

Abstract
Glucocorticoids regulate gene expression by binding and activating the glucocorticoid receptor (GR). While ligand affinity determines the global sensitivity of the response, additional proteins act on the genome to tune sensitivity of some genes. However, the genomic extent and specificity of dose-specific glucocorticoid responses are unknown. We show that dose-specific glucocorticoid responses are extraordinarily specific at the genomic scale, able to distinctly express a single gene, the circadian rhythm gene for Period 1 (PER1), at concentrations consistent with the nighttime nadir of human cortisol. We mapped the PER1 response to a single GR binding site. The specific GR binding sequence did not impact sensitivity, and we instead attributed the response to a combination of additional transcription factors and chromatin accessibility acting in the same locus. The PER1 hypersensitive response element is conserved in the mouse, where we found similar upregulation of Per1 in pituitary cells. Targeted and transient overexpression of PER1 led to regulation of additional circadian rhythm genes hours later, suggesting that hypersensitive expression of PER1 impacts circadian gene expression. These findings show that hypersensitive GR binding occurs throughout the genome, drives targeted gene expression, and may be important to endocrine mediation of peripheral circadian rhythms.
INTRODUCTION
The glucocorticoid receptor (GR) directs the transcriptional response to the steroid hormone cortisol (reviewed in reference 64). Glucocorticoids bind the receptor with high affinity, leading to nuclear translocation, direct binding to DNA, and ultimately regulation of gene expression. The affinity of glucocorticoid for the GR is a primary determinant of activation. Many studies have characterized GR binding and transcriptional regulatory activity at saturating concentrations of hormone at which most of the GR is bound (7, 38, 43, 48, 50, 56). While these studies have revealed hundreds of genes that are differentially expressed in response to treatment with high concentrations of hormone, endogenous cortisol concentrations vary throughout the day as part of circadian rhythms (70) and are often near or below the dissociation constant (Kd) for the GR (46).
Low doses of glucocorticoids that are well below the Kd of the GR have been shown to elicit GR-mediated gene regulation for a small number of genes (47, 55). Such findings indicate that GR activity can be tuned to different doses of glucocorticoids in a tissue-specific manner, potentially in order to regulate expression of specific genes at different points in the circadian cycle. For example, subsaturating doses of the synthetic glucocorticoid dexamethasone (DEX) can drive expression of the tyrosine aminotransferase (TAT) gene in FU5-5 cells but not in the HTC rat hepatoma cell line (44). In addition, doses of DEX as low as 0.001 nM, well below the Kd of DEX for the GR (Kd, ∼3 to 5 nM [31, 66]), can act in a GR- and protein synthesis-dependent manner to inhibit the activity of vasoactive intestinal peptide (VIP) in primary rat pituitary cells (52). Numerous studies have shown that long-term and pulsatile glucocorticoids regulate expression of the gene for Period 1 (PER1), a major component of the mammalian circadian clock, in humans and rodents (3, 9, 14, 32, 57). Glucocorticoids also regulate PER1 in primary tissues in response to acute physical stress (72) and as part of circadian rhythms (58). We also recently showed that expression of PER1 is sensitive to subnanomolar doses of DEX in the human lung epithelial cell line A549 (50).
A prominent mechanism for tuning the glucocorticoid response involves interaction between the GR and cofactors. Szapary and colleagues reported that increasing the concentration of the GR, the nuclear receptor coactivators 1 and 2 (NCOA1 and NCOA2; also known as SRC-1 and TIF2, respectively), or of the CREB binding protein (CREBBP), can dramatically sensitize the GR-mediated response (47, 61, 63). More recently, a tubulin tyrosine ligase-like family member (TTLL5; also known as STAMP) was found to cooperate with the NCOA proteins and further enhance sensitivity to glucocorticoids (22). Nearby DNA sequence elements can also tune glucocorticoid responses. For example, a DNA sequence known as the glucocorticoid modulatory element (GME) associates with the GMEB-1 and -2 proteins and, depending on the presence of additional factors, increases or decreases the sensitivity of the local glucocorticoid response (28, 49, 62). The modulation of sensitivity typically applies equally well to gene induction and repression, indicating that the mechanisms of tuning the corticosteroid response may be shared (59). Dose-specific responses are general to other nuclear receptor-mediated hormone responses, and similar mechanisms can alter expression responses to estrogens and mineralocorticoids as well (69).
Based on these results, we hypothesized that the dose at which GR binds DNA and regulates gene expression varies throughout the genome. To investigate, we measured low-dose specific changes in GR occupancy and related changes in gene expression across the human genome. We found genome-wide sensitivity of GR occupancy to be associated both with open chromatin and also with cooccupancy of numerous cofactors. We mapped in detail the minimal enhancer element responsible for driving the GR-mediated regulation of the expression of PER1, the most sensitive glucocorticoid-responsive gene in A549 cells. Doing so revealed that a complex set of nucleotide signals across a 274-bp minimal enhancer cooperate to tune the glucocorticoid response. Finally, we show that targeted expression of PER1 regulates expression of other circadian rhythm genes many hours later, suggesting a role in circadian biology.
MATERIALS AND METHODS
Cell growth.
A549 cells were grown at 37°C and 5% CO2 in F-12K medium (Gibco) with 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco). AtT-20 cells were grown in F-12K medium with 2.5% fetal bovine serum, 15% horse serum, and 1% penicillin-streptomycin (Gibco). Dexamethasone and cortisol were dissolved in ethanol at a 5,000× final concentration, and all treatments were performed in parallel with an equal volume (0.02% [vol/vol]) of ethanol to control for solvent effects.
ChIP-seq.
Chromatin immunoprecipitation (ChIP) was performed as described previously with minor modifications (50). Briefly, for each immunoprecipitation mixture, we used 2 × 107 cells, 5 μg antibody (see Table S4 in the supplemental material), and 200 μl Dynal sheep anti-rabbit or sheep anti-mouse antibody beads (Invitrogen). DNA was prepared for sequencing on an Illumina Genome Analyzer 2 as described previously (50) but without PCR prior to agarose gel size selection and with 15 rounds of PCR for final library amplification. Reads were aligned to the hg19 version of the human genome sequence by using Bowtie (35) with the parameter “-m 1 –best –strata.” To evaluate reproducibility between experiments and biological replicates, we correlated the number of reads from each experiment that aligned within 1,000 bp of a transcription start site. For GR ChIP with sequencing (ChIP-seq) experiments, binding sites were called independently for each biological replicate by using the QuEST Peak Caller (65) with the “stringent” threshold setting. We then defined the final set of binding sites for each experiment by requiring each peak summit (i.e., the position of maximal ChIP-seq signal as determined by QuEST) in one replicate to be within 50 bp of a peak summit in the other replicate. We further classified GR binding sites as 0.5 nM, 5 nM, or 50 nM, according to the lowest dose at which the binding site occurred. For all other ChIP-seq experiments, reads from two biological replicates were combined into a single data set, and binding sites were called using QuEST as described above.
Transcription factor binding site motif detection.
Motif detection was performed by using BioProspector (39), followed by BioOptimizer (26). First, we extracted the genomic sequence for each binding site and masked low-complexity sequence by using the DUST program. To investigate the consensus GR DNA binding motif, we ran BioProspector with a motif width of 18 on the 150 strongest binding sites in each class of GR binding sites and optimized each motif by running BioOptimizer with the initial motif on the complete set of DUST-masked sequences for each class. Motif prevalence was calculated by using FIMO (2), with a significance threshold of P < 1 × 10−4.
To identify additional DNA binding motifs common to identified binding sites, we masked GR binding sites identified with FIMO and then used BioProspector to search for additional motifs. Motifs matching AP-1 were commonly identified in this second round of motif detection. For a third round of motif searching, AP-1 sites were masked as before and BioProspector was rerun at various motif widths. This third round yielded motifs matching that of the FOXA factors, at motif widths of >10 bp, and motifs matching that of CREB, at a motif width of 7 bp.
Measuring the gene expression response to DEX.
To evaluate the gene expression response to DEX, we treated A549 cells for 1 h with 0.1 nM, 0.5 nM, 1 nM, or 5 nM DEX or with ethanol as a vehicle control and measured gene expression with RNA sequencing as described previously (45, 50). Briefly, after treatment, we washed cells with phosphate-buffered saline (PBS, pH 7.4), lysed them in buffer RLT (Qiagen) with 1% β-mercaptoethanol, and extracted total RNA using Qiagen RNeasy Mini columns according to the manufacturer's protocol, including the DNase-mediated DNA digestion step. Total RNA was then poly(A) selected, fragmented, reverse transcribed to cDNA, and sequenced to a depth of >20 million 36-bp reads. We then aligned reads to RefSeq transcripts by using Bowtie (35), with parameters “-n 2 -k 1 -m 10 –best,” and calculated expression in units of aligned reads per kilobase of exon and per million aligned reads (RPKM).
To calculate differential expression for each gene at each dose point, we filtered and normalized the expression measurements and identified genes with expression changes greater than would be expected by chance according to biological replicates. To do so, we first filtered transcripts shorter than 150 bp and also lowly expressed genes (average RPKM < 2 across all measurements), because they were unsatisfactorily noisy in our results. Then we normalized expression values by using variance stabilization as implemented in the vsn package for the R statistical language. After variance stabilization, we further filtered out genes with a coefficient of variation between replicates that was greater than 0.1, removing 83 of 11,225 genes. To model noise between biological replicates, we used a normal distribution fit to the difference in transformed RPKM values between biological replicate experiments (μ = −2.5 × 10−3; σ = 0.12). We then calculated the statistical significance of differential expression as the probability that the change in expression between treated and untreated cells occurred in the background noise model by using a two-sided comparison. To correct for multiple hypotheses, we calculated the false discovery rate (FDR) for each experiment by using the p.adjust function implemented in R. Finally, to identify differentially expressed genes, we required that differential expression be significant in both biological replicates with an FDR of <5% and that expression be significantly correlated (P < 0.1) between biological replicates across the dose response.
Glucocorticoid reporter constructs.
Reporter plasmids were constructed either by traditional cloning or by custom synthesis. For the reporters containing the endogenous promoter as well as for many of the enhancer constructs, we used PCR with Pfu Ultra II DNA polymerase (Stratagene) to amplify the indicated regions from the A549 genome and to tail amplified regions with unique restriction enzyme sites. We then used restriction enzyme digestion of PCR products and plasmid, followed by ligation to insert PCR products into versions of the pGL4-luc2p luciferase reporter plasmids (Promega). For the 160-bp, 135-bp, 100-bp, and the PER1-intronic enhancer constructs that were modified to match regions of the PER1 upstream enhancer region, the DNA sequence was directly synthesized (Blue Heron Biotechnology) and subcloned into the luciferase reporter plasmid. Reporters containing the endogenous transcription start site were inserted into the pGL4.24 vector, whereas enhancer regions were inserted in the pGL4.14 vector, which contains a minimal promoter. Lastly, we confirmed the oligonucleotide sequence of all reported plasmids with Sanger sequencing. The mouse mammary tumor virus (MMTV) reporter we used was pGL4.36 from Promega.
Site-directed mutagenesis.
To introduce 2-bp mutations into the nucleotide sequence of enhancer regions, we performed site-directed mutagenesis with the Stratagene QuikChange kit, with the modification of propagating plasmid in TOP10 chemically competent cells (Invitrogen). We verified correct mutations by Sanger sequencing.
Transient transfection and reporter dose response.
To measure activity of each construct, 5,000 A549 cells were seeded into each well of a 96-well plate. After incubating cells overnight, we transiently transfected cells with 100 ng luciferase reporter and 4 ng Renilla-simian virus 40 control plasmid (Promega) by using a 6:1 ratio of FuGene to DNA, according to the manufacturer's protocol. After 48 h, medium was removed and replaced with medium containing corticosteroids or ethanol as a vehicle control. After 4 h of incubation at 37°C, cells were lysed using Promega Dual-GLO reagent, and luciferase expression was measured using a LMAX384 plate reader adapted for 96-well plates. To control for transfection efficiency, luciferase signal was normalized to the Renilla signal in each well, and log2 ratios over ethanol-treated control wells were determined. All experiments were repeated in 8 replicates and at 12 doses. To measure the DEX dose response, we treated cells with DEX at 0.05 nM, 0.1 nM, 0.25 nM, 0.5 nM, 1 nM, 2.5 nM, 5 nM, 10 nM, 25 nM, 50 nM, or 100 nM. To measure the cortisol dose response, we treated cells with cortisol at 1 nM, 2.5 nM, 5 nM, 10 nM, 25 nM, 50 nM, 100 nM, 250 nM, 500 nM, 1 μM, and 2.5 μM.
Measuring gene expression in response to transient PER1 induction.
To measure the long-term effects of glucocorticoid-mediated induction of PER1 expression, we seeded A549 cells into wells of a 6-well plate. Twenty-four hours later, we began treating individual wells for 1 h with 0.5 nM DEX. After a 1-h treatment, cells were washed twice with 5 ml of fresh medium and incubated in 2 ml of fresh medium for 0 to 48 h, as indicated below. Treatments were scheduled such that all time points for a biological replicate concluded at the same time, thus limiting effects due to differing times in culture. Cells were then lysed with buffer RLT (Qiagen) with 1% β-mercaptoethanol, and total RNA was extracted using Qiagen RNeasy Mini columns according to the manufacturer's protocol and including an on-column DNase digestion. RNA was reverse transcribed using a SuperScript VILO cDNA synthesis kit (Invitrogen), and relative transcript abundance of each transcript was measured in TaqMan gene expression assays (Invitrogen) in a 384-well plate and with an assay for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), used as an endogenous control. We performed 4 technical replicates per measurement, and the average threshold cycle (CT) was used for downstream calculations. Expression measurements were then normalized to GAPDH to control for differences in cDNA amount, and finally expression was calculated relative to cells harvested at the same time but that were never treated with DEX. For the 12-h time course (see Fig. 7E), the procedure was repeated five times on different days, and we report the relative expression as the means and standard deviations of those five biological replicates. For the 48-h time course (see Fig. S6 in the supplemental material), six biological replicates were performed.
Fig 7.

Hypersensitive PER1 expression is general to the cell line and conserved in the mouse. (A) Dose-response curves for enhancer elements, as described for Fig. 5A, but transfected into the human endometrial cell line ECC-1, using the same protocol as used for A549 cells. As in Fig. 5, points indicate mean responses, with error bars indicating standard errors of the means. The two enhancer regions that contained the GR binding site upstream of PER1 (green and blue) showed a more sensitive dose response than an enhancer derived from the GR binding site in the first intron of PER1. (B) EC50s, calculated for the enhancer elements presented in panel A, with error bars indicating 95% confidence intervals. The hypersensitive GR binding sites (green and blue) both resulted in EC50s close to 2 nM DEX, whereas the nonhypersensitive GR binding site was about half as sensitive (EC50, ∼4 nM). (C) Comparison of Hill coefficients for activities of hypersensitive reporters between A549 cells and ECC-1 cells. (D) EC50 for each of 20 genes responsive to 5 nM DEX in the mouse pituitary cell line AtT-20 and as measured using RNA sequencing. The lines indicates the means, and error bars indicate standard deviations of the distribution. (E) Time course of gene expression response of circadian rhythm genes after transient induction of PER1 expression. A549 cells were treated for 1 h with 0.5 nM DEX, after which cells were rinsed and returned to DEX-free medium (zero on the x axis). Cells were collected over the following 12 h, and expression was measured in a TaqMan reverse transcriptase quantitative PCR. For each of the five biological replicates, threshold cycles were calculated as the average over four technical replicates, and expression was normalized to that of GAPDH and plotted relative to A549 cells prior to any DEX treatment. Points indicate means, and error bars indicate standard deviations over the five biological replicates. (F) Expression of circadian rhythm genes after 4 h of continuous exposure to 500 pM DEX. Columns indicate mean relative expression levels, and error bars indicate ranges observed across 3 biological replicates.
Data availability.
Results from all high-throughput sequencing experiments are available both on the UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgGateway; human genome version hg19) as well as at the HudsonAlpha website, http://hudsonalpha.org/sites/default/files/DataSets/Myerslab/Low_Dose_Glucocorticoid_Responses.
RESULTS
The GR binds the genome in a dose-dependent manner.
To understand the activity of the GR at physiologically low doses of glucocorticoids, we used ChIP-seq to measure glucocorticoid receptor binding in response to 0.5 nM (sub-Kd), 5 nM (near the Kd), and 50 nM (saturating) DEX in the human lung epithelial cell line A549 (Fig. 1A; see also Table S1 in the supplemental material). At the highest dose (50 nM), we found 5,898 GR binding sites, similar to our previous studies of GR binding with 100 nM DEX (50). Of the sites we found at the highest dose of DEX, the GR bound 145 (2.5%) and 1,449 (24.5%) at 0.5 nM and 5 nM DEX, respectively (Fig. 1A), indicating that the sensitivity of GR binding sites varies throughout the human genome by at least 3 orders of magnitude. We classified sites by the lowest dose of DEX at which we observed GR binding, and we refer to those sites in the remainder of the manuscript as hypersensitive (bound at 0.5 nM DEX), medium sensitive (bound at 5 nM DEX), or low sensitive (bound at 50 nM DEX) GR binding sites (Fig. 1B; see also Table S2 in the supplemental material). It is important to point out that we expect the sensitivity of GR binding sites to follow a continuum that would become evident with the study of many more dose points.
Fig 1.

Dose-specific GR binding across the human genome. (A) Intensities of ChIP-seq signals across all GR binding sites. Briefly, A549 cells were treated in biological duplicate for 1 h with doses of DEX as indicated in the columns. Each row represents 1 of the 5,898 GR binding sites observed at the highest (50 nM) dose of DEX. Binding sites were classified and grouped by the lowest dose of DEX at which a binding site was called, and that class is indicated on the right as hypersensitive (white), medium sensitive (orange), and low sensitive (blue). The color within the ChIP-seq data indicates the ChIP-seq signal intensity, expressed in units of aligned reads per kilobase of the binding site and per million aligned ChIP-seq reads (RPKM). (B) The total number of binding sites in each class, using the same color scheme as in panel A. (C) The consensus GR binding motifs identified in each sensitivity class of GR binding sites.
The hypersensitive GR binding sites had stronger binding signals overall (see Fig. S1 in the supplemental material). Therefore, in some cases, we may have erroneously classified hypersensitive sites, because they are also easier to detect, even when weakly bound. However, many of the medium and low sensitive sites ultimately had stronger ChIP-seq signals after treatment with 50 nM DEX. Examples of sites from each class are in shown in Fig. S2 in the supplemental material.
The GR commonly binds to a consensus DNA binding motif known as the glucocorticoid response element (GRE). The specific sequence of the bound GRE can drive conformational changes in the GR on the DNA and affect association with cofactors at certain enhancers (43). To investigate whether specific subsets of the GR-DNA recognition sequences are bound at very low concentrations of corticosteroids, we used de novo DNA binding motif detection to identify the consensus GR binding motif in each group (39). While we found a clear match to the known GRE in each class of sites, the motif did not substantially differ between the classes of GR binding sites or from that of previous reports (33, 50). The result indicated that dose sensitivity of GR binding is not driven by a specific version of the GRE, as might be expected in light of recent studies (43) (Fig. 1C). An alternative hypothesis is that multiple GR binding sequences clustered in a composite GR binding site may explain the high sensitivity of some sites. That is unlikely, however, because the fraction of GR binding sites with a consensus GR binding sequence did not differ significantly between the different classes (59%, 62%, and 57% of sites in the 0.5 nM, 5 nM, and 50 nM class, respectively; P > 0.01, Kruskal-Wallis test), nor did the number of GR binding sequences per binding site differ between classes, with binding sites in each class containing a median of 1 GRE (see Table S3 in the supplemental material). Together, these results suggest that the sensitivity of GR binding is not determined by the GR binding sequence itself, and we instead sought to identify additional features responsible for modulating the corticosteroid sensitivity of GR binding sites across the genome.
Chromatin accessibility is associated with sensitivity of GR binding.
Sequences outside the core GR binding motif may increase sensitivity by recruiting proteins that establish a more available open chromatin state or that may act as cofactors to stabilize GR-DNA interactions. We reasoned that the regions of open chromatin prior to treatment with glucocorticoid would be more available to the GR, thus enabling GR binding at lower doses of DEX. To test this hypothesis, we measured genomic accessibility to DNase I in A549 cells in the absence of steroid, and we defined open chromatin regions as those with significantly increased DNase I accessibility. Overall, we found that open chromatin accounted for 1.8% of the A549 genome and that 4,098 (69%) of the 5,898 GR binding sites overlapped regions of DNase I hypersensitivity, similar to observations in mouse cells (27). However, when we evaluated each class of GR binding site individually, we found a strong association between chromatin availability and binding site sensitivity, with nearly all (144 of 145, or 99%) of the hypersensitive GR binding sites occurring in already open chromatin (Fig. 2A). Chromatin accessibility appeared important for the less-sensitive GR binding sites as well, as over 75% of the medium sensitive sites also occurred in already open chromatin (27). However, the degree of open chromatin as estimated by the total DNase I hypersensitivity signal in each GR binding site tracked closely with the DEX sensitivity of the GR binding (Fig. 2B). Earlier work showing that open chromatin largely predetermines GR occupancy suggested that cobinding transcription factors (TFs) are important contributors to that sensitivity (27). For example, it may be that cobinding TFs sensitize hypersensitive sites, while the pioneer factor activity of the GR is limited to sites that are only active at high concentrations of glucocorticoids (13, 67).
Fig 2.

Overlap of hypersensitive GR binding with open chromatin and transcriptional cofactors (A) Fraction of GR binding sites in each sensitivity class that overlap regions of DNase I hypersensitivity. (B) For each GR binding site, we estimated the degree of open chromatin by calculating the DNase I signal as the RPKM. Plotted is the distribution of DNase I signal across each class of GR binding site. The inset shows the mean ± standard deviation of each class of GR binding site. (C) Fraction of GR binding sites in each sensitivity class that had overlapped calls of binding sites for cofactors, as indicated on the x axis. As a control, we also performed ChIP-seq for USF1, a factor not known to interact with the GR, and dashed lines indicate the fraction of overlap with USF1 for each class. (D) Receiver operating characteristic (ROC) curve, showing the sensitivity and specificity of predicting hypersensitivity of a GR binding site based on the occupancy of each cofactor. The ROC was calculated for the observed enrichment for the ability to predict a hypersensitive GR binding site versus predicting a medium sensitive GR binding site.
Transcription factors synergistic with the GR are prebound to regions of increased GR sensitivity.
Numerous additional DNA binding proteins may also contribute to modulation of the sensitivity of GR binding by acting synergistically with GR to stabilize DNA interactions. To identify such factors, we searched for DNA binding motifs that were enriched in hypersensitive GR binding sites (39). We found motifs matching the forkhead box (FOX), the AP-1 binding motif, and cyclic AMP response elements (CRE) in sites bound by GR at low doses of DEX. Members of each class are known to interact with the GR at binding sites. Of the FOX family, the FOXA1 and FOXA2 TFs, both expressed in A549 cells, have been associated previously with modulating GR binding to DNA (5, 12, 53, 73). In addition, the AP-1 complex is a well-documented factor known to enhance GR binding (1, 6, 17, 63), and evidence also suggests interactions occur between GR and the CRE binding protein (CREB) (19, 25).
To investigate the role of the FOXA, AP-1, and CREB transcription factors in sensitizing the glucocorticoid response, we used ChIP-seq to measure genomic occupancy of FOXA1, FOXA2, the AP-1 component JUND, and CREB1 in the absence of DEX (see Table S4 in the supplemental material), and the binding sites for each factor are listed in Table S5 of the supplemental material. Occupancy of each factor around transcription start sites was highly correlated between biological replicates as well as between the related FOXA1 and FOXA2 factors, but less correlated between pairs of unrelated factors (see Fig. S3 in the supplemental material).
To examine the association between the binding of synergistic transcription factors and the DEX sensitivity of GR binding, we evaluated the fraction of GR binding sites in each sensitivity class that showed overlapping occupancy of the additional factors. As a negative control, we also performed ChIP-seq for USF1, a factor not known to interact with the glucocorticoid receptor. We found stronger enrichment for GR binding sites for each factor studied, compared to USF1 (Fig. 2C). Occupancy of additional TFs was associated with increased DEX sensitivity of the GR binding site, indicating that these factors may contribute to genome-wide GR recruitment. While FOXA1 and FOXA2 had the greatest overlap with both hypersensitive and medium sensitive GR binding sites, CREB1 occupancy was specific for the hypersensitive sites. We evaluated if the factors were able to distinguish hypersensitive from medium sensitive GR binding and found that, of the factors evaluated, occupancy levels of CREB1 and JUND were the best predictors of hypersensitive GR binding (Fig. 2D). Therefore, CREB1 and AP-1 occupancy may play an important role in establishing hypersensitive GR binding sites, whereas the FOXA factors may more moderately sensitize GR binding. Our work confirmed that GR binding sites in preopen chromatin are enriched for occupancy of other TFs, whereas GR binding sites that require opening of chromatin (i.e., the low-sensitivity GR binding sites in our study) are more likely to act through GR binding alone (27). Specifically, the FOXA proteins are known pioneer factors that play a role to open chromatin in advance of nuclear receptor occupancy (40, 54), and these factors may assist in the occupancy of the GR hypersensitive sites (13, 67).
PER1 expression is uniquely sensitive to DEX in A549 cells.
Having shown that the GR binds throughout the genome in response to subsaturating DEX concentrations, we next sought to determine the genome-wide effects of low doses of DEX on gene expression. We treated A549 cells for 1 h with 0.1 nM, 0.5 nM, 1 nM, or 5 nM DEX and used RNA-seq to measure the gene expression response compared to a mock treatment. At the lowest dose (0.1 nM DEX), we identified no genes with a significant gene expression response. In contrast, at the highest dose (5 nM DEX), we identified 28 genes with significant changes in expression (Fig. 3A; see also Table S6 in the supplemental material). Of the genes responding to 5 nM DEX, expression levels of 23 (79%) were enhanced rather than repressed. For comparison, earlier genome-wide screens showed that saturating (100 nM) DEX led to increases in transcript levels of 59% of the responsive genes (50, 68). That low concentrations of DEX enhanced expression more often than high concentrations of DEX (P = 0.006, binomial test) suggests that hypersensitive and medium sensitive GR binding are more likely to be activating events, whereas low sensitive GR binding can also repress gene expression. Genes responding to low doses of DEX play a role in diverse functions, including inflammation (e.g., NFKBIA and TNFAIP3) and adipogenesis (e.g., CEBPB and CEBPD), but we observed no significant enrichment for any gene ontology (GO) categories, likely due to the small number of genes (FDR, > 0.05 after Bonferroni correction [23, 24]).
Fig 3.

Expression of PER1 is uniquely sensitive to low doses of glucocorticoids. (A) Dexamethasone dose-response curves for all 28 genes responsive to 5 nM DEX, as determined by RNA sequencing. Cells were treated for 1 h with increasing doses of DEX (x axis), and gene expression relative to control-treated cells was plotted (y axis). Of the responsive genes, PER1 (indicated in blue) was particularly sensitive and showed the strongest response to ≤5 nM DEX. (B) Immunoblot of PER1 and actin (as a loading control) after 4-h treatment with the indicated dose of DEX. (C) GR occupancy, as measured by ChIP-seq, in the region surrounding the PER1 transcription start site. Absent DEX, no occupancy was detected in the region (top line), whereas binding upstream of PER1 occurred after addition of 0.5 nM DEX (second line), and binding in the first intron of PER1 only occurred after addition of 5 nM DEX.
Of all DEX-responsive genes in A549 cells, PER1 was the most sensitive to low doses of DEX. We estimated 50% of the PER1 expression response (EC50) occurred at 0.47 nM DEX, a concentration more than 6-fold lower than the mean EC50 of 3.1 nM for all other identified genes. PER1 also had the greatest change in expression across the doses tested, with a 2.8-fold induction of mRNA expression at 0.5 nM DEX and a 5.8-fold induction at 5 nM DEX, as well as a corresponding increase in PER1 protein production after 4 h of DEX treatment (Fig. 3B). For comparison, the next most sensitive gene, for angiopoietin-related protein 4 (ANGPTL4), was expressed at a 26% higher level with 0.5 nM DEX and 3.3-fold higher at 5 nM DEX. Notably, numerous genes that do not respond to low doses are induced more strongly than PER1 at saturating doses of DEX (e.g., PTGER4, SOCS1, SPRY1, RASD1, and PSORS1C2) (50), showing that the sensitivity of PER1 expression to DEX is independent of the magnitude of expression response.
We found two GR binding sites near the PER1 transcription start site (TSS), one located in the first intron and the second located 2 kb upstream of the TSS. Both the intronic and upstream sites contain a GR binding motif near the site of maximal ChIP-seq signal. Both sites are conserved in the mouse genome, where the syntenic binding sites are thought to regulate glucocorticoid-mediated Per1 expression in peripheral mouse tissues (36) (see Fig. S4 in the supplemental material). While both sites are bound by the GR at 5 nM DEX, only the upstream site is bound with 0.5 nM DEX (Fig. 3C), suggesting that the upstream site is responsible for the hypersensitivity of PER1 expression to DEX.
A single GR binding site is sufficient for the hypersensitive response to DEX and cortisol.
To test whether the low-dose PER1 response is driven by DNA sequence alone, we cloned regions containing the endogenous PER1 promoter and either one or both of the nearby GR binding sites into a destabilized luciferase reporter vector. As a control, we also cloned the promoter and GR binding site for the gene for stomatin (STOM), a gene that is expressed only in response to high doses of DEX (Fig. 4A). After transient transfection of each construct into A549 cells, the cells were exposed for 4 h to doses of DEX ranging from 0.05 nM to 100 nM. In all experiments, the reporter expression clearly reached saturation by the 100 nM dose. Regions containing the hypersensitive GR binding site drove luciferase expression, with an EC50 of ∼0.25 nM, whereas regions lacking the hypersensitive GR binding site upstream of PER1 were substantially less responsive to DEX and responded similarly to the mouse mammary tumor virus promoter (Fig. 4B). These results indicate that the DNA sequence of the GR binding region upstream of PER1 is sufficient to drive hypersensitive expression of PER1.
Fig 4.

The GR binding sites flanking the PER1 transcription start site are sufficient to drive gene expression outside the genome. (A) Diagram of the genomic regions that were cloned into a promoterless luciferase reporter vector. Three regions were cloned from the PER1 locus. Two regions (“Full” and “Upstream,” shown in red and blue, respectively) contain the GR binding site upstream of the PER1 transcription start site. A third region (“Intron,” in purple) only contains the intronic GR binding site. For both the full and intron reporter, luciferase was maintained in phase with the PER1 start codon; for the upstream reporter, cloning included the endogenous promoter and stopped at the first nucleotide of the annotated transcription start site. As an additional control, a pair of GR binding sites upstream of the STOM gene were also cloned into the same reporter vector. (B) Luciferase expression, as a percentage of maximal response (y axis), across increasing doses of DEX (x axis) for each reporter construct. Each point indicates mean ± the standard error of the mean (SEM) for 8 biological replicate measurements, and the line indicates a fitted dose-response curve with fixed minimum and maximum but variable Hill slope. (C) Response to DEX of the PER1 reporter fragments with the upstream GR binding sequence converted to match the intronic GR binding sequence (inset). As for panel B, points indicate means ± SEM for 8 biological replicates, and lines indicates the fitted dose-response curve with variable Hill slopes.
The specific GR binding sequence at the site upstream of the PER1 TSS differs from the downstream intronic binding sequence by 3 nucleotides (1 nucleotide in the GR consensus sequence and 2 nucleotides in the spacer between the GR half-sites). To confirm our earlier results, that the specific GR binding is not important for hypersensitive GR binding, we interconverted the two binding sequences while leaving the local DNA context intact and looked for an effect on reporter sensitivity. Converting the upstream binding sequence to the intronic sequence did not diminish the hypersensitivity of the response (Fig. 4C). Therefore, consistent with the lack of a GR binding sequence specific to hypersensitive GR binding sites, the particular version of the GR binding sequence is not responsible for the observed differences in the DEX sensitivities between the two binding sites flanking the PER1 TSS.
Having confirmed that the specific sequence of the GR DNA recognition motif did not alter sensitivity to DEX, it may instead be that the sensitivity to DEX results either from the location of the binding site relative to the TSS or from interaction with the endogenous promoter. To test this hypothesis, we cloned DNA flanking each of the PER1 GR binding sites in front of a minimal promoter driving luciferase (Fig. 5A). We found that a minimal 274-bp region flanking the upstream GR binding site was sufficient to drive expression, with an EC50 of 0.25 nM DEX (Fig. 5B). Cortisol produced a similar response, with an EC50 of 21 nM for the GR binding site upstream of PER1 and 59 nM for the site in the first intron of PER1 (Fig. 5C). The result was important, as it showed that the difference in sensitivity between the two sites is not an artifact of DEX. Enhancer regions shorter than 274 bp incrementally reduced the sensitivity of the reporter to DEX, and the response of the shortest region tested (69 bp) was indistinguishable from that of either the intronic PER1 binding site or the MMTV promoter (Fig. 5D).
Fig 5.

Minimal enhancer region sufficient to drive a hypersensitive glucocorticoid response. (A) Regions surrounding the hypersensitive GR binding site upstream of the PER1 transcription start site were cloned in front of luciferase with a minimal promoter. Also, regions surrounding the nonhypersensitive GR binding site in the first intron of PER1 were cloned into the same reporter (not shown) and are referred to as the “intronic enhancer.” (B) Reporter plasmids were transiently transfected into A549 cells and treated for 1 h with doses of DEX (x axis). For each construct, the response (log fold change) was calculated compared to control-treated cells and then normalized to the maximal response observed. Error bars indicate standard errors of the means, and curves indicate the fitted dose-response curve with a fixed maximum and minimum and a variable slope. Each line indicates a different enhancer, as indicated, with regions containing the hypersensitive GR binding site upstream of PER1 shown in the same colors as in panel A. MMTV results show the dose-response curve of the MMTV promiter, driving the same luciferase. (C) Dose-response curves of the PER1 enhancer regions in response to cortisol treatment. (D) EC50s, estimated by fitting dose-response curves to each experiment's results. Error bars indicate 95% confidence intervals of the estimates. (E) Hill coefficients of the fitted dose-response curves for each enhancer, with error bars indicating 95% confidence intervals. Values close to 1 indicate first-order response kinetics, whereas values close to 2 indicate second-order kinetics.
We also evaluated whether there was evidence for cooperativity that might be particular to the hypersensitive GR binding sites. To do so, we evaluated the maximal slope of the dose-response curve, known as the Hill coefficient. A Hill coefficient of 1 indicates monomeric association of GR with DNA, while greater Hill coefficients imply the presence of an allosteric activator that increases the affinity of corticosteroids to the GR (47). While the normally responsive enhancers had a Hill coefficient close to 1, the hypersensitive enhancers responded with a Hill coefficient near 2 (Fig. 5E), suggesting that proteins may interact with the GR away from the DNA as part of effecting the more sensitive response. In this case, it may be that the hypersensitive GR enhancer is both prebound by cofactors that establish and maintain an open chromatin state and also contains a composite recognition sequence specific to a particular GR-containing complex that has a greater affinity to corticosteroids.
To probe for key nucleotides within the minimal necessary hypersensitive enhancer and that are important for tuning the sensitivity of GR binding, we used site-directed mutagenesis to introduce 2-bp mutations proximal to the GR binding site (Fig. 6A). Mutation of one site located 13 nucleotides upstream of the GR binding sequence had a mild effect on dose sensitivity. Independently mutating a second site, located 4 nucleotides upstream of the GR binding sequence, entirely ablated both hypersensitivity and the higher-order dose-response kinetics observed with the wild-type enhancer (Fig. 6C and D), indicating the presence of a nearby cofactor binding site essential for hypersensitivity. While we observed binding of CREB and FOXA1 in the same enhancer region, the introduced mutation appeared to disrupt a consensus DNA binding motif for the NFI family of transcription factors. Members of the NFI family have been shown to interact with the GR, and it is not yet clear if that interaction may contribute to sensitizing the PER1 response to low concentrations of glucocorticoids (37). To test if the mutated sequences were sufficient to sensitize a GR binding site, we introduced select sequences flanking the GR binding sequence from the hypersensitive enhancer element into the corresponding region of the intronic binding site. However, we observed no increase in sensitivity in the hybrid enhancer (see Fig. S4 in the supplemental material). It is therefore likely that within the minimal 274-bp enhancer identified, additional unidentified protein recognition sequences also contribute to the enhancement of gene expression at low doses of corticosteroids.
Fig 6.
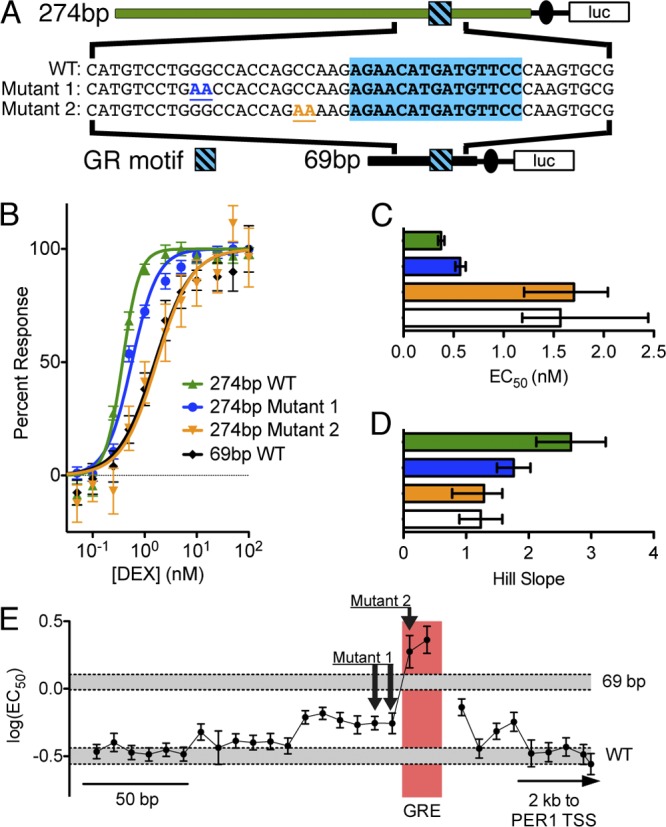
Select nucleotides within the minimal hypersensitive enhancer are necessary for the low-dose response. (A) Diagram of mutations introduced into the hypersensitive GR response element via site-directed mutagenesis. The blue box indicates the GR binding motif in the enhancer. WT indicates the wild-type sequence and, for both mutated sequences, the changed bases are colored and underlined. (B) A549 cells were transiently transfected with mutated plasmids and then treated for 4 h with increasing doses of DEX (x axis), and luciferase activity relative to a control treatment was calculated (y axis) and plotted, with error bars representing standard errors of the means. Lines indicate dose-response curves fit to each reporter response. The wild-type sequence (green) had the most sensitive response, whereas the mutated sequences (blue and orange) had less hypersensitive and normally sensitive responses, respectively. Black indicates a 69-bp enhancer element derived from the same binding site but which lacks hypersensitivity. (C) EC50 values were calculated from dose-response curves for wild-type and mutated enhancer elements, with error bars indicating 95% confidence intervals. (D) Hill coefficients for all four enhancer elements presented in panel B, with error bars indicating 95% confidence intervals. While the wild-type enhancer had second-order kinetics, one mutant (blue) had intermediate kinetics, and the second mutant (orange) had first-order kinetics that were indistinguishable from the 69-bp enhancer region. (E) Mutation scanning results for the enhancer region. Each point represents the effect on sensitivity to DEX of replacing 10 bp of wild-type DNA with (AC)5. Error bars represent 95% confidence intervals of the fit to the log10(EC50). WT and 69 bp indicate 95% CIs of the wild-type sequence of the 235-bp enhancer and of the truncated 69-bp enhancer, respectively.
To map key nucleotides throughout the hypersensitive enhancer region, we introduced mutations along the length of the hypersensitive region (see the methods described in the supplemental material) and measured sensitivity to DEX (Fig. 6E). The mutation scanning confirmed the effects of the 2-bp mutations tested in Fig. 6A, as well as identifying numerous other regions where mutations affected sensitivity. While nucleotides important to hypersensitivity were found throughout the enhancer region, many had modest effects on the EC50. These results indicated that the sensitivity of PER1 expression is unlikely to result from a single additional recognition motif, such as the previously reported GME (28). Instead, it is more likely that a combination of DNA sequences contribute to the expression of PER1 at low concentrations of glucocorticoids.
Hypersensitive PER1 expression is general to cell line and conserved in the mouse.
To determine if the sensitivity of PER1 expression to glucocorticoids was specific to the A549 cell line or a more general phenomenon, we measured enhancer activity in the human endometrial cell line ECC-1. Overall, higher doses of DEX were needed to drive reporter expression in ECC-1 cells, consistent with expectations, given the >20-fold-lower expression of GR in ECC-1 cells (J. Gertz, unpublished data). Despite the overall diminished glucocorticoid sensitivity in ECC-1 cells, the hypersensitive enhancer element derived from the region upstream of PER1 still responded to lower concentrations of DEX than the intronic enhancer elements (Fig. 7A and B). The 3-fold difference in sensitivities of the upstream versus intronic enhancer elements in ECC-1 cells was less than the 6-fold increase in sensitivity we found in A549 cells and may indicate intermediate sensitivity of PER1 expression in ECC-1 cells. In addition, evidence of a cooperative response that we observed in A549 cells was not recapitulated in ECC-1 cells (Fig. 7C). Therefore, we hypothesize that the hypersensitive expression of PER1 results from the combination of at least two mechanisms, one that increases sensitivity without altering response kinetics and another that increases both sensitivity and kinetics, and that only the former mechanism is active in ECC-1 cells.
The GR binding region that drives hypersensitive PER1 expression in A549 cells is conserved in the mouse genome, suggesting that mouse Per1 expression may also be more sensitive to glucocorticoids (36). To test whether the PER1 response is conserved in mice, we used RNA sequencing to measure genome-wide gene expression after administration of low doses of DEX to the mouse pituitary cell line AtT-20. Confirming our hypothesis, we found that four genes were unusually sensitive to DEX in the mouse pituitary cells, including Per1 (Fig. 7D). The other genes (Irs2, Klf15, and Necap2) play a role in the insulin response (30), glucose homeostasis (20, 21), and clathrin-mediated endocytosis (51), albeit primarily in tissues other than mouse pituitary. Studies of neuroendocrine function of the insulin response and Irs2 in particular have pointed to a possible role of insulin in regulating gonadotropin levels that may be linked with infertility resulting from diet-induced obesity (8, 10, 71). Many have noted that gonadotropin release varies diurnally during and after puberty, and while primarily due to hypothalamic control of gonadotropin-releasing hormone (42), corticosteroids may offer an additional level of regulation. In A549 cells, we have found that the GR binding sites near PER1 are conserved in the mouse and likely explain the observed sensitivity of Per1 expression (36) (see Fig. S4 in the supplemental material). We did not find GR binding sites near Irs2, Klf15, or Necap2 in A549 cells, indicating that their regulation is either tissue or species specific. Together, these results show both that that the regulation of PER1 expression by low concentrations of glucocorticoids is general to multiple cell types, conserved in the mouse, and also part of a larger set of similar responses in other tissues.
Targeted expression of PER1 by low-dose glucocorticoids drives circadian gene expression.
As shown above, a 1-h subsaturating low-dose corticosteroid treatment was sufficient to enhance expression of PER1 in mouse and human cells. The concentration at which we observed PER1 overexpression in A549 cells was similar to the low levels of free serum cortisol during the day (14). This suggests that triggering PER1 expression at low levels of cortisol may have a specific impact on circadian rhythms in peripheral tissues. To test if targeted induction of PER1 expression by corticosteroids affects circadian gene expression, we treated A549 cells for 1 h with 0.5 nM DEX to induce expression of PER1. We then washed the cells thoroughly with fresh medium and followed the expression of a panel of additional circadian rhythm genes over the following 12 h.
The transcriptional regulation of both the core and peripheral circadian rhythms consists of a negative feedback loop involving both regulatory and physical interactions between the Period genes PER1 and PER2, the cryptochrome genes CRY1 and CRY2, and the complex between CLOCK and ARNTL (also called BMAL1). Over the 12 h following withdrawal of 0.5 nM DEX, we observed significant changes in mRNA levels of all of these genes except for CLOCK (Fig. 7E). The Period genes followed a similar pattern of expression, with both elevated PER1 and PER2 expression over the 3 h immediately following DEX treatment, followed by significantly lower expression from 4 to 8 h after withdrawal of DEX treatment. The cryptochrome genes exhibited significantly increased expression for the first 6 h after DEX removal. For ARNTL, expression was significantly decreased between 1 and 4 h after treatment, reaching a minimum expression 3 h after treatment. Expression of ARNTL then began to rise, and by 12 h, we found significant overexpression compared to untreated cells. Interestingly, increased expression of CRY1 and CRY2 coincides with repression of PER1, PER2, and ARNTL expression. A recent study showing that the CRY genes repress GR activity may explain the result (34). Together, our results show that transient induction of PER1 expression by glucocorticoids leads to expression changes in many circadian rhythm genes many hours later and in a pattern consistent with circadian rhythms (3). The regulation of other circadian rhythm genes did not appear to result from long-term retention of DEX in the cell culture medium, as PER1 remained overexpressed after 4 h of continuous exposure to 500 pM DEX (Fig. 7F). We also observed repression of CRY1, CRY2, and ARNTL after continuous treatment with 500 pM DEX. The repression is not likely to arise from direct regulation by GR, as our previous has showed that high doses of DEX increased expression of those genes (50), and it may instead result from continuous PER1 overexpression.
To determine if transient induction of PER1 expression was sufficient to establish oscillating patterns of gene expression, we repeated the experiment over a 48-hour period. The results confirmed our initial findings for the first 12 h, but we found no evidence for circadian oscillations in gene expression (see Fig. S6 in the supplemental material). These results showed that low doses of glucocorticoids are sufficient to regulate expression of circadian genes in a manner consistent with circadian rhythms, but not sufficient to entrain circadian rhythms. It may therefore be that endogenous cortisol acts through PER1 to shift already-established circadian rhythms. A recent study in adrenalectomized rodents supported our hypothesis, showing both that endogenous cortisol was required for circadian expression of rat rPer1 and that daily cortisol administration restored rPer1 expression and entrained circadian rhythms in lung and kidney (58). Combined with our study, these results suggest that GR-mediated responses to low doses of corticosteroids are important for mammalian peripheral circadian physiology.
DISCUSSION
The glucocorticoid receptor is pivotal to the physiological response to stress and peripheral circadian rhythms. The GR is broadly expressed, and nearly all cells have ready access to cortisol provided by the bloodstream. Therefore, how the GR separates stress and circadian effects remains an enigma. Here we have shown that, in A549 cells, the GR can drive expression of many glucocorticoid-responsive genes in a dose-dependent manner. Of the genes responding to <5 nM DEX, PER1 is uniquely sensitive in A549 cells, exhibiting 2-fold overexpression at 0.5 nM DEX, a dose at which no other genes have a significant response. To the best of our knowledge, our study is the first to show that in some tissue or cell types, low doses of glucocorticoids can directly regulate the expression of one and only one gene. A 274-bp enhancer upstream of PER1 is sufficient for a hypersensitive response to DEX or to cortisol, and the endogenous promoter or proximity to additional GR binding sites nearby does not affect the response. The region does not contain an instance of a previously reported glucocorticoid modulatory element (62). Instead, our results suggest that a complex recognition motif likely coordinates the binding of numerous additional factors, or perhaps even additional GR molecules through noncanonical binding motifs (60), that tune the glucocorticoid response. Furthermore, we have found that those prebound factors are associated with open chromatin. However, the identity of those factors, and their specific roles in remodeling chromatin or otherwise modulating the glucocorticoid responses, remains unclear.
Current models of GR binding suggest that chromatin remodeling is an integral component of GR activity (e.g., references 27 and 67). Pioneer factors, such as the FOXA family of TFs, actively open regions of chromatin and in turn promote GR occupancy (5, 13, 40). The GR itself can also act to remodel chromatin at some sites, and in doing so it appears to assist in the loading of additional factors into the same region (67). Our data revealed that increased chromatin availability prior to GR occupancy is strongly associated with GR occupancy at low doses of glucocorticoids and significantly more so than for medium sensitive GR binding sites (P = 1 × 10−12, Fisher's exact test). In contrast, sites where the GR directs chromatin remodeling in response to DEX are uniformly bound at higher doses of DEX. The distinction is not absolute, and open chromatin is generally associated with GR binding regardless of sensitivity, suggesting that open chromatin only partially contributes to tuning glucocorticoid responses and that genetic mechanisms are also likely to be important. For example, mutation mapping of the hypersensitive PER1 enhancer revealed that a number of DNA sequences outside the core DNA binding motif contribute to the sensitivity of the corticosteroid response. These sites may contribute both to the occupancy of pioneer factors as well as to the prior recruitment of additional interacting transcription factors and cofactors that would in turn decrease the Kd of the GR for DNA upstream of PER1. In this model, low concentrations of DEX would result in a low concentration of active GR in the nucleus, which would only be able to productively bind the hypersensitive sites. Fully confirming the role of open chromatin will require additional experiments to determine if disrupting the chromatin state near hypersensitive GR binding sites affects the sensitivity of those sites.
Given the potential importance of open chromatin for hypersensitive GR binding, it is interesting that we see similar expression responses originating from plasmid reporters. Transiently transfected plasmids are known to have incomplete nucleosome structure and often to not faithfully model chromosomal DNA structure (49, 62). It may be that the minimal enhancer element contains enough information to establish an open chromatin state on the plasmid, or that incomplete nucleosome structure on transfected plasmids may be permissive to GR binding. Our observed associations with open chromatin in the genome may therefore reflect prebound TFs that help to recruit the GR rather than a strict prerequisite for gene expression responses to low glucocorticoid concentrations. Alternatively, while our motif analysis did not indicate specific GR binding sequences associated with increased or decreased sensitivity, we cannot exclude the possibility that some GR binding motifs may limit remodeling and influence sensitivity. Resolution of whether indeed plasmid chromatin structure matches that seen on the genome at the PER1 response elements may therefore provide further insights into if and how chromatin is established near hypersensitive GR binding sites and the extent to which that chromatin may tune glucocorticoid responses. One possible approach is to determine how stably integrated versions of hypersensitive GR binding sites respond relative to transfected plasmids. Alternatively, it is now possible to directly modify genomic GR binding regions, allowing study of the genetics and epigenetics of glucocorticoid responses in the native chromatin context (11, 37).
As described recently, monomeric GR interacting with DNA (even at a high-affinity site) is expected to follow a first-order dose response (47, 63). In that model, increased affinity of the GR for DNA at hypersensitive sites would shift the dose-response curve toward lower concentrations of DEX but would not cause an increase in the steepness of the dose-response curve (i.e., the Hill coefficient). The model further predicts, as we see in many of our nonhypersensitive response curves, that a Hill coefficient of 1 is characteristic of diffusion-mediated interactions of the GR with DNA. In our results, however, the PER1 enhancer elements that respond to a low concentration of DEX follow a steeper dose-response curve in A549 cells. That change in the shape of the dose-response curve suggests that cooperative binding of the GR with other molecules also contributes to the sensitivity of the response (47). It may be that a fraction of nuclear GR binds effector molecules prior to binding chromatin and that that complex then binds specific regions of the genome at a composite DNA binding motif.
Consistent with the model showing that both increased affinity to DNA and cooperative interactions with other molecules contribute to the hypersensitive gene expression response, deletion constructs that respond to higher DEX concentrations in A549 cells also lack the higher-order response. Meanwhile, in ECC-1 cells, the higher-order dose response is not evident despite increased sensitivity, indicating that the two mechanisms may be distinct and tissue specific. Together, the results point to a model where a combination of chromatin state, occupancy of additional transcription factors and cofactors, and cooperative interactions with other molecules work together to tune GR binding at specific genomic loci, including a locus responsible for the regulation of PER1.
The level of cortisol required for expression from the GR binding site upstream of PER1 was 21 nM, below the normal range of plasma cortisol. However, a substantial fraction of plasma cortisol is bound by proteins, such as corticosteroid binding globulin (32), and is unavailable to activate the GR. Estimates of free cortisol levels are as low as 5 nM in blood (3, 14, 57) and in tissues (57), similar to the range that we expect would be required to dynamically regulate PER1 expression through the day.
Cortisol released from the adrenal gland plays an important role in the regulation of PER1 in some but not all mouse peripheral tissues and is an important messenger in peripheral circadian rhythms (36, 57). Some peripheral clocks can be entrained independent of neuronal signals by restricted feeding times, suggesting additional clock mechanisms (9, 58). Researchers have shown that either a serum shock or a DEX shock can induce circadian patterns of gene expression in tissue culture cells (3, 4) and that glucocorticoids can also influence circadian rhythms in peripheral tissues (3). Our work suggests that hypersensitive PER1 expression may contribute to peripheral circadian timing by controlling the time of day when PER1 is first expressed. Supporting our hypothesis, a recent study of adrenalectomized rats showed that glucocorticoids are essential for oscillation in rPer1 expression and that daily injection of cortisol into the adrenalectomized rodents entrained circadian rhythms (58). These results suggest that cortisol-mediated regulation of PER1 is an important component of circadian gene expression in peripheral tissues. Our time course study also showed that triggering of PER1 by low-dose glucocorticoids is sufficient to regulate expression of other circadian rhythm genes many hours later. That regulation reveals coordinated expression of PER1 and PER1, as well as of CRY1 and CRY2. In our study, PER1/2 expression gave way to CRY1/2 gene expression, which was followed by expression of ARNTL. While we did not observe clear circadian oscillation after specific induction of PER1 expression, the order of expression matched that of in vivo studies of circadian gene expression (3, 58). Together, these studies suggest that GR regulation of PER1 plays an important role in maintaining circadian rhythms in peripheral tissues and that specific induction of PER1 may help better dissect the regulatory network controlling circadian gene expression.
The ability to regulate circadian rhythms via a highly targeted PER1 response may ultimately be pharmacologically useful. Abnormal circadian rhythms, including abnormal cortisol levels, are involved in many diseases, including depression (29), schizophrenia (15), and metabolic syndrome (41). Correcting the circadian oscillation of cortisol presents a treatment option in some patients (18). In such cases, the use of low doses of corticosteroids to target expression of PER1 and to renormalize circadian rhythms and related metabolic oscillations may provide a novel treatment option that is free of typical and serious side effects of high-dose exogenous glucocorticoids (16). It may be that different cortisol levels regulate different physiological functions. Testing that hypothesis will require studies in diverse primary tissues to understand if the genes sensitive to low doses of glucocorticoids coordinately regulate specific pathways or functions.
Supplementary Material
ACKNOWLEDGMENTS
We thank Greg Barsh, Chris Gunter, and members of the Myers lab for useful discussions and advice.
This work was funded by NHGRI ENCODE grant 5U54HG004576. Support for T.E.R. was from NIH/NIAMS fellowship 5T32AR007450.
Footnotes
Published ahead of print 16 July 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Almawi WY, Melemedjian OK. 2002. Negative regulation of nuclear factor-κB activation and function by glucocorticoids. J. Mol. Endocrinol. 28:69–78 [DOI] [PubMed] [Google Scholar]
- 2. Bailey TL, et al. 2009. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 37:W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balsalobre A, et al. 2000. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science 289:2344–2347 [DOI] [PubMed] [Google Scholar]
- 4. Balsalobre A, Damiola F, Schibler U. 1998. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 93:929–937 [DOI] [PubMed] [Google Scholar]
- 5. Belikov S, Astrand C, Wrange O. 2009. FoxA1 binding directs chromatin structure and the functional response of a glucocorticoid receptor-regulated promoter. Mol. Cell. Biol. 29:5413–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Biddie SC, et al. 2011. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell 43:145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blind RD, Garabedian MJ. 2008. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J. Steroid Biochem. Mol. Biol. 109:150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brothers KJ, et al. 2010. Rescue of obesity-induced infertility in female mice due to a pituitary-specific knockout of the insulin receptor. Cell. Metab. 12:295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burioka N, et al. 2005. Dexamethasone influences human clock gene expression in bronchial epithelium and peripheral blood mononuclear cells in vitro. Chronobiol. Int. 22:585–590 [DOI] [PubMed] [Google Scholar]
- 10. Burks DJ, et al. 2000. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature 407:377–382 [DOI] [PubMed] [Google Scholar]
- 11. Cermak T, et al. 2011. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39:e82 doi:10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christoffels VM, et al. 1998. Glucocorticoid receptor, C/EBP, HNF3, and protein kinase A coordinately activate the glucocorticoid response unit of the carbamoylphosphate synthetase I gene. Mol. Cell. Biol. 18:6305–6315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cirillo LA, et al. 2002. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9:279–289 [DOI] [PubMed] [Google Scholar]
- 14. Conway-Campbell BL, et al. 2010. Glucocorticoid ultradian rhythmicity directs cyclical gene pulsing of the clock gene period 1 in rat hippocampus. J. Neuroendocrinol. 22:1093–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Costa e Silva JA. 2006. Sleep disorders in psychiatry. Metabolism 55:S40–S44 [DOI] [PubMed] [Google Scholar]
- 16. Debono M, et al. 2009. Modified-release hydrocortisone to provide circadian cortisol profiles. J. Clin. Endocrinol. Metab. 94:1548–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Bosscher K, Vanden Berghe W, Haegeman G. 2003. The interplay between the glucocorticoid receptor and nuclear factor-κB or activator protein-1: molecular mechanisms for gene repression. Endocr. Rev. 24:488–522 [DOI] [PubMed] [Google Scholar]
- 18. Gorwood P. 2010. Restoring circadian rhythms: a new way to successfully manage depression. J. Psychopharmacol. 24:15–19 [DOI] [PubMed] [Google Scholar]
- 19. Govindan MV. 2010. Recruitment of cAMP-response element-binding protein and histone deacetylase has opposite effects on glucocorticoid receptor gene transcription. J. Biol. Chem. 285:4489–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gray S, et al. 2007. Regulation of gluconeogenesis by Kruppel-like factor 15. Cell. Metab. 5:305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gum RJ, et al. 2003. Antisense protein tyrosine phosphatase 1B reverses activation of p38 mitogen-activated protein kinase in liver of ob/ob mice. Mol. Endocrinol. 17:1131–1143 [DOI] [PubMed] [Google Scholar]
- 22. He Y, Simons SS., Jr 2007. STAMP, a novel predicted factor assisting TIF2 actions in glucocorticoid receptor-mediated induction and repression. Mol. Cell. Biol. 27:1467–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang da W, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4:44–57 [DOI] [PubMed] [Google Scholar]
- 25. Imai E, Miner JN, Mitchell JA, Yamamoto KR, Granner DK. 1993. Glucocorticoid receptor-cAMP response element-binding protein interaction and the response of the phosphoenolpyruvate carboxykinase gene to glucocorticoids. J. Biol. Chem. 268:5353–5356 [PubMed] [Google Scholar]
- 26. Jensen ST, Liu JS. 2004. BioOptimizer: a Bayesian scoring function approach to motif discovery. Bioinformatics 20:1557–1564 [DOI] [PubMed] [Google Scholar]
- 27. John S, et al. 2011. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 43:264–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaul S, Blackford JA, Jr, Cho S, Simons SS., Jr 2002. Ubc9 is a novel modulator of the induction properties of glucocorticoid receptors. J. Biol. Chem. 277:12541–12549 [DOI] [PubMed] [Google Scholar]
- 29. Kennaway DJ. 2010. Clock genes at the heart of depression. J. Psychopharmacol. 24:5–14 [DOI] [PubMed] [Google Scholar]
- 30. Kido Y, et al. 2000. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J. Clin. Invest. 105:199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koubovec D, Ronacher K, Stubsrud E, Louw A, Hapgood JP. 2005. Synthetic progestins used in HRT have different glucocorticoid agonist properties. Mol. Cell. Endocrinol. 242:23–32 [DOI] [PubMed] [Google Scholar]
- 32. Koyanagi S, et al. 2006. Chronic treatment with prednisolone represses the circadian oscillation of clock gene expression in mouse peripheral tissues. Mol. Endocrinol. 20:573–583 [DOI] [PubMed] [Google Scholar]
- 33. La Baer J, Yamamoto KR. 1994. Analysis of the DNA-binding affinity, sequence specificity and context dependence of the glucocorticoid receptor zinc finger region. J. Mol. Biol. 239:664–688 [DOI] [PubMed] [Google Scholar]
- 34. Lamia KA, et al. 2011. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature 480:552–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25 doi:10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Le Minh N, Damiola F, Tronche F, Schutz G, Schibler U. 2001. Glucocorticoid hormones inhibit food-induced phase-shifting of peripheral circadian oscillators. EMBO J. 20:7128–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li T, et al. 2011. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 39:6315–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li X, Wong J, Tsai SY, Tsai MJ, O'Malley BW. 2003. Progesterone and glucocorticoid receptors recruit distinct coactivator complexes and promote distinct patterns of local chromatin modification. Mol. Cell. Biol. 23:3763–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu X, Brutlag DL, Liu JS. 2001. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac. Symp. Biocomput. 2001:127–138 [PubMed] [Google Scholar]
- 40. Lupien M, et al. 2008. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 132:958–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maury E, Ramsey KM, Bass J. 2010. Circadian rhythms and metabolic syndrome: from experimental genetics to human disease. Circ. Res. 106:447–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McCartney CR. 2010. Maturation of sleep-wake gonadotrophin-releasing hormone secretion across puberty in girls: potential mechanisms and relevance to the pathogenesis of polycystic ovary syndrome. J. Neuroendocrinol. 22:701–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meijsing SH, et al. 2009. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 324:407–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mercier L, Thompson EB, Simons SS., Jr 1983. Dissociation of steroid binding to receptors and steroid induction of biological activity in a glucocorticoid-responsive cell. Endocrinology 112:601–609 [DOI] [PubMed] [Google Scholar]
- 45. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621–628 [DOI] [PubMed] [Google Scholar]
- 46. Naray-Fejes-Toth A, Fejes-Toth G. 1997. 11-β-Hydroxysteroid dehydrogenase-2 is a high affinity corticosterone-binding protein. Mol. Cell. Endocrinol. 134:157–161 [DOI] [PubMed] [Google Scholar]
- 47. Ong KM, Blackford JA, Jr, Kagan BL, Simons SS, Jr, Chow CC. 2010. A theoretical framework for gene induction and experimental comparisons. Proc. Natl. Acad. Sci. U. S. A. 107:7107–7112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Onica T, et al. 2008. Dexamethasone-mediated up-regulation of human CYP2A6 involves the glucocorticoid receptor and increased binding of hepatic nuclear factor 4 alpha to the proximal promoter. Mol. Pharmacol. 73:451–460 [DOI] [PubMed] [Google Scholar]
- 49. Oshima H, Simons SS., Jr 1992. Modulation of transcription factor activity by a distant steroid modulatory element. Mol. Endocrinol. 6:416–428 [DOI] [PubMed] [Google Scholar]
- 50. Reddy TE, et al. 2009. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 19:2163–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ritter B, Blondeau F, Denisov AY, Gehring K, McPherson PS. 2004. Molecular mechanisms in clathrin-mediated membrane budding revealed through subcellular proteomics. Biochem. Soc. Trans. 32:769–773 [DOI] [PubMed] [Google Scholar]
- 52. Rotsztejn WH, Dussaillant M, Nobou F, Rosselin G. 1981. Rapid glucocorticoid inhibition of vasoactive intestinal peptide-induced cyclic AMP accumulation and prolactin release in rat pituitary cells in culture. Proc. Natl. Acad. Sci. U. S. A. 78:7584–7588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Roux J, Pictet R, Grange T. 1995. Hepatocyte nuclear factor 3 determines the amplitude of the glucocorticoid response of the rat tyrosine aminotransferase gene. DNA Cell Biol. 14:385–396 [DOI] [PubMed] [Google Scholar]
- 54. Sekiya T, Muthurajan UM, Luger K, Tulin AV, Zaret KS. 2009. Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 23:804–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Simons SS, Jr, Oshima H, Szapary D. 1992. Higher levels of control: modulation of steroid hormone-regulated gene transcription. Mol. Endocrinol. 6:995–1002 [DOI] [PubMed] [Google Scholar]
- 56. So AY, Chaivorapol C, Bolton EC, Li H, Yamamoto KR. 2007. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet. 3:e94 doi:10.1371/journal.pgen.0030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stavreva DA, et al. 2009. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat. Cell Biol. 11:1093–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sujino M, et al. 2012. Differential entrainment of peripheral clocks in the rat by glucocorticoid and feeding. Endocrinology. 153:2277–2286 [DOI] [PubMed] [Google Scholar]
- 59. Sun Y, Tao YG, Kagan BL, He Y, Simons SS., Jr 2008. Modulation of transcription parameters in glucocorticoid receptor-mediated repression. Mol. Cell Endocrinol. 295:59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Surjit M, et al. 2011. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 145:224–241 [DOI] [PubMed] [Google Scholar]
- 61. Szapary D, Huang Y, Simons SS., Jr 1999. Opposing effects of corepressor and coactivators in determining the dose-response curve of agonists, and residual agonist activity of antagonists, for glucocorticoid receptor-regulated gene expression. Mol. Endocrinol. 13:2108–2121 [DOI] [PubMed] [Google Scholar]
- 62. Szapary D, Oshima H, Simons SS., Jr 1992. Modulation of glucocorticoid induction of stably transfected tyrosine aminotransferase gene constructs involves elements up-stream of the glucocorticoid-responsive element. Endocrinology 130:3492–3502 [DOI] [PubMed] [Google Scholar]
- 63. Szapary D, Xu M, Simons SS., Jr 1996. Induction properties of a transiently transfected glucocorticoid-responsive gene vary with glucocorticoid receptor concentration. J. Biol. Chem. 271:30576–30582 [DOI] [PubMed] [Google Scholar]
- 64. Tsai MJ, O'Malley BW. 1994. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem. 63:451–486 [DOI] [PubMed] [Google Scholar]
- 65. Valouev A, et al. 2008. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods 5:829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vedder H, Weiss I, Holsboer F, Reul JM. 1993. Glucocorticoid and mineralocorticoid receptors in rat neocortical and hippocampal brain cells in culture: characterization and regulatory studies. Brain Res. 605:18–24 [DOI] [PubMed] [Google Scholar]
- 67. Voss TC, et al. 2011. Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell 146:544–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang JC, et al. 2004. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc. Natl. Acad. Sci. U. S. A. 101:15603–15608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang Q, Anzick S, Richter WF, Meltzer P, Simons SS., Jr 2004. Modulation of transcriptional sensitivity of mineralocorticoid and estrogen receptors. J. Steroid Biochem. Mol. Biol. 91:197–210 [DOI] [PubMed] [Google Scholar]
- 70. Weitzman ED. 1976. Circadian rhythms and episodic hormone secretion in man. Annu. Rev. Med. 27:225–243 [DOI] [PubMed] [Google Scholar]
- 71. Withers DJ. 2001. Insulin receptor substrate proteins and neuroendocrine function. Biochem. Soc. Trans. 29:525–529 [DOI] [PubMed] [Google Scholar]
- 72. Yamamoto T, et al. 2005. Acute physical stress elevates mouse Period1 mRNA expression in mouse peripheral tissues via a glucocorticoid-responsive element. J. Biol. Chem. 280:42036–42043 [DOI] [PubMed] [Google Scholar]
- 73. Zhang L, Rubins NE, Ahima RS, Greenbaum LE, Kaestner KH. 2005. Foxa2 integrates the transcriptional response of the hepatocyte to fasting. Cell. Metab. 2:141–148 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Results from all high-throughput sequencing experiments are available both on the UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgGateway; human genome version hg19) as well as at the HudsonAlpha website, http://hudsonalpha.org/sites/default/files/DataSets/Myerslab/Low_Dose_Glucocorticoid_Responses.