

Abstract
Sp1 is a ubiquitously expressed transcription factor that is phosphorylated by ataxia telangiectasia mutated kinase (ATM) in response to ionizing radiation and H2O2. Here, we show by indirect immunofluorescence that Sp1 phosphorylated on serine 101 (pSp1) localizes to ionizing radiation-induced foci with phosphorylated histone variant γH2Ax and members of the MRN (Mre11, Rad50, and Nbs1) complex. More precise analysis of occupancy of DNA double-strand breaks (DSBs) by chromatin immunoprecipitation (ChIP) shows that Sp1, like Nbs1, resides within 200 bp of DSBs. Using laser microirradiation of cells, we demonstrate that pSp1 is present at DNA DSBs by 7.5 min after induction of damage and remains at the break site for at least 8 h. Depletion of Sp1 inhibits repair of site-specific DNA breaks, and the N-terminal 182-amino-acid peptide, which contains targets of ATM kinase but lacks the zinc finger DNA binding domain, is phosphorylated, localizes to DSBs, and rescues the repair defect resulting from Sp1 depletion. Together, these data demonstrate that Sp1 is rapidly recruited to the region immediately adjacent to sites of DNA DSBs and is required for DSB repair, through a mechanism independent of its sequence-directed transcriptional effects.
INTRODUCTION
Transcription factor Sp1 regulates the expression of genes involved in cell proliferation, DNA repair, and apoptosis/senescence (9). DNA binding of Sp1 is mediated through three zinc fingers in the C-terminal region, which recognize GC-rich elements in a large number of promoters that are frequently found in euchromatic CpG islands. Posttranslational modifications throughout Sp1, including phosphorylation, acetylation, O-linked glycosylation, and sumoylation, modulate its interaction with chromatin remodeling factors, DNA, transcription machinery, and other transcription factors to enhance or repress gene expression (13, 35, 36, 40, 51, 53, 56, 69, 82). Our group and others have shown that transcription factor Sp1, which contains two S/TQ cluster domains (SCDs), characteristic of phosphoinositide 3-kinase-like kinase (PI3KK) substrates, is phosphorylated by the ataxia telangiectasia mutated kinase (ATM) in response to ionizing radiation, H2O2 (64), and other DNA-damaging agents (unpublished data), as well as herpesvirus infection (33).
Genomic stability is maintained by the cellular response to DNA damage. In response to DNA double-strand breaks (DSBs), ATM is activated (80) and initiates a cascade of DNA damage signals by phosphorylation of hundreds of proteins involved in cell cycle checkpoint activation, DNA repair, and apoptosis, including p53, Chk2, γH2Ax, BRCA1, and Nbs1 (48, 57). Mutations in the ATM gene result in the autosomal recessive disorder ataxia telangiectasia (AT), which is characterized by radiation sensitivity, immunodeficiency, neurodegeneration, and cancer predisposition (79). Cells derived from AT patients exhibit increased chromosome breaks, defects in cell cycle checkpoints, and increased sensitivity to ionizing radiation (IR) (66, 78). Inactive ATM forms homodimers and is activated by intermolecular phosphorylation at serine 1981 and dissociation into monomers (4). The complex of Mre11, Rad50, and Nbs1 (MRN) binds to DSBs, facilitates the recruitment of ATM through direct interaction with the C terminus of Nbs1 (18, 50, 93), and enhances phosphorylation of ATM substrates (45, 48, 49, 87). The MRN complex enhances ATM activation and thereby affects phosphorylation of several ATM substrates, including Chk2 and p53 (11, 24, 25, 83).
Many of the proteins involved in the DNA damage response can be visualized by indirect immunofluorescence in discrete foci known as ionizing radiation-induced foci (IRIF). Each of the foci is thought to correspond to a single DSB, in which multiple proteins have assembled around a DNA lesion to facilitate repair processes. The recruitment of many DNA repair proteins to IRIF is dependent on γH2Ax, a variant of H2A, which is phosphorylated in the vicinity of DSBs and is detected by an antibody that recognizes pSer139 (73). Colocalization at foci with γH2Ax is indicative of proteins playing various roles in the DNA damage response (19). Many key components of the DNA damage response, including ATM, Mdc1, 53BP1, Smc1, Rad51, and the MRN complex (5, 26, 29, 55, 84), colocalize with γH2Ax in IRIF (79). In addition to factors known to play a role in DNA repair, several transcription factors, like Sp1, are substrates of ATM, including CREB, p53, E2F1, and ATF2; with the exception of CREB, these transcription factors localize to damage-induced foci (8, 54, 61, 76), although the timing and function of their recruitment is not entirely understood (8, 12, 21). Here, we demonstrate that Sp1 is recruited in close proximity to DSBs, where it promotes repair, independent of sequence-specific DNA binding.
Materials and Methods
Cell lines.
Normal human diploid fibroblast (NHDFs; Clonetics) and human osteosarcoma cell line U2OS (ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM; Cellgro, Mediatech, Inc.) with 10% fetal bovine serum (FBS; Gemini), 2 mM l-glutamine (Cellgro, Mediatech, Inc.), 110 mg/ml sodium pyruvate (Sigma), 0.1 mg/ml penicillin, and 60 μg/ml streptomycin (Pen-Strep) in a 37°C humidified atmosphere of 5% CO2. During infection of U2OS cells with I-PpoI endonuclease fusion construct and treatment with 4-hydroxytamoxifen (4-OHT), the cells were maintained in phenol-free DMEM with 10% charcoal-stripped FBS (Gemini). The viral packaging cell line 293-GPG (VSV-G) was maintained in DMEM containing 10% heat-inactivated FBS, 2 mM l-glutamine, 110 mg/ml sodium pyruvate, Pen-Strep, 1 μg/ml tetracycline, 2 μg/ml puromycin, and 0.3 mg/ml G418 at 37°C in a humidified atmosphere of 5% CO2. During production of short hairpin RNA (shRNA)-expressing lentiviruses or pLKO retrovirus constructs, 293-GPG and 293T cells were maintained in DMEM containing 10% heat-inactivated FBS, 2 mM l-glutamine, 110 mg/ml sodium pyruvate, and Pen-Strep.
Cell treatment.
For the experiments using ionizing radiation, exponentially growing cells were irradiated at room temperature using a Siemens Primus linear accelerator (Radiation Oncology Department, Hahnemann University Hospital) at 6 MV energy at a dose rate of 3 Gy/min.
Antibodies and Western blot analysis.
For Western blot analysis, cells were directly lysed in 2× SDS sample buffer (12.5 mM Tris [pH 6.8], 20% glycerol, 4% [wt/vol] SDS, 0.01 mg/ml bromophenol blue). Proteins were separated by traditional SDS-PAGE, transferred to a polyvinylidene difluoride (PVDF) membrane, and analyzed by immunoblotting with the following antibodies: Sp1 (pAb581) (53), Sp1 (pS101) (64), γH2Ax (pS139) (Millipore), α-tubulin (Sigma), Nbs1 (pS343) (Millipore), Nbs1 (generously provided by John Petrini, Sloan-Kettering Cancer Center), ATM (pS1985) (Rockland), HA (Cell Signaling), and nucleolin (Santa Cruz). Licor infrared imaging was used for secondary detection.
Plasmid constructs.
The hemagglutinin (HA)–estrogen receptor (ER)–I-PpoI construct was kindly provided by M. Kastan (St. Jude Children's Hospital, Memphis, TN). pLKO-shRNA vectors for Sp1 and Nbs1 shRNA sequences were acquired from Sigma, and targets begin at nucleic acid 1805 (coding region, number 1) and 7276 (3′ long terminal repeat [LTR], number 2) of the Sp1 mRNA sequence and nucleic acid 705 of the Nbs1 sequence. Viral packaging vectors, pCMV-VSV-G, pRSV-Rev, and pMDLg/pRRE, were generously donated by M. Reginato (Drexel University College of Medicine, Philadelphia, PA). pLXSN-Sp1-damage response domain (DRD) was constructed from pLXSN-FLAG-Sp1-HA (3). SalI restriction sites were inserted adjacent to codon 183 of the Sp1 sequence using PCR and the following primers: Forward (5′ CCC ACA GTT CCA GAC CGT CGA CGG GCA ACA GCT GCA G 3′) and Reverse (5′ CTG CAG CTG TTG CCC GTC GAC GGT CTG GAA CTG TGG G 3′). An additional SalI site was inserted along with a nuclear localization signal (38) since the region reportedly required for nuclear localization of Sp1 is not in the first 182 amino acids (77). This was done using PCR and the following primers: Forward (5′ CAG TGG CAA TGG CTT CGT CGA CCC AAA GAA GAA GCG CAA GGT CTA CCC ATA CGA TGT TCC AG 3′) and Reverse (5′ CTG GAA CAT CGT ATG GGT AGA CCT TGC GCT TCT TCT TTG GGT CGA CGA AGC CAT TGC CAC TG 3′). Mutated Sp1 vector was then cut with SalI and self-ligated to make pLXSN-Flag-Sp1Δ183-785-NLS-HA (pLXSN-Sp1-DRD). For ChIP experiments in which Sp1-DRD is transduced, TET-inducible shRNA constructs, expressing Sp1 shRNA 1, were constructed as previously described (88, 89). The TET vector was virally packaged like other pLKO vectors and expressed in U2OS cells. These cells were then clonally selected to identify those that gave the largest induction of shRNA in response to doxycycline. shRNA inductions were done using 50 μg/ml doxycycline for 72 h, with fresh doxycycline added after 48 h.
Viral production.
293-GPG cells were transfected with 10 μg of HA–ER–I-PpoI (7), pLXSN-Sp1-DRD, or pLXSN plasmids using Genedrill transfection reagent (Bamagin) by following the manufacturer's instructions. Virus was collected on days 5 to 7 after transfection. 293T cells were cotransfected for 6 h with pLKO vectors containing the shRNA sequences along with viral packaging components. Virus was collected 48 h posttransfection and stored frozen at −80°C.
Laser-induced DNA DSBs.
Laser microirradiation was done as previously described (81). Cells were cultured on glass coverslips and incubated with 10 μM bromodeoxyuridine (10 mM stock BrdU; Sigma) for 24 to 36 h prior to laser irradiation by P.A.L.M. microbeam laser microdissection (Carl Zeiss MicroImaging). Laser output was set to 45%, with laser power set to 59%. Cells were visualized at ×40 magnification, and an average of 100 cells were exposed to the laser over a period of 1 to 3 min, each cell being exposed to the laser only for 500 milliseconds. Cells were immediately returned to the incubator until fixation in 2% paraformaldehyde and 3% sucrose.
Immunofluorescence.
U2OS cells were washed twice in ice-cold phosphate-buffered saline (PBS) and preextracted for 5 min in 0.2% Triton X-100–30% double-distilled water (ddH2O)–2.0 mM phenylmethylsulfonyl fluoride (PMSF) in PBS at 4°C. Cells were then washed with PBS and fixed in 2% paraformaldehyde and 3% sucrose in PBS for 10 min at room temperature. Cells were washed twice in PBS at room temperature and then permeabilized in 0.5% Triton X-100 in PBS for 5 min at 4°C. Cells were washed 2 times in room-temperature PBST (0.1% Tween 20, 0.02% sodium azide in PBS) and incubated in primary antibody overnight at 4°C. Antibodies used were γH2Ax (Millipore), pSp1, pNbs1 (Millipore), Nbs1 (Millipore), 53BP1 (Novus), and Sp1 (Santa Cruz). Cells were washed in PBST three times followed by the addition of secondary antibodies, Alexa Fluor 488–donkey anti-rabbit and Alexa Fluor 594–donkey anti-mouse antibodies (both from Invitrogen, diluted 1:1,000 in PBST), for 1 h at room temperature in the dark. Cells were stained with 0.5 μg/ml DAPI (4′,6-diamidino-2-phenylindole) in PBST for 5 min and then washed 4 times with PBST. Slides were mounted using Vecta Mount mounting medium (Vector Labs).
Chromatin immunoprecipitation.
Briefly, 1 × 106 U2OS cells were seeded on 150-cm dishes and infected 12 h later with retrovirus expressing HA-ER-I-PpoI for 6 h and then placed in charcoal-stripped FBS. The ER fusion allows for nuclear localization of the enzyme after 4-OHT treatment of cells. Seventy-two hours postinfection, I-PpoI was induced by the addition of 8 μM 4-OHT (Sigma) to cell medium for 6 h. For chromatin immunoprecipitations, the protocol from Nelson et al. (62) was followed. Briefly, cells were cross-linked with 1.5% formaldehyde in medium for 15 min. Reactions were quenched by the addition of 1 M glycine to a final concentration of 125 mM. Cells were then scraped and collected in 1× PBS. Cell pellets were then lysed in 1 ml ChIP lysis buffer (50 mM Tris [pH 7.5], 5 mM EDTA, 150 mM NaCl, 0.5% NP-40, 1% Triton X-100, and protease and phosphatase inhibitors). Chromatin was sheared by sonication using a Branson sonicator at 50% duty for a total of 50 s in 5 pulsed intervals, with 2 min on ice between each interval. Protein was quantified in each treatment using a standard bicinchoninic acid (BCA) assay, and samples were normalized to the same concentration. A total of 20% of the lysate was reserved, and genomic DNA was isolated as immunoprecipitation (IP) input. The remaining lysate was then equally aliquoted and used for immunoprecipitation with Sp1 preconjugated agarose beads (Santa Cruz catalog number sc-59AC), Nbs1, γH2Ax (Millipore catalog number 07-164), and IgG (Abcam catalog number ab46540) antibodies as a control. Antibodies other than Sp1 were preincubated with protein A/G agarose beads (Santa Cruz catalog number 2003). All beads were preblocked with 1 mg/ml low IgG bovine serum albumin (BSA) (Gemini Bio-Products) and 1 mg/ml tRNA (Sigma) prior to IP. For Sp1-DRD ChIP, lysates were immunoprecipitated with anti-Flag-M2-conjugated agarose (Sigma), preblocked as described above. Identical IPs were done from cells expressing the LXSN control vector as an IP control. Immunoprecipitated DNA was isolated overnight, and beads were washed 5 times with lysis buffer. DNA was purified using Chelex-100 resin as described previously (62). Analysis of protein binding around the break site was assessed by SYBR green (Bio-Rad) quantitative PCR (qPCR) with previously published primers (7) by using a Bio-Rad CFX-96 real-time PCR detection system. Fold induction of binding was calculated using a modified ΔΔCT method in which untreated and treated IP samples values are normalized to the difference in input DNA. Threshold cycle (CT) values for control IPs and GAPDH control primers were used to set gates for background amplification.
Coimmunoprecipitation.
The immunoprecipitation protocol was adapted from a previously described method (41). Cells were treated with 200 μM H2O2 for 1 h. At the conclusion of treatment, cells were washed twice with 1× PBS on ice and collected in 500 μl cold TGN buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1% Tween 20, 0.3% NP-40, and protease and phosphatase inhibitors). Cells were dispersed 5× with a tuberculin syringe and then sonicated twice at 40% duty for 10 s on ice. Protein was quantified and 1.8 to 2.5 mg of protein lysate was used for each IP. A total of 10% of the lysate was saved for input. Cells were immunoprecipitated with preconjugated Sp1 beads (Santa Cruz) preblocked for 1 h with 1 mg/ml BSA. IPs were completed overnight at 4°C. Beads were washed twice with TGN buffer and twice with 1× Tris-buffered saline (TBS). Protein was eluted with 2× SDS sample buffer. Precipitates were assessed by Western blotting as described above. Ethidium bromide (10 mg/ml stock) was added to lysate at a final concentration of 100 μg/ml prior to binding to antibody and beads (43).
DNA damage repair experiments.
For damage repair assays, U2OS cells were seeded and transduced overnight with medium containing I-PpoI retrovirus. Twelve hours postseeding, cells were placed in charcoal-stripped medium. Eight additional hours later, cells were transduced with shRNA virus, as described above, to knock down various targets. A total of 72 h after I-PpoI infections, I-Ppol was induced with 4-OHT. For measuring DNA breaks, cells were lysed in DNA isolation buffer (50 mM Tris [pH 8.0], 50 mM EDTA, 10 mM NaCl, and 1% SDS) at various time points after the addition of 4-OHT. Lysates were sonicated at 20% duty for 20 s to complete lysis and fragment chromatin. DNA was then RNase and proteinase K treated and purified as described above for ChIP. Sample concentrations were normalized, and analysis of DNA breaks was assessed by qPCR with previously published primers flanking the DNA break (7). The percentage of damaged DNA was calculated using the ΔΔCT method. The change in product from primers flanking the DAB1 break site was normalized to the change in product of primers that amplify an adjacent region 280 bp away from the cut site; the value calculated represents the fold change in product. This fold change was then converted to percentage of cut DNA by subtracting the value from 1 and multiplying by 100. For experiments in which the effect of Sp1-A on repair was addressed, cells transduced with a control vector (pLXSN) or the Sp1 peptide (pLXSN-Sp1-DRD) and neomycin selected were infected with virus packaged with Sp1 shRNA 1 to knock down endogenous protein, and experiments were completed as described above with I-PpoI. Experiments compare cells expressing LXSN control plasmid with nontargeting or Sp1 shRNA to those expressing Sp1 shRNA and Sp1-DRD.
RESULTS
Phosphorylated Sp1 localizes to ionizing radiation-induced foci.
We and others have previously shown that Sp1 is phosphorylated by ATM with kinetics similar to those of H2Ax (33, 64) and that phosphorylation at serine 101 acts as a priming event required for additional phosphorylation in response to hydrogen peroxide or ionizing radiation (33, 64). To further characterize the phosphorylation of Sp1 in response to DNA damage, we sought to establish the relationship between Sp1 phosphorylation and dose of ionizing radiation (IR). Western blots show increased phosphorylation of both Sp1 and H2Ax in response to increasing doses of IR. Total cellular Sp1 undergoes an electrophoretic mobility shift from predominately 95 kDa to 105 kDa (Fig. 1A), which corresponds to increased immune reactivity with an antibody that specifically recognizes Sp1 phosphorylated on S101 (pSp1). To determine if Sp1 localized to IRIF, U2OS cells were exposed to low levels of IR (0.5 Gy) followed by immunofluorescence using the pSp1 antibody. pSp1 was localized to discrete foci that are largely colocalized with γH2Ax (Fig. 1B). The specificity of the pSp1 antibody is demonstrated by the absence of pSp1 signal by IF in cells depleted of Sp1 by shRNA (Fig. 1B).
Fig 1.

Colocalization of pSp1 with ionizing radiation-induced foci. (A) Normal human diploid fibroblasts (NHDF) were exposed to various doses of ionizing radiation and harvested 30 min after exposure. Immunoblots were performed with antibodies directed to Sp1, pSp1, γH2Ax, or α-tubulin as indicated. (B) Immunofluorescence micrographs of U2OS expressing nontargeting or Sp1 shRNA exposed to 0.5 Gy of IR and fixed 60 min after exposure and immunostained as described for γH2Ax and pSp1.
Phosphorylated Sp1 is rapidly localized to DSB sites.
To further investigate the localization of Sp1 to sites of DNA DSBs and determine the kinetics of recruitment, we used laser microirradiation to induce DSBs in defined areas of the nucleus (39). Cells were processed for immunofluorescence at various time points after laser treatment. Five minutes after laser irradiation, γH2Ax was detected at the sites of damage (stripes), while only a faint signal was detected with pSp1 (Fig. 2). At 7.5 min postirradiation, localization of pSp1 was visualized and appeared to coalesce at the stripe, and the signal continued to become brighter up to 30 min.
Fig 2.

Temporal association of pSp1 at sites of DSBs. U2OS cells were prelabeled for 24 to 36 h with 10 μM BrdU. Cells were then subjected to laser microirradiation and fixed at the indicated time points after laser exposure. Laser stripe localization of pSp1 and γH2Ax was assessed by immunofluorescence.
Sp1 is localized in close proximity to DNA DSBs.
We next examined whether pSp1 colocalizes with members of the MRN complex. The MRN complex has been shown to bind double-strand break ends and enhance the cascade of phosphorylation of ATM targets and recruitment of repair factors (16, 24, 32, 49). Immunofluorescence analysis shows that pSp1 is colocalized with Nbs1 30 min after microirradiation (Fig. 3A).
Fig 3.
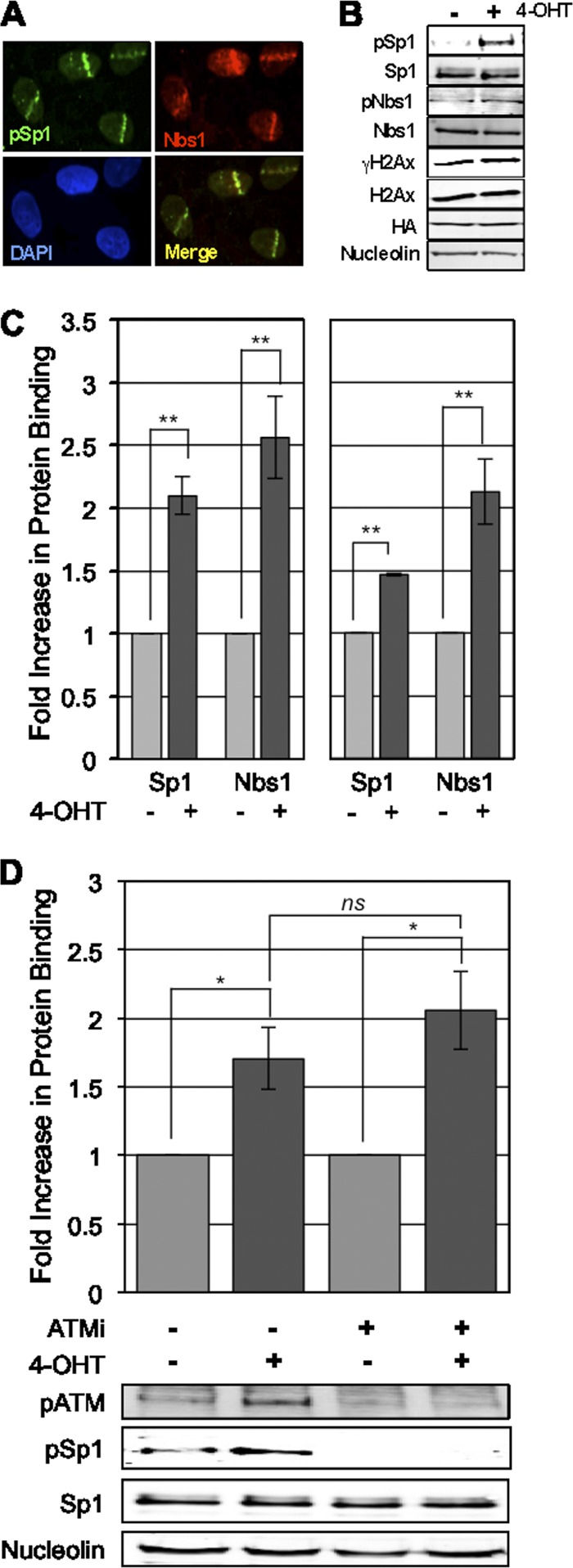
Sp1 localizes adjacent to break sites with members of the MRN complex. (A) U2OS cells were laser microirradiated and fixed 30 min after exposure. Localization of pSp1 and Nbs1 was performed by immunofluorescence using the indicated antibody and staining with DAPI. (B) Cells expressing I-PpoI homing endonuclease were induced with 8 μM 4-OHT to promote enzyme-induced double-strand breaks. Six hours postinduction cells were lysed and assessed for activation of DNA damage response. (C) Cells treated as described in the legend to panel B were isolated, and chromatin immunoprecipitations were performed for Sp1, Nbs1, and normal rabbit IgG; immunoprecipitated chromatin was assessed by SYBR green qPCR; CT values for IPs were compared to CT values for input DNA using the ΔΔCT method described in Materials and Methods. Binding to sites 280 bp from the DAB1 cut site and 489 bp from the rDNA cut sites is presented. IPs for IgG and primers for GAPDH control target were used to set gates for real CT values in tested IPs (values not shown here). Data represent means and standard errors of the means (SEM) from 3 independent experiments assessed in triplicate. (D) Experiments in panel C were repeated with cells pretreated with 10 μM KU-55933, an ATM-specific inhibitor, 1 h prior to 4-OHT inductions. Data graphed are the means and SEM from 2 independent experiments assessed in triplicate. Western blots below show reduced ATM autophosphorylation and phosphorylation of Sp1 in cells treated with the ATM inhibitor.
More direct association with DNA double-strand break sites has been shown through induction of sequence-specific DSBs by endonucleases, such as HO (74, 86), I-SceI (70, 71), and I-PpoI. I-PpoI can induce sequence-specific DNA DSBs in the mammalian genome (6, 20, 22, 59), and binding of proteins around the breaks can be mapped by chromatin immunoprecipitation and qPCR using different primer sets. I-PpoI endonuclease has a 15-bp recognition sequence (CTCTCTTAA cut GGTAGC), with approximately 300 endogenous targets sites within the human genome. I-PpoI's recognition sequence is found in the coding region of the 28S ribosomal DNA (rDNA) sequence, which is tandemly repeated at 5 chromosomal loci (30), as well as in intron 1 of the DAB1 gene on chromosome 1 (7). The enzyme produces about 20 to 30 DSBs per cell (20, 60). Using this system and others, γH2Ax has been shown by chromatin immunoprecipitation (ChIP) to be present up to a megabase away from the break site but absent from the actual break site and regions in close proximity to the break. In contrast, Nbs1 and ATM are bound within 200 to 500 bp of the break site (7, 72, 75). However, at the resolution of IF microscopy, all three proteins appear to colocalize to IRIF (7).
To more precisely determine the localization of Sp1, we used the I-PpoI endonuclease system. I-PpoI is fused with an estrogen responsive element for inducible expression and nuclear localization of the enzyme. Induction of I-PpoI by 4-hydroxytamoxifen (4-OHT), an estrogen analog, activates the DNA damage response as shown by increased phosphorylation of Sp1 and Nbs1 (Fig. 3B). Next, ChIP was performed using antibodies specific for Sp1 and Nbs1. The Sp1 antibody used recognizes both phosphorylated and unphosphorylated forms of Sp1, to bring down Sp1 regardless of phosphorylation at S101. The association of Sp1 and Nbs1 with the DNA break sites was detected by performing qPCR of the immunoprecipitates using primers 280 bp 5′ of the DSB in the DAB1 intron and 489 bp 3′ of the DSBs in the rDNA genomic regions (Fig. 3C). Sp1 ChIP was repeated in cells treated with the ATM inhibitor (KU-55933) (31) to determine whether Sp1 was recruited independently of ATM activity (Fig. 3D). Sp1 phosphorylation, as well as that of other ATM targets, is not required for Sp1 binding at break sites.
Further analysis of association of these proteins with the region surrounding the chromosome 1 cut site, including up to 9,000 bp distal to the break, shows that like Nbs1, Sp1 occupancy is maximal immediately adjacent to the break site (Fig. 4A). In contrast, γH2Ax is present in the surrounding regions up to 9,000 bp away from DSBs but is not detected immediately adjacent to the break site, perhaps as a result of nucleosome remodeling and clearance to accommodate binding of repair proteins.
Fig 4.
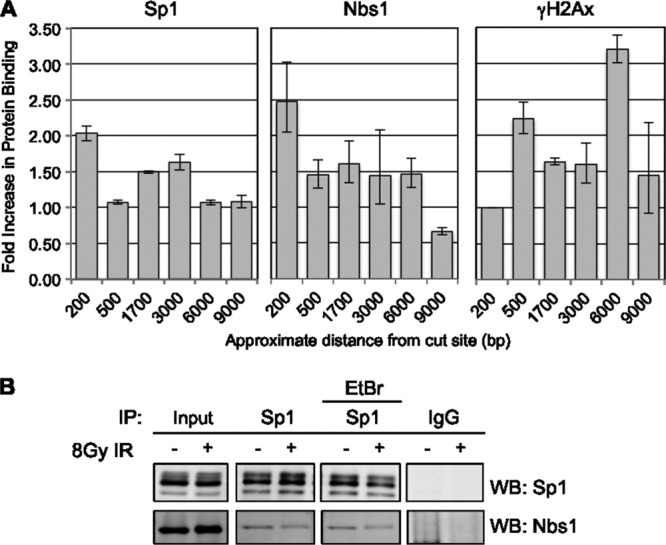
Sp1 and Nbs1 localize adjacent to breaks, and the two factors interact. (A) Chromatin immunoprecipitations for Sp1, Nbs1, and γH2Ax were done 6 h after 4-OHT treatment as previously described; binding to regions various distances from the DAB1 break site was assessed by qPCR. Data represent means and SEM from a single independent experiment, assessed in triplicate. Independent experiments were repeated in triplicate with similar trends. (B) Following 8 Gy of IR treatment to induce damage, U2OS lysates were immunoprecipitated for Sp1, followed by Western blotting with Nbs1 antibody to determine a potential interaction with Sp1. Addition of 100 μg/ml ethidium bromide was used to determine whether DNA was required for the interaction.
Based on the finding that Sp1 and Nbs1 are both localized in close proximity to the break site, we sought to determine whether there was an interaction between Sp1 and Nbs1. Immunoprecipitations were performed using Sp1 antibody in cells untreated or treated with 8 Gy of IR. Nbs1 was detected in the Sp1 immunoprecipitate from both undamaged cells and cells 2 h after DNA damage, suggesting a damage-independent interaction between the two proteins (Fig. 4B). Ethidium bromide did not preclude this interaction, suggesting that the interaction is between the proteins and is not bridged by DNA. Small amounts of Sp1 were detectable in Nbs1 IPs (results not shown). Nbs1 was depleted from cells to determine if it is required for Sp1 recruitment to damage sites; surprisingly, Nbs1 depletion led to a large decrease in Sp1 protein level (data not shown), precluding examining the possible role of Nbs1 in Sp1 recruitment.
Sp1 depletion does not affect recruitment of important damage response factors.
Previous work demonstrated that in response to IR, the number of γH2AX foci in Sp1-depleted cells is quantitatively greater and the foci persist longer (64). To address whether this apparent effect on repair is due to a possible role of Sp1 in the recruitment of immediate DNA damage response factors, cells were treated with nontargeting (control) or Sp1 shRNA and subjected to IR. Immunofluorescence using antibodies to 53BP1 and Nbs1 shows that Sp1 does not affect the recruitment of these factors (data not shown).
Sp1 is required for effective repair of site-specific DNA DSBs.
Based on the prolonged occupancy of the break site by Sp1, we sought to determine Sp1's effect on DNA double-strand break repair utilizing I-PpoI cleavage and qPCR with primers that are located on either side of the I-PpoI cut site in the DAB1 gene. This assay avoids potential differential susceptibility to damage between cells with and without Sp1 and allows measurement of the amount of DNA repair by quantifying the PCR product (6, 7). DSBs were induced by the addition of 4-OHT to I-PpoI-expressing cells depleted of Sp1 or Nbs1 by shRNA (Fig. 5). Total DNA was isolated at various time points after I-PpoI induction, and qPCR was used to amplify two DNA products, the product produced by the primers located on either side of the cut site and the product from a primer pair 280 bp away from the cut site, which serves as a control. The amount of product produced from the primers that flank the cut site is directly proportional to the net amount of cutting and repair occurring, i.e., the PCR product is decreased when the DNA target is cut and increased with repair across the site. Fold decreases calculated by the ΔΔCT method were converted to percentages to simplify the data. The requirement for Nbs1, as well as that of ATM, in DSB repair was previously demonstrated with this assay (7). The percentage of unrepaired DNA was significantly increased in cells depleted of Sp1 relative to that in control cells, and the inhibition of repair was not significantly different from that observed in cells depleted of Nbs1 (Fig. 5).
Fig 5.
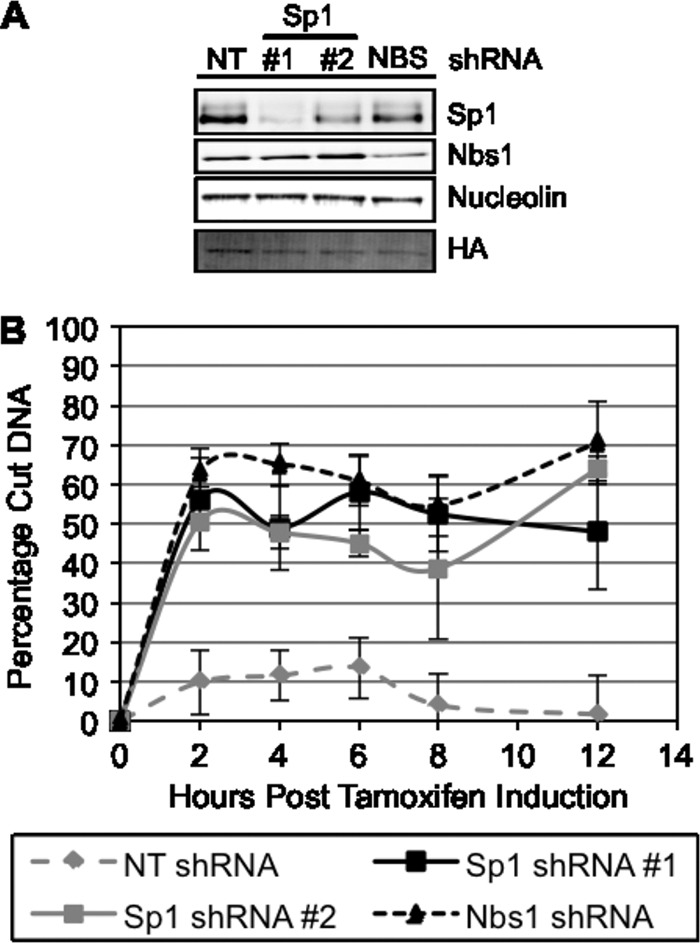
Depletion of Sp1 affects site-specific DNA double-strand break repair. (A) U2OS cells expressing I-PpoI were depleted of Sp1 or Nbs1 with RNAi, and cells were lysed 48 h after shRNA introduction and used for Western blot analysis of protein expression. (B) Cells prepared as described in the legend to panel A were induced with 4-OHT, and DNA was isolated from cell lysates at various time points postinduction of I-PpoI. Percentage of damaged DNA was calculated as described in Materials and Methods. Data represent means and SEM from 3 independent experiments assessed in triplicate. Data points for Sp1 shRNA 1 and shRNA 2 and Nbs1 shRNA at 2 to 12 h after tamoxifen are significantly different from nontargeting shRNA and not significantly different from one another. P < 0.05.
The N-terminal 182-amino-acid peptide is sufficient for phosphorylation and localization to damage-induced foci.
Sp1 has 2 transactivation domains, A and B, which both contain serine/threonine-rich cluster domains (64); C-terminal to these transactivation domains is a highly charged region, domain C, followed by three zinc finger motifs which mediate sequence-specific DNA binding (14, 37). At the C terminus, there is a D domain, which has been shown to mediate the multimerization of Sp1 (Fig. 6A) (14, 89). The peptide comprising the first 182 amino acids contains most of the first transactivation domain (A domain) of Sp1 and is here referred to as the “damage response domain” (Sp1-DRD). It contains 6 SQ/TQ sites that are potential targets for PI3KK, including the known S101 site. It has no known structural domains and has been determined to be intrinsically unstructured by several protein disorder prediction tools (e.g., GlobPlot, DisEMBL, PrDOS, RONN, and IUPred). Accumulating evidence suggests that intrinsically unstructured regions can be highly modified, adapt to interactions with multiple binding partners, and undergo disorder to order transitions in their structure that may be an important part of their function (88). Previous studies have shown that deletion of this domain decreases Sp1's sequence-specific transcriptional activity by only 10% and that the transactivation domains alone (A or B), in the absence of the Zinc-finger binding region, have less than 1% of the transcriptional activity of the full-length protein (14). In order to determine whether the DNA binding domain is required for recruitment to break sites, a vector coding Flag-Sp1-DRD-HA was expressed in cells that were depleted of endogenous Sp1 by shRNA. Western blots with antibodies to HA (to detect expression of Sp1-DRD) and pSp1 (to detect phosphorylated Sp1-DRD) show that Sp1-DRD is phosphorylated on S101 in response to DNA damage (Fig. 6B). Additionally, this domain alone is sufficient for localization to IRIF as well as site-specific DSBs (Fig. 6C and D), indicating that the sequence-specific DNA binding domain of Sp1 is not required for localization to DSBs.
Fig 6.

The N-terminal peptide comprising amino acids 1 to 182 of Sp1 is sufficient for phosphorylation and colocalization to IRIF. (A) The Sp1 protein is composed of several domains; A and B are two highly similar transactivation domains containing serine-, threonine-, and glutamine-rich regions and S/TQ cluster domains (SCD 1 and 2). Domain C is a charged domain followed by 3 highly homologous zinc fingers. Domain D is important for Sp1 oligomerization. (B) Cells were depleted of endogenous Sp1, and a retrovirus expressing a Sp1 truncation (Sp1-DRD) encoding the N-terminal 182 amino acids of the protein (marked in panel A) or an empty control vector were transduced. Cells were damaged with 8 Gy of IR and assessed by Western blotting with pSp1 and HA antibodies. (C) Cells prepared as described in the legend to panel B were also damaged with 0.5 Gy of radiation and processed for immunofluorescence with HA, pSp1, and γH2Ax antibodies. (D) Cells depleted of wtSp1 and expressing the Sp1-DRD protein were infected with I-PpoI, induced with 4-OHT, and processed for ChIP as in experiments shown in Fig. 3 and 4. Data graphed are the means and SEM from two independent experiments assessed in triplicate.
The N-terminal 182-amino-acid peptide is sufficient to rescue the repair defect in cells depleted of Sp1.
To rule out a transcriptional role for Sp1 in DSB repair, we tested the ability of Sp1-DRD to rescue the decreased repair observed in cells depleted of endogenous Sp1. Sp1-DRD was expressed in cells depleted of endogenous Sp1 (Fig. 7A). DSB repair was monitored by detection of PCR product using primers on either side of the DSB at the indicated time points after I-PpoI induction. Cells depleted of Sp1 showed decreased repair, and expression of Sp1-DRD restored the repair to the level seen in cells infected with the nontargeting shRNA (Fig. 7B). These data demonstrate that Sp1 can modulate repair of DSBs through its N-terminal region independently of its sequence-specific transcriptional effects.
Fig 7.

Sp1-DRD is sufficient to rescue the repair defect due to Sp1 depletion. Cells stably expressing Sp1-DRD were transduced for expression of I-PpoI and shRNA-targeting Sp1. (A) Western blotting was used to assess KD and reexpression levels of endogenous Sp1 and the mutant Sp1. (B) Seventy-two hours after I-PpoI infection and Sp1 KD, cells were treated with 4-OHT, and damage was assessed by qPCR at various time points after induction, as described in the legend to Fig. 5. Data represent means and SEM from three independent experiments assessed in triplicate. Data points for Sp1 shRNA are significantly different from nontargeting shRNA and Sp1 shRNA plus Sp1-DRD, which are not significantly different from one another at 2 to 12 h after tamoxifen. P < 0.05.
DISCUSSION
Our previous reports showing that cells depleted of Sp1 are more sensitive to DNA damage, have delayed resolution of γH2Ax foci (64), and accumulate chromosomal abnormalities (3) suggest that Sp1 plays a role in DSB repair. Taken together with the finding that the sensitivity could not be rescued by phosphorylation-deficient Sp1, S101A (64), the results here suggest that Sp1 phosphorylation and recruitment to double-strand breaks are important for their repair.
IRIF are dynamic structures that represent the assembly of multiple proteins in the vicinity of double-strand break sites (5, 67), including the phosphorylated histone variant, γH2Ax. Although each individual IRIF is thought to represent one double-strand break, many H2Ax molecules are phosphorylated in the vicinity of a DSB to form the visible focus (67). While the exact mechanism by which the cell senses a DSB is unknown, previous data suggest that alterations in chromatin structure result in the autophosphorylation and monomerization of ATM (4), which phosphorylates H2Ax on serine 139 near the break site (10). The spread of the signal to multiple H2Ax molecules is thought to occur through the subsequent recruitment of additional ATM (68, 84). Once the MRN complex is recruited to a DSB, Nbs1 can interact with and recruit additional ATM (18, 92), which then phosphorylates additional H2Ax, as well as other proteins, many of which are involved in DNA repair.
Our data demonstrate that phosphorylated Sp1 colocalizes with γH2Ax and members of the MRN complex at IRIF induced by ionizing radiation and microirradiated stripes. Localization of Sp1 occurs within minutes of damage induction and is maintained over several hours. Chromatin immunoprecipitation data demonstrate that Sp1, like Nbs1, localizes predominantly in close proximity to the break site rather than in the surrounding chromatin regions. This suggests that Sp1 may play a role in the recruitment of important repair factors to DSBs and may be essential to facilitate repair processes occurring at break sites. The interaction between Sp1 and Nbs1 is consistent with recruitment of Sp1 to break sites by Nbs1. Experiments to knock down Nbs1 and address its effect on Sp1 localization led to the observation that depletion of Nbs1 resulted in a large decrease in Sp1 levels, making it impossible to address the role of Nbs1 in Sp1 recruitment and suggesting that Nbs1 may stabilize Sp1 (data not shown). The lack of an effect of Sp1 depletion on Nbs1 recruitment suggests that MRN is upstream of Sp1 recruitment or, if Sp1 is complexed with MRN in the absence of damage, it is not necessary for MRN recruitment. Further analysis of the phenomenon is needed to address the role of Nbs1 in potentially stabilizing Sp1 and the implications this may have on the NBS disease. Sp1 depletion had no effect on formation of IRIF containing 53BP1 or γH2Ax, suggesting that Sp1 is not required for the initial recognition events in the damage response.
While there is likely a transcriptional role for Sp1 in the damage response, especially given the presence of Sp1 sites in the promoters of many genes transcriptionally activated in response to DNA damage, e.g., ATM, Gadd45, DDB1/2, and XRCC1 (15, 23, 27, 63, 94), it is highly unlikely that its function in localization to and repair of DSBs is transcriptional. We (unpublished) and others have determined that Sp1 transcriptional activity is not affected by phosphorylation of Sp1 on serine 101 (34), and the rapid DNA repair response is unlikely to result from altered transcription. Importantly, although Sp1 is a member of the Kruppel-like transcription factor family, members of which have common DNA binding domains and interact with similar DNA motifs, Sp1 is the only family member with SCDs, suggesting that unlike transcription, its role in the DNA damage response may not be redundant.
Based on the observed decrease in the repair of site-specific DSBs when Sp1 was depleted (by two different shRNAs), Sp1 is likely playing a role in promoting repair at the break site. Sp1 is thought to regulate transcription of several damage response factors. Interestingly, very few mRNAs for known damage response proteins showed significant (>2-fold) increases or decreases by microarray analysis in cells depleted of Sp1 by shRNA for 10 days. In addition, these cells were treated with IR for up to 8 h and showed no significant Sp1-dependent changes after radiation in any known DNA repair genes (K. Beishline, J. Azizkhan-Clifford, C. Innes, and R. Paules, unpublished data). In all of the in vitro experiments presented here, Sp1 was knocked down for no more than 72 h, which would likely produce even fewer transcriptional changes. A peptide comprising the N-terminal 182 amino acids, which includes the S/TQ cluster domain A, as illustrated in Fig. 7A (64), localized to break sites and rescued the DSB repair deficiency resulting from depletion of endogenous full-length Sp1. Sp1-DRD lacks both the DNA binding and the multimerization domains. These are required for sequence-specific transcriptional activation, and Sp1-DRD is unlikely to rescue any transcriptional changes resulting from Sp1 depletion. Taken together, these data show that the role of Sp1 in repair of DNA double-strand breaks is independent of its transcriptional regulation of other repair factors and is mediated through its N-terminal region.
Several other transcription factors have been shown to be involved in the DNA damage response. There is strong evidence that both E2F1 and ATF2 localize to IRIF in cells damaged with IR or the radiomimetic neocarzinostatin (8, 12). E2F1 has been shown to localize to damage sites in response to UV damage and is required for recruitment of the chromatin remodeling factor GCN5, a histone acetyltransferase responsible for remodeling of chromatin to facilitate nucleotide excision repair (28). GCN5 has also been implicated in DSB repair and has been shown to be responsible for the acetylation of γH2Ax-containing nucleosomes in response to ionizing radiation (46). It also promotes the recruitment of Swi/Snf components to sites of DNA damage (47). The first association of GCN5 with E2F1 was made when it was shown that GCN5 was required for the stimulation of E2F1- and E2F4-dependent transcriptional changes (44). Like E2F1, Sp1 reportedly interacts with chromatin remodeling enzymes and complexes that decrease or increase Sp1 sequence-specific transcriptional activity through remodeling of chromatin in promoters (17, 52, 65, 90). Acetyltransferases, such as p300/CBP and histone deactylases, as well as proteins such as BRCA1, BRG1, and the high-mobility group (HMG) protein 1, have been shown to play a role in chromatin remodeling in response to DNA damage and are interacting partners of Sp1, as is E2F1 (1, 2, 17, 53, 85, 91). An interesting possibility is that phosphorylated Sp1 may function at sites of DNA damage to recruit chromatin-remodeling proteins to facilitate nucleosome clearance around DSBs.
In summary, we have shown that the N-terminal portion of Sp1, the DRD, functions as a mediator of DNA DSB repair. The N-terminal region, in the absence of the remainder of the protein, is phosphorylated by ATM and can localize to DSBs and facilitate DSB repair. Further studies are under way to determine whether Sp1 mediates chromatin remodeling at DSBs.
ACKNOWLEDGMENTS
Special thanks go to Michael Kastan (St. Jude Children's Hospital, Memphis, TN) and John Petrini (Sloan-Kettering Cancer Center, NY) for the generous donation of plasmid constructs and antibodies which were essential in the completion of this study. In addition, we thank Behzad Torabi for construction and use of the pLXSN-Sp1-DRD construct.
Footnotes
Published ahead of print 23 July 2012
REFERENCES
- 1. Abramovitch S, Glaser T, Ouchi T, Werner H. 2003. BRCA1-Sp1 interactions in transcriptional regulation of the IGF-IR gene. FEBS Lett. 541:149–154 [DOI] [PubMed] [Google Scholar]
- 2. Aiello A, et al. 2010. HMGA1 protein is a positive regulator of the insulin-like growth factor-I receptor gene. Eur. J. Cancer 46:1919–1926 [DOI] [PubMed] [Google Scholar]
- 3. Astrinidis A, et al. 2010. The transcription factor SP1 regulates centriole function and chromosomal stability through a functional interaction with the mammalian target of rapamycin/raptor complex. Genes Chromosomes Cancer 49:282–297 [DOI] [PubMed] [Google Scholar]
- 4. Bakkenist CJ, Kastan MB. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499–506 [DOI] [PubMed] [Google Scholar]
- 5. Bekker-Jensen S, et al. 2006. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 173:195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berkovich E, Monnat RJ, Jr, Kastan MB. 2008. Assessment of protein dynamics and DNA repair following generation of DNA double-strand breaks at defined genomic sites. Nat. Protoc. 3:915–922 [DOI] [PubMed] [Google Scholar]
- 7. Berkovich E, Monnat RJ, Jr, Kastan MB. 2007. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 9:683–690 [DOI] [PubMed] [Google Scholar]
- 8. Bhoumik A, et al. 2005. ATM-dependent phosphorylation of ATF2 is required for the DNA damage response. Mol. Cell 18:577–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Black AR, Black JD, Azizkhan-Clifford J. 2001. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell. Physiol. 188:143–160 [DOI] [PubMed] [Google Scholar]
- 10. Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. 2001. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276:42462–42467 [DOI] [PubMed] [Google Scholar]
- 11. Buscemi G, et al. 2001. Chk2 activation dependence on Nbs1 after DNA damage. Mol. Cell. Biol. 21:5214–5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen J, et al. 2011. E2F1 promotes the recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle 10:1287–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chu S, Ferro TJ. 2005. Sp1: regulation of gene expression by phosphorylation. Gene 348:1–11 [DOI] [PubMed] [Google Scholar]
- 14. Courey AJ, Tjian R. 1988. Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell 55:887–898 [DOI] [PubMed] [Google Scholar]
- 15. Daino K, Ichimura S, Nenoi M. 2003. Comprehensive search for X-ray-responsive elements and binding factors in the regulatory region of the GADD45a gene. J. Radiat. Res. 44:311–318 [DOI] [PubMed] [Google Scholar]
- 16. Difilippantonio S, et al. 2005. Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat. Cell Biol. 7:675–685 [DOI] [PubMed] [Google Scholar]
- 17. Doetzlhofer A, et al. 1999. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol. Cell. Biol. 19:5504–5511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Falck J, Coates J, Jackson SP. 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434:605–611 [DOI] [PubMed] [Google Scholar]
- 19. Fernandez-Capetillo O, Celeste A, Nussenzweig A. 2003. Focusing on foci: H2AX and the recruitment of DNA-damage response factors. Cell Cycle 2:426–427 [PubMed] [Google Scholar]
- 20. Flick KE, Jurica MS, Monnat RJ, Jr, Stoddard BL. 1998. DNA binding and cleavage by the nuclear intron-encoded homing endonuclease I-PpoI. Nature 394:96–101 [DOI] [PubMed] [Google Scholar]
- 21. Frame FM, Rogoff HA, Pickering MT, Cress WD, Kowalik TF. 2006. E2F1 induces MRN foci formation and a cell cycle checkpoint response in human fibroblasts. Oncogene 25:3258–3266 [DOI] [PubMed] [Google Scholar]
- 22. Galburt EA, et al. 1999. A novel endonuclease mechanism directly visualized for I-PpoI. Nat. Struct. Biol. 6:1096–1099 [DOI] [PubMed] [Google Scholar]
- 23. Gartel AL, Shchors K. 2003. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp. Cell Res. 283:17–21 [DOI] [PubMed] [Google Scholar]
- 24. Gatei M, et al. 2003. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J. Biol. Chem. 278:14806–14811 [DOI] [PubMed] [Google Scholar]
- 25. Girard PM, Riballo E, Begg AC, Waugh A, Jeggo PA. 2002. Nbs1 promotes ATM dependent phosphorylation events including those required for G1/S arrest. Oncogene 21:4191–4199 [DOI] [PubMed] [Google Scholar]
- 26. Goldberg M, et al. 2003. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 421:952–956 [DOI] [PubMed] [Google Scholar]
- 27. Gueven N, et al. 2001. Epidermal growth factor sensitizes cells to ionizing radiation by down-regulating protein mutated in ataxia-telangiectasia. J. Biol. Chem. 276:8884–8891 [DOI] [PubMed] [Google Scholar]
- 28. Guo R, Chen J, Mitchell DL, Johnson DG. 2011. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res. 39:1390–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haaf T, Golub EI, Reddy G, Radding CM, Ward DC. 1995. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. U. S. A. 92:2298–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Henderson AS, Warburton D, Atwood KC. 1972. Location of ribosomal DNA in the human chromosome complement. Proc. Natl. Acad. Sci. U. S. A. 69:3394–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hickson I, et al. 2004. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 64:9152–9159 [DOI] [PubMed] [Google Scholar]
- 32. Horejsi Z, et al. 2004. Distinct functional domains of Nbs1 modulate the timing and magnitude of ATM activation after low doses of ionizing radiation. Oncogene 23:3122–3127 [DOI] [PubMed] [Google Scholar]
- 33. Iwahori S, et al. 2007. Enhanced phosphorylation of transcription factor sp1 in response to herpes simplex virus type 1 infection is dependent on the ataxia telangiectasia-mutated protein. J. Virol. 81:9653–9664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iwahori S, et al. 2008. Identification of phosphorylation sites on transcription factor Sp1 in response to DNA damage and its accumulation at damaged sites. Cell Signal. 20:1795–1803 [DOI] [PubMed] [Google Scholar]
- 35. Jackson S, Gottlieb T, Hartley K. 1993. Phosphorylation of transcription factor Sp1 by the DNA-dependent protein kinase. Adv. Second Messenger Phosphoprotein Res. 28:279–286 [PubMed] [Google Scholar]
- 36. Jackson SP, Tjian R. 1988. O-glycosylation of eukaryotic transcription factors: implications for mechanisms of transcriptional regulation. Cell 55:125–133 [DOI] [PubMed] [Google Scholar]
- 37. Kadonaga JT, Courey AJ, Ladika J, Tjian R. 1988. Distinct regions of Sp1 modulate DNA binding and transcriptional activation. Science 242:1566–1570 [DOI] [PubMed] [Google Scholar]
- 38. Kalderon D, Roberts BL, Richardson WD, Smith AE. 1984. A short amino acid sequence able to specify nuclear location. Cell 39:499–509 [DOI] [PubMed] [Google Scholar]
- 39. Kim JS, et al. 2007. In situ analysis of DNA damage response and repair using laser microirradiation. Methods Cell Biol. 82:377–407 [DOI] [PubMed] [Google Scholar]
- 40. Kim K, Barhoumi R, Burghardt R, Safe S. 2005. Analysis of estrogen receptor alpha-Sp1 interactions in breast cancer cells by fluorescence resonance energy transfer. Mol. Endocrinol. 19:843–854 [DOI] [PubMed] [Google Scholar]
- 41. Kim ST, Lim DS, Canman CE, Kastan MB. 1999. Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem. 274:37538–37543 [DOI] [PubMed] [Google Scholar]
- 42. Reference deleted.
- 43. Lai JS, Herr W. 1992. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl. Acad. Sci. U. S. A. 89:6958–6962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lang SE, McMahon SB, Cole MD, Hearing P. 2001. E2F transcriptional activation requires TRRAP and GCN5 cofactors. J. Biol. Chem. 276:32627–32634 [DOI] [PubMed] [Google Scholar]
- 45. Lavin MF. 2007. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 26:7749–7758 [DOI] [PubMed] [Google Scholar]
- 46. Lee HS, Park JH, Kim SJ, Kwon SJ, Kwon J. 2010. A cooperative activation loop among SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J. 29:1343–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee HS, Park JH, Kim SJ, Kwon SJ, Kwon J. 2010. A cooperative activation loop among SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J. 29:1434–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee JH, Paull TT. 2007. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 26:7741–7748 [DOI] [PubMed] [Google Scholar]
- 49. Lee JH, Paull TT. 2004. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304:93–96 [DOI] [PubMed] [Google Scholar]
- 50. Lee JH, Paull TT. 2006. Purification and biochemical characterization of ataxia-telangiectasia mutated and Mre11/Rad50/Nbs1. Methods Enzymol. 408:529–539 [DOI] [PubMed] [Google Scholar]
- 51. Li L, Davie JR. 2010. The role of Sp1 and Sp3 in normal and cancer cell biology. Ann. Anat. 192:275–283 [DOI] [PubMed] [Google Scholar]
- 52. Li L, He S, Sun JM, Davie JR. 2004. Gene regulation by Sp1 and Sp3. Biochem. Cell Biol. 82:460–471 [DOI] [PubMed] [Google Scholar]
- 53. Lin SY, et al. 1996. Cell cycle-regulated association of E2F1 and Sp1 is related to their functional interaction. Mol. Cell. Biol. 16:1668–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu K, Lin FT, Ruppert JM, Lin WC. 2003. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol. Cell. Biol. 23:3287–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lowndes NF, Toh GW. 2005. DNA repair: the importance of phosphorylating histone H2AX. Curr. Biol. 15:R99–R102 [DOI] [PubMed] [Google Scholar]
- 56. Majumdar G, et al. 2004. Insulin stimulates and diabetes inhibits O-linked N-acetylglucosamine transferase and O-glycosylation of Sp1. Diabetes 53:3184–3192 [DOI] [PubMed] [Google Scholar]
- 57. Matsuoka S, et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316:1160–1166 [DOI] [PubMed] [Google Scholar]
- 58. Reference deleted.
- 59. Monnat RJ, Jr, Hackmann AF, Cantrell MA. 1999. Generation of highly site-specific DNA double-strand breaks in human cells by the homing endonucleases I-PpoI and I-CreI. Biochem. Biophys. Res. Commun. 255:88–93 [DOI] [PubMed] [Google Scholar]
- 60. Muscarella DE, Ellison EL, Ruoff BM, Vogt VM. 1990. Characterization of I-Ppo, an intron-encoded endonuclease that mediates homing of a group I intron in the ribosomal DNA of Physarum polycephalum. Mol. Cell. Biol. 10:3386–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nakagawa K, Taya Y, Tamai K, Yamaizumi M. 1999. Requirement of ATM in phosphorylation of the human p53 protein at serine 15 following DNA double-strand breaks. Mol. Cell. Biol. 19:2828–2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nelson JD, Denisenko O, Bomsztyk K. 2006. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 1:179–185 [DOI] [PubMed] [Google Scholar]
- 63. Nichols AF, Itoh T, Zolezzi F, Hutsell S, Linn S. 2003. Basal transcriptional regulation of human damage-specific DNA-binding protein genes DDB1 and DDB2 by Sp1, E2F, N-myc and NF1 elements. Nucleic Acids Res. 31:562–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Olofsson BA, Kelly CM, Kim J, Hornsby SM, Azizkhan-Clifford J. 2007. Phosphorylation of Sp1 in response to DNA damage by ataxia telangiectasia-mutated kinase. Mol. Cancer Res. 5:1319–1330 [DOI] [PubMed] [Google Scholar]
- 65. Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. 1998. Progesterone regulates transcription of the p21(WAF1) cyclin-dependent kinase inhibitor gene through Sp1 and CBP/p300. J. Biol. Chem. 273:10696–10701 [DOI] [PubMed] [Google Scholar]
- 66. Petrini JH. 2000. The Mre11 complex and ATM: collaborating to navigate S phase. Curr. Opin. Cell Biol. 12:293–296 [DOI] [PubMed] [Google Scholar]
- 67. Pilch DR, et al. 2003. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem. Cell Biol. 81:123–129 [DOI] [PubMed] [Google Scholar]
- 68. Polo SE, Jackson SP. 2011. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Devel. 25:409–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pugh BF, Tjian R. 1990. Mechanism of transcriptional activation by Sp1: evidence for coactivators. Cell 61:1187–1197 [DOI] [PubMed] [Google Scholar]
- 70. Richardson C, Jasin M. 2000. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature 405:697–700 [DOI] [PubMed] [Google Scholar]
- 71. Rodrigue A, et al. 2006. Interplay between human DNA repair proteins at a unique double-strand break in vivo. EMBO J. 25:222–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rogakou EP, Boon C, Redon C, Bonner WM. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146:905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273:5858–5868 [DOI] [PubMed] [Google Scholar]
- 74. Rudin N, Haber JE. 1988. Efficient repair of HO-induced chromosomal breaks in Saccharomyces cerevisiae by recombination between flanking homologous sequences. Mol. Cell. Biol. 8:3918–3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Savic V, et al. 2009. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol. Cell 34:298–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shi Y, et al. 2004. Direct regulation of CREB transcriptional activity by ATM in response to genotoxic stress. Proc. Natl. Acad. Sci. U. S. A. 101:5898–5903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shields JM, Yang VW. 1997. Two potent nuclear localization signals in the gut-enriched Kruppel-like factor define a subfamily of closely related Kruppel proteins. J. Biol. Chem. 272:18504–18507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shiloh Y. 1997. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu. Rev. Genet. 31:635–662 [DOI] [PubMed] [Google Scholar]
- 79. Shiloh Y. 2003. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3:155–168 [DOI] [PubMed] [Google Scholar]
- 80. Shiloh Y. 2006. The ATM-mediated DNA-damage response: taking shape. Trends Biochem. Sci. 31:402–410 [DOI] [PubMed] [Google Scholar]
- 81. Sobhian B, et al. 2007. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316:1198–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Solomon SS, Majumdar G, Martinez-Hernandez A, Raghow R. 2008. A critical role of Sp1 transcription factor in regulating gene expression in response to insulin and other hormones. Life Sciences 83:305–312 [DOI] [PubMed] [Google Scholar]
- 83. Stiff T, et al. 2005. Nbs1 is required for ATR-dependent phosphorylation events. EMBO J. 24:199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Su TT. 2006. Cellular responses to DNA damage: one signal, multiple choices. Annu. Rev. Genet. 40:187–208 [DOI] [PubMed] [Google Scholar]
- 85. Suzuki T, Kimura A, Nagai R, Horikoshi M. 2000. Regulation of interaction of the acetyltransferase region of p300 and the DNA-binding domain of Sp1 on and through DNA binding. Genes Cells 5:29–41 [DOI] [PubMed] [Google Scholar]
- 86. Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. 2005. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 438:379–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Uziel T, et al. 2003. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 22:5612–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wee S, et al. 2008. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. U. S. A. 105:13057–13062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wiederschain D, et al. 2009. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8:498–504 [DOI] [PubMed] [Google Scholar]
- 90. Won J, Yim J, Kim TK. 2002. Sp1 and Sp3 recruit histone deacetylase to repress transcription of human telomerase reverse transcriptase (hTERT) promoter in normal human somatic cells. J. Biol. Chem. 277:38230–38238 [DOI] [PubMed] [Google Scholar]
- 91. Xu YZ, et al. 2010. Brg-1 mediates the constitutive and fenretinide-induced expression of SPARC in mammary carcinoma cells via its interaction with transcription factor Sp1. Mol. Cancer 9:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. You Z, Chahwan C, Bailis J, Hunter T, Russell P. 2005. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 25:5363–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhou J, Lim CU, Li JJ, Cai L, Zhang Y. 2006. The role of NBS1 in the modulation of PIKK family proteins ATM and ATR in the cellular response to DNA damage. Cancer Lett. 243:9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhou ZQ, Walter CA. 1998. Cloning and characterization of the promoter of baboon XRCC1, a gene involved in DNA strand-break repair. Somat. Cell Mol. Genet. 24:23–39 [DOI] [PubMed] [Google Scholar]