

Abstract
Cellular levels of NAD+ and NADH are thought to be controlled by de novo and salvage mechanisms, although evidence has not yet indicated that they are regulated by NAD+ degradation. Here we show that the conserved nudix hydrolase isozyme NdxA hydrolyzes and decreases cellular NAD+ and NADH in Aspergillus nidulans. The NdxA-deficient fungus accumulated more NAD+ during the stationary growth phase, indicating that NdxA maintains cellular NAD+/NADH homeostasis. The deficient strain also generated less of the secondary metabolites sterigmatocystin and penicillin G and of their gene transcripts than did the wild type. These defects were associated with a reduction in acetylated histone H4 on the gene promoters of aflR and ipnA that are involved in synthesizing secondary metabolites. Thus, NdxA increases acetylation levels of histone H4. We discovered that the novel fungal sirtuin isozyme SirA uses NAD+ as a cosubstrate to deacetylate the lysine 16 residue of histone H4 on the gene promoter and represses gene expression. The impaired acetylation of histone and secondary metabolite synthesis in the NdxA-deficient strain were restored by eliminating functional SirA, indicating that SirA mediates NdxA-dependent regulation. These results indicated that NdxA controls total levels of NAD+/NADH and negatively regulates sirtuin function and chromatin structure.
INTRODUCTION
Nudix (nucleoside diphosphates linked to moiety X) hydrolases are ubiquitous in viruses, bacteria, and eukaryotes, and they hydrolyze NADH, NAD+, ADP-ribose, and other nucleotide sugars, which allows their classification into subfamilies (6, 22). One important function of nudix hydrolases is the hydrolytic degradation of oxidatively damaged nucleotides to prevent spontaneous mutations (39). Saccharomyces cerevisiae Ysa1p and Arabidopsis thaliana AtNUDX2 and AtNUDX7 hydrolyze ADP-ribose and respond to oxidative stress (17, 43). Nudix hydrolases that hydrolyze NAD+ and NADH [NAD(H)] exist throughout the biological kingdom and include A. thaliana AtNUDX1 (12) and yeast peroxisomal Npy1p (1). The physiological role of NAD(H) hydrolases is unknown, especially that in epigenetic gene regulation.
Gene expression is regulated by specific transcription regulators and by posttranslational modifications of nucleosomal histones. Acetylation is this type of modification that correlates with conformational changes in chromatin. Two groups of histone deacetylases (HDAC) deacetylate acetylated histones. One is classical HDAC, and the other is sirtuin, which deacetylates lysine residues of histones H3 and/or H4 using NAD+ as a cosubstrate (10, 16, 45). Yeast Sir2p is the prototype sirtuin, and it silences genes at mating type, ribosomal DNA (rDNA), and subtelomeric loci (14, 37). Its mammalian counterparts control aging, stress responses, and circadian rhythms (15, 25). Sirtuin activity is controlled by cellular NAD+ production (4, 19) that links cellular metabolic status and gene regulation, since NAD+ is a crucial coenzyme for biological redox reactions and energy conservation.
Filamentous fungi often activate secondary metabolites, including antibiotics of pharmacological importance and lethal mycotoxins. The Ascomycetes genus Aspergillus includes large numbers of strains that produce secondary metabolites. The production of secondary metabolites by the opportunistic pathogen Aspergillus fumigatus is associated with its virulence (23). Aspergillus nidulans is a classical model eukaryote, and it produces the antibiotic penicillin G as well as toxic and carcinogenic sterigmatocystin, which is related to the agricultural contaminant aflatoxin (9, 20). Secondary metabolite production is regulated by cyclic AMP (cAMP) and light through the activities of protein kinase A and the transcription factor LaeA (3, 8). Classical HDAC are also regulators of secondary metabolite synthesis (36, 44), whereas no known sirtuin regulates such synthesis in response to cellular NAD(H) levels. Here we investigated fungal nudix hydrolases that hydrolyze NAD(H). Our findings showed that a nudix hydrolase combined with a novel fungal sirtuin constitutes a novel epigenetic mechanism that degrades cellular NAD(H) and negatively regulates sirtuin function and chromatin structure.
MATERIALS AND METHODS
Strains, cultures, and media.
Aspergillus nidulans strains A26 (biA1) and A89 (biA1, argB2) were purchased from the Fungal Genetic Stock Center (FGSC; University of Missouri). Aspergillus nidulans ABPUN (biA1, argB2; pyrG; yA2; pyroA4) was the meiotic progeny of ABPU1 (biA1, pyrG89; wA3, argB2; pyroA4) and A713 (FGSC). Conidia (108) were transferred to 500-ml flasks containing 100 ml of GMM (40) and incubated at 30°C for 16 h at 120 rpm. Hypoxic conditions were maintained by aerating 1% O2 at 0.2 liters min−1 using a 1-liter jar fermentor (BMJ-01NC; Able & Biott Co., Tokyo, Japan) at 30°C and 180 rpm. Arginine (0.2 mg liter−1), pyridoxine (0.1 mg liter−1), uracil and uridine (1.12 g liter−1 and 1.2 g liter−1, respectively), and biotin (0.25 mg liter−1) were added to the culture medium for auxotrophic mutants. Nicotinamide riboside (NR; 0.3 mM final concentration) was added to the medium at 60 h after seeding, and cultures were further incubated for 12 h in some experiments.
Production of Ndx-GFP fusion proteins.
The DNA fragment corresponding to the gene promoter of gpdA was amplified using primers (Table 1), digested with BstXI and NotI, and cloned into the same restriction sites of pBSarg1 containing the argB gene to generate pBSarg2. DNA fragments for ndxA and ndxB were amplified and inserted into NotI-XbaI sites of pBSarg2, and then the XbaI-BamHI DNA fragment of the gfp gene amplified using the primers (Table 1) was inserted into the same restriction sites of the resulting plasmids. These plasmids were designated pNdxAgfp1 and pNdxBgfp1, respectively. To construct plasmid pGfp1ndxC to produce the green fluorescent protein (GFP)-NdxC fusion protein, the DNA fragment for gfp was amplified and cloned into the NotI-XbaI site of pBSarg2. The resulting plasmid was spliced with XbaI and BamHI and then ligated with the XbaI- and BamHI-digested DNA fragment for ndxC. Plasmids for producing DsRed-SKM protein were constructed as follows. The cDNA for DsRed harboring the SKM sequence was amplified using the primers listed in Table 1 and cloned into pBSpyrG (40) at a unique SmaI site to generate pDsRed-SKM1. Plasmids pNdxAgfp1 and pNdxBgfp1 were introduced into A. nidulans A89, and then pGfp1ndxC and pDsRed-SKM1 were introduced into A. nidulans ABPUN. Aspergillus nidulans strains were transformed as described previously (40).
Table 1.
Oligonucleotide primers used in this study
| Primer | Gene | Nucleotide sequencea | Use |
|---|---|---|---|
| Plasmids for gene disruption | |||
| ndxA5-f | ndxA | 5′-GCGGCCGCCAAGTGCTGGACCTGTACGA-3′ | 5′ region of ndxA |
| ndxA5-r | 5′-GCGGCCGCGGTATCGGGTGGAGATGAGA-3′ | ||
| ndxA3-f | 5′-GAATTCACCCGCTACGATGTTACCTG-3′ | 3′ region of ndxA | |
| ndxA3-r | 5′-CTCGAGAATATGGAAGCGTCCTCACG-3′ | ||
| ndxB5-f | ndxB | 5′-GCGGCCGCGTTAGATCCGCTCACCGAAG-3′ | 5′ region of ndxB |
| ndxB5-r | 5′-GCGGCCGCAAGCGAATCTCAAGCGACAT-3′ | ||
| ndxB3-f | 5′-GAATTCCCGTAAGGACTGCCCATTTA-3′ | 3′ region of ndxB | |
| ndxB3-r | 5′-CTCGAGCAAAGGTCAGCGCACATCTA-3′ | ||
| ndxC5-f | ndxC | 5′-GCGGCCGCCTCGAAAGGGAATGAACCAA-3′ | 5′ region of ndxC |
| ndxC5-r | 5′-GCGGCCGCTCTTGCCTGCAGAAAACTCA-3′ | ||
| ndxC3-f | 5′-GAATTCCGTGGACCCAACTTCTGTATATC-3′ | 3′ region of ndxC | |
| ndxC3-r | 5′-CTCGAGGAACTCCAACTGTGGTTGATAGG-3′ | ||
| sirA5-f | sirA | 5′-GCGGCCGCGCTCGATCTCAAAACGGAAG-3′ | 5′ region of sirA |
| sirA5-r | 5′-TCTAGATCTTCGACATTGCTGTCGTC-3′ | ||
| sirA3-f | 5′-GAATTCATCCTTGCCCAACAGATCAC-3′ | 3′ region of sirA | |
| sirA3-r | 5′-CTCGAGGGAAGCTCCGTCCATCAATA-3′ | ||
| ndxA3-f2 | ndxA | 5′-CTGCAGACCCGCTACGATGTTACCTG-3′ | 3′ region of ndxA (for insertion into pPyrG plasmid) |
| ndxA3-r2 | 5′-GAATTCAATATGGAAGCGTCCTCACG-3′ | ||
| Plasmids for GFP-fused protein production | |||
| gpdpro-f | gpdA | 5′-CCAATGCATTGGAAAAGTCACACAACACAAGC-3′ | gpdA gene promoter |
| gpdpro-r | 5′-GCGGCCGCTGTGATGTCTGCTCAAGC-3′ | GFP-NdxC fusion | |
| gfpN-f | gfp | 5′-GCGGCCGCATGGTGAGCAAGGGCGAGGAGCTG-3′ | |
| gfpN-r | 5′-TCTAGACTTGTACAGCTCGTCCAT-3′ | NdxA- or NdxB-GFP fusion | |
| gfpC-f | gfp | 5′-TCTAGAATGGTGAGCAAGGGCGAGGAGCTG-3′ | NdxA-GFP fusion |
| gfpC-r | 5′-GGATCCTTACTTGTACAGCTCGTCCAT-3′ | ||
| gndxA-f | ndxA | 5′-GCGGCCGCATGCCCACAGAGACAAAATCCGTTCGCGTT-3′ | NdxB-GFP fusion |
| gndxA-r | 5′-TCTAGAGTTCAAAACAGGCTGAAAA-3′ | ||
| gndxB-f | ndxB | 5′-GCGGCCGCATGTCAGATCCAATGTACGCCAGGTCAAA-3′ | GFP-NdxC fusion |
| gndxB-r | 5′-TCTAGAGAGCTTCAGTTTCTTCGCC-3′ | ||
| gndxC-f | ndxC | 5′-TCTAGAATGTCCAATAATTCACAAATCC-3′ | DsRed-SKM |
| gndxC-r | 5′-GGATCCCTACATCTTCGACTTGTCTGCAAG-3′ | ||
| ds-red-f | ds-red | 5′-GCGGCCGCATGGCCTCCTCCGAGGACGT-3′ | |
| ds-red-r | 5′-GGATCCCTACATCTTCGACAGGAACAGGTGGTGGCGG-3′ | ||
| Plasmids for recombinant protein production | |||
| rndxA-f | ndxA | 5′-CATATGCCGACCGAAACCAAATCCGTTCGCGTT-3′ | pETndxA |
| rndxA-r | 5′-AAGCTTTCAGTTCAAAACAGGCTGAAAA-3′ | ||
| rndxB-f | ndxB | 5′-CATATGTCAGATCCGATGTACGCCCGCTCAAA-3′ | pETndxB |
| rndxB-r | 5′-AAGCTTTCAGAGCTTCAGTTTCTTCGCC-3′ | ||
| rndxC-f | ndxC | 5′-CATATGTCCAATAATTCACAAATCC-3′ | pETndxC |
| rndxC-r | 5′-AAGCTTCTACATCTTCGACTTGTCTGCAAG-3′ | ||
| rndxD-f | ndxD | 5′-GGATCCGTGGACCAGG TTCGTTCAAT-3′ | pETndxD |
| rndxD-r | 5′-GCGGCCGCCTATCTCTTCATGGAACTTC-3′ | ||
| rSirA-f | sirA | 5′-CGGGATCCATGGACCTTGCTTCAGCGCCCCGTGGGGAGA-3′ | pETsirA |
| rSirA-r | 5′-GCGGCCGCTCCGCTAACCTTGAATACATGTCGTGA-3′ | ||
| Site-directed mutagenesis | |||
| E57Q-f | ndxA | 5′-TGCGCGGCTCGCCAATTAATAGAAGAA-3′ | pETndxA-E57Q |
| E57Q-r | 5′-TTCTTCTATTAATTGGCGAGCCGCGCA-3′ | ||
| E61Q-f | ndxA | 5′-GAATTAATAGAACAAACGGGGGTACAT-3′ | pETndxA-E61Q |
| E61Q-r | 5′-ATGTACCCCCGTTTGTTCTATTAATTC-3′ | ||
| H286N-f | sirA | 5′-ATTGTACAGTGCAACGGCTCTTTTGCC-3′ | pETsirA-H286N |
| H286N-r | 5′-GGCAAAAGAGCCGTTGCACTGTACAAT-3′ | ||
| Plasmids for expressing ndxA and sirA in A. nidulans | |||
| ndxA-f | ndxA | 5′-GCGGCCGCTGACAGGGTACCAATGCAAA-3′ | pBSndxA, pBSndxA-E57Q |
| ndxA-r | 5′-GCGGCCGCAGCTCGCAGTTTTGTCCAAT-3′ | pBSsirA, pBSsirA-H286N | |
| sirA-f | sirA | 5′-GCGGCCGCTCTCCCGAGCCTCTTATCAA-3′ | |
| sirA-r | 5′-GCGGCCGCTTCAGTATAGCCCCGCACTC-3′ | ||
| Quantitative PCR | |||
| RTactA-f | actA | 5′-GAAGTCCTACGAACTGCCTGATG-3′ | |
| RTactA-r | 5′-AAGAACGCTGGGCTGGAA-3′ | ||
| RTndxA-f | ndxA | 5′-GGACGGGAAGCACTACGTTA-3′ | |
| RTndxA-r | 5′-CAGCTTCGACCTGCTTTTTC-3′ | ||
| RTndxB-f | ndxB | 5′-CGTGAGCTCAAGGAGGAGAC-3′ | |
| RTndxB-r | 5′-AGTTGTGGCTTGGGATTTTG-3′ | ||
| RTndxC-f | ndxC | 5′-AGCAACAACACAATGCGAAG-3′ | |
| RTndxC-r | 5′-AGTAGCTGTTGCGGGTGTTC-3′ | ||
| RTaflR-f | aflR | 5′-CTGCCTTGCGAGTATATGGTTTC-3′ | |
| RTaflR-r | 5′-TTGGTGATGGTGCTGTCTTG-3′ | ||
| RTstcU-f | stcU | 5′-CATTTCCATTCAAGCCGATGT-3′ | |
| RTstcU-r | 5′-CCAGGTATCCGAAGTGCTCAA-3′ | ||
| RTipnA-f | ipnA | 5′-CAGCGTGATTGATCCATTTG-3′ | |
| RTipnA-r | 5′-CTAAATCCAACTCCCGTCCA-3′ | ||
| RTpenD-f | penDE | 5′-AATGGGCGGATAGTGCATAC-3′ | |
| RTpenD-r | 5′-CCGTCAAAACCAGAGAGGAG-3′ | ||
| RTsirA-f | sirA | 5′-CGCATTCAGGAATCAAACCT-3′ | |
| RTsirA-r | 5′-GCGTGTTCCCTGAAAACATT-3′ | ||
| ChIP assay | |||
| ipnA #1-f | ipnA #1 | 5′-TAATCCACGCAATCCACTGA-3′ | 5′ region of ipnA |
| ipnA #1-r | 5′-CCGGTGCATAATGACAAGTG-3′ | ||
| ipnA #2-f | ipnA #2 | 5′-TGAGAGCTACGCTTCCCATT-3′ | 5′ region of ipnA |
| ipnA #2-r | 5′-AGTTCTCCAAAGCTGGCTCA-3′ | ||
| ipnA #3-f | ipnA #3 | 5′-CGTGCCTACAACAAAGAGCA-3′ | ipnA |
| ipnA #3-r | 5′-AGTTGTGGCTTGGGATTTTG-3′ | ||
| aflR #4-f | aflR #4 | 5′-GACTCCTAGACCCCGACAGG-3′ | 5′ region of aflR |
| aflR #4-r | 5′-TACTGCGGGCTAGAACTGGT-3′ | ||
| aflR #5-f | aflR #5 | 5′-ATCTCCTCATGGCGAATCTC-3′ | 5′ region of aflR |
| aflR #5-r | 5′-TATTCCCGCAGGGATTACAG-3′ | ||
| aflR #6-f | aflR #6 | 5′-GCTCCAGATCCAAGGTCAAG-3′ | aflR |
| aflR #6-r | 5′-CGTATTCGTCGGTGTTGTTG-3′ | ||
| actApro-f | actA | 5′-TACTCCGCCTACCGCTACAA-3′ | 5′ region of actA |
| actApro-r | 5′-GGAAGGGAGGAGGAGAGATG-3′ | ||
| Producing SirA-HA protein | |||
| SirAHA-f | sirA-HA | 5′-ATAAGAATGCGGCCGCATGGACCTTGCTTCAGCGCCCCGT-3′ | pSirA-HA |
| SirAHA-r | 5′-CTCGAGCTACGCATAGTCCGGGACGTCATAGGGATAGGATCCCGCATAGTCAGGAACATCGTATGGGTAATAGCCCGCATAGTCAGGAACATCGTATGGGTATCCGCTAACCTTGAATACATGTCGTGA-3′ |
Restriction sites are underlined. Silent mutations are double underlined.
Fluorescence microscopy.
Conidia of the transformants harboring pNdxAgfp1, pNdxBgfp1, pGfp1ndxC, and pDsRed-SKM were incubated on glass coverslips in GMM medium at 37°C for 10 h and then analyzed using the DMLB fluorescence microscope (Leica, Heidelberg, Germany) with a BP 450-to-490 filter for fluorescence excitation of GFP and a BP 515-to-560 filter for excitation of DsRed.
Preparation of recombinant proteins.
We prepared ndxA cDNA by PCR using the A. nidulans cDNA and a set of oligonucleotide primers (Table 1). The PCR products were purified, digested by NdeI and HindIII, and then ligated to pET28a (Novagen, Darmstadt, Germany) that had been digested with the same restriction enzymes (pETndxA). Plasmids for producing recombinant NdxB, NdxC, NdxD, and SirA were constructed essentially by the same strategy using the primers (Table 1) and pET28a and designated pETndxB, pETndxC, pETndxD, and pETsirA, respectively. Mutations were introduced into the plasmids using the QuikChange site-directed mutagenesis kit (Stratagene, CA) and primers for overlap PCR (Table 1) to generate pETndxA-E57Q, pETndxA-E61Q, and pETsirA-H286N.
The plasmids were introduced into Escherichia coli BL21(DE3)pLysS (Novagen) and cultured in LB containing 50 μg ml−1 kanamycin sulfate for 12 h, and then a portion (3 ml) was agitated at 120 rpm in 200 ml of LB containing 50 μg ml−1 kanamycin sulfate at 30°C. After the optical density reached 1.0, isopropyl-thio-β-d-galactoside (0.1 mM) was added to the medium and the flasks were further incubated for 12 h at 120 rpm at 30°C. The E. coli cells were harvested, suspended in 50 ml of 20 mM potassium phosphate (pH 7.5), and disrupted by sonication. The sonicate was centrifuged at 6,000 × g for 15 min, and the resulting cell extract was centrifuged at 100,000 × g for 60 min to obtain soluble fractions. These fractions were passed through a column containing nickel-nitrilotriacetic acid agarose (5 by 20 mm; Qiagen, Hilden, Germany). The column was washed with 10 ml of 20 mM potassium phosphate (pH 7.5) containing 20 mM imidazole, and proteins were eluted with the same buffer containing 200 mM imidazole. All manipulations proceeded at temperatures below 4°C.
Enzyme assays.
Fungal cells were homogenized, and cell extract (100,000 × g supernatant) was prepared as described previously (40). Nudix hydrolase was assayed in reaction mixtures (50 μl) containing 50 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 25 to 100 μM substrate, and 0.2 to 1.0 mg recombinant proteins or fungal cell extract. Reaction mixtures were incubated at 37°C for 10 min, terminated by adding 10 μl of 100 mM EDTA, and analyzed by high-performance liquid chromatography (HPLC) through a Cosmosil C18 column (4.6 by 250 mm; Nacalai Tesque, Kyoto, Japan) with buffer (73 mM KH2PO4, 5 mM tetrabutylammonium dihydrogen phosphate, 20% methanol) at a flow rate of 0.6 ml min−1. We quantified NAD+-dependent HDAC using a SIRT/Sir2 NAD+-dependent HDAC assay kit (Cyclex Co. Ltd., Nagano, Japan) according to the manufacturer's protocol. Reaction mixtures containing 0.25 μg of recombinant SirA were monitored every 30 s for 1 h. Fluorescence was monitored using a microplate reader (Beckman Coulter, Inc., CA).
Determination of nucleotides.
We quantified NAD(H) in mycelia (20 mg) that were ground in liquid nitrogen and suspended with extraction buffer supplied in NAD+ and NADH quantitation kits (BioVision, Mountain View, CA). ADP-ribose was extracted from 0.5 g of mycelia as described previously (11) and determined using an HPLC system equipped with a YMC-ODS column (250 by 4.6 mm; particle size, 5 μm; Waters, Milford, MA) and a guard column (17 by 4.6 mm; Waters). Nucleotides were monitored by measuring absorption at 260 nm.
Quantitative PCR.
Total RNA was extracted from disrupted fungal cells. Single-strand cDNA was synthesized, and then real-time quantitative PCR proceeded as described previously (34). Table 1 shows the gene-specific primers. The expression of each gene was normalized against that of the actin gene (actA). Results are shown as relative expression.
Gene disruption of ndxA, ndxB, ndxC, and sirA.
Plasmids for disrupting the ndxA, ndxB, ndxC, and sirA genes were constructed by inserting DNA fragments carrying the 5′ and 3′ regions of each gene into the upstream and downstream regions of the argB gene in pBSarg1, respectively (40). The DNA fragments carrying the 5′ regions fused with appropriate restriction sites were amplified using primers (Table 1), digested with restriction enzymes, and ligated with pBSarg1 that had already been spliced with the same restriction enzymes. The 3′ region of each gene was amplified using primers (Table 1), digested with restriction enzymes, and inserted into the same restriction sites of the resulting plasmids to generate pNdxA1, pNdxB, pNdxC, and pSirA1. These plasmids were introduced into A. nidulans A89 as described previously (40), and the resulting gene disruptants were designated ΔNdxA, ΔNdxB, ΔNdxC, and ΔSirA, respectively. We introduced pSirA1 into the ABPUN strain and constructed the gene disruptant for sirA (ΔSirA2) using the strategy described above. A double gene disruptant (ΔSirA ΔNdxA) was constructed by introducing pNdxA2 to ΔSirA2. Fragments of DNA carrying the 5′ and 3′ regions of ndxA were amplified using the primers listed in Table 1 and inserted into the upstream NotI-XbaI sites and downstream EcoRI-XhoI sites of the pyrG gene in pBSpyrG (40) to construct pNdxA2. We confirmed targeted gene disruptions by Southern blotting of fungal total DNA using a digoxigenin (DIG) DNA labeling and detection kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions (Fig. 1A).
Fig 1.

Disruption of ndx genes in A. nidulans and introduction of ndxA and sirA alleles to A. nidulans. (A) Strategy for homologous recombination into ndxA, ndxB, and ndxC loci to construct ndxA, ndxB, and ndxC gene disruptants. Total DNA from strains was digested with BstXI (B), SphI (S), and PstI (P) before Southern blotting. Bars indicate positions and sizes of hybridization probes. Lanes 1, 3, and 5, A. nidulans wild type (WT); lane 2, ΔNdxA; lane 4, ΔNdxB; lane 6, ΔNdxC. (B) Strategy for introducing ndxA and sirA to ΔNdxA2 and ΔSirA2. Plasmids harboring wild-type or mutated ndxA and sirA were integrated to chromosomal pyrG regions by single crossover. Total DNA from strains was digested with PstI (P) before Southern blotting. Bars indicate positions and sizes of hybridization probes. Lanes 1 and 5, A. nidulans WT; lane 2, ΔNdxA2; lane 3, ΔNdxA2 plus pBSndxA; lane 4, ΔNdxA2 plus pBSndxA-E57Q; lane 6, ΔSirA2; lane 7, ΔSirA2 plus pBSsirA; lane 8, ΔSirA2 plus pBSsirA-H286N.
Introducing ndxA and sirA to gene disruptants.
Genomic DNA fragments carrying the ndxA and sirA genes with additional NotI recognition sequences were amplified using primers (Table 1). Mutations of ndxA-E57Q and sirA-H286N were introduced using the QuikChange site-directed mutagenesis kit (Stratagene) and primers (Table 1). The DNA fragments were digested with NotI and ligated with pBSpyrG (40) that had been spliced with the same restriction enzyme to generate pBSndxA, pBSndxA-E57Q, pBSsirA, and pBSsirA-H286N. The plasmids were introduced into strains ΔNdxA2 and ΔSirA2. The ΔNdxA2 strain was a gene disruptant for ndtxA constructed by introducing pNdxA1 into the ABPUN strain using the strategy described above. Southern blot analysis confirmed recombination of the plasmids and chromosome at the pyrG genes (Fig. 1B).
Determination of secondary metabolites.
We determined sterigmatocystin as described previously (8) with the following modifications. Conidia (108) of the fungal strains were shaken at 120 rpm in 100 ml of GMM for 72 h at 30°C. Mycelia were collected by filtration, extracted with chloroform, and dried in vacuo. The dried materials were resuspended in 100 μl of chloroform, and a portion (5 μl) was separated by thin-layer chromatography on silica gel plates using toluene-ethyl acetate-acetic acid (8:1:1) as the mobile phase. Spots for sterigmatocystin were quantified by densitometry. To quantify penicillin G, mycelia (typically 1 g [wet weight]) were collected by filtration, immediately frozen in liquid nitrogen, and ground to a powder, which was suspended in 3 ml of ice-cold 20 mM phosphate buffer (pH 7.6) and separated by centrifugation at 15,000 × g for 10 min at 4°C. The resulting supernatant was filtered through a 0.22-μm filter. The filtrates were dissolved in 0.2 M benzoic anhydride, incubated with 1-hydroxybenzotriazole and HgCl2 as described previously (31, 32), and separated on an HPLC equipped with a Cosmosil C18 column (4.6 by 250 mm; Nacalai Tesque). Penicillin G was determined by absorption at 323 nm.
Western blotting.
Nuclear fractions were prepared as described previously (18), resolved by Tricine-SDS-PAGE (30), blotted onto a nitrocellulose membrane, and incubated with polyclonal anti-acetyl histone H4 (Lys16) (YO51215; Applied Biological Materials, Richmond, BC, Canada) and anti-tetra-acetyl histone H4 (YO51212; Applied Biological Materials) and also monoclonal anti-histone H3 acetylated K9 (1328-1; Upstate, Billerica, MA), anti-histone H4 (ab10158; Epitomics Inc., Burlingame, CA), and anti-histone H3 (LS-C67991-100; Epitomics Inc.) antibodies at a 1:1,000 dilution. Cell extract (50 μg) was resolved by SDS-PAGE, blotted onto nitrocellulose membranes, and incubated with an antihemagglutinin (anti-HA) antibody diluted 1:1,000 (1583816; Roche, Basel, Switzerland) to detect SirA-HA. Goat anti-rabbit IgG conjugated with horseradish peroxidase (Pierce, Rockford, IL) was used as a secondary antibody at a 1:20,000 dilution. Chemiluminescence was detected and quantified using the ECL detection system and ImageQuant LAS4000 Mini (GE Healthcare, Waukesha, WI).
Construction of strain producing HA-tagged SirA.
A DNA fragment carrying the 3′ region of sirA with additional nucleotides encoding the HA tag and a NotI site was amplified using primers (Table 1), digested with NotI, and ligated with pBSarg1 that had been spliced with the same restriction enzyme. The 3′ region of sirA was amplified using primers (Table 1); digested with EcoRI, XhoI, and NotI; and inserted into the same restriction sites of the resulting plasmid to generate pSirA-HA, which was introduced into A. nidulans A89 as described previously (40).
Other methods.
Mycelial dry weight was determined (40), and nicotinamide riboside (NR) was prepared (7) as described previously. Antibacterial activity was analyzed by measuring the diameters of inhibitory zones of bacterial growth on agar plates (8). Standard DNA manipulation techniques proceeded according to the method of Sambrook et al. (29). Amino acid sequences were aligned using CLUSTAL W (42). Phylogenetic analyses of full-length amino acid sequences proceeded using MEGA version 4.0.2 software (41) by adapting the neighbor-joining method. Chromatin immunoprecipitation (ChIP) proceeded as described previously (5, 27) using the antibodies described above. DNA purified using the QIAquick PCR purification kit (Qiagen) was analyzed by quantitative real-time PCR as described previously (34). Relative amounts of DNA were calculated by dividing the amount of immunoprecipitated DNA by that of the input DNA.
RESULTS
Nudix hydrolases that hydrolyze NAD+ and NADH.
Twelve genes in the A. nidulans genome encoding predicted nudix hydrolases were characterized by a highly conserved motif (22) for catalytic and nucleotide binding sites (Fig. 2A and B). Among them, NdxA, NdxB, NdxC, and NdxD were included in the same branches as A. thaliana AtNUDX1 and the respective yeast isozymes (Fig. 2B). The recombinant NdxA and NdxC (Fig. 2C) hydrolyzed NAD+, NADH, NADPH, and ADP-ribose (Table 2), and the recombinant NdxA produced NMN+ and AMP from NAD+ and NMNH and AMP from NADH (Fig. 2D and E). Their apparent Km values for NADH and ADP-ribose were 4- to 12-fold lower than those for NAD+ (Table 3), indicating their preference for NADH and ADP-ribose over NAD+, like AtNUDX1 and Npy1p (1, 12). Recombinant NdxB and NdxD hydrolyzed ADP-ribose and diadenosine 5′,5′-P1,P6-hexaphosphate (Ap6A), respectively, but neither NAD(H) nor NADPH (Table 2).
Fig 2.

Identifying fungal nudix hydrolase isozymes. (A) Alignment of partial amino acid sequences among Ndx proteins of A. nidulans, A. thaliana AtNUDX1, and human HsNUDT1. Conserved residues are highlighted, and numbers indicate mutated residues. U, Ile, Leu, or Val. (B) Phylogenetic tree of nudix hydrolases of A. nidulans (prefixed with AN), S. cerevisiae (Sc), and A. thaliana (At). Proteins with reported enzyme activity are in blue. (C) SDS-PAGE of purified recombinant NdxA (lane 1), NdxB (lane 2), NdxC (lane 3), and NdxD (lane 4). Purified enzymes (1 μg) were resolved by SDS-PAGE and stained with Coomassie brilliant blue. Lanes M, Bio-Rad Precision protein standard kit. (D) Determination of reaction products of NAD+ hydrolysis catalyzed by recombinant NdxA. Reaction mixtures were analyzed by HPLC. (E) Similar experiments were performed using NADH as a substrate. (F) Fluorescence microscopy of visualized GFP-Ndx fusion proteins in A. nidulans: NdxA-GFP (a), NdxB-GFP (b), and GFP-NdxC (c). (d) DsRed-SKM produced in the same strain as that in subpanel c. Bars, 10 μm.
Table 2.
Substrate specificity of Ndx proteins
| Protein | Sp act (μmol min−1 mg−1)a |
|||||
|---|---|---|---|---|---|---|
| NADH | NAD+ | NADPH | ADP-ribose | dATP | Ap6A | |
| NdxA | 0.88 ± 0.09 | 0.14 ± 0.02 | 0.40 ± 0.06 | 0.35 ± 0.02 | 0.34 ± 0.08 | 0.03 ± 0.01 |
| NdxA-E57Q | <0.01 | <0.01 | ND | <0.01 | ND | ND |
| NdxA-E61Q | <0.01 | <0.01 | ND | <0.01 | ND | ND |
| NdxB | <0.01 | <0.01 | <0.01 | 0.15 ± 0.01 | <0.01 | <0.01 |
| NdxC | 1.3 ± 0.03 | 0.39 ± 0.01 | 0.44 ± 0.06 | 0.26 ± 0.03 | <0.01 | <0.01 |
| NdxD | <0.01 | <0.001 | <0.01 | <0.01 | <0.01 | 0.06 ± 0.01 |
Data are means of three experiments ± standard deviations. ND, not determined.
Table 3.
Apparent kinetic parameters of recombinant NdxA, NdxB, and NdxCa
| Protein | Substrate | Km (μM) | kcat (s−1) |
|---|---|---|---|
| rNdxA | NAD+ | 1,700 ± 600 | 0.15 ± 0.4 |
| NADH | 140 ± 12 | 0.35 ± 0.05 | |
| ADP-ribose | 180 ± 30 | 0.27 ± 0.04 | |
| rNdxB | ADP-ribose | 280 ± 40 | 0.11 ± 0.02 |
| rNdxC | NAD+ | 390 ± 26 | 0.14 ± 0.01 |
| NADH | 71 ± 9 | 0.72 ± 0.06 | |
| ADP-ribose | 100 ± 20 | 0.39 ± 0.05 |
Data are means of three experiments ± standard deviations.
Fusing NdxA or NdxB to the amino terminus of green fluorescent protein resulted in fluorescence signal dispersion within A. nidulans cells, indicating the cytosolic location of the two enzymes (Fig. 2Fa and -b). GFP fused to the amino terminus of NdxC, which contains a putative peroxisome targeting sequence (SKM) at the carboxy terminus, showed clear punctate patterns and colocalized with peroxisomes visualized by the SKM sequence fused to DsRed (Fig. 2Fc and -d). These findings imply that NdxA and NdxC are cytosolic and peroxisomal NAD(H) hydrolases, respectively.
Quantitative PCR analyses showed that the fungus generated 8-fold-more ndxA transcripts at the stationary (72-h) than at the early growth phase (Fig. 3A). This accompanied a 2.7-fold increase in cellular NAD+ hydrolase activity (Fig. 3B), indicating that NdxA is upregulated during the stationary growth phase. Disrupting the ndx genes did not affect fungal growth rates (Fig. 3C). A strain lacking ndxA (ΔNdxA) during the early growth phase produced as much NAD+ hydrolase activity as did the wild-type strain, and the amount of NAD+ hydrolase activity minimally increased in the stationary phase compared with the wild type (Fig. 3B). The wild-type strain cultured for 24 h contained 0.54 and 0.44 μmol g−1 of NAD+ and NADH, respectively, which remained unchanged during further incubation (Fig. 3D and E), whereas ΔNdxA accumulated more NAD+ (0.84 μmol g−1) during the stationary growth phase than did the wild type (Fig. 3D), which coincided with the decreasing NAD+ hydrolase level. Total amounts of NAD(H) increased 1.5-fold in the absence of NdxA, suggesting that A. nidulans in the stationary phase upregulates NAD(H) synthesis, which NdxA counteracts to balance the NAD+ level. Less ndxB and ndxC expression was induced during the incubation (Fig. 3A), and their depletion did not affect the NAD(H) level (Fig. 3D and E). Cellular NAD+ hydrolase activity significantly decreased in stationary ΔNdxC cells (Fig. 3B), indicating a partial contribution of NdxC to the activity although it minimally affected the cellular NAD(H) level (Fig. 3D and E). A comparison of the amino acid sequences of nudix hydrolases predicted that the conserved Glu residues 57 and 61 of NdxA are catalytic (Fig. 2A). Replacing these residues with Gln impaired the NAD(H) hydrolase activities of the recombinant proteins (Table 2). Introducing wild-type ndxA to ΔNdxA2 (ΔndxA) restored the defective cellular NAD+ hydrolase activity and the high NAD+ level, whereas the mutant ndxA-E57Q did neither (Fig. 3F and G), indicating that the enzymatic function of NdxA is required to decrease the NAD+ level. These results indicated that NdxA is the predominant nudix hydrolase involved in the homeostatic mechanism of NAD+ in A. nidulans, especially during the stationary growth phase.
Fig 3.

NdxA regulates cellular NAD(H). (A) Transcripts of ndxA, ndxB, and ndxC in A. nidulans wild type (WT) cultured at 30°C and quantified by PCR. (B) Intracellular NAD+ hydrolase activity. (C) Time-dependent changes in dry weight of cells. (D) Intracellular NAD+. (E) Intracellular NADH. (F and G) Intracellular NAD+ hydrolase activity and NAD(H) after a 72-h culture. WT, wild type; ΔndxA, ΔNdxA; ΔndxB, ΔNdxB; ΔndxC, ΔNdxC; ΔndxA+ndxA, ΔNdxA2 strain transformed with pNdxA; ΔndxA+E57Q, ΔNdxA2 strain transformed with pNdxA-E57Q. Data are means ± standard deviations (n = 3). *, P < 0.005; **, P < 0.03 versus WT.
Adding azide or antimycin A to the culture medium inhibited respiratory NADH oxidization and decreased NAD+ in both ΔNdxA and the wild type (Fig. 4). In the presence of these inhibitors, ΔNdxA accumulated 1.4-fold more NADH than did the wild type (Fig. 4A and B), indicating that NdxA affects cellular NADH as well as NAD+. That more NADH than NAD+ accumulated in ΔNdxA than in the wild type is consistent with the NdxA preference for NADH over NAD+ in vitro (Table 3). Disrupting ndxA decreased cellular ADP-ribose hydrolase activity but not the cellular ADP-ribose level (Fig. 5A and B), indicating that NdxA specifically maintains cellular NAD(H), in contrast to its isozymes from A. thaliana and from the most closely related species, S. cerevisiae, that regulate ADP-ribose (17, 43). The ndxA gene affected cellular NADH hydrolase activity (Fig. 5C). These results indicated that NdxA controls cellular NAD(H) by hydrolyzing NAD+ and/or NADH.
Fig 4.
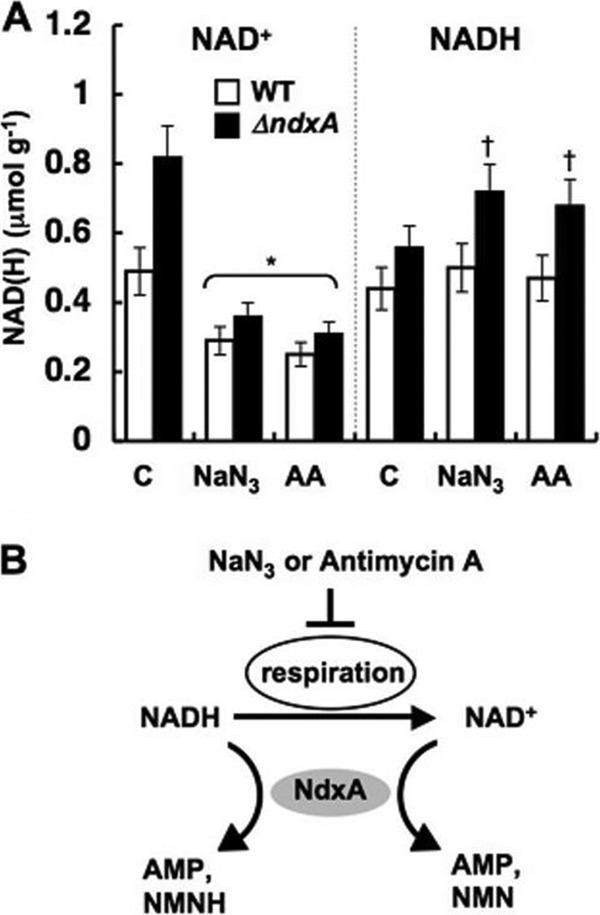
NdxA affects cellular NAD(H) by respiratory activity. (A) NAD(H) in cells incubated with 4 mM NaN3 and 30 μM antimycin A (AA) for 24 h. Untreated controls (C) are shown. Data are means ± standard deviations (n = 3). *, P < 0.03 versus control. †, P < 0.03 versus wild type (WT). (B) Model for modulation of cellular NAD(H) levels by NdxA through respiratory activity.
Fig 5.

Hydrolyzing activity of ADP-ribose and NADH and ADP-ribose contents in A. nidulans. (A) Time-dependent changes in intracellular ADP-ribose hydrolase activity. (B) ADP-ribose contents in the A. nidulans cells after a 72-h culture. (C) Time-dependent changes in intracellular NADH hydrolase activity. WT, wild type; ΔndxA, ΔNdxA; ΔndxB, ΔNdxB; ΔndxC, ΔNdxC. Data are means of three experiments. Error bars indicate standard deviations.
NdxA upregulates fungal secondary metabolite production.
We found that the ΔNdxA strain produced less penicillin G and sterigmatocystin than did the wild type (Fig. 6A to C); these are typical secondary metabolites of this fungus. ΔNdxA accumulated fewer transcripts of penicillin G cluster genes encoding isopenicillin-N synthetase (ipnA) and its acyltransferase (penDE) (20) and of aflR and stcU encoding a transcription factor and an enzyme for sterigmatocystin biosynthesis (9) (Fig. 6D and E). Introducing the ndxA gene eliminated these defects arising from the ΔndxA mutation, whereas the mutant ndxA gene encoding the catalytically inactive NdxA-E57Q was inert for the secondary metabolite production and expression of the associated genes (Fig. 6A to E). These results further implied that NdxA lowered cellular NAD+ levels, through which NdxA upregulates the synthesis of these secondary metabolites.
Fig 6.

NdxA requirement for maximal secondary metabolite production. (A) Penicillin G (PN) production after 72 h in culture. (B) Bacterial growth inhibition assay shows penicillin production by A. nidulans. (C) Sterigmatocystin production after 72 h in culture. (D and E) Transcripts of secondary metabolite genes in cells quantified by PCR after 72 h in culture. WT, wild type; ΔndxA, ΔNdxA; ΔndxA+ndxA, ΔNdxA2 strain transformed with pNdxA; ΔndxA+E57Q, ΔNdxA2 strain transformed with pNdxA-E57Q; ΔsirA, ΔSirA; ΔsirA+sirA, ΔSirA2 strain transformed with pSirA; ΔsirA+H286N, ΔSirA2 strain transformed with pSirA-H286N; ΔsirAΔndxA, ΔSirA ΔNdxA. Data are means ± standard deviations (n = 3). *, P < 0.005 versus WT. (F) NAD+ synthesis in A. nidulans. Predicted genes for nicotinamide riboside (NR) salvage pathways for NAD+ synthesis and for de novo synthetic pathway of NAD+ are presented as gene identities. NaMN, nicotinic acid mononucleotide; NaAD, nicotinic acid adenine dinucleotide; NMN, nicotinamide mononucleotide; Na, nicotinic acid; Nam, nicotinamide. NdxA counteracts NMN adenylyltransferase (AN1745) in that they catalyze NMN+ and NAD+ interconversion.
Nicotinamide riboside (NR) is a salvage substrate of NAD+ synthesis (4). The A. nidulans genome carries a set of predicted NR salvage genes (Fig. 6F), and adding NR (>0.3 mM) to fungal cultures increased cellular NAD+ levels as it does in yeast (4) (Fig. 7A) without affecting ndxA transcription and cellular NAD(H) hydrolase activity (Fig. 7B and C). The secondary metabolite production (Fig. 7D and G) and the expression of their biosynthetic genes (Fig. 7F and I) decreased under these conditions, indicating that increased NAD+ represses secondary metabolite production.
Fig 7.

Cellular NAD(H) regulates secondary metabolite synthesis. Aspergillus nidulans wild type (WT), ΔNdxA (ΔndxA), ΔSirA (ΔsirA), and ΔSirA ΔNdxA (ΔsirA ΔndxA) were cultured for 72 h under normoxic and hypoxic (1% O2) conditions. +NR, WT cultured with 0.3 mM nicotinamide riboside. (A) Intracellular NAD(H). (B) PCR-quantified ndxA transcripts. (C) Intracellular NAD(H) hydrolase activity. (D and E) Penicillin G production. (F) Penicillin G gene transcripts quantified by PCR. (G and H) Sterigmatocystin (ST) production. (I) Sterigmatocystin gene transcripts quantified by PCR. Data are means ± standard deviations (n = 3). *, P < 0.005; **, P < 0.03 (versus WT); †, P < 0.03 (versus WT with 1% O2).
Cellular NAD(H) levels were decreased when the fungus was cultured under hypoxic (low-oxygen) conditions, compared with normoxic conditions (Fig. 7A). The wild type produced 2.6- and 2.0-fold more ndxA transcripts (Fig. 7B) and NAD(H) hydrolase activity (Fig. 7C) under hypoxic than under normoxic conditions. The constant level of NADH in ΔNdxA under hypoxia (Fig. 7A) indicated that NdxA decreases cellular NADH under this condition. Additional factors might lower the NAD+ level, since ΔNdxA still contained less NAD+ than did the wild type under hypoxic conditions. Hypoxia limits the availability of electron acceptors (oxygen) for oxidizing NADH and decreases cellular NAD+ levels (35). The wild type produced more penicillin G and sterigmatocystin, as well as transcripts of their biosynthetic genes, under such conditions (Fig. 7D, F, G, and I). These results indicate that decreased NAD+ enhances the production of secondary metabolites.
Novel A. nidulans sirtuin represses secondary metabolite production.
The NAD+-independent HDAC of A. nidulans HdaA regulates histone H3 acetylation and the expression of secondary metabolite cluster genes (26), whereas the sirtuin isozyme HstA has predicted obscure enzymatic and physiological functions (36). We found a fungal gene encoding a sirtuin isozyme (SirA) with an amino acid identity of 47% to yeast Sir2p. Phylogenetically, SirA is more closely related to known sirtuins than is HstA (Fig. 8A). Recombinant SirA exhibited nicotinamide-sensitive NAD+-dependent HDAC activity (Fig. 8B and C), indicating that SirA is a fungal sirtuin counterpart. Chromatin immunoprecipitation (ChIP) analyses of a strain producing hemagglutinin-tagged SirA (SirA-HA) indicated that SirA-HA was enriched at the ipnA and aflR gene promoters (Fig. 9A and B). During the stationary growth phase, the sirA-depleted strain ΔSirA (Fig. 8D) accumulated more acetylated lysine residue 16 of histone H4 (H4K16) and tetra-acetyl histone H4 than did the wild type, whereas acetylated lysine residue 9 of histone H3 (H3K9) remained unchanged (Fig. 9C). ΔSirA accumulated more acetylated H4K16 at the ipnA and aflR loci (Fig. 9D) and their transcripts (Fig. 6D and E) and produced more penicillin G and sterigmatocystin (Fig. 6A to C), indicating that SirA removes H4K16 acetylation and represses the expression of these secondary metabolite genes. Levels of SirA did not change during culture (Fig. 9B), indicating that the repression mechanism is posttranslational. Recombinant SirA protein with Asn substituted for His286 lost NAD+-dependent HDAC activity (Fig. 8C), and the mutated gene could not complement these defects of ΔSirA, in contrast to the wild-type sirA (Fig. 6A to E and 9D). These findings indicated that SirA regulated the level of H4K16 acetylation and the secondary metabolite production through its H4K16-deacetylating activity. The increased cellular NAD+ and the decreased secondary metabolite production induced by adding nicotinamide riboside (Fig. 7) accompanied decreased levels of H4K16 acetylation at the secondary metabolite gene promoters, a finding which was not evident in ΔSirA (Fig. 7E and H and 9E). These results indicate that cellular NAD+ modulates the HDAC reaction by SirA.
Fig 8.

Identification of sirtuin isozyme SirA from A. nidulans. (A) Phylogenetic relationship among sirtuin isozymes. All amino acid sequences of proteins similar to sirtuins collected from A. nidulans (prefixed with AN), S. cerevisiae (Sc), Schizosaccharomyces pombe (Sp), and human genes are shown as their corresponding gene identifier and/or gene names. SIRT and Hdac represent human proteins. Proteins described in the text are highlighted by boldface and underlining. Classes of HDAC are shown on the right. (B) SDS-PAGE of purified recombinant SirA (rSirA) (lane 1) and rSirA-H286N (lane 2) (1 μg each). (C) NAD+-dependent HDAC activity of rSirA with (solid bars) or without (open bars) 2 mM nicotinamide. Reactions proceeded with NAD+ (200 μM), rSIRT1, rSirA, and rSirA-H286N (25 μg each). Data are means of three experiments. Error bars indicate standard deviations. P < 0.005. (D) Strategy for gene disruption of sirA and Southern blot analysis of A. nidulans WT (lane 1) and ΔSirA2 (lane 2). Total DNA from strains was digested with HindIII (H) before blotting. The position and size of the hybridization probe are shown.
Fig 9.

NdxA represses histone deacetylation. (A) ChIP analyses indicate SirA association with ipnA and aflR gene promoters in A. nidulans cells cultured for 48 h. Top panel, positions (1 to 6) of immunoprecipitated DNA. Data are means ± standard deviations (n = 3). *, P < 0.005, and **, P < 0.01, versus ΔsirA. (B) Western blot analysis of cell extracts of the strain producing SirA-HA. Anti-HA antibody detected levels of SirA-HA similar between early stationary (48-h) and stationary (72-h) growth phases. Arrowhead, SirA-HA. (C) Western blotting detected acetylated histones in nuclear fractions of cells cultured for 72 h. H3 and H4, total histones H3 and H4. Representative results of four repeated experiments are presented with relative signal intensity under bands. (D and E) ChIP findings of effects of ndxA and sirA (D) and of nicotinamide riboside and hypoxia (E) on H4K16 acetylation at ipnA, aflR, and actA gene promoters in strains cultured for 72 h. Relative IP represents signal ratio between precipitated and input DNA. WT, wild type; ΔndxA, ΔNdxA; ΔndxA+ndxA, ΔNdxA2 strain transformed with pNdxA; ΔndxA+E57Q, ΔNdxA2 strain transformed with pNdxA-E57Q; ΔsirA, ΔSirA; ΔsirA+sirA, ΔSirA2 strain transformed with pSirA; ΔsirA+H286N, ΔSirA2 strain transformed with pSirA-H286N; ΔsirAΔndxA, ΔSirA ΔNdxA. Data are means ± standard deviations (n = 3). *, P < 0.005, and **, P < 0.01, versus WT. (F) Model of negative epigenetic control by NdxA through NAD(H) hydrolysis.
NdxA negatively regulates sirtuin function.
The NdxA-dependent increase in the gene expression of secondary metabolites and their SirA-dependent repression indicated that NdxA and SirA cooperate to control histone acetylation and secondary metabolite gene expression. ChIP analyses showed that ΔNdxA accumulated less acetylated H4K16 at the ipnA and aflR loci than did the wild type (Fig. 9D). The defective H4K16 acetylation evoked by ndxA depletion was restored by introducing wild-type ndxA without the nonfunctional mutant ndxA with the mutation at Glu57 (Fig. 9D). The defects were not identified in the double-gene disruptant of sirA and ndxA (ΔsirA ΔndxA in Fig. 9C and D), indicating that NdxA increases H4K16 acetylation through repressing the HDAC activity of SirA. The defective production of secondary metabolites and the transcription of their synthetic genes in ΔNdxA were restored by depleting sirA (Fig. 6A to E). These results indicated that fungal NdxA decreases NAD+, represses the HDAC activity of SirA, increases H4K16 acetylation, and derepresses secondary metabolite gene expression (Fig. 9F). Under hypoxic conditions with low NAD+ (Fig. 7A), both the wild type and ΔNdxA accumulated more acetylated H4K16 at the ipnA and aflR loci (Fig. 9E) and produced more secondary metabolites (Fig. 7D and G). These findings imply that significantly higher levels of cellular NAD+ are required before NdxA can downregulate SirA, which agrees with the model of SirA inactivation by NdxA-dependent NAD+ degradation (Fig. 9F).
DISCUSSION
NAD(H) is a classical hydride-transferring cofactor, and its ratios are controlled to maintain cellular reduction-oxidation reactions. Repression of the glycolytic mechanism by NADH is an example of a response to environmental and nutrient conditions that lowers cellular levels of NADH compared with NAD+ (15). The present study demonstrated that NdxA hydrolyzes NAD(H) in vivo and in vitro, decreases the total amount of NAD(H), and controls cellular NAD(H) homeostasis. These findings indicate a hitherto-undiscovered mechanism of NAD(H) regulation that is governed by the novel NAD(H) regulator NdxA. We also found that the mechanism downregulates sirtuin-dependent gene silencing. Recent studies have demonstrated that de novo and salvage NAD+ biosynthesis mechanisms upregulate available NAD+ (4, 7). These are also likely to function in A. nidulans since this fungus carries sets of the genes involved in these mechanisms (Fig. 6F), and a mutation of ndxA increased the NAD+ concentration at the stationary phase. The NdxA-mediated downregulation mechanisms of total NAD(H) are considered to counteract biosynthetic mechanisms and fine-tune NAD(H), which in turn regulates levels of histone acetylation.
Reports have shown that histone H3and H4 acetylation regulates the aflatoxin biosynthetic genes of Aspergillus parasiticus and the secondary metabolite cluster genes of A. nidulans, respectively (27, 28). Besides these processes involving NAD+-independent class II HDAC, the present study is the first to demonstrate that both a sirtuin-type HDAC and the NAD(H) state are involved in the fungal production of secondary metabolites. Most known secondary metabolites of filamentous fungi are produced by stationary-phase mechanisms, although the initiation mechanism(s) has not been defined. Microorganisms at the stationary phase encounter stress caused by low nutrition and oxidative damage, both of which affect cytosolic NAD(H) (13). The missing link between NAD+ metabolism and secondary metabolite production presented here therefore suggests that an altered NAD(H) state is one trigger for secondary metabolite production at the stationary phase. Functional interaction between the NAD(H) state and nutrient signaling through the cAMP-protein kinase A pathway (33) will be important for understanding fungal secondary metabolite production.
The A. nidulans biosynthetic gene clusters for penicillin G and sterigmatocystin are located within regions that are 30 and 90 kb apart from subtelomeric regions, respectively, as they are in other aspergilli (21, 36). Increased H4K16 acetylation at the subtelomeric regions of S. cerevisiae and Neurospora crassa accompanies decreased sirtuin (Sir2p and NST-1, respectively) levels (10, 38). The two deacetylases apparently share the same substrate (acetylated H4K16), indicating that the process of telomere silencing by deacetylation is conserved among these fungi and A. nidulans. The yeast Sir2 interacts with Sir3/Sir4, which are not found in A. nidulans and N. crassa. This suggests that the yeast silences telomeric genes via a different mechanism than that of the filamentous fungi and probably also of higher eukaryotes. Besides functions on subtelomeres, yeast Sir2p silences genes at specific loci concerned with mating type, rRNA production, and aging. Our preliminary screening found no defect in the growth rate, hyphal morphology, and sexual/asexual development of the sirA gene disruptant. This is in contrast to other HDAC genes in this fungus (26, 36), suggesting that SirA plays a specific role in secondary metabolite production. However, a detailed study will be required to prove this notion.
Nudix hydrolases are ubiquitous among eukaryotes. The distribution of nudix hydrolase orthologs in most analyzed fungal genomes and of histone modifications on the secondary metabolite genes of mycotoxigenic fungi (26) indicates that nudix hydrolase inhibitors could be used to diminish mycotoxin contamination of cereals. Upregulated sirtuins increase energy-state-dependent ribosomal DNA (rDNA) silencing (24) and extend life span (19). Known mammalian nudix hydrolases that hydrolyze NAD(H) (2, 22) might have therapeutic potential as novel targets in the treatment of metabolic disorders and life span.
ACKNOWLEDGMENTS
We thank Akiko Murayama and Norma Foster for helpful discussion and critical reading of the manuscript.
This study was supported by a Grant-in-Aid for Scientific Research (21380055 to N.T.).
Footnotes
Published ahead of print 16 July 2012
REFERENCES
- 1. AbdelRaheim SR, Cartwright JL, Gasmi L, McLennan AG. 2001. The NADH diphosphatase encoded by the Saccharomyces cerevisiae NPY1 nudix hydrolase gene is located in peroxisomes. Arch. Biochem. Biophys. 388: 18–24 [DOI] [PubMed] [Google Scholar]
- 2. Abdelraheim SR, Spiller DG, McLennan AG. 2003. Mammalian NADH diphosphatases of the Nudix family: cloning and characterization of the human peroxisomal NUDT12 protein. Biochem. J. 374: 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bayram O, et al. 2008. VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 320: 1504–1506 [DOI] [PubMed] [Google Scholar]
- 4. Belenky P, et al. 2007. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 129: 473–484 [DOI] [PubMed] [Google Scholar]
- 5. Bernreiter A, et al. 2007. Nuclear export of the transcription factor NirA is a regulatory checkpoint for nitrate induction in Aspergillus nidulans. Mol. Cell. Biol. 27: 791–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bessman MJ, Frick DN, O'Handley SF. 1996. The MutT proteins or “Nudix” hydrolases, a family of versatile, widely distributed, “housecleaning” enzymes. J. Biol. Chem. 271: 25059–25062 [DOI] [PubMed] [Google Scholar]
- 7. Bieganowski P, Brenner C. 2004. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117: 495–502 [DOI] [PubMed] [Google Scholar]
- 8. Bok JW, Keller NP. 2004. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot. Cell 3: 527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown DW, et al. 1996. Twenty-five coregulated transcripts define a sterigmatocystin gene cluster in Aspergillus nidulans. Proc. Natl. Acad. Sci. U. S. A. 93: 1418–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dang W, et al. 2009. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459: 802–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Di Pierro D, et al. 1995. An ion-pairing high-performance liquid chromatographic method for the direct simultaneous determination of nucleotides, deoxynucleotides, nicotinic coenzymes, oxypurines, nucleosides, and bases in perchloric acid cell extracts. Anal. Biochem. 231: 407–412 [DOI] [PubMed] [Google Scholar]
- 12. Dobrzanska M, Szurmak B, Wyslouch-Cieszynska A, Kraszewska E. 2002. Cloning and characterization of the first member of the Nudix family from Arabidopsis thaliana. J. Biol. Chem. 277: 50482–50486 [DOI] [PubMed] [Google Scholar]
- 13. Fabrizio P, Longo VD. 2003. The chronological life span of Saccharomyces cerevisiae. Aging Cell 2: 73–81 [DOI] [PubMed] [Google Scholar]
- 14. Guarente L. 1999. Diverse and dynamic functions of the Sir silencing complex. Nat. Genet. 23: 281–285 [DOI] [PubMed] [Google Scholar]
- 15. Haigis MC, Guarente LP. 2006. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev. 20: 2913–2921 [DOI] [PubMed] [Google Scholar]
- 16. Imai S, Armstrong CM, Kaeberlein M, Guarente L. 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403: 795–800 [DOI] [PubMed] [Google Scholar]
- 17. Ishikawa K, et al. 2009. Modulation of the poly(ADP-ribosyl)ation reaction via the Arabidopsis ADP-ribose/NADH pyrophosphohydrolase, AtNUDX7, is involved in the response to oxidative stress. Plant Physiol. 151: 741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee I, Shwab EK, Dagenais TR, Andes D, Keller NP. 2009. HdaA, a class 2 histone deacetylase of Aspergillus fumigatus, affects germination and secondary metabolite production. Fungal Genet. Biol. 46: 782–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin SJ, Defossez PA, Guarente L. 2000. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289: 2126–2128 [DOI] [PubMed] [Google Scholar]
- 20. MacCabe AP, Riach MB, Unkles SE, Kinghorn JR. 1990. The Aspergillus nidulans npeA locus consists of three contiguous genes required for penicillin biosynthesis. EMBO J. 9: 279–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McDonagh A, et al. 2008. Sub-telomere directed gene expression during initiation of invasive aspergillosis. PLoS Pathog. 4: e1000154 doi: 10.1371/journal.ppat.1000154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McLennan AG. 2006. The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 63: 123–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mullbacher A, Eichner RD. 1984. Immunosuppression in vitro by a metabolite of a human pathogenic fungus. Proc. Natl. Acad. Sci. U. S. A. 81: 3835–3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murayama A, et al. 2008. Epigenetic control of rDNA loci in response to intracellular energy status. Cell 133: 627–639 [DOI] [PubMed] [Google Scholar]
- 25. Nakahata Y, et al. 2008. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 134: 329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Palmer JM, Keller NP. 2010. Secondary metabolism in fungi: does chromosomal location matter? Curr. Opin. Microbiol. 13: 431–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reyes-Dominguez Y, et al. 2010. Heterochromatic marks are associated with the repression of secondary metabolism clusters in Aspergillus nidulans. Mol. Microbiol. 76: 1376–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roze LV, Arthur AE, Hong SY, Chanda A, Linz JE. 2007. The initiation and pattern of spread of histone H4 acetylation parallel the order of transcriptional activation of genes in the aflatoxin cluster. Mol. Microbiol. 66: 713–726 [DOI] [PubMed] [Google Scholar]
- 29. Sambrook J, Fritch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 30. Schagger H. 2006. Tricine-SDS-PAGE. Nat. Protoc. 1: 16–22 [DOI] [PubMed] [Google Scholar]
- 31. Shah AJ, Adlard MW, Holt G. 1988. Determination of natural penicillins in fermentation media by high-performance liquid chromatography using pre-column derivatisation with 1-hydroxybenzotriazole. Analyst 113: 1197–1200 [DOI] [PubMed] [Google Scholar]
- 32. Shah AJ, Tilburn J, Adlard MW, Arst HN. 1991. pH regulation of penicillin production in Aspergillus nidulans. FEMS Microbiol. Lett. 61: 209–212 [DOI] [PubMed] [Google Scholar]
- 33. Shimizu K, Keller NP. 2001. Genetic involvement of a cAMP-dependent protein kinase in a G protein signaling pathway regulating morphological and chemical transitions in Aspergillus nidulans. Genetics 157: 591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shimizu M, Fujii T, Masuo S, Fujita K, Takaya N. 2009. Proteomic analysis of Aspergillus nidulans cultured under hypoxic conditions. Proteomics 9: 7–19 [DOI] [PubMed] [Google Scholar]
- 35. Shimizu M, Fujii T, Masuo S, Takaya N. 2010. Mechanism of de novo branched-chain amino acid synthesis as an alternative electron sink in hypoxic Aspergillus nidulans cells. Appl. Environ. Microbiol. 76: 1507–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shwab EK, et al. 2007. Histone deacetylase activity regulates chemical diversity in Aspergillus. Eukaryot. Cell 6: 1656–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smeal T, Claus J, Kennedy B, Cole F, Guarente L. 1996. Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 84: 633–642 [DOI] [PubMed] [Google Scholar]
- 38. Smith KM, et al. 2008. The fungus Neurospora crassa displays telomeric silencing mediated by multiple sirtuins and by methylation of histone H3 lysine 9. Epigenetics Chromatin 1: 5 doi:10.1186/1756-8935-1-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Taddei F, et al. 1997. Counteraction by MutT protein of transcriptional errors caused by oxidative damage. Science 278: 128–130 [DOI] [PubMed] [Google Scholar]
- 40. Takasaki K, et al. 2004. Fungal ammonia fermentation, a novel metabolic mechanism that couples the dissimilatory and assimilatory pathways of both nitrate and ethanol. Role of acetyl CoA synthetase in anaerobic ATP synthesis. J. Biol. Chem. 279: 12414–12420 [DOI] [PubMed] [Google Scholar]
- 41. Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24: 1596–1599 [DOI] [PubMed] [Google Scholar]
- 42. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tong L, Lee S, Denu JM. 2009. Hydrolase regulates NAD+ metabolites and modulates cellular redox. J. Biol. Chem. 284: 11256–11266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tribus M, et al. 2010. A novel motif in fungal class 1 histone deacetylases is essential for growth and development of Aspergillus. Mol. Biol. Cell 21: 345–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang T, Kraus WL. 2010. SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim. Biophys. Acta 1804: 1666–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]