

Abstract
The lymphoid enhancer factor 1/T cell factor (LEF/TCF) family of transcription factors are downstream effectors of the WNT signaling pathway, which drives colon tumorigenesis. LEF/TCFs have a DNA sequence-specific high-mobility group (HMG) box that binds Wnt response elements (WREs). The “E tail” isoforms of TCFs are alternatively spliced to include a second DNA binding domain called the C-clamp. We show that induction of a dominant negative C-clamp version of TCF1 (dnTCF1E) induces p21 expression and a stall in the growth of DLD1 colon cancer cells. Induction of a C-clamp mutant did not efficiently induce p21, nor did it stall cell growth. Microarray analysis revealed that induction of p21 by wild-type dnTCF1E (dnTCF1EWT) correlated with a decrease in expression of multiple p21 suppressors that act at multiple levels from transcription (SP5, YAP1, and RUNX1), RNA stability (MSI2), and protein stability (CUL4A). We show that the C-clamp is a sequence-specific DNA binding domain that can make contacts with 5′-RCCG-3′ elements upstream or downstream of WREs. The C-clamp–RCCG interaction was critical for TCF1E-mediated transcriptional control of p21-connected target gene promoters. Our results indicate that a rapid-response WNT/p21 circuit is driven by C-clamp target gene selection.
INTRODUCTION
Overactive Wnt signaling is causally linked to carcinogenesis in many different tumor types, with colon tumors having an especially strong link to this pathway (18, 21). Over 80% of colon tumors derive from aberrantly activated Wnt signals, which are most often created by familial or sporadic mutation of the tumor suppressor adenomatous polyposis coli (63). Loss of tumor suppressor function results in elevated levels of β-catenin protein which, in turn, drives a Wnt transcriptional program essential to the generation of colon cancer (7, 67, 72). The Wnt signaling transcriptional program is directed by interactions between β-catenin and the lymphoid enhancer factor 1/T cell factor (LEF/TCF) family of DNA binding transcription factors (2). Two family members, TCF1 and TCF4, are expressed in normal adult colon epithelial cells where they mediate Wnt-directed control of stem cell homeostasis and differentiation (1, 46, 59, 66). TCF1 and TCF4 are also expressed in colon cancer, and in this setting they direct aberrant proliferation and survival (2, 59, 72).
Extensive alternative splicing of LEF/TCFs generates considerable isoform diversity; however, the functional importance of this is not well understood (70, 74). The “E tail” isoforms of TCF1 and TCF4 (referred to as TCF1E and TCF4E, respectively) are generated by alternative splicing at the 3′ end of the pre-mRNA that encodes a long C-terminal tail (3, 4, 70, 74). The E tails of TCF1 (3) and TCF4 (74) are known to be required for the activation of two WNT target genes. TCF1E is uniquely suited for activation of the LEF1 promoter (3, 4), and TCF4E is the only isoform of TCF4 that can activate the CDX1 promoter (32, 74). The E-tail of human TCF1 was recently shown to contain a DNA binding domain called the C-clamp that was required for activation of the LEF1 promoter (4). The C-clamp was also shown to harbor sequence specificity for GC-rich elements in mammals (4) and Drosophila (15). In Drosophila the C-clamp of the TCF ortholog pangolin/dTCF interacts with an extended GC-rich element referred to as a “Helper” site, which was shown to be required for WNT (Wg)-induced transcription of several target genes (15). In addition, a mutation in the Drosophila C-clamp causes embryonic lethal Wg signaling defects (71). These data suggest that the C-clamp is required for Wg signal regulation of target gene expression in vivo. However, despite its initial discovery in a mammalian system, the importance of C-clamp DNA binding activities to the regulation of WNT target genes in mammals is largely unknown. It is also unknown whether the C-clamp is important for regulation of a broad range of WNT target genes or whether it is important for a functional subset of targets.
There have been several efforts to define the Wnt transcriptome in colon epithelial cells and identify target genes that are important for normal intestinal epithelium homeostasis and colon cancer (47, 69, 72). These efforts have been quite successful; for example, they led to the identification of the adult stem cell marker LGR5 (48, 69, 72). To identify target genes in colon cancer cells, expression systems were established with inducible dominant negative isoforms of TCFs. These dominant negative isoforms lack the β-catenin binding domain and compete with endogenous full-length isoforms to downregulate WNT target gene expression (72). The most notable phenotype from these experiments was that overexpression of dominant negative TCF1E or TCF4E (dnTCF1E or dnTCF4E, respectively) caused a p21-dependent stall in the G1 phase of the cell cycle (72). p21 is a cyclin-dependent kinase inhibitor and master regulator of the G1-to-S-phase transition. Here, we show that the previously observed p21-dependent growth arrest is dependent on C-clamp activity because induction of a C-clamp mutant defective for DNA binding did not induce p21 expression and did not cause a stall in cell growth. We demonstrate through microarray analysis that the C-clamp enables transcriptional regulation of a distinct set of WNT target genes that does not include cMYC, AXIN2, LGR5, and other classic Wnt targets. Instead, a significant subset of the C-clamp targets are important for the regulation of p21. Biochemical analysis of the WREs in the promoters of C-clamp targets shows that the C-clamp can make contacts with Helper sites upstream of Wnt response elements (WREs) in addition to downstream of WREs, as was previously reported (4). We show that C-clamp–Helper interactions are required for TCF1E-mediated activation of several mammalian promoters, including the promoters of C-clamp-specific, p21 transcriptional regulators. Thus, we have linked colon cancer cell proliferation with in vitro observations of the C-clamp's DNA binding activities.
MATERIALS AND METHODS
CASTing.
Cyclic amplification and selection of targets (CASTing) was performed as described previously (4, 76).
Establishment of stable cell lines and Dox concentrations.
To establish inducible wild-type and mutant dnTCF1E (dnTCF1EWT and dnTCF1Emut, respectively)-inducible cell lines, a parental DLD1 clonal cell line which constitutively expresses a tetracycline repressor (generously provided by van de Wetering et al. [72]) was transfected with plasmids encoding dnTCF1EWT and dnTCF1Emut (with the mutation CRARF → VALAL). Selection and expansion of clones were carried out essentially as described previously (4, 72). To start, the starter T-Rex cell line was cotransfected with linearized plasmids encoding an expression vector for the neomycin resistance gene and a tetracycline-regulated promoter/dnTCF transgene plasmid. Hundreds of clonal isolates were expanded and analyzed for transgene expression in the absence of the inducer doxycycline (Dox). Multiple clonal isolates (∼20 to 30) were compared for tight doxycycline induction. Once pairs of cell lines were chosen, doxycycline titrations were carried out in parallel to ensure identical induction levels of dnTCF1EWT and dnTCF1Emut. We determined that, for the chosen clonal isolates, different amounts of doxycycline are needed to induce the same amount of each transgene. The Dox concentrations used for the experiments are 0.0005 μg/ml for dnTCF1EWT and 1.0 μg/ml for dnTCF1Emut. The large difference in doxycycline concentrations used for dnTCF1EWT- and dnTCF1Emut-expressing cells was not a reflection of differential protein stability. Northern blotting, reverse transcription-PCR (RT-PCR), and microarray data confirm that the chosen Dox concentrations produce nearly identical levels of these transgenes. For example, the mean robust multiarray average (RMA) intensity values for TCF7 mRNA (probe set 205254_x_at; Hu133 Affymetrix array) were 13.19 for dnTCF1EWT and 13.13 dnTCF1Emut. Similarly, the calculated fold inductions for TCF7 were 5.7-fold for the wild type and 5.6-fold for the mutant (data are available in the Gene Expression Omnibus [GEO] microarray data set). Thus, the large difference in doxycycline concentrations to induce similar levels of wild-type and mutant proteins likely reflect differences in the chromatin conformation at the site of transgene integration.
Transient transfections.
Cos1 cells were transiently transfected with BioT transfection reagent according to the manufacturer's protocol (Bioland Scientific LLC). Colo320, Cos1, or DLD1 cells were plated at a density of 200,000 cells/well in six-well plates ∼20 h before transfection. Luciferase reporter constructs (0.4 μg) were cotransfected with β-catenin (0.4 μg), β-galactosidase (0.1 μg), and an LEF/TCF expression vector (0.005 μg to 0.1 μg). Cells were harvested after ∼20 h, and luciferase and β-galactosidase activities were determined as described by Atcha et al. (3).
Plasmids.
Construction of TCF1EWT, TCF1Emut, and LEF1 expression plasmids was described previously (3, 33). The TCF4EWT expression plasmid was previously described and generously provided by Weise et al. (74). TCF4Emut was generated from a TCF4EWT expression plasmid using the following primers (mutant sequences are in bold): 5′-CCTTGATCAACAGAATAACTGGGCCGGCCCTTGC-3′ (sense) and 5′-GCAAGGGCCGGCCCAGTTATTCTGTTGATCAAGG-3′ (antisense).
Helper Downstream, Topmod, Helper Upstream, Helper2, and TOP2 sequences (Fig. 1C) were cloned into the BamHI site in the TK100 luciferase reporter. The TOPTK plasmid was a generous gift of Hans Clevers. The SP5 promoter luciferase reporter plasmid was previously described and generously provided by Fujimura et al. (23). The SP5 promoter luciferase reporter was mutated with the following primers (mutations are in boldface): site E sense, 5′-GCGCGAGTCTCCAGTCTATAAGGCCCCCTTTGATCAGG-3′; site E antisense, 5′-CCTGATCAAAGGGGGCCTTATAGACTGGAGACTCGCGC-3′; site G sense, 5′-CGCTTCTGAAAGAGACAATATTCTTTGATGATTGGGTAGCGGC-3′; site G antisense, 5′-GCCGCTACCCAATCATCAAAGAATATTGTCTCTTTCAGAAGCG-3′; site H sense, 5′-GCCGCTATTCTTTGATGATTGGGTAGAGTTAAACTTCAAAGCC-3′; and site H antisense, 5′-GGCTTTGAAGTTTAACTCTACCCAATCATCAAAGAATAGCGGC-3′.
Fig 1.

The C-clamp functionally interacts with Helper sites. (A) Cyclic amplification and selection of targets (CASTing) was performed with TCF1EWT and TC1Emut (C-clamp mutant). Sequence logo alignment of oligonucleotides revealed a short GC-rich Helper site that was pulled down by TCF1EWT but not TCF1Emut. TCF1EWT and TCF1Emut had the same sequence specificities for the WRE (CTTTGATSTT). (B) Percentage of independent TCF1EWT and TCF1Emut sequences that contained WREs alone, Helper sequences downstream from the WRE, and Helper sequences upstream from the WRE. (C) Oligonucleotides were cloned into the TK100 luciferase reporter under control of the minimal thymidine kinase promoter. (D) Reporters described in panel C were transfected into Colo320 cells along with increasing concentrations of TCF1EWT and TCF1Emut expression plasmids (25 ng, 50 ng, and 100 ng). TCF1EWT activated the Helper Downstream and Helper Upstream reporters, whereas TCF1Emut could not activate these reporters over the high basal levels of WNT signaling in Colo320 cells. Western analysis shows that equal levels of TCF1EWT and TCF1Emut were expressed with 50 ng of expression plasmid. (E) Helper2, TOP2, and TOPTK reporters were transfected into Cos1 cells along with β-catenin and TCF1EWT or TCF1Emut. TCF1EWT could activate the Helper2 construct, whereas neither TCF1EWT nor TCF1Emut could activate the TOP2 construct. TCF1EWT and TCF1Emut showed similar activation levels of the TOPTK construct. The Western blot panel shows that equal levels of TCF1EWT and TCF1Emut were expressed.
The CDX1 promoter luciferase reporter plasmid was previously reported and generously provided by A. Hecht and M. P. Stemmler (32). CDX1 promoter mutants were generated with the following primers (mutations are in boldface): site 1 sense, 5′-CGACGGGCTTCCCCCTTTGATTCTATTATCCGAGGCTTCCCCCCG-3′; site 1 antisense, 5′CGGGGGGAAGCCTCGGATAATAGAATCAAAGGGGGAAGCCCGTCG-3′; site 2 sense, 5′-GGCTTCCCCCCGCTTTGAAATGCAAAGCATTATGGCTGGGGCCGC-3′; site 2 antisense, 5′-GCGGCCCCAGCCATAATGCTTTGCATTTCAAAGCGGGGGGAAGCC-3′; site 3 sense, 5′-GCAAAGCATTATGGCTGGGTACTAGGACGGCCCGCGGCTA-3′; site 3 antisense, 5′-TAGCCGCGGGCCGTCCTAGTACCCAGCCATAATGCTTTGC-3′; site 4 sense, 5′-CTCCTTTTGAACCCCCTCATACGACGGGCTTCCCC-3′; site 4 antisense, 5′-GGGGAAGCCCGTCGTATGAGGGGGTTCAAAAGGAG-3′; site 5 sense, 5′-CCACCTCCCGCTTAGGGTATCAATTTGTCTCCTTTTGAACC-3′; and site 5 antisense, 5′-GGTTCAAAAGGAGACAAATTGATACCCTAAGCGGGAGGTGG-3′.
The LEF1 promoter luciferase reporter plasmid has been previously described (36). LEF1 promoter mutants were generated with the following primers (mutations are in boldface): site 1 sense, 5′-GCTTTGACAGAGCTGTACTGTGGAGGCGTGCAGAGCGGC-3′; site 1 antisense, 5′-GCCGCTCTGCACGCCTCCACAGTACAGCTCTGTCAAAGC-3′; site 2 sense, 5′-CGAGCCAGGCTGAGAAACTCGAGAATTGAACAAAGAGGGGTCGG-3′; site 2 antisense, 5′-CCGACCCCTCTTTGTTCAATTCTCGAGTTTCTCAGCCTGGCTCG-3′; site 3 sense, 5′-CGGGGCGTCCCCTCCCCTCTGTAGTACTAACTCAAGGGGCGCAGC-3′; site 3 antisense, 5′-GCTGCGCCCCTTGAGTTAGTACTACAGAGGGGAGGGGACGCCCCG-3′; site 4 sense, 5′-GGAGGCGTGCAGATCATCTAGCCGGCGAGCCAGG-3′; site 4 antisense, 5′-CCTGGCTCGCCGGCTAGATGATCTGCACGCCTCC-3′; site 5 sense, 5′-CGAGAATTGAACAAAGAGGAGTAGTACTGAGTGTGTGTGT-3′; and site 5 antisense, 5′-GCCGACACACACACTCAGTACTACTCCTCTTTGTTCAATTCTCG-3′.
Growth analysis.
dnTCF1EWT and dnTCF1Emut cell lines were induced with doxycycline, and cell number was monitored by the sulforhodamine B (SRB) cell proliferation assay as described by Atcha et al. (4).
Microarray.
dnTCF1EWT and dnTCF1Emut cell lines were induced with doxycycline or treated with a water control. The induction protocol was repeated independently three times, and the total RNA from each of the three independent inductions was pooled. The pooled RNA samples were sent to the DNA microarray facility at the University of California, Irvine (Irvine, CA). The pooled RNA was used for probe synthesis. The probe was applied to the Affymetrix human HG-U133 plus 2.0 array, and hybridization proceeded overnight. The entire microarray protocol was repeated three times, making a total of nine independent RNA preparations for each condition. RNA was harvested from triplicate independent cultures at 8 and 22 h of induction. Genes that were differentially regulated by dnTCF1EWT and dnTCF1Emut were determined by comparing the Dox-treated with untreated gene expression profiles for each cell line and by comparison to the parental DLD1 cell line. Although the microarray analysis cannot definitively assign any particular gene to be a direct TCF1E target, early time points for induction and cell harvest conditions were used to ensure an enrichment of direct target genes. In addition, low concentrations of doxycycline were used to induce relatively low levels of the dominant negative transgenes. Expression summaries of the 44 Affymetrix GeneChip measurements were obtained using the robust multiarray average (RMA) method (39, 40). This method adjusts the background and normalizes and log-transforms the probe level data prior to summarization. Since each of the 14 conditions is replicated only three to four times, we addressed the issue of obtaining reliable variance by performing a regularization of the observed variance. This regularization was performed using a Bayesian framework (6), which estimates variance of each probe set by taking into account the variance of neighboring probe sets, i.e., genes with similar expression levels. Probe sets were ranked according to expression levels, and then a group of neighboring probe sets was defined in terms of a window size (for this data set, window size was set to 101 genes). The resulting regularized variance estimates derived from this ranking were then used to perform one-way analysis of variance (ANOVA) to investigate group differences, an approach that has been previously reported and independently validated (6, 17, 28, 37). Due to the large number of hypotheses being tested, an additional false-discovery rate (FDR) and receiver operating characteristic (ROC) curve analysis was performed. This analysis uses previously described methods (37) to establish that a false-discovery rate of 1% corresponds to a P value cutoff of 0.016 in the regularized one-way ANOVA test among the 14 groups. Probe sets found significant (P < 0.016) were further analyzed using Tukey-Kramer posthoc pairwise comparisons to determine which pairs of groups showed differential expression using the same P value cutoff. A cutoff fold change of 1.3 for upregulated and downregulated probe sets was used to generate lists of differentially regulated genes because CDKN1A (p21) was detected as upregulated by 1.34-fold. Since genes downregulated by dnTCFs are more likely to be direct Wnt target genes than upregulated ones, our analysis focused on genes downregulated >1.3-fold. The entire data set is provided in Table S1 in the supplemental material.
Western analysis.
Stable dnTCF1EWT and dnTCF1Emut cell lines were cultured in RPMI medium with 1:1,000 blasticidin and 1:200 zeocin. dnTCF1EWT- and dnTCF1Emut-expressing cells were seeded at 400,000/well in six-well plates. dnTCF1EWT was induced with 0.0005 μg/ml doxycycline, and dnTCF1Emut was induced with 1 μg/ml doxycycline in RPMI medium. Cells were collected at preinduction and at 3, 5, 7, and 9 h postinduction and resuspended in 60 μl of SDS with 10% β-mercaptoethanol. Fifteen microliters of lysate was run on SDS–10% polyacrylamide gels. Blots were probed with 1:2,000 anti-Flag antibody (Sigma), 1:1,000 p21 antibody (Cell Signaling), 1:500 SP5 antibody (Abcam), 1:1,000 cMYC antibody (Cell Signaling), and 1:2,000 lamin AC antibody (Cell Signaling).
Semiquantitative RT-PCR.
The semiquantitative RT-PCR experiments were performed using the residual RNA generated for the microarray experiments. RNA samples were reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's protocol. Semiquantitative RT-PCR was performed on cDNAs using an RT real-time SYBR green PCR Master Mix (SuperArray) and commercial primers from SuperArray. The signals of Dox-treated and untreated samples for each gene were normalized to the signal for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Relative fold change was calculated by dividing the normalized signals of Dox-treated samples by the normalized signals of untreated samples.
ChIP.
Three million DLD1 cells were seeded in 12 15-cm plates. The next day six plates were transfected with 10 μg of FLAG-SP5 expression construct, and six plates were transfected with EVR2. After 24 h, transfection was repeated. The next day cells were cross-linked with 1% formaldehyde in phosphate-buffered saline (PBS) for 12 min. Nuclear extracts were made and sonicated to generate DNA fragments of ∼200 bp. Immunoprecipitation was performed with FLAG antibody-conjugated agarose beads or IgG-conjugated agarose beads (Sigma) according to the manufacturer's protocol. Three plates' worth of cells (∼20 million) were used for each immunoprecipitation. The following primers were used for chromatin immunoprecipitation (ChIP) at the p21 locus: primer set 1, 5′-AGCTGGCTCGGCGCTGGGCAG-3′ (sense) and 5′-TTCGGCAGCTGCTCACACCTC-3′ (antisense); primer set 2, 5′-TCAGTTCCTTGTGGAGCCGG-3′ (sense) and 5′-CCTGCCGCAGAAACACCTGT-3′ (antisense); primer set 3, 5′-GAATGACGGGCGTGGGTCGG-3′ (sense) and 5′-ACTGCGCCTGGGGCCTGGAG-3′ (antisense); primer set 4, 5′-TGCTCGCGGCGTGGGGATGA-3′ (sense) and 5′-CTGGCACATTCCCAAGGGCC-3′ (antisense); control primer, 5′-GTAGAGACAAGGTCTCACCA-3′ (sense) and 5′-ATCACTCTACCTCTCTGAGC-3′ (antisense).
EMSA.
Electrophoretic mobility shift assays (EMSAs) were carried out with 1 ng (approximately 200 cps) of radioactive oligonucleotide (see sequences listed in Fig. 5) in a final reaction volume of 20 μl containing 10 mM HEPES (pH 8.0), 2.5 mM EDTA, 10% glycerol, 20 mM KCl, 5 mM MgCl2, 0.024 μg/μl salmon sperm DNA, and 20 mM dithiothreitol (DTT). Cos1 cells were transiently transfected with EVR2 and expression vectors for full-length human TCF1EWT and TCF1Emut. Lysates from Cos1 cells were prepared 48 h after transfection by swelling cells on ice, immersing them for 15 min in hypotonic lysis buffer (10 mM Tris [pH 7.9], 50 mM KCl, 10 mM MgCl2, 0.01 mM EDTA, 1 mM DTT, 0.01 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail), and douncing. For competition experiments increasing concentrations of cold probe were added to the reaction mixture (50 pmol, 100 pmol, 200 pmol, 400 pmol, and 600 pmol).
Fig 5.

Electrophoretic mobility shift assays confirm the flexibility of the C-clamp–Helper interaction. (A) Probes used for EMSAs. SP5G Helper Up is a radioactive probe taken from the SP5 promoter with an upstream Helper site. SP5G Helper Down is a modified version of SP5G Helper Up, with the Helper site placed downstream the WRE. Helper WT and Helper mut are unlabeled oligonucleotides used for the competition experiments shown in panels C and E. (B) His-TCF1EWT-transfected Cos1 lysates were incubated with the SP5G Helper Up radioactive probe and 1 μg of anti-FLAG antibody (Sigma) or anti-TCF1 antibody (Cell Signaling) for 15 min at room temperature. A clear supershift was observed with the anti-TCF1 antibody but not with the anti-FLAG antibody. (C) Mock- and TCF1EWT-transfected Cos1 lysates (left) were used to shift SP5G Helper Up probes. The SP5G Helper Up radioactive probe was used in an EMSA with TCF1EWT lysates (middle). Increasing concentrations of Helper WT, but not Helper mut, displaced TCF1E from the SP5G Helper Up radioactive probe. An SP5G Helper Down radioactive probe was used for the experiment shown in the right-hand panel. The asterisk marks a lane with less radioactive probe due to a pipetting error. (D) Quantification of bands shown in panel C. The shifted band signal was normalized to the free probe signal in each lane. (E) Increasing amounts of mock-transfected and TCF1Emut-transfected Cos1 cell lysates were used to shift the SP5G Helper Down radioactive probe (left). In order to observe a shift of the probe, a greater amount of TCF1Emut lysate was needed (minimum, 8 μl) than TCF1EWT lysate (2 μl), despite equal protein expression levels (Fig. 1E). Increasing amounts of cold Helper WT and cold Helper mut did not displace TCF1Emut from the radioactive probe (right). (F) Model for DNA binding of TCF1E. The HMG box binds to WREs and bends the DNA ∼90° toward the C-terminal C-clamp, which makes contacts with Helper sites on either side of the WRE.
Microarray data accession number.
The microarray data developed in this study were deposited in the GEO database under accession number GSE37964.
RESULTS
The C-clamp functionally interacts with GC-rich Helper sites.
TCF1E contains two evolutionarily conserved DNA binding domains: a sequence-specific high-mobility group (HMG) box that binds Wnt response elements (WREs) and the C-clamp, which derives its name from conserved spacing of cysteine residues (4). Cyclic amplification and selection of targets (CASTing), an approach based on the systematic evolution of ligands by exponential enrichment (SELEX), was previously performed to determine the sequence specificity of the C-clamp (4). CASTing was performed with TCF1EWT and a C-clamp mutant (TCF1Emut) that contains a 5-amino-acid substitution in the C-clamp, rendering it null for DNA binding (4). Our previously published CASTing analysis indicated that TCF1EWT and TCF1Emut could bind the same WRE (CTTTGATSTT, where S is G or C) through the HMG box. However, only TCF1EWT was able to bind an additional short sequence (5′-RCCG-3′, where R is G or A) downstream of WREs, whereas TCF1Emut could not (4). After the publication of the downstream 5′-RCCG-3′ sequence, additional analysis, including both analysis of original CASTing results and a repeat CASTing experiment to expand the number of independent, TCF1EWT CASTing sequences, revealed that TCF1EWT was also enriched for the short sequence 5′-CGGY-3′ (Y is C or T) upstream of WREs (Fig. 1A). Note that this sequence is the same as the 5′-RCCG-3′ downstream element but in the opposite orientation. These results suggest that the C-clamp can make contacts with Helper sites on either side of the WRE, a feature consistent with what has been observed for the C-clamp of Drosophila dTCF (15). Experiments with the C-clamp in dTCF show that this domain can make contacts with an extended GC-rich element on either side of a WRE (GCCGCCR) (15). Chang et al. call this extended GC-rich element a Helper sequence, which is how we refer to the RCCG element due to the similarity between Drosophila and human C-clamp specificity.
To determine if the Helper sites play a role in transcriptional activation by TCF1E, several versions of this sequence were cloned into the TK100 luciferase reporter. Helper Downstream contains three Helper sites downstream of three multimerized WREs (Fig. 1C). TCF1EWT was able to activate this construct in the colon cancer cell line Colo320 by ∼8 to 10-fold over mock-transfected cells (Fig. 1D). Colo320 cells have high levels of endogenous Wnt signaling, and TCF1Emut could not activate this reporter construct over these high levels. TCF1EWT could also activate the Helper Upstream construct, which has the Helper sites upstream of the WREs. As with the Helper Downstream construct, TCF1Emut could not activate Helper Upstream. In contrast, TCF1EWT and TCF1Emut both showed no activation of the Topmod reporter, which has multimerized WREs without Helper sites (Fig. 1C and D). Importantly, TCF1EWT and TCF1Emut were expressed at equal levels in these experiments (Fig. 1D). The results indicate that the C-clamp is required to confer a Helper site contribution to TCF1E-mediated transcriptional activation.
Previous work has shown that modification of a strong WRE (CTTTGAT) can create a weaker WRE (CTTTGTT) that decreases HMG box binding (26, 52). To determine whether C-clamp–Helper site interactions allow TCF1E to compensate for weak WREs, two more sequences were cloned into the TK100 luciferase reporter. Helper2 (Fig. 1C) contains multimerized weak WREs (CTTTGTT) and an extended Helper site (GCCGCCG), which was isolated in a subset of the CASTing sequences and perfectly matches Drosophila Helper sites. Cos1 cells do not have endogenous Wnt signaling in that LEF/TCFs are only very weakly expressed, and endogenous β-catenin is not stabilized. Coexpression of β-catenin and either wild-type TCF1E or C-clamp mutant TCF1E in Cos1 cells revealed that TCF1EWT/β-catenin activated the Helper2 construct (∼11-fold), whereas TCF1Emut did not (Fig. 1E). In contrast, TCF1EWT only weakly activated the TOP2 construct (∼2-fold), which contains the same weak WREs but lacks Helper sites. These two proteins are expressed at equivalent levels (Fig. 1E), and similar results were found in Colo320 cells (data not shown). These results indicate that Helper sites allow TCF1E to compensate for weak WREs in a C-clamp-dependent manner. As a control for these experiments, TCF1EWT and TCF1Emut were assessed for their ability to activate the TOPTK reporter, which contains multimerized strong WREs without Helper sites (Fig. 1C). TOPTK was similarly activated by TCF1EWT and TCF1Emut (∼13- to 15-fold) in Cos1 cells transfected with β-catenin (Fig. 1E). Taken together, the results shown in Fig. 1 reveal that a combined C-clamp–Helper site interaction makes an important contribution to the transcriptional output of TCF1E.
The C-clamp regulates DLD1 cell growth.
To identify genes regulated by the C-clamp, we used a system developed by van de Wetering and colleagues that allows induction of transgenes in the DLD1 colon cancer cell line. In this system, doxycycline treatment induces the expression of dnTCF1E (72). Since dnTCF1E interferes with Wnt signaling by competing with endogenous TCF/β-catenin complexes for binding to WREs, its overexpression causes downregulation of the WNT transcriptome. It was previously reported that overexpression of either dnTCF1E or dnTCF4E causes DLD1 cells to stall in the G1 phase of the cell cycle. This stall was dependent on induction of the cyclin-dependent kinase inhibitor, p21 (CDKN1A) (72). We engineered a companion stable cell line where doxycycline can induce the expression of a C-clamp mutant version of dominant negative TCF1E (dnTCF1Emut).
We compared the ability of dnTCF1EWT and dnTCF1Emut to regulate DLD1 colon cancer cell growth (Fig. 2A). Doxycycline titrations were carried out to ensure that equal levels of dnTCF1E protein were induced. Using these established concentrations, doxycycline inductions of dnTCF1EWT, dnTCF1Emut, and the parental DLD1 (control) cell line were carried out for 10 days, and each day the number of cells was determined by the SRB quantitative color assay (see Materials and Methods). Western analysis was performed to ensure that similar levels of dnTCF1EWT and dnTCF1Emut were induced over the course of the experiment (Fig. 2B). Doxycycline treatment of the parental DLD1 cells had no effect on their growth, indicating that doxycycline treatment has no adverse effects on the cells (Fig. 2A). Doxycycline induction of dnTCF1EWT caused a cessation of proliferation starting at day 3, an effect that lasted for the duration of the 10-day experiment. As previously reported, the cells remained viable throughout the experiment (72). In contrast, induced expression of dnTCF1Emut did not lead to a stall in cell growth. Cell proliferation was no different than that in mock-treated cells until day 4, when the rate of growth slowed (Fig. 2A). A time course and Western blot analysis of dnTCF1E and p21 expression confirmed that dnTCF1EWT and dnTCF1Emut continued to be expressed throughout the experiment. This analysis also revealed that p21 protein expression appeared in the dnTCF1Emut-expressing culture but with delayed kinetics (between days 4 and 5) (data not shown). While it is possible that the slowing rate of proliferation between day 4 and day 5 in this culture was due to a late-stage induction, the cells never stalled, and they continued to proliferate, surpassing confluence on day 6.
Fig 2.
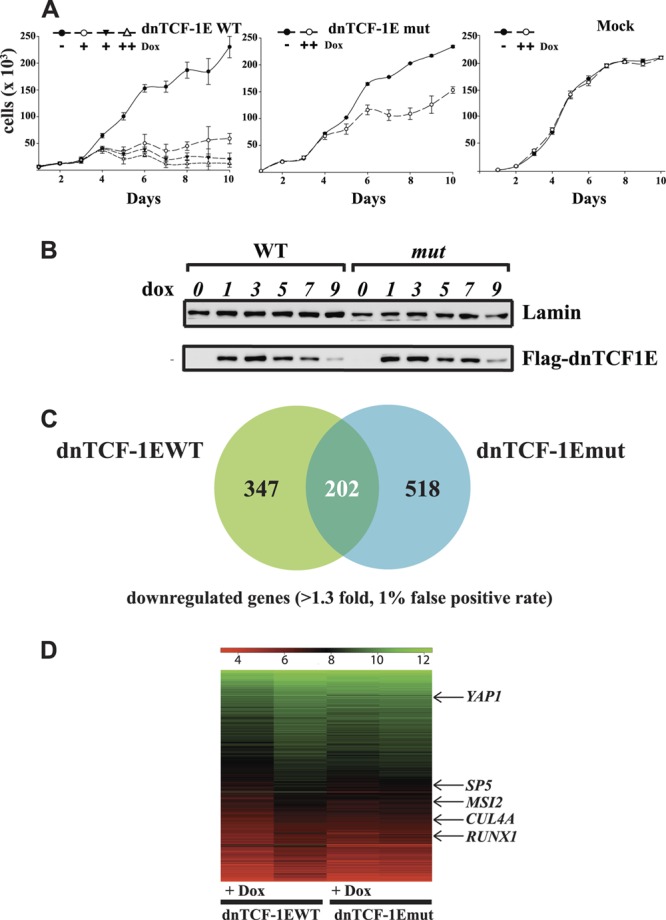
The C-clamp controls the growth of DLD1 colon cancer cells. (A) SRB growth profile of dnTCF1EWT-expressing stable cells, dnTCF1Emut-expressing stable cells, and DLD1 cells (mock) after induction of doxycycline. Induction of dnTCF1EWT without doxycycline (filled circle) and with doxycycline concentrations of 0.0005 μg/ml (open circle), 0.0006 μg/ml (filled triangle), and 1 μg/ml (open triangle) show that all three doses caused a stall in cell growth starting at day 4 postinduction. Induction of dnTCF1Emut (open circle; 1 μg/ml Dox) slowed growth starting at day 5 postinduction, but cells continued to divide; and the culture surpassed confluence starting at day 6. Induction of DLD1 cells (Mock) with doxycycline (open circle; 1 μg/ml) had no effect on their growth. (B) dnTCF1EWT was induced with 0.0005 μg/ml doxycycline, and dnTCF1Emut was induced with 1 μg/ml doxycycline for 9 days. Western analysis revealed that similar levels of dnTCF1EWT and dnTCF1Emut were expressed through the course of the growth curve. (C) Microarray analysis after induction of dnTCF1EWT and dnTCF1Emut revealed the total number of genes uniquely downregulated by dnTCF1EWT (347) and by dnTCF1Emut (518) and commonly regulated (202). (D) Heat map representation of dnTCF1EWT and dnTCF1Emut microarrays with selected wild-type-specific target genes labeled (see Table S1 in the supplemental material for full gene lists).
Microarray analysis identifies a subset of genes regulated by the C-clamp.
Microarray experiments were performed to identify dnTCF1E target genes involved in growth regulation (Fig. 2C and D). Using the same doxycycline conditions described above for cells expressing dnTCF1EWT and dnTCF1Emut and DLD1 parental cells, RNA was harvested from triplicate independent cultures after 8 h. The entire experiment was repeated three times for each condition. Genes that were differentially regulated by dnTCF1EWT and dnTCF1Emut were determined by comparing the gene expression profiles of doxycycline-treated cells, untreated cells, and the parental DLD1 cell line. Early time points for induction and cell harvest conditions were used to ensure an enrichment of direct target genes. Affymetric probe set measurements were analyzed using the robust multiarray average (RMA) and Bayesian methods (6, 39, 40) to estimate the variance of the data. One-way ANOVA, false-discovery rate, and receiver operating characteristic analyses were then performed to establish that a 1% false-discovery rate corresponded to a P value cutoff of 0.016 (see Materials and Methods; see Table S1 in the supplemental material for the data). Of the 662 probe sets (549 genes) downregulated by wild-type dnTCF1EWT, 275 (202 genes) were also downregulated by C-clamp-defective dnTCF1Emut (Fig. 2C). Many genes within this common subset are known Wnt target genes, such as ASCL2 (41), LGR5 (69), AXIN2 (53), cMYC (31), JAG1 (42), BMP4 (43), MET (11), TBX3 (65), and TNFRSF19 (TROY) (13) (for a complete list of genes, see Table S1). These common downregulated genes do not require the C-clamp DNA binding domain for their regulation because both dnTCF1EWT and the C-clamp-defective dnTCF1Emut could downregulate their expression. A total of 387 probe sets (347 genes) were specifically downregulated by dnTCF1EWT. SP5 (23), SOX9 (10), PITX2 (44), and SGK1 (20) are the only previously reported WNT target genes that were downregulated specifically by dnTCF1EWT (see Table S1). Since these genes are uniquely downregulated by dnTCF1EWT, we predict that WNT regulation of these genes is C-clamp dependent.
We used semiquantitative RT-PCR to validate a representative set of genes from the C-clamp-specific (SP5 and CDKN1A; regulated by dnTCF1EWT only) and common (AXIN2 and cMYC; downregulated by both dnTCF1EWT and dnTCF1Emut) gene lists. RT-PCR levels were normalized to GAPDH in each of the Dox-treated and mock-treated samples, and fold change was calculated by comparing the normalized signals (Fig. 3A). In agreement with the microarray, semiquantitative RT-PCR detected downregulation of AXIN2 and cMYC by both dnTCF1EWT and dnTCF1Emut, while SP5 was significantly downregulated only by dnTCF1EWT. CDKN1A (p21) expression was upregulated by dnTCF1EWT but not by dnTCF1Emut. Overall, the RT-PCR results validate the microarray data.
Fig 3.
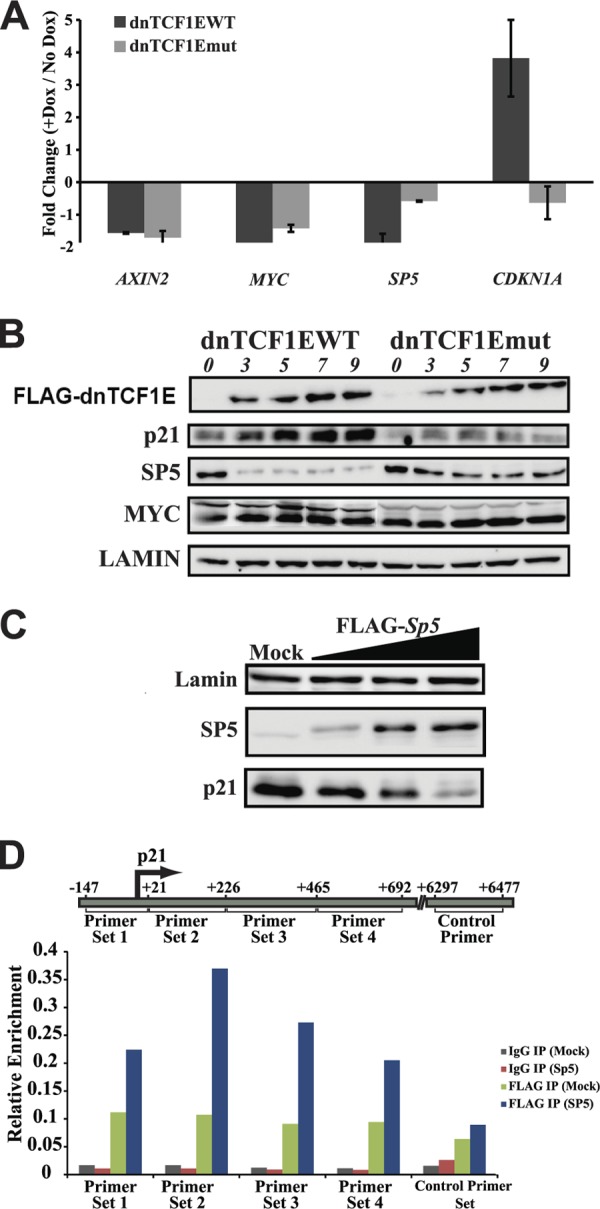
RT-PCR and Western blot validation of microarrays. (A) RT-PCR analysis of selected genes. Induction of dnTCF1EWT and dnTCF1Emut decreased AXIN2 and cMYC mRNA expression. Induction of dnTCF1EWT but not dnTCF1Emut caused a decrease in SP5 mRNA and an increase in CDKN1A (p21) mRNA levels. (B) Stable cell lines were induced with doxycycline, and lysates were collected over the course of 9 h. Induction of dnTCF1EWT with 0.0005 μg/ml doxycycline caused an increase in p21, a decrease in SP5, and no change in cMYC protein levels. Induction of dnTCF1Emut with 1 μg/ml doxycycline caused little or no change in p21, SP5, and cMYC protein levels. Western blot analysis shows that induced protein levels of dnTCF1EWT and dnTCF1Emut were equivalent. (C) dnTCF1EWT-expressing stable cells were transfected with EVR2 (mock) and increasing amounts of FLAG-SP5 expression plasmid (500 ng, 1 μg, and 2 μg). After 24 h, mock- and SP5-transfected cells were induced with doxycycline, and lysates were collected after an additional 9 h to allow induction of dnTCF1EWT. (D) SP5 binds near the p21 transcription start site. ChIP was performed with mock- or FLAG-SP5-transfected DLD1 cells. FLAG-SP5 was immunoprecipitated from cellular lysates with IgG-agarose or FLAG-agarose beads. Quantitative PCR was performed with the indicated primer set. FLAG-SP5 was enriched near the GC-rich p21 transcription start site but not at a downstream intragenic region.
Western analysis was performed to assess the protein levels of cMYC after induction of dnTCF1E. cMYC is a p21 repressor, and downregulation of cMYC by dnTCF1E was reported to be responsible for the p21-dependent G1 phase stall in DLD1 cells (72). In contrast to the microarray (see Table S1 in the supplemental material) and RT-PCR (Fig. 3A) experiments, Western analysis revealed that induction of dnTCF1EWT and dnTCF1Emut did not decrease cMYC protein levels (Fig. 3B). cMYC RNA and protein levels have been shown to be decoupled in other systems due to complex posttranscriptional regulation, and it is possible that these other modes of regulation mask the decrease in cMYC RNA levels (38, 55). We assessed the pattern of SP5 protein expression since it is another known repressor of p21 transcription (23), and it was selectively downregulated by dnTCF1EWT. In agreement with the microarray and RT-PCR data, SP5 protein was strongly decreased 3 h after induction of dnTCF1EWT; induction of dnTCF1Emut caused little change in SP5 expression (Fig. 3B). Also in agreement with the microarray and RT-PCR data, p21 protein levels were strongly induced by dnTCF1EWT but not dnTCF1Emut (Fig. 3B). Western analysis over a longer time course showed that dnTCF1Emut induction can lead to increases in p21 protein levels, but such increases are delayed and not evident until day 5 (data not shown). Since the growth phenotype caused by induction of dnTCF1EWT paralleled the selective decrease in expression of SP5 and not cMYC, we tested whether reintroduction of SP5 alone was sufficient to repress p21 expression. Increasing amounts of a FLAG-SP5 expression plasmid were transfected into cells expressing dnTCF1EWT. Figure 3C data show that reintroduction of SP5 restored p21 repression, confirming that SP5 is a key p21 repressor in DLD1 cells. It was previously suggested that SP5 exerts its repressive activity on p21 through binding to SP1 sites just upstream of the transcription start site (23). However, in this study, binding of SP5 to the GC-rich SP1 sites was shown only by EMSA. We therefore performed chromatin immunoprecipitation experiments with an anti-FLAG antibody in DLD1 cells with overexpressed FLAG-SP5. This experiment confirmed that SP5 binds the p21 promoter upstream of the transcription start site as well as GC-rich regions just downstream of the transcription start site (Fig. 3D). As a control, we tested for binding to an intragenic region 6,300 bp down from the transcription start site and observed no enrichment of FLAG-SP5 at this site.
The C-clamp–Helper interaction regulates Wnt target promoters.
We examined the ability of TCF1E to directly regulate the SP5 promoter. We focused specifically on SP5 because of its strong downregulation of p21 and because its expression is elevated in primary human colon cancers (16, 23). SP5 expression is directly regulated by Wnt signaling, and multiple Wnt response elements have been identified in the promoter (23). We tested whether the C-clamp-specific regulation observed in our experiments could derive from actions on the delimited promoter (Fig. 4A). A transient-transfection assay was performed in Cos1 cells in which expression plasmids for full-length TCF1EWT or TCF1Emut were cotransfected with an expression plasmid for β-catenin and an SP5 luciferase reporter plasmid (Fig. 4B). SP5 was strongly activated by TCF1EWT (27-fold) but was not by TCF1Emut or LEF1 (Fig. 4B). These data show that regulation of the SP5 promoter is direct and that the C-clamp DNA binding domain greatly facilitates this regulation. Inspection of the DNA sequence surrounding the WREs in the SP5 promoter revealed several matches to Helper site elements (labeled E, G, and H in Fig. 4A). Mutation of the Helper sites strongly decreased TCF1EWT activation of the SP5 promoter (Fig. 4B). Mutation of individual Helper sites caused a variable decrease in activation, with the site G mutant showing the greatest decrease in activation (∼2-fold). Mutation of Helper sites E and G together caused a moderate decrease in activation (∼3-fold), and mutation of all three Helper sites caused the strongest decrease (∼7-fold) (Fig. 4C). This suggests that multiple Helper sites contribute toward activation of the Sp5 promoter by TCF1E/β-catenin. None of the WREs were altered in the promoter; only Helper sites were inactivated. A similar trend was found in parallel luciferase assays with the SP5 promoter in Colo320 (Fig. 4D) and DLD1 cells (Fig. 4E) although there was a lower fold activation of the SP5 promoter by TCF1EWT in these cells. This confirms that Cos1, Colo320, and DLD1 cells are reliably interchangeable for luciferase assays that measure C-clamp activity. We conclude that the DNA binding specificity of the C-clamp makes an essential contribution to a gene uniquely regulated by TCF1EWT.
Fig 4.

The SP5 promoter is regulated by the C-clamp–Helper site interaction. (A) Schematic of the SP5 promoter with Helper sites represented as black boxes and in bold and WREs highlighted in green. (B) Transfection of Cos1 cells with the SP5 promoter reporter, β-catenin, and TCF1EWT, TCF1Emut, and LEF1 expression plasmids. TCF1EWT/β-catenin could activate the promoter, whereas TCF1Emut/β-catenin and LEF1/β-catenin could not. Mutation of three Helper sites in the promoter strongly decreased activation by TCF1EWT/β-catenin. (C) Cos1 cells were transfected with TCF1EWT, β-catenin, and the indicated SP5 promoter reporters. Individual Helper site mutations caused a variable decrease in activation by TCF1EWT/β-catenin, and a triple Helper mutation (EGH mut) caused the greatest decrease in activation. Colo320 (D) and DLD1 (E) cells were transfected with SP5 WT or SP5 EGH mut and 20 ng of TCF1EWT or TCF1Emut expression construct. β-Catenin was not included because these cells already have high levels of nuclear β-catenin. TCF1EWT activated the SP5 promoter in Colo320 and DL D1 cells as well as Cos1 cells (Fig. 4), whereas TCF1Emut did not activate the promoter. Mutation of three Helper sites in the SP5 promoter significantly decreased activation by TCF1EWT.
To provide further evidence that the C-clamp can make contacts with RCCG elements, electrophoretic mobility shift assays were carried out using a probe from the SP5 promoter that contains one WRE and one upstream Helper site (Fig. 5A, SP5G Helper Up). Mock- and TCF1E-transfected Cos1 lysates were combined with radioactive SP5 probe in a binding reaction (Fig. 5C). TCF1EWT lysates strongly shifted the SP5 probe in comparison with mock lysates, which did not have binding activity. As an additional control, a supershift assay was performed confirming that TCF1E was responsible for shifting the SP5 probe (Fig. 5B).
Competition experiments with cold competitor probes were then performed. Competition with increasing concentrations of a cold probe containing wild-type Helper sites displaced TCF1E from the radioactive probe (Fig. 5C and D, Helper WT). However, cold competition with a probe containing a mutated Helper site did not displace TCF1E (Fig. 5C and D, Helper mut), indicating that TCF1E has a binding preference for Helper sites. To confirm the flexibility of the C-clamp–Helper interaction, a second radioactive probe was created with the Helper site placed downstream of the WRE (Fig. 5A, SP5G Helper Down), and a similar competition experiment was performed. Again, increasing concentrations of the wild-type Helper cold probe displaced TCF1E in the binding reaction, whereas very little competition occurred with increasing concentrations of the mutant Helper cold probe. A similar set of experiments was performed with TCF1Emut (Fig. 5E). Increasing concentrations of cold wild-type Helper and cold mutant Helper did not compete TCF1Emut from the radioactive probe, indicating that the interaction between TCF1E and the Helper site is C-clamp specific.
Taken together, the CASTing, luciferase assays, and EMSA data suggest that the C-clamp of TCF1E can make contacts with RCCG elements on either side of the WRE (Fig. 5F). This unusual mode of binding may be facilitated by the strong DNA bend created by the HMG domain (∼90° for TCF1) (4). Bending is oriented away from the HMG box and envelopes the C-terminal extension of the protein—a conformation that could position upstream and downstream sequences close to the C-terminal C-clamp domain (52).
CASTing revealed that C-clamp–Helper interactions could occur as far as 9 bp upstream of a WRE and up to 11 bp downstream of a WRE. However, the random portion of the DNA library used in CASTing was 30 bp, which may have constrained the distance between the Helper site and the WRE. Since the C-clamp of Drosophila pangolin/dTCF can functionally interact with Helper sites as far as 70 bp from the WRE (15), we analyzed other Wnt target genes that contain putative C-clamp–Helper elements at longer distances from the WRE. CDX1 is a homeobox transcription factor, a possible p21 repressor (58), and a known Wnt target gene (32, 49) with several Helper sites adjacent to WREs in its promoter (Fig. 6A). While CDX1 expression is often lost in colon cancer and while we found no evidence of its expression in DLD1 cells (19), the CDX1 promoter has been shown to be selectively activated by the C-clamp isoform of TCF4 (TCF4E) (74). We assessed the ability of TCF1EWT and TCF1Emut to activate a luciferase reporter plasmid driven by the CDX1 promoter (Fig. 6B). TCF1EWT could robustly activate the promoter, whereas TCF1Emut could not. While mutation of the nearest two neighboring Helper sites did not have a significant effect on the ability of TCF1EWT to activate the CDX1 promoter (Fig. 6C), mutation of additional putative Helper sites positioned 10 nucleotides (nt), 26 nt, and 28 nt distant significantly decreased activation by TCF1EWT (Fig. 6B). The observed partial dependence on the indicated Helper sites for activation may be due to incomplete mutation of all possible Helper sites since the CDX1 promoter is highly GC rich and since many partial matches to the Helper sequence are present (Fig. 6A). Since TCF4E contains a C-clamp, we tested whether its activation of the CDX1 promoter was also C-clamp dependent. Mutation of a single cysteine residue in the C-clamp of TCF4E weakened its ability to drive transcription of the CDX1 promoter (Fig. 6B), despite equal levels of TCF4EWT and TCF4Emut expression (Fig. 6B). Mutation of the five Helper sites in the CDX1 promoter also decreased activation by TCF4E. Our results indicate that CDX1 is a C-clamp-specific target gene (for both TCF1E and TCF4E) and that this regulation depends on Helper sites with flexible distance and orientation with respect to the WRE.
Fig 6.

The CDX1 promoter is regulated by the C-clamp–Helper interaction. (A) Schematic of the CDX1 promoter, which contains WREs with adjacent Helper sites at variable distances. (B) Activation of the CDX1 promoter is fully (TCF1E) or partially (TCF4E) C-clamp dependent. The CDX1 promoter was transfected in Cos1 cells along with β-catenin and increasing amounts of TCF1EWT, TCF1Emut (CRARF → VALAL), TCF4EWT, or TCF4Emut (WC → WA) expression plasmids (20 ng, 40 ng, and 80 ng). Mutation of five Helper sites (see Materials and Methods) decreased activation by TCF1EWT/β-catenin and TCF4EWT/β-catenin. (C) Double Helper site mutations (asterisks) had no effect on activation of the CDX1 promoter by TCF1E.
We also analyzed the LEF1 promoter (Fig. 7A) since the first reported C-clamp activity was its requirement for TCF regulation of LEF1 promoter 1 (3). Our previous studies identified TCF1E as the only LEF/TCF isoform capable of activating the LEF1 promoter, and mutation of the C-clamp eliminated activation (3). Like SP5 and CDX1, the LEF1 promoter contains matches to the Helper site adjacent to WREs (Fig. 7A). Mutation of the two Helper sites closest to the WREs had no effect on TCF1E-mediated activation of the promoter (Fig. 7C). However, additional mutation of more distant Helper sites in the LEF1 promoter destroyed activation by TCF1E (Fig. 7B). This again underscores the greater degree of flexibility in Helper site orientation and distance relative to the WRE than was initially revealed by CASTing.
Fig 7.

The LEF1 promoter is regulated by the C-clamp–Helper interaction. (A) Schematic of the LEF1 promoter which has two WREs and several adjacent Helper sites at variable distances. (B) Activation of the LEF1 promoter by TCF1E/β-catenin is Helper dependent. Increasing amounts of TCF1EWT expression plasmid (20 ng, 40 ng, and 80 ng) were transfected into Cos1 cells along with β-catenin and a LEF1 promoter luciferase construct. Mutation of five of the identified Helper sites in the promoter destroyed activation by TCF1E/β-catenin. (C) Double Helper site mutations (asterisks) had no effect on activation of the LEF1 promoter.
Many dnTCF1EWT-specific genes are p21 regulators.
Since dnTCF1EWT increases p21 protein expression rapidly and forces a stall in cell growth, we asked whether other genes besides SP5 are direct, C-clamp-specific targets whose actions are important contributors to the regulation of p21. Table 1 illustrates that dnTCF1EWT downregulated the expression of eight known regulators of p21. Seven of eight genes have been shown to repress p21 function at the level of transcription (RUNX1, SMARCA4, SP5, TGIF, and YAP1), RNA stability (MSI2), or protein stability (CUL4A). This suggests that dnTCF1EWT may induce p21 expression through a multileveled reduction in p21 repressor activity. Since we observed p21 protein levels to increase within 3 h of doxycycline induction, we asked which of these eight genes are direct targets of dnTCF1EWT as these would be the most likely key, early regulators of p21. To identify direct target genes, we used results reported from recent chromatin immunoprecipitation studies on TCF4 in LS174T colon cancer cells (29) and β-catenin in HCT116 colon cancer cells (12). Hatzis et al. used a genome-wide chromatin immunoprecipitation study to identify 6,858 high-confidence TCF4 binding sites. We used this binding pattern as a guide to the probable binding locations of TCF1 since TCF4E and TCF1E are nearly identical in amino acid sequence throughout both the HMG and C-clamp DNA binding domains, and they have been shown to regulate similar sets of genes in DLD1 and LS174T cells (69). Table 2 shows that four of the eight C-clamp-specific p21 regulators are occupied by TCF4 in the LS174T colon cancer cell line (RUNX1, SP5, TGIF, and YAP1), and three of these are occupied by β-catenin in HCT116 colon cancer cells (RUNX1, SP5, and YAP1) (12). Genes that were occupied by β-catenin (within 2.5 kb of their protein coding boundaries) and TCF4 (within 100 kb of their promoters) are listed in Table 2. Three of the genes occupied by TCF4 and β-catenin (SP5, YAP1, and RUNX1) contain WREs with adjacent Helper sites within 10 kb of their promoters, but it is possible that regulation of these genes may occur by WRE-Helper sites in unmapped long-range enhancers. Overall, ChIP studies in collaboration with our microarray results suggest that TCF1E and TCF4E are potent growth factors because the C-clamp DNA binding domain enables specific, direct regulation of a group of genes that control p21 transcription, translation, and protein stability.
Table 1.
C-clamp specific and common regulated genes: connections to p21
| p21 regulator | Fold change by gene groupa |
Known direct Wnt target gene | Known action on p21 | Relative expression in colon cancer | Reference(s) | ||
|---|---|---|---|---|---|---|---|
| C-clamp specific | Common |
||||||
| WT | Mutant | ||||||
| CUL4A | −1.7 | Yes | Ubiquitin-dependent degradation | ? | 56, 61 | ||
| FOXO3A | −1.6 | No | Activates transcription | Increased | 30 | ||
| MSI2 | −1.5 | No | Represses translation | ? | 8 | ||
| RUNX1 (AML1) | −1.5 | No | Represses transcription | ? | 34 | ||
| SMARCA4 (Brg1) | −1.2 | No | Activates/represses transcription | ? | 27 | ||
| SP5 | −1.6 | Yes | Represses transcription | Increased | 16, 23 | ||
| TGIF | −1.5 | No | Represses transcription | Increased | 51 | ||
| YAP1 | −1.3 | Yes | Represses transcription | Increased | 45, 73 | ||
| BCL6 | −1.5 | −1.6 | No | Represses transcription | ? | 62 | |
| CDX2 | −1.8 | −1.5 | No | Activates transcription | Increased | 5, 57 | |
| MYC | −1.7 | −2.5 | Yes | Represses transcription | Increased | 22, 72 | |
| NR2F2 (COUP-TFII) | −1.7 | −1.4 | No | Activates transcription | Increased | 60, 68 | |
| RUNX2 | −1.5 | −1.4 | Yes | Represses transcription | ? | 24, 75 | |
| TBX3 | −2.2 | −2.0 | Yes | Prevents expression | Increased | 35, 65 | |
C-clamp specific p21 regulators were identified in a literature search. p21 regulators that were regulated by both dnTCF1EWT and dnTCF1Emut were also identified (common regulated genes). Fold change of each gene in the microarray experiment is indicated. “?” indicates that no data are available.
Table 2.
C-clamp-specific target genes: matches to ChIP experiments
| Binding pattern (experiment type)a | Proposed direct Wnt target genesb |
|---|---|
| β-Catenin (ChIP-Seq) | ARHGEF3, CDC2L5, EIF4E, EZH2, FRMD5, GRHL3, HOXA3, IL17RD, ITGA2, MARCKS, MCCC2, MID1, MSI2, MYO1B, NXN, P4HB, PLCXD2, PTCH1, RUNX1, SERINC5, SP5, TSPAN5, UBE2E2, YAP1, ZBED5, ZBTB20 |
| TCF4 (ChIP-chip) | ACOT11, ATF7IP, BAMBI, BDNF, BTG1, CDC14A, CLK2, CRIM1, CRIPAK, DOCK8, ECT2, EIF4E, FAM63A, FASLG, FRMD5, GRHL3, GUSBP1, HAS2, HIST1H4H, HMGA2, HOXA11, HOXC11, IL17RD, IL-8, IRF2BP2, ITGB4, JHDM1D, KIAA1718, KLF5, LEF1, LIPG, LOC286297, LOC643367, MARCKS, MID1, MIRHG1, MYLIP, MYO1B, NT5E, OSBPL5, PITX2, PKP4, PMEPAI, RUNX1, SCARB2, SEPP1, SERPINA1, SIPA1L2, SLC38A2, SP5, TBX3, TGIF, TLE4, TPCN1, YAP1, ZNF703 |
| β-Catenin and TCF4 | EIF4E, FRMD5, GRHL3, GRIN2B, IL17RD, MARCKS, MID1, MYO1B, RUNX1, SP5, YAP1 |
Target genes were assessed for β-catenin occupancy (within 2.5 kb of the protein coding region) by ChIP-Seq and for TCF4 occupancy (within 100 kb of the transcription start site) by ChIP with microarray technology (ChIP-chip) in HCT116 (12) and LS174T (27) cells, respectively.
Genes in bold are known direct regulators of p21 expression.
DISCUSSION
It is essential that sequence-specific transcription factors find their target sites for precise regulation of target genes. We have described a mechanism by which C-clamp isoforms of LEF/TCFs regulate a subset of WNT target genes through interaction with Helper sites adjacent to WREs. Microarray experiments performed after inducing expression of dnTCF1EWT and dnTCF1Emut revealed that the majority of known WNT target genes do not require C-clamp activity (see Table S1 in the supplemental material). However, our results indicate that a subset of WNT target genes require C-clamp activity for their regulation. Many of the genes that changed expression in a dnTCF1EWT-specific manner are known regulators of p21 expression (Table 1), a cell cycle inhibitor that is critical for the dnTCF1EWT-dependent stall in cell growth (Fig. 2A) (72). p21 is a marker of intestinal differentiation, and expression of p21 in intestinal tumors is a good prognostic indicator as its levels are higher in well-differentiated tumors (78). Most of the dnTCF1EWT-specific p21 regulators have not yet been shown experimentally to be direct WNT target genes, with the exception of CUL4A and SP5. YAP1 was listed as a direct WNT target gene because we have cloned an intronic enhancer from the YAP1 locus into a luciferase reporter and have demonstrated that it is activated by TCF1EWT/β-catenin (data not shown). In addition, it has very recently been shown that YAP1 is a direct target of Wnt/β-catenin in colon cancer cells (45). YAP1 is a nuclear component of the Hippo signaling pathway, which is involved in the maintenance of organ size in Drosophila and mammals. Like TCF1, YAP1 has been shown to control intestinal stem cell proliferation in Drosophila (64) and mice (14). Our data provide evidence for a potential direct link between the WNT pathway, which controls stem cell maintenance and differentiation, and the Hippo pathway, suggesting that the two pathways may cooperate to integrate stem cell proliferation with responses to injury and maintenance of organ size.
TCF1 is expressed predominantly as full-length B tail and dominant negative E tail isoforms in normal human intestinal epithelium (59). During the progression to colon cancer, dnTCF1E expression is lost, and full-length TCF1B is exclusively expressed (59). We propose that dnTCF1E plays an important role in suppressing adenoma formation and promoting differentiation through induction of p21. Homozygous knockout of TCF1 in mice leads to adenoma formation in the mouse small intestine, an effect that is compounded by introduction of a mutant APC allele (66). Our data indicate that loss of the dnTCF1E-p21 circuit likely plays a role in the development of these adenomas as knockout of p21 also enhances adenoma formation in APC mutant mice (77). The role of dnTCF1E in intestinal stem cells may be to enforce appropriate levels of p21 expression to modulate stem cell cycle kinetics. There is evidence that under certain conditions, p21 does play a role in intestinal stem cell division as knockout of p21 led to increased intestinal cell proliferation (25). In addition, irradiation of p21 knockout mice led to enhanced stem cell survival compared to wild-type mice (25).
We were interested to find that more probe sets were uniquely downregulated by dnTCF1Emut than by dnTCF1EWT (709 versus 387). We speculate that the C-clamp restricts the number of sites bound by dnTCF1E by facilitating rapid binding to WREs, although we have yet to test this experimentally. Interestingly, the tumor suppressor p53 has also been shown to have a second C-terminal basic DNA binding domain. Unlike the C-clamp, this domain is strictly nonspecific in its DNA binding, but, similar to the C clamp, it is enriched for basic amino acid residues, it is an alternatively spliced domain, and it elevates DNA binding affinity. DNA binding studies with long (>50 bp) DNA oligonucleotides and ChIP experiments showed that this second DNA binding domain is responsible for rapid and specific binding of p53 to the p21 locus and for facilitated sliding along the DNA template (50, 54). As such, the auxiliary domain discourages prolonged binding to inappropriate sites and rapid, strong binding to correct sites. Additional biochemical and ChIP experiments will determine whether the C-clamp plays a similar role for TCF1E in rapid identification of WREs.
We were surprised that induction of dnTCF1EWT and dnTCF1Emut did not decrease cMYC protein levels even though both caused a decrease in cMYC RNA levels. A decrease in cMYC expression was previously shown to be responsible for the dnTCF1E-dependent induction of p21 in DLD1 cells (72). However, cMYC has also been shown to activate p21 expression in intestinal crypts when expressed at high levels (22). Since, in our study, cMYC protein expression did not change after induction of dnTCF1EWT, we conclude that the Wnt/p21 network must involve additional target genes.
One such target gene, SP5, is a p21 regulator and C-clamp-specific target gene. Regulation of the SP5 promoter by TCF1E is dependent on C-clamp interactions with multiple Helper sites (Fig. 4 and 5). SP5 was previously reported to be a Wnt target gene in the mouse embryonic telencephalon (23). It is not clear which isoforms of LEF/TCFs are present in the developing telencephalon although TCF4E isoforms have been detected in the mouse brain (74). It was originally reported that LEF1, which does not contain a C-clamp, could recruit β-catenin to activate the SP5 promoter in 293T cells. This contrasts with our observations that LEF1 could not activate the SP5 promoter in any cell type tested (Fig. 4B; also data not shown), whereas TCF1E could always activate the promoter, sometimes up to ∼100-fold with higher levels of TCF1E expression plasmid (data not shown). It is possible that SP5 is a C-clamp-preferred target gene, with the C-clamp providing a strong contribution to transcriptional control of the promoter.
The same may be said of CDX1, which we demonstrate to be completely dependent on the C-clamp for activation in the case of TCF1E and moderately dependent on the C-clamp in the case of TCF4E (Fig. 6B). This difference in C-clamp dependence may be due to the completeness of the mutation in TCF1E versus TCF4E. TCF1Emut has a 5-amino-acid substitution in the C-clamp (CRARF → VALAL), whereas TCF4Emut contains a single residue mutation (C → A). Regardless of the TCF family member, however, the C-clamp seems to be an important player in the transcriptional control of CDX1. Interestingly, LEF1 seems to override the need for a C-clamp in control of the CDX1 promoter in P19 embryonal carcinoma cells through interaction with CDX1 protein itself (9). This type of cooperative regulation may explain how LEF1 could override a C-clamp requirement. Indeed, LEF/TCFs are known to be context-dependent regulators, a feature consistent with the themes of redundancy and flexibility in the control of gene expression. Mutation of five Helper sites in the CDX1 promoter strongly reduced, but did not eliminate, activation by TCF1E/β-catenin. This may be due to C-clamp-specific protein-protein interactions, interactions with non-Helper sites, or incomplete mutation of Helper sites in the GC rich CDX1 promoter. Indeed, a major stumbling block in discovering new C-clamp–Helper target sites is that it is unknown how far the C-clamp–Helper site interaction can occur from the anchoring HMG box-WRE interaction (Fig. 5F). Experiments in Drosophila have indicated that a functional interaction can occur up to 70 bp away from the WRE (15), which is much longer than we report with CASTing (Fig. 1A). However, our analysis of the CDX1 and LEF1 reporters suggests that significant separation of the WRE and Helper site are tolerated. This degree of flexibility in C-clamp–Helper interactions will make binding site predictions for TCFs challenging.
It is also unknown whether the C-clamp–Helper site interaction contributes toward transcriptional activation strictly by strengthening overall protein-DNA interactions or through some other mechanism. Studies by Weise et al. showed that mutation of Helper sites in EMSAs with probes made from the CDX1 promoter decreased binding by TCF4E when only one WRE was present but not when multiple WREs were present (74). However, in this experiment Helper site mutations were point mutations, and it is likely that the C-clamp is a flexible sequence-specific DNA binding domain as not all TCF1EWT CASTing sequences contained perfect matches to Helper sites (Fig. 1B; see also Fig. S1 in the supplemental material). In the context of a luciferase assay, mutation of Helper sites caused a significant but moderate decrease in activation by TCF4E (Fig. 6B). It may be that subtle differences in binding strength as revealed by EMSA make a large contribution to activation of transcription in cells.
We have found that the C-clamp directs a dnTCF1E-p21 circuit in colon cancer cells that is likely dependent on C-clamp–Helper interactions. Our data and that of other investigators indicate that the dnTCF1E-p21 circuit plays an important role in modulating the cell cycle of intestinal cancer cells and normal intestinal stem cells. Experiments in knockout mice and colon organoids will clarify the role of the dnTCF1E-p21 circuit in intestinal stem cells, and experiments with ChIP and high-throughput sequencing (ChIP-Seq) will elucidate the role of the C-clamp in target site selection and constraints on the C-clamp–Helper site interaction.
Supplementary Material
ACKNOWLEDGMENTS
We thank Zbynek Kozmik for generously providing the SP5 reporter and expression constructs. We thank Hans Clevers and Marc van de Wetering for providing the TR7 parental DLD1 cell line. We thank Andreas Hecht for providing the CDX1 reporter plasmid. We thank Beibei Wu for assistance generating the stable cell lines. We thank Ricardo Ramirez for technical assistance with plasmid cloning. Finally, we thank members of the Waterman laboratory for discussions and critique.
The work of S.S. and P.B. was supported by NIH grants LM010235-01A1 and 5T15LM007743, NSF grant 0513376, and a Microsoft Research Award to P.B. The work of N.P.H., J.-H.T., and M.L.W. was supported by NIH grants CA096878, CA108697, and P30CA062203 from the National Cancer Institute.
N.P.H., J.-H.T., and M.L.W. designed the experiments. N.P.H. and J.-H.T. generated stable cell lines and plasmid constructs and performed the experiments. S.S. and P.B. performed statistical analysis of the array data. N.P.H. and M.L.W. wrote the paper, and all authors contributed to editing.
Footnotes
Published ahead of print 9 July 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Angus-Hill ML, Elbert KM, Hidalgo J, Capecchi MR. 2011. T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. U. S. A. 108: 4914–4919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arce L, Yokoyama NN, Waterman ML. 2006. Diversity of LEF/TCF action in development and disease. Oncogene 25: 7492–7504 [DOI] [PubMed] [Google Scholar]
- 3. Atcha FA, Munguia JE, Li TW, Hovanes K, Waterman ML. 2003. A new beta-catenin-dependent activation domain in T cell factor. J. Biol. Chem. 278: 16169–16175 [DOI] [PubMed] [Google Scholar]
- 4. Atcha FA, et al. 2007. A unique DNA binding domain converts T-cell factors into strong Wnt effectors. Mol. Cell Biol. 27: 8352–8363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bai YQ, Miyake S, Iwai T, Yuasa Y. 2003. CDX2, a homeobox transcription factor, upregulates transcription of the p21/WAF1/CIP1 gene. Oncogene 22: 7942–7949 [DOI] [PubMed] [Google Scholar]
- 6. Baldi P, Long AD. 2001. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics 17: 509–519 [DOI] [PubMed] [Google Scholar]
- 7. Barker N, et al. 2009. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457: 608–611 [DOI] [PubMed] [Google Scholar]
- 8. Battelli C, Nikopoulos GN, Mitchell JG, Verdi JM. 2006. The RNA-binding protein Musashi-1 regulates neural development through the translational repression of p21WAF-1. Mol. Cell Neurosci. 31: 85–96 [DOI] [PubMed] [Google Scholar]
- 9. Beland M, et al. 2004. Cdx1 autoregulation is governed by a novel Cdx1-LEF1 transcription complex. Mol. Cell. Biol. 24: 5028–5038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blache P, et al. 2004. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J. Cell Biol. 166: 37–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boon EM, van der Neut R, van de Wetering M, Clevers H, Pals ST. 2002. Wnt signaling regulates expression of the receptor tyrosine kinase met in colorectal cancer. Cancer Res. 62: 5126–5128 [PubMed] [Google Scholar]
- 12. Bottomly D, Kyler SL, McWeeney SK, Yochum GS. 2010. Identification of β-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res. 38: 5735–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Buttitta L, Tanaka TS, Chen AE, Ko MS, Fan CM. 2003. Microarray analysis of somitogenesis reveals novel targets of different WNT signaling pathways in the somitic mesoderm. Dev. Biol. 258: 91–104 [DOI] [PubMed] [Google Scholar]
- 14. Camargo FD, et al. 2007. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 17: 2054–2060 [DOI] [PubMed] [Google Scholar]
- 15. Chang MV, Chang JL, Gangopadhyay A, Shearer A, Cadigan KM. 2008. Activation of wingless targets requires bipartite recognition of DNA by TCF. Curr. Biol. 18: 1877–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Y, et al. 2006. Elevated expression and potential roles of human Sp5, a member of Sp transcription factor family, in human cancers. Biochem. Biophys. Res. Commun. 340: 758–766 [DOI] [PubMed] [Google Scholar]
- 17. Choe SE, Boutros M, Michelson AM, Church GM, Halfon MS. 2005. Preferred analysis methods for Affymetrix GeneChips revealed by a wholly defined control dataset. Genome Biol. 6: R16 doi:10.1186/gb-2005-6-2-r16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clevers H. 2006. Wnt/beta-catenin signaling in development and disease. Cell 127: 469–480 [DOI] [PubMed] [Google Scholar]
- 19. Crissey MA, et al. 2008. The homeodomain transcription factor Cdx1 does not behave as an oncogene in normal mouse intestine. Neoplasia 10: 8–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dehner M, Hadjihannas M, Weiske J, Huber O, Behrens J. 2008. Wnt signaling inhibits Forkhead box O3a-induced transcription and apoptosis through up-regulation of serum- and glucocorticoid-inducible kinase 1. J. Biol. Chem. 283: 19201–19210 [DOI] [PubMed] [Google Scholar]
- 21. Fearon ER, Vogelstein B. 1990. A genetic model for colorectal tumorigenesis. Cell 61: 759–767 [DOI] [PubMed] [Google Scholar]
- 22. Finch AJ, Soucek L, Junttila MR, Swigart LB, Evan GI. 2009. Acute overexpression of Myc in intestinal epithelium recapitulates some but not all the changes elicited by Wnt/beta-catenin pathway activation. Mol. Cell. Biol. 29: 5306–5315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fujimura N, et al. 2007. Wnt-mediated down-regulation of Sp1 target genes by a transcriptional repressor Sp5. J. Biol. Chem. 282: 1225–1237 [DOI] [PubMed] [Google Scholar]
- 24. Gaur T, et al. 2005. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 280: 33132–33140 [DOI] [PubMed] [Google Scholar]
- 25. George RJ, et al. 2009. Loss of p21Waf1/Cip1/Sdi1 enhances intestinal stem cell survival following radiation injury. Am. J. Physiol. Gastrointest. Liver Physiol. 296: G245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giese K, Pagel J, Grosschedl R. 1997. Functional analysis of DNA bending and unwinding by the high mobility group domain of LEF-1. Proc. Natl. Acad. Sci. U. S. A. 94: 12845–12850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giraud S, Hurlstone A, Avril S, Coqueret O. 2004. Implication of BRG1 and cdk9 in the STAT3-mediated activation of the p21waf1 gene. Oncogene 23: 7391–7398 [DOI] [PubMed] [Google Scholar]
- 28. Hatfield GW, Hung SP, Baldi P. 2003. Differential analysis of DNA microarray gene expression data. Mol. Microbiol. 47: 871–877 [DOI] [PubMed] [Google Scholar]
- 29. Hatzis P, et al. 2008. Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol. Cell. Biol. 28: 2732–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hauck L, et al. 2007. Critical role for FoxO3a-dependent regulation of p21CIP1/WAF1 in response to statin signaling in cardiac myocytes. Circ. Res. 100: 50–60 [DOI] [PubMed] [Google Scholar]
- 31. He TC, et al. 1998. Identification of c-MYC as a target of the APC pathway. Science 281: 1509–1512 [DOI] [PubMed] [Google Scholar]
- 32. Hecht A, Stemmler MP. 2003. Identification of a promoter-specific transcriptional activation domain at the C terminus of the Wnt effector protein T-cell factor 4. J. Biol. Chem. 278: 3776–3785 [DOI] [PubMed] [Google Scholar]
- 33. Higuchi R, Krummel B, Saiki RK. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16: 7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hoi CS, et al. 2010. Runx1 directly promotes proliferation of hair follicle stem cells and epithelial tumor formation in mouse skin. Mol. Cell. Biol. 30: 2518–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoogaars WM, et al. 2008. TBX3 and its splice variant TBX3 + exon 2a are functionally similar. Pigment Cell Melanoma Res. 21: 379–387 [DOI] [PubMed] [Google Scholar]
- 36. Hovanes K, et al. 2001. Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat. Genet. 28: 53–57 [DOI] [PubMed] [Google Scholar]
- 37. Hung SP, Baldi P, Hatfield GW. 2002. Global gene expression profiling in Escherichia coli K-12: the effects of leucine-responsive regulatory protein. J. Biol. Chem. 277: 40309–40323 [DOI] [PubMed] [Google Scholar]
- 38. Ioannidis P, et al. 1999. The polyadenylation inhibitor cordycepin (3′dA) causes a decline in c-MYC mRNA levels without affecting c-MYC protein levels. Oncogene 18: 117–125 [DOI] [PubMed] [Google Scholar]
- 39. Irizarry RA, et al. 2003. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Irizarry RA, et al. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264 [DOI] [PubMed] [Google Scholar]
- 41. Jubb AM, et al. 2006. Achaete-scute like 2 (ascl2) is a target of Wnt signalling and is upregulated in intestinal neoplasia. Oncogene 25: 3445–3457 [DOI] [PubMed] [Google Scholar]
- 42. Katoh M. 2006. Notch ligand, JAG1, is evolutionarily conserved target of canonical WNT signaling pathway in progenitor cells. Int. J. Mol. Med. 17: 681–685 [PubMed] [Google Scholar]
- 43. Kim JS, et al. 2002. Oncogenic beta-catenin is required for bone morphogenetic protein 4 expression in human cancer cells. Cancer Res. 62: 2744–2748 [PubMed] [Google Scholar]
- 44. Kioussi C, et al. 2002. Identification of a Wnt/Dvl/beta-catenin → Pitx2 pathway mediating cell-type-specific proliferation during development. Cell 111: 673–685 [DOI] [PubMed] [Google Scholar]
- 45. Konsavage WM, Jr, Kyler SL, Rennoll SA, Jin G, Yochum GS. 2012. Wnt/beta-catenin signaling regulates Yes-associated protein (YAP) gene expression in colorectal carcinoma cells. J. Biol. Chem. 287: 11730–11739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Korinek V, et al. 1998. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 19: 379–383 [DOI] [PubMed] [Google Scholar]
- 47. Kosinski C, et al. 2007. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc. Natl. Acad. Sci. U. S. A. 104: 15418–15423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li L, Clevers H. 2010. Coexistence of quiescent and active adult stem cells in mammals. Science 327: 542–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lickert H, et al. 2000. Wnt/(beta)-catenin signaling regulates the expression of the homeobox gene Cdx1 in embryonic intestine. Development 127: 3805–3813 [DOI] [PubMed] [Google Scholar]
- 50. Liu Y, Lagowski JP, Vanderbeek GE, Kulesz-Martin MF. 2004. Facilitated search for specific genomic targets by p53 C-terminal basic DNA binding domain. Cancer Biol. Ther. 3: 1102–1108 [DOI] [PubMed] [Google Scholar]
- 51. Liu ZM, Huang HS. 2008. Inhibitory role of TGIF in the As2O3-regulated p21 WAF1/CIP1 expression. J. Biomed. Sci. 15: 333–342 [DOI] [PubMed] [Google Scholar]
- 52. Love JJ, et al. 1995. Structural basis for DNA bending by the architectural transcription factor LEF-1. Nature 376: 791–795 [DOI] [PubMed] [Google Scholar]
- 53. Lustig B, et al. 2002. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell. Biol. 22: 1184–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McKinney K, Mattia M, Gottifredi V, Prives C. 2004. p53 linear diffusion along DNA requires its C terminus. Mol. Cell 16: 413–424 [DOI] [PubMed] [Google Scholar]
- 55. Meyer N, Penn LZ. 2008. Reflecting on 25 years with MYC. Nat. Rev. Cancer 8: 976–990 [DOI] [PubMed] [Google Scholar]
- 56. Miranda-Carboni GA, et al. 2008. A functional link between Wnt signaling and SKP2-independent p27 turnover in mammary tumors. Genes Dev. 22: 3121–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Moskaluk CA, et al. 2003. Cdx2 protein expression in normal and malignant human tissues: an immunohistochemical survey using tissue microarrays. Mod. Pathol. 16: 913–919 [DOI] [PubMed] [Google Scholar]
- 58. Moucadel V, et al. 2002. The homeobox gene Cdx1 belongs to the p53-p21(WAF)-Bcl-2 network in intestinal epithelial cells. Biochem. Biophys. Res. Commun. 297: 607–615 [DOI] [PubMed] [Google Scholar]
- 59. Najdi R, et al. 2009. A Wnt kinase network alters nuclear localization of TCF-1 in colon cancer. Oncogene 28: 4133–4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nakshatri H, et al. 2000. The orphan receptor COUP-TFII regulates G2/M progression of breast cancer cells by modulating the expression/activity of p21(WAF1/CIP1), cyclin D1, and cdk2. Biochem. Biophys. Res. Commun. 270: 1144–1153 [DOI] [PubMed] [Google Scholar]
- 61. Nishitani H, et al. 2008. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J. Biol. Chem. 283: 29045–29052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R. 2005. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat. Immunol. 6: 1054–1060 [DOI] [PubMed] [Google Scholar]
- 63. Polakis P. 2000. Wnt signaling and cancer. Genes Dev. 14: 1837–1851 [PubMed] [Google Scholar]
- 64. Ren F, et al. 2010. Hippo signaling regulates Drosophila intestine stem cell proliferation through multiple pathways. Proc. Natl. Acad. Sci. U. S. A. 107: 21064–21069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Renard CA, et al. 2007. Tbx3 is a downstream target of the Wnt/beta-catenin pathway and a critical mediator of beta-catenin survival functions in liver cancer. Cancer Res. 67: 901–910 [DOI] [PubMed] [Google Scholar]
- 66. Roose J, et al. 1999. Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science 285: 1923–1926 [DOI] [PubMed] [Google Scholar]
- 67. Segditsas S, et al. 2008. Putative direct and indirect Wnt targets identified through consistent gene expression changes in APC-mutant intestinal adenomas from humans and mice. Hum. Mol. Genet. 17: 3864–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shin SW, et al. 2009. Clinical significance of chicken ovalbumin upstream promoter-transcription factor II expression in human colorectal cancer. Oncol. Rep. 21: 101–106 [PubMed] [Google Scholar]
- 69. Van der Flier LG, et al. 2007. The intestinal Wnt/TCF signature. Gastroenterology 132: 628–632 [DOI] [PubMed] [Google Scholar]
- 70. van de Wetering M, Castrop J, Korinek V, Clevers H. 1996. Extensive alternative splicing and dual promoter usage generate Tcf-1 protein isoforms with differential transcription control properties. Mol. Cell. Biol. 16: 745–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. van de Wetering M, et al. 1997. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell 88: 789–799 [DOI] [PubMed] [Google Scholar]
- 72. van de Wetering M, et al. 2002. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111: 241–250 [DOI] [PubMed] [Google Scholar]
- 73. Watt KI, et al. 2010. Yap is a novel regulator of C2C12 myogenesis. Biochem. Biophys. Res. Commun. 393: 619–624 [DOI] [PubMed] [Google Scholar]
- 74. Weise A, et al. 2010. Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/beta-catenin targets. Nucleic Acids Res. 38: 1964–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Westendorf JJ, et al. 2002. Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol. Cell. Biol. 22: 7982–7992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wright WE, Binder M, Funk W. 1991. Cyclic amplification and selection of targets (CASTing) for the myogenin consensus binding site. Mol. Cell. Biol. 11: 4104–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yang WC, et al. 2001. Targeted inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated tumor formation and the tumor-promoting activity of a Western-style high-risk diet by altering cell maturation in the intestinal mucosal. Cancer Res. 61: 565–569 [PubMed] [Google Scholar]
- 78. Zirbes TK, et al. 2000. Prognostic impact of p21/waf1/cip1 in colorectal cancer. Int. J. Cancer 89: 14–18 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.