

Abstract
Kinase suppressor of Ras 1 (KSR1) and KSR2 are scaffolds that promote extracellular signal-regulated kinase (ERK) signaling but have dramatically different physiological functions. KSR2−/− mice show marked deficits in energy expenditure that cause obesity. In contrast, KSR1 disruption has inconsequential effects on development but dramatically suppresses tumor formation by activated Ras. We examined the role of KSR2 in the generation and maintenance of the transformed phenotype in KSR1−/− mouse embryo fibroblasts (MEFs) expressing activated RasV12 and in tumor cell lines MIN6 and NG108-15. KSR2 rescued ERK activation and accelerated proliferation in KSR1−/− MEFs. KSR2 expression alone induced anchorage-independent growth and synergized with the transforming effects of RasV12. Similarly, RNA interference (RNAi) of KSR2 in MIN6 and NG108-15 cells inhibited proliferation and colony formation, with concomitant defects in AMP-activated protein kinase (AMPK) signaling, nutrient metabolism, and metabolic capacity. While constitutive activation of AMPK was sufficient to complement the loss of KSR2 in metabolic signaling and anchorage-independent growth, KSR2 RNAi, MEK inhibition, and expression of a KSR2 mutant unable to interact with ERK demonstrated that mitogen-activated protein (MAP) kinase signaling is dispensable for the transformed phenotype of these cells. These data show that KSR2 is essential to tumor cell energy homeostasis and critical to the integration of mitogenic and metabolic signaling pathways.
INTRODUCTION
The kinase suppressor of Ras (KSR) proteins are molecular scaffolds that coordinate Raf/MEK/extracellular signal-regulated kinase (ERK) kinase cascade signaling (10, 31, 44). KSR1 is an essential component for Ras signaling, as loss-of-function alleles restore the hyperactive Ras phenotype in Drosophila melanogaster and Caenorhabditis elegans (28, 55, 57). In C. elegans, KSR2 is required for Ras-mediated signaling during germ line meiotic progression and functions redundantly with KSR1 during development of the excretory system, hermaphrodite vulva, and male spicules (46). Subsequent studies in mammalian systems showed that KSR1 and KSR2 interact with Raf, MEK, and ERK to coordinate the intensity and duration of ERK signaling (3, 9, 10, 29, 31, 54, 62, 63). Manipulation of KSR1 in mouse embryo fibroblasts (MEFs) revealed the intricate regulation of ERK signaling to enhance the oncogenic potential of Ras, adipogenic differentiation, and replicative senescence (29–31). Studies with mammalian KSR2 showed that it uniquely coordinates calcium-mediated Ras-to-ERK signaling (10). KSR scaffolds have also been implicated in regulating cellular metabolism, as both KSR1−/− and KSR2−/− mice exhibit metabolic defects. KSR1−/− mice have hypertrophic adipocytes (29). Disruption of KSR2 in mice led to reduced expression of genes responsible for oxidative phosphorylation (OXPHOS) and deregulated 5′ AMP-activated protein kinase (AMPK)-mediated processes, which led to spontaneous obesity and insulin resistance. KSR2 interacts with all AMPK subunits and promotes AMPK phosphorylation in vitro and in vivo (6). KSR1 mediates the Ras-dependent upregulation of peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) and estrogen-related receptor α (ERRα), transcription factors that regulate mitochondrial biogenesis in MEFs, and is essential to promote anchorage-independent growth (12).
AMPK is a trimeric enzyme regulating the cell energy homeostasis that is activated during nutrient deprivation due to increased intracellular AMP/ATP ratios, as the allosteric activator AMP promotes AMPK activity during energy stress (16, 51). AMP and ADP promote the binding of α and γ subunits to protect dephosphorylation of Thr172 in the α subunit (45, 61). As ATP levels increase, AMPK activity is suppressed (45, 51). Activation of AMPK promotes the activation of catabolic pathways that generate ATP and the inhibition of anabolic pathways that consume ATP. AMPK activation promotes glucose and fatty acid uptake, glycolysis, and fatty acid oxidation and enhances mitochondrial biogenesis while inhibiting fatty acid synthesis, gluconeogenesis, glycogen storage, and cholesterol biosynthesis (53).
KSR1 is the most extensively characterized of the KSR scaffolds involved in the regulation of cell proliferation and tumorigenesis. T cells isolated from KSR1−/− mice exhibited lower proliferative rates (44). Elevating KSR1 protein to levels that optimize ERK activity enhanced the proliferative rate of MEFs (31). In vitro and in vivo studies have demonstrated that KSR1 is required for Ras-induced transformation. The deletion of KSR1 in MEFs abolished the ability of H-RasV12 to promote anchorage-independent growth due to decreased ERK activation (31, 44). KSR1−/− mice are resistant to tumor formation in mammary epithelial cells expressing polyomavirus middle T or Ras-induced skin tumorigenesis (35, 44). We tested the role of KSR2 in cell proliferation and anchorage-independent growth to determine the extent to which KSR2 contributes to the transformed phenotype. Our data show that KSR2 expression enhances the proliferative rate of MEFs and KSR2 depletion reduces tumor cell growth. KSR2 expression in MEFs promotes anchorage-independent growth and synergizes with H-RasV12 to promote anchorage-independent growth. KSR2 depletion attenuated the ability of tumor cells to grow in an anchorage-independent manner. Enhancement of cell proliferation and anchorage-independent growth by KSR2 is not dependent on ERK activation. However, the ability of KSR2 disruption to suppress the transformed phenotype is rescued by the introduction of constitutively active AMPK (AMPK-CA). Furthermore, KSR2 regulates glycolytic and oxidative metabolism in tumor cells via AMPK. These data implicate KSR2 as a regulator of cellular metabolism affecting the proliferative rate and tumorigenic potential of cells.
MATERIALS AND METHODS
Cell culture and cell lines.
KSR1−/− MEFs were generated from 13.5-day-old embryos as described previously and immortalized using a 3T9 protocol (31). MEFs were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals), 2 mM l-glutamine (Gemini Bioproducts), 0.1 mM minimum essential medium with nonessential amino acids (Life Technologies), and 1% penicillin-streptomycin (Pen/Strep) (Gemini Bioproducts). Cells were incubated at 37°C in 5% carbon dioxide (CO2). MIN6 cells were maintained in DMEM supplemented with 10% FBS, 4 mM l-glutamine, 1 mM sodium pyruvate, 10 mM HEPES (Thermo Scientific), 50 μM β-mercaptoethanol (Thermo Scientific), and 1% Pen/Strep. NG108-15 cells were maintained in DMEM supplemented with 10% FBS, 2 mM l-glutamine, 1× HAT (hypoxanthine, aminopterine, and thymidine) solution (Life Technologies), and 1% Pen/Strep. MIN6 and NG108-15 cells were incubated at 37°C in 10% CO2. 293T cells were transfected with Lipofectamine 2000 per the manufacturer's protocol (Life Technologies). The transfection of small interfering RNA (siRNA) in MIN6 and NG108-15 cells was performed with Lipofectamine 2000, DharmaFECT 1, or DharmaFECT 3 (Dharmacon) per the manufacturer's protocol. All reagents were obtained from Sigma, unless otherwise indicated. MIN6 cells were a gift from Melanie Cobb (University of Texas—Southwestern). NG108-15 cells were obtained from ATCC.
Construction and production of recombinant retroviruses and lentiviruses.
KSR2-FLAG, KSR2.FIFP/AAAP-FLAG, and KSR2.C907Y-FLAG were subcloned from pCDNA3.1 into murine stem cell virus-internal ribosome entry site-green fluorescent protein (MSCV-IRES-GFP). MSCV-KSR1-IRES-GFP has been previously described (31). MSCV-IRES-GFP, MSCV-KSR2-IRES-GFP, MSCV-KSR2.FIFP/AAAP-GFP, MSCV-KSR1-GFP, pBabepuroH-RasV12, or pBabepuro retroviruses were produced as previously described (31). The short hairpin targeting the nucleotides for amino acids 686 to 692 of mouse KSR2 (5′-CCGGGCCATCCGGTTGATTGACATACTCGAGTATGTCAATCAACCGGATGGCTTTTT-3′) and the short hairpin targeting the nucleotides for amino acids 375 to 382 of mouse KSR1 (5′-CCGGGTGCCAGAAGAGCATGATTTTCTCGAGAAAATCATGCTCTTCTGGCACTTTTTG-3′) were cloned into the lentiviral vector MISSION pLKO.1-puro, and lentivirus was produced as previously described (6). Puromycin-resistant KSR1−/− MEFs (4 μg/ml) and MIN6 or NG108-15 cells (2 μg/ml) were selected after 2 days of treatment.
Generation of cell lines.
For the production of KSR1−/− MEFs expressing various concentrations of KSR2, KSR2.FIFP/AAAP, KSR2.C907Y, or KSR1, KSR1−/− MEFs were infected with the respective retrovirus and populations were isolated using flow cytometry as previously described (30, 31). The expression levels of KSR1, KSR2, KSR2.FIFP/AAAP, and KSR2.C907Y were assessed by Western blotting.
Western blotting.
Cycling cells were lysed in buffer containing 20 mM Tris (Thermo Scientific) (pH 8.0), 137 mM NaCl (Thermo Scientific), 10% glycerol (Thermo Scientific), 1% Igepal, 10 μg/ml aprotinin, 20 mM leupeptin, 0.5 mM sodium orthovanadate (Thermo Scientific), 2 mM EDTA, 10 mM sodium fluoride, 2 mM EGTA, 10 mM β-glycerophosphate, and 1 mM phenylmethylsulfonyl fluoride (PMSF). Soluble lysates were isolated by centrifugation at 11,000 to 15,000 × g for 10 min. Protein concentrations were quantified using the bicinchoninic acid (BCA) protein assay (Promega). Proteins were resolved using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Li-COR). Membranes were blocked in Odyssey blocking buffer (Li-COR) for 1 to 12 h and probed with primary antibodies (1 to 16 h) and secondary antibodies (45 min) in a 1:1 mixture of Odyssey blocking buffer and Tris-buffered saline with 0.1% Tween 20 and hybridized proteins detected using the Odyssey imaging system (Li-COR). All reagents were obtained from Sigma, unless specified.
Immunoprecipitations.
To detect KSR2-ERK association, transfected 293T cells were subjected to serum starvation for 4 h and stimulated with human epidermal growth factor (EGF) (Life Technologies) for 5 min. Cells were lysed in lysis buffer containing 25 mM Tris (pH 7.4), 125 mM NaCl, 1 mM MgCl2, and 1% Brij 98 with 10 μg/ml aprotinin, 20 mM leupeptin, 0.5 mM sodium orthovanadate, 2 mM EDTA, 10 mM sodium fluoride, 2 mM EGTA, 10 mM β-glycerophosphate, and 1 mM PMSF. Lysates were cleared and protein concentrations determined. To detect KSR2-MEK association, transfected cycling 293T cells were lysed in Igepal lysis buffer as described above. Total protein (2 mg) was immunoprecipitated overnight with 40 μl of a 1:1 slurry of anti-FLAG-conjugated agarose in phosphate-buffered saline (PBS). The beads were washed three times in lysis buffer, and protein was eluted using the FLAG peptide. To detect KSR proteins in tumor cells, where indicated, total protein (1 mg) was incubated with 4 μg of KSR1 or KSR2 antibodies overnight, subjected to a 1:1 slurry of protein G-PBS for 30 min, washed three times with lysis buffer, and boiled in sample buffer. The collected supernatants were analyzed via Western blotting. All reagents were obtained from Sigma, unless otherwise specified.
Reagents.
The following primary antibodies were purchased from Cell Signaling, unless otherwise indicated: FLAG (M2; Sigma), KSR2 (rabbit polyclonal raised against a recombinant protein containing amino acids 168 to 262 of mouse KSR2 [for the detection of ectopic proteins]), KSR2 (1G4 [lot 08344-1G4]; Abnova), KSR2 (K75; Santa Cruz), α-tubulin (B-5-1-2; Santa Cruz), c-H-Ras (Ab-1; EMD Biosciences), phospho-Thr202/Tyr204 ERK1/2 (9106), ERK1/2 (9102), ERK1 (sc-93; Santa Cruz), KSR1 (H-70; Santa Cruz), KSR1 (C-19; Santa Cruz), Myc (4A6; Thermo Scientific), phospho-Thr172 AMPKα (2531), AMPKα (2532), AMPKα1 (2795), AMPKα2 (R&D Systems), AMPKγ1 (4187), phospho-Ser79 acetyl coenzyme A (CoA) carboxylase (ACC) (3661), ACC (3676), phospho-Ser792 Raptor (2083), and Raptor (2280). Anti-mouse and anti-rabbit antibodies conjugated with Infrared Dye 680LT (Li-COR), Alexa Fluor 680 (Life Technologies), or Infrared Dye 800CW (Rockland or Li-COR) were used to detect primary antibodies. 5-Aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR) was purchased from Toronto Research Chemicals. U0126 was purchased from Cell Signaling. ON-TARGETplus siRNA preparations targeting AMPKα1, AMPKα2, and AMPKγ1 were obtained from Thermo Scientific. Constitutively active AMPKα2 (AMPKα2Δ312) and kinase dead/dominant-negative AMPKα2 (AMPKα2-K45R) constructs of rat origin were gifts from Morris Birnbaum (University of Pennsylvania). Mutation of amino acid 172 from Thr to Asp was carried out via site-directed mutagenesis [AMPKα2Δ312(T172D)] (52).
In situ ERK activation assay.
The in situ ERK activation assay was performed as previously described (30, 31). Briefly, MEFs were seeded at 5 × 103 cells per well in a 96-well black-wall plate, in triplicate, 16 h prior to analysis and subjected to an in situ plate assay using a Li-COR Odyssey infrared imaging system to quantify ERK activation. Cells at 80% confluence were subjected to serum starvation for 4 h and treated with 25 ng/ml platelet-derived growth factor (PDGF) in DMEM–1% bovine serum albumin at the indicated time points. Primary antibodies and secondary antibodies were used to detect phosphorylated and total ERK, respectively. The microplates were imaged and measurements acquired using a Li-COR Odyssey imaging system.
Cell proliferation assays.
Cells (4 × 104 MEF, 2.5 × 104 MIN6, or 5 × 103 NG108-15) were seeded in 35-mm-diameter dishes. In triplicate, cells were treated with trypsin at the indicated time points, collected, and counted using a Beckman Coulter counter. To account for discrepancies in plating, the first count was performed 4 h after plating.
Anchorage-independent growth assays.
A total of 5 × 103 cells were suspended in Iscove's modified Dulbecco's medium (IMDM) (Life Technologies) supplemented with 15% FBS, 2 mM l-glutamine, 1% Pen/Strep, 2 μg/ml puromycin, and 0.65% NuSeive GTG agarose (Thermo Scientific) and seeded on a layer of IMDM supplemented with 0.32% NuSeive GTG agarose in 35-mm-diameter dishes. MIN6 cells were supplemented with an additional mixture of 2 mM l-glutamine, 1 mM sodium pyruvate, 10 mM HEPES, and 50 μM β-mercaptoethanol. NG108-15 cells were supplemented with HAT solution. Colonies were counted, and representative photomicrographs were taken at the indicated times. For short-term anchorage-independent growth assays, 96-well microplates were coated with poly(2-hydroxyethyl methacrylate) [poly(HEMA)] (Sigma) to prevent cell attachment. A total of 5 × 103 cells were seeded in full media, and the viability of cells was assessed using CellTiter-Glo reagent (Thermo Scientific) at the indicated time points.
Metabolic assays.
The rate of nutrient consumption and the maximal capacity for OXPHOS and aerobic glycolysis were determined by measuring the oxygen consumption rate (OCR) or extracellular acidification rate (ECAR) using an XF24 Analyzer (Seahorse Biosciences). To determine glucose metabolism levels, 4 × 104 MIN6 cells were seeded in a 24-well XF microplate in bicarbonate-free low-buffer DMEM containing 2.5 mM glucose. OCR measurements were performed prior to and after an injection of DMEM or 22.5 mM glucose. Similarly, glutamine consumption was assessed in 4 × 104 MIN6 and 2 × 104 NG108-15 cells after they were seeded in bicarbonate-free low-buffer DMEM. OCR measurements were performed prior to and after a 10 mM l-glutamine (Life Technologies) injection. To determine fatty acid oxidation, 4 × 104 MIN6 cells were seeded in unbuffered modified KHB media (111 mM NaCl, 4.7 mM KCl, 2 mM MgSO4, 1.2 mM Na2HPO4) containing 2.5 mM glucose and 0.5 mM carnitine. OCR measurements were performed prior to and after injections of BSA or 200 μM palmitate–BSA conjugate. The maximal capacity for OXPHOS and aerobic glycolysis in 4 × 104 MIN6 and 5 × 104 NG108-15 cells was assessed by seeding in bicarbonate-free low-buffer DMEM supplemented with 25 mM glucose, 4 mM l-glutamine, and 2 mM sodium pyruvate. OCR and ECAR measurements were performed prior to and after three successive injections of carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) at the indicated concentrations.
Statistics.
Two-tailed Student's t tests were performed using GraphPad Prism 5 (GraphPad Software).
RESULTS
KSR2 is a scaffold for the ERK signaling pathway and regulates cell proliferation.
KSR1 is required for maximal activation of ERK and facilitates signaling by growth factors and oncogenic Ras by spatially and temporally regulating ERK signaling to affect cell fate (29–31). The KSR1 paralog, KSR2, modulates calcium-induced processes by mediating Ras with respect to ERK signaling (10). Extensive studies of the role of KSR1 in cell proliferation and oncogenic potential have been performed, but little is known about the regulation of these cell fates by KSR2. Previous work in our laboratory used KSR1−/− MEFs to characterize KSR1 function (12, 29–31). MEFs do not express KSR2 mRNA or protein, making KSR1−/− MEFs effectively doubly null for KSR1 and KSR2 (29). To assess distinct and overlapping functions of the KSR scaffolds, KSR2 was ectopically expressed in KSR1−/− MEFs. The levels of KSR2 expression were modulated using a bicistronic vector encoding FLAG-tagged KSR2 and green fluorescent protein (GFP). MEFs expressing GFP were separated via flow cytometry into two populations. One population expressed the lowest detectable level of GFP. A second population expressed high levels of GFP. The level of KSR2 expression relative to that of GFP was then confirmed via Western blotting (Fig. 1A). For comparison, FLAG-tagged KSR1 was reintroduced to KSR1−/− MEFs at a level that significantly enhanced growth factor-induced ERK phosphorylation above wild-type levels (31). To characterize the role of KSR2 in growth factor-mediated ERK activation, each population was stimulated with PDGF and the level of ERK phosphorylation was assessed over a 30-min time course. In comparison to KSR1−/− MEFs expressing GFP only, KSR1−/− MEFs expressing low levels of KSR2 restored growth factor-induced ERK activation to an intensity similar to that seen with ectopic KSR1 (Fig. 1C). However, the level of ectopic KSR2 that restored ERK phosphorylation was one-ninth the level of KSR1 that induced a similar effect (Fig. 1B). A level of KSR2 approximately three times higher than that of ectopic KSR1 (Fig. 1B) impaired ERK phosphorylation at a level below that observed in KSR1−/− MEFs expressing GFP alone (Fig. 1C). Strikingly, intermediate levels of KSR2 expression inhibited growth factor-induced ERK activation in a concentration-dependent manner (data not shown). These data indicate that KSR2 rescues ERK activation debilitated by the disruption of KSR1 but also that KSR1−/− MEFs are more sensitive to KSR2 than KSR1.
Fig 1.

KSR2 differentially mediates growth factor-induced ERK activation and cell proliferation. Bicistronic recombinant retroviruses that encompass genes for KSR2 and GFP, KSR1 and GFP, or GFP alone were expressed in KSR1−/− MEFs. (A) Whole-cell extracts (WCEs) were immunoblotted (IB) with the indicated antibodies to confirm the expression of KSR proteins after FACS. (B) The optical density (OD) values for FLAG and α-tubulin immunoreactive bands of three distinct membranes from the experiment described for panel A were quantified using the Li-COR Odyssey system. (C) In situ analysis of PDGF-induced ERK activation. (D) Proliferation of KSR1−/− MEFs expressing KSR2 at low or high levels, KSR1, or GFP only. All data shown are the results of triplicate measurements and representative of at least three independent experiments. Data represent means ± standard deviations. *, P ≤ 0.001; **, P ≤ 0.0001 (two-tailed Student's t test).
Moderately increasing the levels of KSR1 enhanced the growth rate of KSR1−/− MEFs. However, this phenotype was restored when KSR1 levels were elevated to levels 21-fold above those found in wild-type MEFs. Since the effects on cell proliferation by KSR1 paralleled the biological effects on ERK activation, it was postulated that KSR1 mediates cell proliferation through regulation of ERK activity (31). To assess the role of KSR2 in cell proliferation and determine if it is dependent on ERK activity, the growth rate of KSR1−/− MEFs expressing low or high levels of KSR2 was compared to those of GFP-only cells and cells expressing KSR1. Levels of KSR1 that enhanced ERK activation also accelerated MEF proliferation. KSR2 at low levels that restored ERK activity (Fig. 1C) also enhanced cell proliferation, albeit not as efficiently as KSR1 (Fig. 1D). Surprisingly, high levels of KSR2 further increased proliferation of KSR1−/− MEFs beyond that seen with low KSR2 levels, mirroring the growth rate of KSR1−/− MEFs expressing ectopic KSR1. This was an unexpected result, since the high level of KSR2 expression did not mediate ERK activation (Fig. 1C and D). These data show that KSR1 and KSR2 regulate cell proliferation; however, they suggest that KSR2 asserts its effect on cell proliferation through a mechanism independent of the Raf/MEK/ERK kinase cascade.
KSR2 regulates cell proliferation independently of ERK signaling.
The interaction between ERK and some of its effectors is mediated via a DEF domain, an FXFP motif, located on KSR homologs (20). Mutating this domain on KSR1 incapacitates KSR1-mediated ERK phosphorylation by MEK (30, 38). To further examine the ability of KSR2 to regulate cell proliferation and its dependence on the Raf/MEK/ERK pathway, the DEF domain of KSR2 was mutated (570 FIFP/AAAP 573) to abolish the KSR2-ERK interaction (Fig. 2A). The KSR2.AAAP mutant was transduced into KSR1−/− MEFs using the bicistronic vector described previously. KSR1−/− MEFs expressing either low or high levels of KSR2.AAAP were isolated using the same gates used for wild-type KSR2, and the level of expression was verified by Western blot analysis (Fig. 2B). The KSR2.AAAP mutant was then examined for its ability to mediate ERK activation in response to growth factors and promote cell proliferation. After assessing ERK activity in situ after PDGF stimulation, abolishing the KSR2-ERK interaction decreased the level of ERK activity mediated by KSR2 expressed at low levels in KSR1−/− MEFs (Fig. 2C). Furthermore, the level of ERK activity was suppressed in comparison to the level seen with GFP-only infected cells. Expressing KSR2.AAAP at high levels suppressed ERK activity in a manner similar to that seen with expression at low levels, but the effect was not as severe as that seen with expression at high levels of wild-type KSR2 (Fig. 1C and 2C). As expected, the KSR2-ERK interaction is necessary for KSR2 to mediate growth factor-induced ERK activation.
Fig 2.

KSR2 mediates cell proliferation through an ERK1/2-independent pathway. (A and E) 293T cells transfected with the indicated plasmids were lysed and evaluated via immunoprecipitation (IP) using anti-FLAG-agarose to detect KSR2-ERK and KSR2-MEK association, respectively. Precipitates were eluted, resolved by electrophoresis, and probed on Western blots with the indicated antibodies. WCEs were analyzed to show the uniform expression of FLAG-tagged KSR2 proteins MEK and ERK. (B to D and F to H) Bicistronic recombinant retroviruses that encompass genes for KSR2 and GFP, KSR2.AAAP and GFP, KSR2.C907Y and GFP, or GFP alone were expressed in KSR1−/− MEFs. (B and F) Western blots showing the level of KSR2, KSR2.AAAP, and KSR2.C907Y protein expression in KSR1−/− MEFs after FACS. (C and G) In situ analysis of PDGF-induced ERK activation. (D and H) Proliferation of KSR1−/− MEFs expressing KSR2.AAAP, KSR2.C907Y or GFP only. IB, immunoblot. All data shown are the results of triplicate measurements and representative of at least three independent experiments. Data represent means ± standard deviations. *, P ≤ 0.0001; **, P ≤ 0.01 (two-tailed Student's t test).
To determine if the KSR2.AAAP mutant had the ability to enhance cell proliferation, the growth rate of KSR1−/− MEFs expressing low or high levels of KSR2.AAAP was determined and compared to that of cells expressing wild-type KSR2 or GFP-only cells. The results show that, expressed at low levels, KSR2.AAAP increases cell proliferation with a greater effect than wild-type KSR2 (Fig. 1D and 2D). Furthermore, increasing KSR2.AAAP expression modestly elevated cell proliferation in KSR1−/− MEFs (Fig. 2D). These data show that the KSR2-ERK interaction is necessary for growth factor-mediated ERK activation but is dispensable for cell proliferation.
Studies with KSR2.AAAP further demonstrated the inverse correlation between cell proliferation and ERK activity mediated by KSR2. Levels of KSR2 that are able to promote ERK activation do not optimally enhance cell proliferation, and levels of KSR2 that are able to optimally enhance cell proliferation do not promote ERK activation (Fig. 1 and 2A to D). To further examine this correlation, we sought a KSR2 mutant that hyperstimulates ERK activation by growth factors and tested its ability to enhance cell proliferation. Mutating Cys809 to Tyr on KSR1 leads to hyperstimulation of Raf/MEK/ERK by growth factors (30) and disrupts the KSR1-MEK interaction (30, 38, 54). As with the KSR1 mutation, the equivalent mutation on KSR2 (KSR2.C907Y) disrupts the KSR2-MEK interaction (Fig. 2E). KSR1−/− MEFs expressing low and high levels of KSR2.C907Y were isolated via fluorescence-activated cell sorting (FACS), and the ability of KSR2.C907Y to mediate growth factor-induced ERK activation and cell proliferation was tested. In similarity to the KSR1 results (30), abolishing the KSR2-MEK interaction did not disrupt the ability of KSR2 to mediate growth factor-induced ERK activation (Fig. 2G). Moreover, both low and high levels of KSR2.C907Y were capable of mediating ERK activation, indicating that, like that of KSR1, interaction of KSR2 with MEK is not required for ERK activation. However, irrespective of the expression level, KSR2.C907Y did not enhance cell proliferation of KSR1−/− MEFs (Fig. 2H) to the rates observed in MEFs expressing high levels of wild-type KSR2 or KSR2.AAAP. Together, these data demonstrate that the proliferative effects of KSR2 are independent of the Raf/MEK/ERK kinase cascade.
KSR2-mediated anchorage-independent growth is not dependent on ERK signaling.
KSR1 is required for oncogenic Ras-mediated cell transformation and tumorigenesis, a biological action attributed to its role as a scaffold regulating ERK signaling (31, 35, 44). Furthermore, activated Ras requires KSR1 to promote anchorage-independent growth in KSR1−/− MEFs (31). To assess the role of KSR2 in oncogenic Ras-mediated anchorage-independent growth, H-RasV12 or the control vector was introduced in KSR1−/− MEFs simultaneously with KSR2. Low and high levels of KSR2 expression were isolated, and the expression levels of KSR2 and H-RasV12 were confirmed via Western blot analysis (Fig. 3A). The ability of these cells to promote anchorage-independent growth in soft agar was determined. Expressing KSR2 at low levels did not significantly enhance the ability of H-RasV12 to form colonies in soft agar compared to the results seen with cells expressing GFP only. However, expressing high levels of KSR2 did enhance H-RasV12-induced anchorage-independent growth in KSR1−/− MEFs. Interestingly, in the absence of H-RasV12, KSR2 promoted colony formation in a dose-dependent manner in KSR1−/− MEFs (Fig. 3B). Since only low levels of KSR2 expression mediate PDGF-induced ERK activation, these data suggest that ERK signaling is not required for the effects of KSR2 on anchorage-independent growth in the presence or absence of H-RasV12.
Fig 3.
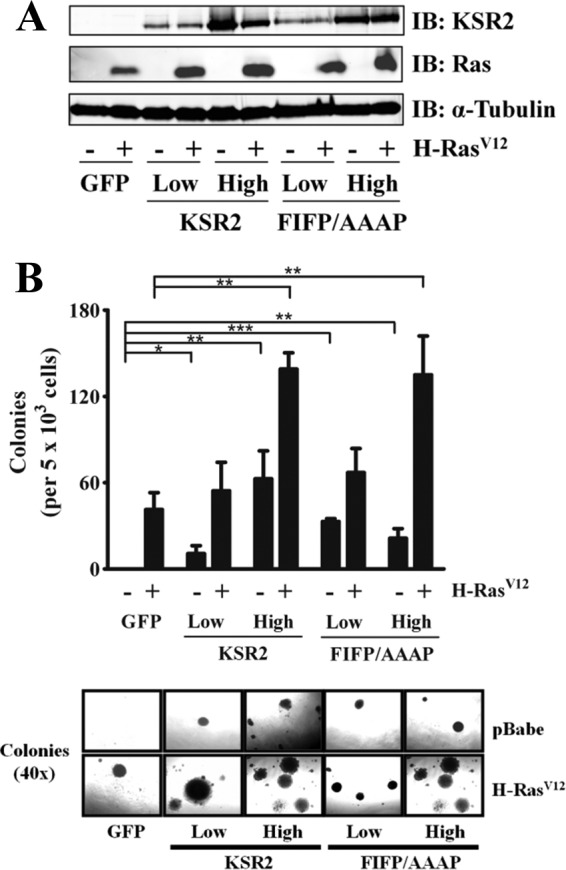
KSR2 promotes anchorage-independent growth in an ERK-independent manner and synergizes H-RasV12-induced anchorage-independent growth of KSR1−/− MEFs. KSR1−/− MEFs were infected with a bicistronic recombinant retrovirus encompassing KSR2 and GFP, KSR2.AAAP and GFP, or GFP only, with either H-RasV12 or the respective vector alone. (A) A representative immunoblot (IB) using the indicated antibodies, illustrating the expression levels of KSR2, KSR2.AAAP, and H-RasV12 after sorting. (B) Anchorage-independent growth was examined by assessing the growth of MEFs expressing the indicated proteins in soft agar. Data are representative of three independent experiments and are illustrated as the number of colonies per 5 × 103 cells present after 4 weeks. The lower panel shows representative photomicrographs of colonies for each sample. Data represent means ± standard deviations of the results determined with triplicate samples. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.0001 (two-tailed Student's t test).
To further examine this possibility, KSR1−/− MEFs expressing low or high levels of the KSR2.AAAP mutant with or without H-RasV12 were isolated (Fig. 3A) and tested for their ability to grow in soft agar. KSR2.AAAP was equal to wild-type KSR2 in its ability to promote anchorage-independent growth in the presence or absence of H-RasV12. Only high levels of KSR2.AAAP expression were capable of enhancing H-RasV12-mediated anchorage-independent growth. However, KSR2.AAAP alone was able to promote anchorage-independent growth to levels similar to those seen with H-RasV12 alone (Fig. 3B). These data suggest that KSR2 does not directly mediate but acts synergistically with H-RasV12 to promote H-RasV12-mediated anchorage-independent growth. Moreover, the effect of KSR2 on anchorage-independent growth is not dependent on ERK signaling and is conceivably mediated through an alternative signaling pathway.
KSR2 knockdown affects tumor cell proliferation but not growth factor-mediated ERK activation.
The tumor cell lines MIN6 and NG108-15 express KSR2 at levels detectable by Western blot analysis. KSR2 depletion by RNA interference (RNAi) was used on each line to test its effect on growth factor-mediated ERK activation and cell proliferation. Knockdown of KSR2 decreased proliferation of both MIN6 and NG108-15 cells (Fig. 4A and B), affirming the importance of KSR2 in regulating cell proliferation. To determine if KSR2 depletion had any effect on growth factor-mediated ERK activation, MIN6 and NG108-15 were treated with epidermal growth factor (EGF) over a time course of 30 min and the levels of phosphorylated ERK analyzed via Western blot analysis. KSR2 depletion modestly decreased EGF-mediated ERK activation in both MIN6 and NG108-15 cells (Fig. 4C and D).
Fig 4.

KSR2 knockdown results in decreased growth rates without affecting growth factor-mediated ERK activation. Tumor cell lines were infected with lentiviruses expressing a nontargeting short hairpin (shCTRL) or short hairpins that target KSR2 (shKSR2) or KSR1 (shKSR1). Cells expressing the indicated short hairpin were selected with puromycin. Growth rates of MIN6 (A and E) and NG108-15 (B and F) cell lines were assessed by seeding 2.5 × 104 and 5 × 103 cells, respectively, in 35-mm-diameter dishes and triplicate counts performed at the indicated time points. MIN6 (C and G) and NG108-15 (D and H) cells were stimulated with EGF for the indicated times, and the phosphorylation and total levels of ERK were assessed by immunoblotting (IB) with the indicated antibodies. KSR1 expression and knockdown were confirmed via KSR1 immunoprecipitations (IP). (A, B, E, and F) *, P ≤ 0.01; **, P ≤ 0.001; ***, P ≤ 0.0001 (two-tailed Student's t test). Data are representative of the results of three independent experiments.
Since MIN6 and NG108-15 cells express both KSR1 and KSR2, it is possible that KSR2 depletion only modestly decreased EGF-mediated ERK activation due to the ability of KSR1 to compensate for the absence of KSR2. To examine this possibility, KSR1 was depleted in these cell lines. Similarly to that of KSR2, knockdown of KSR1 decreased proliferation of MIN6 and NG108-15 cells (Fig. 4E and F). Depletion of KSR1 completely abrogated EGF-stimulated ERK activation in each tumor cell line (Fig. 4G and 4H), indicating that KSR2 alone cannot compensate for the absence of KSR1 and is not sufficient to mediate the EGF response in MIN6 and NG108-15 cells. These data complement the results obtained with MEFs and reinforce the idea that the ability of KSR2 to regulate cell proliferation is mediated primarily through signaling pathways other than the Raf/MEK/ERK kinase cascade.
KSR2 is required for proper activation of AMPK in tumor cells.
KSR2 associates with AMPK subunits α, β, and γ (6, 10). In vivo studies showed that KSR2 deletion led to obesity, insulin resistance, and disruption of AMPK-mediated effects on metabolism. The presence of KSR2 was required for AMPK activation in vivo and in vitro (6). To determine if the depletion of KSR2 affects the activation of AMPK, phosphorylation of Thr172 on AMPKα was assessed in MIN6 cells stimulated with the AMPK agonist AICAR (5) after KSR2 RNAi. Depletion of KSR2 led to a 50% reduction in the levels of phosphorylation on Thr172, which is critical to sustain catalytic activity of the AMPKα subunit (Fig. 5A). To determine if the activity of AMPK directed toward its substrates also required KSR2, phosphorylation on Ser79 of ACC and Ser792 of Raptor was examined. Coincident with decreased phosphorylation of Thr172 on AMPKα, phosphorylation decreased on ACC and Raptor by approximately 50% and 25%, respectively (Fig. 5A). Moreover, depletion of KSR2 led to a reduction in the basal levels of AMPKα and ACC. These results reinforce those observed previously for NG108-15 cells (6) and raise the possibility that AMPK may mediate the effects of KSR2 on cell transformation.
Fig 5.

ERK activity is not necessary for KSR2-mediated AICAR-induced AMPK activation in MIN6 cells. (A) MIN6 cells expressing the nontargeting short hairpin, the KSR2-specific short hairpin, or the KSR2-specific short hairpin and the constitutively active AMPKα2 (AMPKα-CA) were seeded in serum-free media for 4 h prior to stimulation with AICAR for 30 min (*, nonspecific bands). (B) MIN6 cells were subjected to serum starvation for 4 h and treated with the MEK inhibitor U0126 for 30 min prior to treatment with AICAR for 30 min at the indicated concentrations. For both panels, KSR2 knockdown, AMPKα, ACC, Raptor, and ERK activity were examined by immunoblotting (IB) with the indicated antibodies. Immunoblots were quantified using the Li-COR Odyssey system, and the optical densities are illustrated as means ± standard deviations. *, P ≤ 0.05; **, P ≤ 0.01 (two-tailed Student's t test). Data are representative of the results of at least three independent experiments.
To determine if ERK activity is required for AICAR-mediated AMPK activation, MIN6 cells were treated with the MEK inhibitor U0126 prior to stimulation of cells with AICAR. Inhibition of ERK activity did not affect the levels of AMPKα phosphorylation on Thr172 due to AICAR treatment. This indicates that the effects of KSR2 on AICAR-mediated AMPK activation are independent of its scaffolding function for the Raf/MEK/ERK pathway (Fig. 5B).
KSR2 knockdown reduces the rate of nutrient consumption in tumor cells.
Tumor cells alter key metabolic pathways, such as glycolysis and glutaminolysis, to meet the bioenergetic and biosynthetic demands necessary to maintain their rapid cell growth and survival (8, 17, 34). These pathways are regulated by the cellular energy sensor AMPK, which becomes activated when the levels of ATP are low or the AMP/ATP ratio increases (16, 51). AMPK activation leads to the breakdown of nutrients, such as glucose and fatty acids, to produce ATP (53). Since KSR2 knockdown reduces cell growth and AMPK activation in tumor cells, we postulated that KSR2-depleted cells have lost the ability to properly sense their energy status, impairing nutrient metabolism. Nutrient consumption was analyzed using the Seahorse XF24 Analyzer to measure OCR and ECAR, indices for OXPHOS and aerobic glycolysis, respectively. We showed previously that depletion of KSR2 reduced aerobic glycolysis in NG108-15 cells (6). Analysis of MIN6 cells shows that KSR2 knockdown led to the reduced ability of these cells to metabolize glucose, as OCR was suppressed (Fig. 6A). However, in contrast to NG108-15 cell results, pyruvate was diverted to mitochondria in MIN6 cells, as changes in ECAR were not detectable in control and KSR2 knockdown cells (data not shown).
Fig 6.

KSR2 knockdown attenuates the ability of tumor cells to metabolize nutrients. Cells expressing the nontargeting short hairpin, the KSR2-specific short hairpin, or the KSR2-specific short hairpin and the constitutively active AMPKα2 (AMPKα-CA) were seeded in growth media on XF24 microplates 16 h prior to analysis. MIN6 (A to C) and NG108-15 (D) cells were stimulated with 22.5 mM glucose (A), 200 μM palmitate (B), or 10 mM l-glutamine (C and D), after incubation in bicarbonate-free low-buffered media containing low concentrations of nutrients as described in Materials and Methods. The rate of oxygen consumption (OCR) was measured prior to and after nutrient addition. (E) Representative immunoblots illustrating the expression of the indicated proteins represented in panel D. Data are represented as means ± standard errors of the means of percent change from basal values (n = 3 to 5 wells). *, P ≤ 0.05; **, P ≤ 0.01 (two-tailed Student's t test). Data are representative of the results of at least three independent experiments. IP, immunoprecipitation.
In addition to glucose, tumor cells use fatty acids and glutamine as additional energy sources for the production of ATP (24, 34, 59). The rate of palmitate consumption was analyzed in MIN6 cells, and the rate of glutamine consumption was examined in MIN6 and NG108-15 cells. KSR2 knockdown inhibited palmitate oxidation in MIN6 cells (Fig. 6B) and glutamine oxidation in both MIN6 and NG108-15 cells (Fig. 6C and D). These data reveal KSR2 as a regulator of oxidative metabolism in tumor cells.
AMPK regulates glycolysis and fatty acid oxidation via phosphorylation of phosphofructokinase 2 (PFK-2) and ACC, respectively (7, 36, 37, 39). KSR2 is necessary for proper AMPK activation (Fig. 5A). A constitutively active form of AMPK (AMPK-CA) (52) was introduced into KSR2-depleted cells to determine if the role of KSR2 in regulating nutrient consumption is dependent on AMPK activity. Expression of AMPK-CA restored the phosphorylation of ACC and Raptor in the absence of KSR2 (Fig. 5A, lane 5). Furthermore, AMPK-CA restored the ability of MIN6 cells to properly metabolize glucose (Fig. 6A) and palmitate (Fig. 6B). Glutamine starvation has been reported to activate AMPK in K-Ras-transformed fibroblasts and brain tumor cells (14, 42). The ability of AMPK-CA to restore glutaminolysis in both MIN6 and NG108-15 cells was also examined. Although AMPK has not been shown to directly regulate glutamine oxidation, AMPK-CA restored the ability of MIN6 cells (Fig. 6C) and NG108-15 cells (Fig. 6D) to metabolize glutamine in the absence of KSR2. These data demonstrate that KSR2 regulates key metabolic pathways by promoting the proper activation of AMPK.
KSR2 mediates maximal capacity for OXPHOS and aerobic glycolysis in an AMPK-dependent manner.
Tumor cells commonly display the ability to shift bioenergetic demand toward aerobic glycolysis. This shift converts the majority of pyruvate to lactate producing NAD+ and provides a carbon source for the increased synthesis of macromolecules. Although tumor cells obtain most of their ATP from glycolysis, the tricarboxylic acid (TCA) cycle and OXPHOS remain important processes for the production of key intermediates required for biosynthetic pathways (24, 58). KSR2−/− mice exhibit lower expression levels of genes that coordinate OXPHOS, including the transcription factor PGC-1α (6). KSR1 is required for RasV12-mediated upregulation of PGC-1α and ERRα, which maximizes glycolytic and OXPHOS capacity in MEFs (12). AMPK has been implicated in the regulation of mitochondrial biogenesis and OXPHOS by inducing PGC-1α transcriptional activity (21, 56, 64). To further elucidate the role of KSR2 and AMPK in tumor growth and survival, basal and maximal rates of OXPHOS and glycolysis with and without KSR2 and AMPK-CA were determined. Cells were plated in nutrient-rich media, and the OCR and ECAR were assessed before and after stimulation with FCCP, which uncouples the electron transport chain and reveals maximal OXPHOS and glycolytic potential (1, 4, 11, 60). To assess the overall maximal capacity of OXPHOS and aerobic glycolysis, cells were treated with successive doses of FCCP adjusted to a final concentration that maximizes OXPHOS. Knockdown of KSR2 decreased both the basal levels and maximal rates of OCR in both the MIN6 (Fig. 7A) and NG108-15 (Fig. 7B) cell lines. Expression of AMPK-CA after either cell line was depleted of KSR2 restored basal and maximal OXPHOS to levels observed in control cells (Fig. 7A and B). Furthermore, KSR2 knockdown reduced the maximal glycolytic capacity of NG108-15 cells but did not affect basal glycolysis, as measured by ECAR. Maximal glycolytic capacity was restored to NG108-15 cells by the expression of AMPK-CA in the absence of KSR2 (Fig. 7C). ECAR was undetectable prior to and after FCCP treatment in MIN6 cells (data not shown), indicating that these cells need to optimize OXPHOS in order to maintain their tumorigenic properties. These data demonstrate that KSR2 influences maximal glycolytic and OXPHOS potential by regulating the activation of AMPK and emphasize the importance to tumor cells of properly maintaining bioenergetic and biosynthetic demand for sustained cell proliferation.
Fig 7.
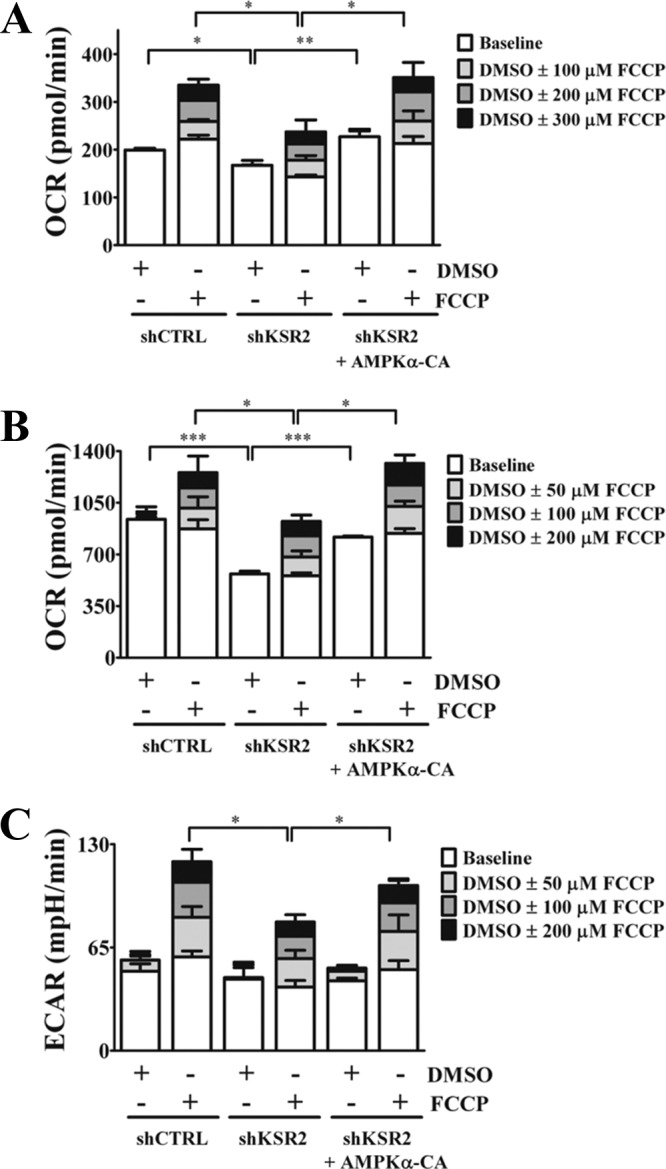
KSR2 knockdown affects the maximal metabolic capacity of tumor cells. Cells expressing the nontargeting short hairpin, the KSR2-specific short hairpin, or the KSR2-specific short hairpin and constitutively active AMPKα2 (AMPKα-CA) were seeded in growth media on XF24 microplates 16 h prior to analysis. MIN6 (A) and NG108-15 (B and C) cells were plated in bicarbonate-free low-buffered media with a full complement of nutrients as described in Materials and Methods. The rates of oxygen consumption (OCR) and extracellular acidification (ECAR) were measured prior to and 15 min after three successive stimulations with FCCP at the indicated concentrations. Data represent means ± standard errors of the means of the results determined for 3 or 4 wells. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 (two-tailed Student's t test). Statistical comparison was performed on data collected after the third stimulation. Data are representative of the results of three independent experiments.
KSR2 mediates anchorage-independent growth via regulation of AMPK.
High levels of KSR2 expression promote ERK-independent anchorage-independent growth in MEFs (Fig. 3B). KSR2 knockdown reduces the growth rate and AMPK-mediated cell metabolism of MIN6 and NG108-15 cells without affecting growth factor-mediated ERK activation (Fig. 4 to 7). These data suggest that the AMPK signaling pathway may regulate the effects of KSR2 on anchorage-independent growth. However, AMPK can have opposing roles in cancer. On the one hand, AMPK has been implicated as a tumor suppressor, as its activation leads to cell cycle arrest through negative regulation of the mTOR signaling pathway and activation of p53 (15, 18, 19, 23). On the other hand, AMPK activity has been shown to have protumorigenic effects. AMPKα1 and AMPKα2 or AMPKβ1 depletion decreased cell proliferation and survival of human prostate cancer cells (47, 50). AMPK also increased the ability of Ras-transformed MEFs and estrogen receptor α (ERα)-negative tumors to grow as xenografts (33, 48). Furthermore, AMPK activity is required for the survival of AKT-transformed cells to promote the activation of fatty acid oxidation during glucose deprivation and for the survival of simian virus 40 small T (SV40 ST) antigen-positive tumor cells to maintain energy homeostasis under low-oxygen and hypoxic conditions (2, 32). A recent study demonstrated that AMPK-regulated NAPDH homeostasis during energy stress is critical to anchorage-independent growth (22).
To assess whether AMPK activity is necessary for KSR2-mediated anchorage-independent growth, AMPK-CA was expressed in MIN6 and NG108-15 cells depleted of KSR2 and the cells were assayed for their ability to grow in an anchorage-independent manner by the use of two distinct methods. Growth on poly(HEMA)-coated plates was used for a quick and quantitative measurement of anchorage-independent viability (13) in addition to the standard, long-term assay, soft-agar colony formation. As expected, KSR2 knockdown reduced the ability of MIN6 (Fig. 8A) and NG108-15 (Fig. 8B) cells to grow in an anchorage-independent manner, whereas the growth phenotype was rescued by expression of AMPK-CA. To further assess the importance of AMPK activity for anchorage-independent growth, a dominant-negative form of AMPK (AMPK-KD) (41) was expressed in MIN6 and NG108-15 cells expressing the nontargeting short hairpin. The importance of AMPK activity to cell transformation was demonstrated by the ability of AMPK-KD to impede anchorage-independent viability in a manner similar to that seen with KSR2 depletion (Fig. 8A and B). Complementing the results observed with the poly(HEMA) assay, KSR2 knockdown suppressed the ability of MIN6 (Fig. 8C) and NG108-15 (Fig. 8D) cells to form colonies in soft agar. AMPK-CA restored the ability of KSR2-depleted cells to form colonies in soft agar, and colonies were similar in size to those observed in control cells. Expression of AMPK-KD in MIN6 cells (Fig. 8C) and NG108-15 cells (Fig. 8D) diminished the ability of the cells to form colonies in soft agar. These data indicate that KSR2 mediates anchorage-independent growth through AMPK regulation.
Fig 8.

AMPK activity is required for KSR2-mediated anchorage-independent growth. MIN6 (A) and NG108-15 (B) cells expressing the nontargeting short hairpin, the KSR2-specific short hairpin, the KSR2-specific short hairpin and the constitutively active AMPKα2 (AMPKα-CA), or the nontargeting short hairpin and dominant-negative AMPKα2 (AMPKα-KD) were seeded in poly(HEMA)-coated plates, and the ability to grow in an anchorage-independent manner was examined by assessing cell viability using CellTiter-Glo at the indicated time points. Data representative of at least three independent experiments are illustrated as means ± standard errors of the means of the results determined for triplicate samples. Additionally, anchorage-independent growth of MIN6 (C) and NG108-15 (D) cells was examined after plating in soft agar. Data are illustrated as the number of colonies per 5 × 103 cells present after 4 weeks (C) and 2 weeks (D). The lower panel shows representative photomicrographs of colonies for each sample. Data are representative of at least three independent experiments and illustrated as means ± standard deviations of the results determined with triplicate samples. Immunoblots (IB) demonstrating the expression of the myc-tagged AMPKα constructs in MIN6 (E) and NG108-15 (F) cells are shown. *, P ≤ 0.01; **, P ≤ 0.001 (two-tailed Student's t test).
To further demonstrate the importance of AMPK signaling for anchorage-independent growth, MIN6 cells (Fig. 9A and C) and NG108-15 cells (Fig. 9B and D) were transfected with siRNA targeting the AMPKγ1 subunit alone or the AMPKα1 and AMPKα2 subunits in combination. Depletion of AMPKγ1 attenuated anchorage-independent growth in MIN6 (Fig. 9A) and NG108-15 (Fig. 9B) cells. Interestingly, AMPKγ1 appears necessary to stabilize expression of the α subunit, as a reduction in the expression levels of AMPKα1 and AMPKα2 was observed upon AMPKγ1 knockdown (Fig. 9C and D). Similar effects on anchorage-independent growth were observed due to simultaneous knockdown of the AMPKα1 and AMPKα2 catalytic subunits. These data indicate that AMPK signaling is necessary to mediate anchorage-independent viability and that KSR2 mediates anchorage-independent viability by promoting the activation of AMPK to supply the energy necessary for cells to sustain their tumorigenic potential.
Fig 9.
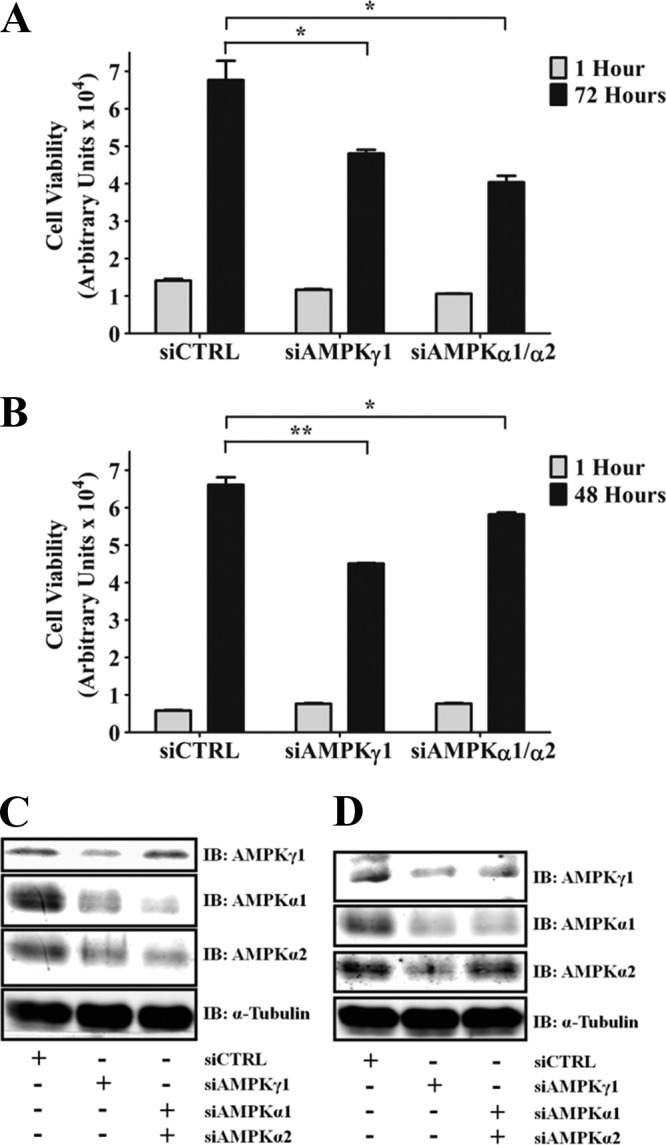
AMPK knockdown attenuates tumor cell anchorage-independent growth. MIN6 cells (A) and NG108-15 cells (B) were transfected with the indicated siRNA and seeded in poly(HEMA)-coated plates. Anchorage-independent viability was examined at the indicated time points using CellTiter-Glo. Data representative of the results of at least three independent experiments are illustrated as means ± standard errors of the means of triplicate samples. *, P ≤ 0.01; **, P ≤ 0.001 (two-tailed Student's t test). (C and D) Representative immunoblots (IB) denoting the expression level of the indicated proteins in MIN6 and NG108-15 cells, respectively.
DISCUSSION
Studies in C. elegans have demonstrated that KSR1 and KSR2 have distinct but overlapping developmental effects (46). Recent work has shown that KSR2, like KSR1, can function as a scaffold to promote ERK signaling (10). However, the phenotypes of KSR1 and KSR2 disruption in mammals are decidedly distinct. While KSR1−/− mice have subtle defects in hair follicle development and metabolism, they are fertile and otherwise normal and yet markedly resistant to Ras-driven tumor formation (27, 29, 35, 44). In contrast, KSR2−/− mice are infertile, obese, and insulin resistant and demonstrate defects in AMPK signaling (6). The current study assessed the role of KSR2 in cell proliferation and transformation. The data show that, independently of its role in ERK scaffold function and ERK signaling, KSR2 alone can induce anchorage-independent growth and also synergize with RasV12 to enhance transformation in MEFs. KSR2 also facilitates the activation of AMPK, which is essential to nutrient metabolism and the maximal glycolytic and OXPHOS capacity of tumor cells.
The use of KSR1−/− MEFs, which do not express KSR2 (29), revealed overlapping and distinct functions of KSR1 and KSR2. MEFs are exquisitely sensitive to the presence of KSR2. Nine times more KSR1 than KSR2 is required to mediate comparable growth factor-induced ERK activation. Expressing KSR2 at levels similar to those of KSR1 inhibits ERK activation. These data suggest that KSR2 may be more potent than KSR1 in promulgating transmission of signals from Raf to MEK and from MEK to ERK. Recent dissection of KSR1 regulation suggests possible explanations for the increased sensitivity of KSR2 in ERK activation. First, compared to KSR1, KSR2 may be more readily or efficiently poised to translocate to the plasma membrane and assemble the kinase cascade in response to growth factor stimulation (10, 38, 43, 49). Second, ERK-dependent feedback loops that negatively regulate KSR1-scaffolded complexes may not be as effective in complexes scaffolded by KSR2 (38). Third, KSR2 may be more efficient than KSR1 in its ability to process MEK-dependent ERK phosphorylation.
Previous data showed a direct, dose-dependent correlation between KSR1 expression, ERK activity, cell proliferation, and RasV12 transformation (31). Moreover, RasV12 transformation demonstrates a strict dependence upon the interaction of KSR1 with ERK (30, 31). With MEFs expressing KSR2, however, the coordination between ERK activation, proliferation, and transformation is disrupted. Although growth factor-induced ERK activity was suppressed by the high levels of KSR2 expression, these cells grew at higher rates than control MEFs and MEFs expressing low levels of KSR2. KSR2.C907Y, which disrupts the interaction of KSR2 with MEK, mediates growth factor-induced ERK activation but does not enhance cell proliferation. Moreover, only the high levels of KSR2 expression, which reduce ERK activation, contribute to RasV12-induced transformation. The conclusion that KSR2-dependent ERK activation is dispensable for KSR2-dependent cell proliferation and transformation is bolstered by the demonstration that disrupting the interaction of KSR2 with ERK did not alter the biological action of KSR2. Furthermore, KSR2 or KSR1 knockdown in MIN6 and NG108-15 cells reduced cell proliferation, but only KSR1 knockdown affected growth factor-mediated ERK activation. These data show that KSR2 mediates its effect on cell proliferation differently from KSR1 (30, 31, 44) despite their strong homology and ability to activate the same kinase cascade. Examination of ksr genes in C. elegans revealed that ksr1 and ksr2 have unique and redundant functions in mediating Ras-dependent development (46). Despite its exclusive contribution to germ line meiotic progression in nematodes, ksr2 function was presumed to be the result of its control over ERK phosphorylation in oocytes because ksr2 disruption caused acute disruption of mpk-1 activation. Manipulation of mammalian cells shows that KSR2 may contribute to Ras signaling via previously unrecognized ERK-independent effects on metabolism.
A role of KSR2 in normal metabolism in KSR2−/− mice was recently identified (6). KSR2 disruption in mice impaired AMPK activation, AMPK-regulated fatty acid oxidation, and thermogenesis, promoting obesity. AMPK activation was also affected by KSR2 knockdown in MIN6 (Fig. 5A) and NG108-15 tumor cell lines (6). These observations run counter to the concept that AMPK promotes catabolism at the expense of anabolic events common to continuously dividing tumor cells (16). AMPK is a regulator of glycolysis and OXPHOS in nontumor tissues, promoting glycolysis via phosphorylation of PFK-2 and mitochondrial metabolism by mediating the activation of PGC-1α (36, 37, 53). AMPK mediates the production of NADPH (22), a biosynthetic intermediate required for the reduction of reactive oxygen species and synthesis of nucleic acids and lipids (8, 26). The current data suggest a prominent role for AMPK in marshalling the nutrients and biosynthetic intermediates necessary for sustained macromolecular biosynthesis and ATP generation required in continuously proliferating tumor cells. Therefore, it is conceivable that the depletion of KSR2 in tumor cells affects these AMPK-mediated processes. Further studies should determine the role of KSR2 in the production of these biosynthetic intermediates and its dependence on AMPK.
KSR2 regulation of tumor cell metabolism is not dependent on the Ras oncogene. The MIN6 cell line was established using SV40 small T antigen, and the NG108-15 cell line was created by a fusion of the N18TG-2 neuroblastoma and C6 BU-1 glioma (25, 40). Ras mutations have not been detected in these cell lines, and yet KSR2 is required for their anchorage-independent growth and metabolic capacity. KSR1 also retains the capacity for ERK-independent increases in glycolytic and OXPHOS capacity through ERK-independent induction of PGC-1α and ERRα. Expression of PGC-1α and ERRα is essential to H-RasV12-dependent anchorage-independent growth (12). The extent to which KSR1 depends on AMPK for the expression of PGC-1α and ERRα is not yet known. However, while proteomic analysis identified the interaction of KSR2 with all three subunits of the AMPK complex, endogenous AMPK was not detected in KSR1 immunoprecipitates (6, 10). Thus, the mechanisms through which these scaffolds affect tumor cell metabolism may differ.
The depletion of KSR2 affected the rates of glycolysis, fatty acid oxidation, and glutaminolysis, all of which were restored with the expression of AMPK-CA. The reduced glycolytic and OXPHOS capacity and the lower rates of nutrient metabolism reduced cell proliferation in MIN6 and NG108-15 cells. Thus, while KSR2 is a scaffold for the Raf/MEK/ERK signaling pathway (10), KSR2 can promote cell proliferation and the transformed phenotype by regulating AMPK activity and cellular metabolism. These data suggest that KSR2 is behaving as a signaling node that can respond to mitogenic signaling while ensuring the availability of a proper energy supply. The identification of KSR1 and KSR2 as regulators of cell metabolism may provide an opportunity to target both mitogenic and metabolic signaling pathways essential to tumor growth and survival.
ACKNOWLEDGMENTS
We thank members of the Lewis laboratory for their constructive comments. We thank Deanna Volle and Kurt Fisher for technical assistance. We thank Charles Kuszynski, Linda Wilke, and Victoria Smith in the UNMC Cell Analysis Facility for their technical expertise in the generation of GFP-expressing cell lines.
This research was supported by NIH grant R01 CA90040 (R.E.L.). M.R.F. was supported by the NCI Minority Supplement through NIH grant R01 CA90040, the Skala Fellowship, and training grant T32 CA09476 to the Eppley Institute for Research in Cancer and Allied Diseases.
Footnotes
Published ahead of print 16 July 2012
REFERENCES
- 1. Abe Y, et al. 2010. Bioenergetic characterization of mouse podocytes. Am. J. Physiol. Cell Physiol. 299:C464–C476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buzzai M, et al. 2005. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 24:4165–4173 [DOI] [PubMed] [Google Scholar]
- 3. Cacace AM, et al. 1999. Identification of constitutive and ras-inducible phosphorylation sites of KSR: implications for 14-3-3 binding, mitogen-activated protein kinase binding, and KSR overexpression. Mol. Cell. Biol. 19:229–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Choi SW, Gerencser AA, Nicholls DG. 2009. Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure. J. Neurochem. 109:1179–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Corton JM, Gillespie JG, Hawley SA, Hardie DG. 1995. 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229:558–565 [DOI] [PubMed] [Google Scholar]
- 6. Costanzo-Garvey DL, et al. 2009. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab. 10:366–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davies SP, Sim AT, Hardie DG. 1990. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur. J. Biochem. 187:183–190 [DOI] [PubMed] [Google Scholar]
- 8. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. 2008. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7:11–20 [DOI] [PubMed] [Google Scholar]
- 9. Denouel-Galy A, et al. 1998. Murine Ksr interacts with MEK and inhibits Ras-induced transformation. Curr. Biol. 8:46–55 [DOI] [PubMed] [Google Scholar]
- 10. Dougherty MK, et al. 2009. KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol. Cell 34:652–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ferrick DA, Neilson A, Beeson C. 2008. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov. Today 13:268–274 [DOI] [PubMed] [Google Scholar]
- 12. Fisher KW, Das B, Kortum RL, Chaika OV, Lewis RE. 2011. Kinase suppressor of ras 1 (KSR1) regulates PGC1alpha and estrogen-related receptor alpha to promote oncogenic Ras-dependent anchorage-independent growth. Mol. Cell. Biol. 31:2453–2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fukazawa H, Mizuno S, Uehara Y. 1995. A microplate assay for quantitation of anchorage-independent growth of transformed cells. Anal. Biochem. 228:83–90 [DOI] [PubMed] [Google Scholar]
- 14. Gaglio D, Soldati C, Vanoni M, Alberghina L, Chiaradonna F. 2009. Glutamine deprivation induces abortive s-phase rescued by deoxyribonucleotides in k-ras transformed fibroblasts. PLoS One 4:e4715 doi:10.1371/journal.pone.0004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gwinn DM, et al. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30:214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hardie DG. 2007. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8:774–785 [DOI] [PubMed] [Google Scholar]
- 17. Hsu PP, Sabatini DM. 2008. Cancer cell metabolism: Warburg and beyond. Cell 134:703–707 [DOI] [PubMed] [Google Scholar]
- 18. Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. 2001. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem. Biophys. Res. Commun. 287:562–567 [DOI] [PubMed] [Google Scholar]
- 19. Inoki K, Zhu T, Guan KL. 2003. TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590 [DOI] [PubMed] [Google Scholar]
- 20. Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. 1999. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 13:163–175 [PMC free article] [PubMed] [Google Scholar]
- 21. Jäger S, Handschin C, St-Pierre J, Spiegelman BM. 2007. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. U. S. A. 104:12017–12022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jeon SM, Chandel NS, Hay N. 2012. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485:661–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jones RG, et al. 2005. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18:283–293 [DOI] [PubMed] [Google Scholar]
- 24. Kaelin WG, Jr, Thompson CB. 2010. Q&A: cancer: clues from cell metabolism. Nature 465:562–564 [DOI] [PubMed] [Google Scholar]
- 25. Klee WA, Nirenberg M. 1974. A neuroblastoma times glioma hybrid cell line with morphine receptors. Proc. Natl. Acad. Sci. U. S. A. 71:3474–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kletzien RF, Harris PK, Foellmi LA. 1994. Glucose-6-phosphate dehydrogenase: a “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J. 8:174–181 [DOI] [PubMed] [Google Scholar]
- 27. Klutho PJ, Costanzo-Garvey DL, Lewis RE. 2011. Regulation of glucose homeostasis by KSR1 and MARK2. PLoS One 6:e29304 doi:10.1371/journal.pone.0029304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kornfeld K, Hom DB, Horvitz HR. 1995. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 83:903–913 [DOI] [PubMed] [Google Scholar]
- 29. Kortum RL, et al. 2005. The molecular scaffold kinase suppressor of Ras 1 (KSR1) regulates adipogenesis. Mol. Cell. Biol. 25:7592–7604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kortum RL, et al. 2006. The molecular scaffold kinase suppressor of Ras 1 is a modifier of RasV12-induced and replicative senescence. Mol. Cell. Biol. 26:2202–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kortum RL, Lewis RE. 2004. The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells. Mol. Cell. Biol. 24:4407–4416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kumar SH, Rangarajan A. 2009. Simian virus 40 small T antigen activates AMPK and triggers autophagy to protect cancer cells from nutrient deprivation. J. Virol. 83:8565–8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laderoute KR, et al. 2006. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol. Cell. Biol. 26:5336–5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Levine AJ, Puzio-Kuter AM. 2010. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330:1340–1344 [DOI] [PubMed] [Google Scholar]
- 35. Lozano J, et al. 2003. Deficiency of kinase suppressor of Ras1 prevents oncogenic ras signaling in mice. Cancer Res. 63:4232–4238 [PubMed] [Google Scholar]
- 36. Marsin AS, et al. 2000. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 10:1247–1255 [DOI] [PubMed] [Google Scholar]
- 37. Marsin AS, Bouzin C, Bertrand L, Hue L. 2002. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J. Biol. Chem. 277:30778–30783 [DOI] [PubMed] [Google Scholar]
- 38. McKay MM, Ritt DA, Morrison DK. 2009. Signaling dynamics of the KSR1 scaffold complex. Proc. Natl. Acad. Sci. U. S. A. 106:11022–11027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Merrill GF, Kurth EJ, Hardie DG, Winder WW. 1997. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol. 273:E1107–E1112 [DOI] [PubMed] [Google Scholar]
- 40. Miyazaki J, et al. 1990. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127:126–132 [DOI] [PubMed] [Google Scholar]
- 41. Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. 2001. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol. Cell 7:1085–1094 [DOI] [PubMed] [Google Scholar]
- 42. Mukherjee P, et al. 2008. Differential effects of energy stress on AMPK phosphorylation and apoptosis in experimental brain tumor and normal brain. Mol. Cancer 7:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Müller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK. 2001. C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol. Cell 8:983–993 [DOI] [PubMed] [Google Scholar]
- 44. Nguyen A, et al. 2002. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol. Cell. Biol. 22:3035–3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oakhill JS, et al. 2011. AMPK is a direct adenylate charge-regulated protein kinase. Science 332:1433–1435 [DOI] [PubMed] [Google Scholar]
- 46. Ohmachi M, et al. 2002. C. elegans ksr-1 and ksr-2 have both unique and redundant functions and are required for MPK-1 ERK phosphorylation. Curr. Biol. 12:427–433 [DOI] [PubMed] [Google Scholar]
- 47. Park HU, et al. 2009. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol. Cancer Ther. 8:733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Phoenix KN, Vumbaca F, Claffey KP. 2009. Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERalpha negative MDA-MB-435 breast cancer model. Breast Cancer Res. Treat. 113:101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Razidlo GL, Kortum RL, Haferbier JL, Lewis RE. 2004. Phosphorylation regulates KSR1 stability, ERK activation, and cell proliferation. J. Biol. Chem. 279:47808–47814 [DOI] [PubMed] [Google Scholar]
- 50. Ros S, et al. 2012. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2:328–343 [DOI] [PubMed] [Google Scholar]
- 51. Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. 2007. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 403:139–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stein SC, Woods A, Jones NA, Davison MD, Carling D. 2000. The regulation of AMP-activated protein kinase by phosphorylation. Biochem. J. 345(Pt 3):437–443 [PMC free article] [PubMed] [Google Scholar]
- 53. Steinberg GR, Kemp BE. 2009. AMPK in health and disease. Physiol. Rev. 89:1025–1078 [DOI] [PubMed] [Google Scholar]
- 54. Stewart S, et al. 1999. Kinase suppressor of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol. Cell. Biol. 19:5523–5534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sundaram M, Han M. 1995. The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell 83:889–901 [DOI] [PubMed] [Google Scholar]
- 56. Suwa M, Nakano H, Kumagai S. 2003. Effects of chronic AICAR treatment on fiber composition, enzyme activity, UCP3, and PGC-1 in rat muscles. J. Appl. Physiol. 95:960–968 [DOI] [PubMed] [Google Scholar]
- 57. Therrien M, et al. 1995. KSR, a novel protein kinase required for RAS signal transduction. Cell 83:879–888 [DOI] [PubMed] [Google Scholar]
- 58. Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Weinberg F, Chandel NS. 2009. Mitochondrial metabolism and cancer. Ann. N. Y. Acad. Sci. 1177:66–73 [DOI] [PubMed] [Google Scholar]
- 60. Wu M, et al. 2007. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 292:C125–C136 [DOI] [PubMed] [Google Scholar]
- 61. Xiao B, et al. 2011. Structure of mammalian AMPK and its regulation by ADP. Nature 472:230–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xing H, Kornfeld K, Muslin AJ. 1997. The protein kinase KSR interacts with 14-3-3 protein and Raf. Curr. Biol. 7:294–300 [DOI] [PubMed] [Google Scholar]
- 63. Yu W, Fantl WJ, Harrowe G, Williams LT. 1998. Regulation of the MAP kinase pathway by mammalian Ksr through direct interaction with MEK and ERK. Curr. Biol. 8:56–64 [DOI] [PubMed] [Google Scholar]
- 64. Zong H, et al. 2002. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. U. S. A. 99:15983–15987 [DOI] [PMC free article] [PubMed] [Google Scholar]