992. Genomic DNA microextraction: a method to screen numerous samples. Anal. Biochem. 201:331–335 [DOI] [PubMed] [Google Scholar]
- 51. Reid LH, Gregg RG, Smithies O, Koller BH. 1990. Regulatory elements in the introns of the human HPRT gene are necessary for its expression in embryonic stem cells. Proc. Natl. Acad. Sci. U. S. A. 87:4299–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rivory LP. 1996. Irinotecan (CPT-11): a brief overview. Clin. Exp. Pharmacol. Physiol. 23:1000–1004 [DOI] [PubMed] [Google Scholar]
- 53. Robertson RB, et al. 2009. Structural transitions within human Rad51 nucleoprotein filaments. Proc. Natl. Acad. Sci. U. S. A. 106:12688–12693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ruksc A, Birmingham EC, Baker MD. 2007. Altered DNA repair and recombination responses in mouse cells expressing wildtype or mutant forms of RAD51. DNA Repair (Amst.) 6:1876–1889 [DOI] [PubMed] [Google Scholar]
- 55. San Filippo J, Sung P, Klein H. 2008. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77:229–257 [DOI] [PubMed] [Google Scholar]
- 56. Schröck E, et al. 1996. Multicolor spectral karyotyping of human chromosomes. Science 273:494–497 [DOI] [PubMed] [Google Scholar]
- 57. Schwab RA, Niedzwiedz W. 2011. Visualization of DNA replication in the vertebrate model system DT40 using the DNA fiber technique. J. Vis. Exp. 56:e3255 doi:10.3791/3255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. She X, et al. 2004. The structure and evolution of centromeric transition regions within the human genome. Nature 430:857–864 [DOI] [PubMed] [Google Scholar]
- 59. Shinohara A, et al. 1993. Cloning of human, mouse and fission yeast recombination genes homologous to RAD51 and recA. Nat. Genet. 4:239–243 [DOI] [PubMed] [Google Scholar]
- 60. Sigurdsson S, Van Komen S, Petukhova G, Sung P. 2002. Homologous DNA pairing by human recombination factors Rad51 and Rad54. J. Biol. Chem. 277:42790–42794 [DOI] [PubMed] [Google Scholar]
- 61. Sirbu BM, et al. 2011. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 25:1320–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sonoda E, et al. 1998. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 17:598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sonoda E, et al. 1999. Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol. Cell. Biol. 19:5166–5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stark JM, et al. 2002. ATP hydrolysis by mammalian RAD51 has a key role during homology-directed DNA repair. J. Biol. Chem. 277:20185–20194 [DOI] [PubMed] [Google Scholar]
- 65. Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. 2004. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell. Biol. 24:9305–9316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stephens PJ, et al. 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144:27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Takata M, et al. 2000. The Rad51 paralog Rad51B promotes homologous recombinational repair. Mol. Cell. Biol. 20:6476–6482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Takata M, et al. 2001. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol. Cell. Biol. 21:2858–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Taniguchi T, et al. 2002. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 100:2414–2420 [DOI] [PubMed] [Google Scholar]
- 70. Tsuzuki T, et al. 1996. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. U. S. A. 93:6236–6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang W. 2007. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 8:735–748 [DOI] [PubMed] [Google Scholar]
- 72. Wilson DM, III, Thompson LH. 2007. Molecular mechanisms of sister-chromatid exchange. Mutat. Res. 616:11–23 [DOI] [PubMed] [Google Scholar]
- 73. Yu DS, et al. 2003. Dynamic control of Rad51 recombinase by self-association and interaction with BRCA2. Mol. Cell 12:1029–1041 [DOI] [PubMed] [Google Scholar]
- 74. Zhang F, Carvalho CM, Lupski JR. 2009. Complex human chromosomal and genomic rearrangements. Trends Genet. 25:298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang F, et al. 2009. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 41:849–853 [DOI] [PMC free article] [PubMed] [Google Scholar]