

Abstract
ThiI has been identified as an essential enzyme involved in the biosynthesis of thiamine and the tRNA thionucleoside modification, 4-thiouridine. In Escherichia coli and Salmonella enterica, ThiI acts as a sulfurtransferase, receiving the sulfur donated from the cysteine desulfurase IscS and transferring it to the target molecule or additional sulfur carrier proteins. However, in Bacillus subtilis and most species from the Firmicutes phylum, ThiI lacks the rhodanese domain that contains the site responsible for the sulfurtransferase activity. The lack of the gene encoding for a canonical IscS cysteine desulfurase and the presence of a short sequence of ThiI in these bacteria pointed to mechanistic differences involving sulfur trafficking reactions in both biosynthetic pathways. Here, we have carried out functional analysis of B. subtilis thiI and the adjacent gene, nifZ, encoding for a cysteine desulfurase. Gene inactivation experiments in B. subtilis indicate the requirement of ThiI and NifZ for the biosynthesis of 4-thiouridine, but not thiamine. In vitro synthesis of 4-thiouridine by ThiI and NifZ, along with labeling experiments, suggests the occurrence of an alternate transient site for sulfur transfer, thus obviating the need for a rhodanese domain. In vivo complementation studies in E. coli IscS- or ThiI-deficient strains provide further support for specific interactions between NifZ and ThiI. These results are compatible with the proposal that B. subtilis NifZ and ThiI utilize mechanistically distinct and mutually specific sulfur transfer reactions.
INTRODUCTION
Posttranscriptional modifications in tRNA are widely represented in all three domains of life (21). In particular, the incorporation of new chemical functionalities to tRNA allows additional intra- and intermolecular interactions that support proper folding to more thermodynamically stable conformations, that guarantee fidelity in translation, and that activate their ability to sense environmental and nutritional changes (1, 17, 31). Nearly a hundred tRNA modifications have been described (22); however, the exact inventory of covalent modifications is not fully established. Even less understood are the identity and mechanisms of modifying enzymes, as well as the impact of these modifications on the various cellular functions performed by individual tRNA molecules.
One of the best characterized tRNA modifications is the C-4 thiolation of the uridine base at position 8 of tRNA (s4U8) (5, 8, 10, 17). This thionucleoside acts as a photosensor of near-UV radiation (18). The absorption of UV light induces a photochemical cross-linking cycloaddition reaction between s4U8 and the cytosine at position 13 of tRNA. This event induces a conformational change that prevents aminoacylation of tRNA molecules, resulting in an accumulation of uncharged tRNA ultimately leading to the stringent response (4). In Escherichia coli, the UV-induced stringent response, manifested as growth delay, is disrupted in strains lacking s4U8 modification. Genetic studies mapped thiI, an essential gene involved in thiamine biosynthesis (29), as the s4U8 biosynthetic and UV-photoprotector gene (18, 29).
In E. coli, the formation of s4U8 is performed by ThiI in conjunction with the cysteine desulfurase IscS (7). This tRNA modification begins with the conversion of cysteine to alanine by IscS with concomitant formation of a cysteine persulfide bond at the enzyme's active-site cysteine residue. The next step consists of a persulfide sulfur transfer from IscS to the Cys456 residue of ThiI and the activation of C-4 of the uridine base at position 8 of tRNA, involving a ThiI-dependent adenylation reaction (32). The proposed catalytic cycle is completed upon transfer of the persulfide sulfur from Cys456 to form s4U8 with the concomitant release of AMP and formation of a disulfide bond between Cys344 and Cys456 (19, 20).
Structural and amino acid sequence analyses show that E. coli and Salmonella enterica ThiI proteins contain three distinct domains: a THUMP domain involved in tRNA binding, a PP-loop pyrophosphatase domain likely involved in the formation of the tRNA adenylated-uridine intermediate, and the rhodanese domain (Rhd domain), which provides the sulfurtransferase site Cys456. In vivo and in vitro data points to the participation of all three domains in the formation of s4U8 (11, 20, 28). A recent report by the Downs' group has demonstrated that, in S. enterica, the Rhd domain alone is sufficient for the sulfur incorporation into the thiazole moiety of thiamine (14). This discovery led to the proposal that ThiI is a bifunctional enzyme carrying distinct tRNA adenylation and sulfurtransferase activities. Interestingly, a subset of species coding for sequences similar to E. coli and S. enterica thiI lacks the Rhd domain (28), thus raising the question of how these species synthesize s4U8 and thiamine.
We have investigated the role of Bacillus subtilis ThiI and NifZ in the biosynthesis of s4U8 and thiamine. In this organism and several other bacteria, the thiI gene encodes for a protein lacking the Rhd domain while retaining the THUMP, the PP-loop pyrophosphatase domains and a Cys residue at an equivalent position of the Cys344 in E. coli ThiI. NifZ is a cysteine desulfurase, which potentially serves as the initial sulfur donor in these pathways. Interestingly, the nifZ gene coding sequence is located four bases upstream from thiI.
In this report, we demonstrate that NifZ and ThiI are sufficient and crucial for the formation of s4U8 modification in tRNA, while their inactivation does not impose a requirement for thiamine upon this bacterium. In addition, in vivo and in vitro experiments described here support the model for NifZ and ThiI interactions in the s4U8 pathway involving a specific sulfur transfer reaction.
MATERIALS AND METHODS
Media, medium additions, and chemicals.
LB medium was used with the following concentrations of antibiotics and media additives, unless otherwise specified: ampicillin (100 μg/ml), kanamycin (40 μg/ml), erythromycin (0.5 μg/ml), X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; 40 μg/ml), IPTG (isopropyl-β-d-thiogalactopyranoside; 10 μg/ml), l-arabinose (2 mg/ml). MS-I medium consists of 17.5 mM NH4SO4, 80.3 mM K2HPO4, 44.1 mM KH2PO4, 3.4 mM trisodium citrate dehydrate, 0.81 mM MgSO4, 0.5% glucose, 0.02% Casamino Acids, and 0.1% yeast extract. MS-II medium consists of MS-I medium with 0.125 mM MgSO4 and 0.0125 mM CaCl2. Spizizen's minimal medium was prepared as previously described (2). Unless specified, all chemicals were purchased from Fisher Scientific and Sigma-Aldrich Inc. Restriction enzymes were purchased from New England BioLabs.
Plasmid construction.
All genes were amplified from B. subtilis PS832 strain genomic DNA prepared by using a commercial DNA extraction kit (QuickExtract; Epicentre). PCR amplification reactions were performed using Fail Safe PCR kit (Epicentre). The PCR products were all previously cloned into TopoTA vector (Invitrogen) for subcloning purposes. The correct sequence of all plasmids used in the present study was confirmed by DNA sequencing (Wake Forest DNA Sequencing Laboratory). A comprehensive list and description of primers and plasmids used in the present study are shown in Table S1 in the supplemental material.
B. subtilis strain construction.
All strains of B. subtilis listed in Table S1 in the supplemental material were derivatives of strain 168 (PS832 strain). Transformation protocol was adapted from previously described (2): B. subtilis PS832 was grown on 2× SG plates for 15 h at 30°C, yielding individual colonies. A single colony was inoculated in 5 ml of MS-I medium at 37°C for 4.5 h. A 10-fold dilution of the “precompetent” cell culture was prepared in 5 ml of MS-II medium and incubated with shaking at 37°C for 1.5 h. Plasmid pDS54 or pDS19 (50 μl) was added to 300 μl of competent cells and incubated with shaking at 37°C for 30 min. Transformed cells were selected for erythromycin resistance and blue/white selection in the presence of X-Gal and IPTG.
Detection of thiI transcript from total RNA samples.
Total RNA purification from B. subtilis strains PS832, DD5, and DD14 grown in the absence or presence of 500 μM IPTG was performed according to the Qiagen RNeasy Kit bacteria protocol, using RNAprotect Bacteria Reagent (Qiagen) and DNase I (Qiagen). Reverse transcription-PCRs (RT-PCRs), using SuperScript III One-Step RT-PCR system with Platinum Taq DNA polymerase (Invitrogen), were performed to amplify thiI with BsthiI-PciI5 and BsthiI-BglII3 primers, sufC with BssufC-HindIII3 and BssufC-BamHI5 primers and suf promoter region with Bssufpro5 and Bssufpro3 primers. RT-PCR products were visualized by agarose ethidium bromide gel electrophoresis.
Thiamine feeding experiments.
B. subtilis wild-type PS832 and DD14 (with or without 500 μM IPTG) and 1A603 strains were inoculated at an initial optical density at 600 nm (OD600) of 0.01 in Spizizen minimal medium (2) in the presence or absence of 0.8 μg of thiamine/ml. Cultures were grown at 300 rpm at 37°C for 14 h.
Sporulation experiments.
B. subtilis strains PS832 and DD14 were inoculated in 5 ml of LB medium at an initial OD600 of 0.01 and incubated at 37°C and 300 rpm. Aliquots (50 μl) were removed at time points between 10 and 24 h. Serial dilutions (10−1 to 10−8) of each aliquot were spotted (5 μl drops) on LB agar plates. Dilutions were incubated at 65°C for 20 min and spotted onto the same LB agar plates. The relative log of spores from heated samples was compared to the total number of viable cells.
B. subtilis PS832 and DD14 sporulation experiments were performed without stress and under UV radiation, iron, and oxidative stress conditions. Cultures at an OD600 of 0.5 were exposed to UV radiation of 350 nm for 30 min. Aliquots were taken at 1, 4, and 16 h after exposure. Cultures were challenged with 1, 1.5, 2, 10, or 100 mM Fe(NO3)3 or 1 mM H2O2 added to the LB medium.
Isolation of tRNA from B. subtilis and E. coli cells.
B. subtilis wild-type strain PS832 was grown in 500 ml LB medium. B. subtilis strains DD5 and DD14 in the presence or absence of 500 μM IPTG were grown in 500 ml of LB medium with erythromycin at 37°C to an OD600 of 1.2–1.5. E. coli strain CL100 transformed with pAra13, pDS21, or pDS115 and E. coli strain JLD26501 transformed with pAra13, pDS108, or pDS115 were grown in LB medium containing ampicillin and arabinose at 37°C to OD600 of 1.2 to 1.5. Cells were harvested by centrifugation at 8,200 × g for 10 min and frozen at −20°C until further use.
Frozen cells were resuspended in 10 ml of 0.3 M sodium acetate–10 mM EDTA (pH 4.5), after which 3.5 ml of Tris-saturated phenol was added. The solution was vortexed for 1 min and after 30 s was vortexed again for 1.5 min and then centrifuged at 3,214 × g for 15 min. The upper layer was transferred to a clean tube, and 3.5 ml of Tris-saturated phenol was added. The resulting solution was vortexed for 1 min and centrifuged at 3,214 × g for 15 min. The upper layer was transferred to a clean tube, followed by addition of 0.5 g of NaCl and 10 ml of chloroform-isoamyl alcohol (24:1). The mixture was vortexed for 5 s and centrifuged at 3,214 × g for 3 min. The upper portion was removed to a clean tube, filled to 50 ml with 100% cold ethanol, and stored at −20°C for 2 h to overnight. The solution was centrifuged at 3,214 × g for 20 min, and the pellet was resuspended in 1.5 ml of 0.3 M sodium acetate–10 mM EDTA (pH 4.5). The solution was vortexed for 3 s, heated at 90°C for 5 min, and centrifuged at 15,700 × g for 5 min. The A260 was measured, and a spectrum from 200 to 800 nm was recorded. An aliquot of the sample was diluted to 500 μl with 0.3 M sodium acetate–10 mM EDTA (pH 4.5) to an equivalent A260 of 40 and then filled to a volume of 1.5 ml with cold ethanol (100%). The sample was stored at −20°C until further use.
Synthesis and isolation of synthetic tRNA.
E. coli tRNAMet was synthesized in vitro as previously described (24) using the following protocol. The E. coli tRNAMet DNA template was synthesized in a 2-ml reaction containing 4 μM EctRNAMet5 and EctRNAMet3 primers, 400 μM deoxynucleoside triphosphates, 50 U of Klenow exo− DNA polymerase, and 1× buffer 2 from New England BioLabs. The reaction mixture was incubated for 20 cycles using a Thermocycler at 10°C for 10 s and 37°C for 30 s. Transcription reaction (8 ml) contained 2 ml of DNA template, 5 mM each nucleoside triphosphates, 20 mM spermidine, 40 mM dithiothreitol (DTT), 250 mM HEPES (pH 7.5), 30 mM MgCl2, 20 μg of bovine serum albumin, 40 μg of T7 RNA polymerase/ml, and 60 U of RNase inhibitor. The reaction was incubated while rotating at 37°C for 8 to 18 h. DNase (81 U) was added to the reaction, followed by 1.5 h of incubation at 37°C while rotating, and centrifuged for 20 min at 2,057 × g. Portions (8 ml) of 3 M ammonium acetate (pH 6) and 32 ml of isopropanol were added to the supernatant and stored at −20°C for 2 h to overnight. Solution was centrifuged for 30 min at 17,555 × g, and isopropanol was removed. Pellet was resuspended in 800 μl of RNase-free water, and 1:1 (vol/vol) of 90% formamide was added to the sample, followed by boiling for 5 min. tRNAMet was isolated using 12% acrylamide (19:1)–8 M urea gel electrophoresis with 100 mM Tris, 100 mM boric acid, and 2 mM EDTA buffer (1× TBE running buffer). Sample was purified from gel using an EluTrap electrophoresis chamber (Schleicher & Schuell) at 150 V. tRNA was precipitated by adding 1:10 (vol/vol) 3 M sodium acetate (pH 5.2) and 2:3 (vol/vol) 100% cold ethanol and stored overnight at −20°C. Each sample was centrifuged for 30 min at 17,555 × g, and the pellet was resuspended in 100 μl 20 mM HEPES (pH 7.5) and stored at −20°C.
In vitro generation of s4U8 in synthetic tRNA.
Assay reactions (150 μl) contained 100 μM l-cysteine, 4 μM NifZ or IscS, 4 μM ThiI, 100 μM synthetic E. coli tRNAMet, 1 mM ATP, and 5 mM MgCl2 in 50 mM Tris-HCl (pH 8). Reactions were incubated at 37°C for 2 h, adding 1 mM DTT after 1 h. To isolate the tRNA, samples were extracted with 45 μl of Tris-saturated phenol and precipitated with 1 ml of cold ethanol.
Analysis of modified tRNA nucleosides.
Unfractionated tRNA isolated from B. subtilis and E. coli cells, or synthetic tRNA from in vitro experiments was digested into individual nucleosides for analysis. Each solution was centrifuged at 15,700 × g for 20 min, and the pellet was resuspended in 150 μl of 40 mM sodium acetate–1 mM ZnCl2 (pH 5.2) and heated at 100°C for 5 min. Once cooled, 20 μl of nuclease P1 (1 mg/ml) was added, followed by incubation at 50°C for 3 h. Then, 20 μl of 1 M Tris (pH 8), 10 μl of shrimp alkaline phosphatase (Thermo), and 10 μl of phosphatase buffer were added, followed by incubation at 37°C for 1.5 h. The sample was centrifuged at 15,700 × g for 5 min and analyzed by HPLC using electron spray ionization mass spectrometry and/or UV detector.
Digested tRNA samples were analyzed by HPLC using a C-18 column (Supelcosil), coupled with a mass spectrometer and diode array detector (1100 Series LC/MSD Trap; Agilent) or a UV/Vis detector (Waters). For analysis of modified tRNA transcripts, a short high-pressure liquid chromatography (HPLC) method was used; method A used Optima LC-MS grade water with 0.1% formic acid as solvent A and 40:60 water-acetonitrile as solvent B. The HPLC conditions for method A included a gradient of solvent B: 5% from 0 to 15 min, 75% at 25 min, 100% at 26 min, and 5% from 28 to 33 min at a flow rate of 0.75 ml/min with 20-μl injections. Analysis of modified tRNA from E. coli and B. subtilis cells were performed using a longer HPLC protocol. Method B used 0.1% (wt/vol) ammonium acetate (pH 5.3) as solvent A and 60:40 water-acetonitrile as solvent B. Method B included a gradient of solvent B: 2 to 5% from 0 to 10 min, 5 to 25% from 10 to 25 min, 25 to 50% from 25 to 30 min, 50 to 75% from 30 to 34%, and 75 to 100% from 34 to 40 min. The flow rate was kept at 0.5 ml/min with 10-μl injections. The diode array detector and UV/Vis detector monitored the UV absorbance at 260 and 330 nm. The mass spectrum was recorded in the negative mode under the following conditions: nebulizer pressure of 60 lb/in2, drying gas flow of 11 liters/min at 350°C, and capillary voltage of 3,500 V. Standards of s4U and pseudouridine (Berry & Associates) were run as controls.
Protein expression and purification.
Plasmid pDS21 containing nifZ was transformed into E. coli CL100 competent cells and selected for ampicillin resistance. Single colonies were inoculated in 3 liters of LB medium containing ampicillin and grown overnight (20 h) at 30°C in the presence of l-arabinose (0.2%). Cells were harvested by centrifugation at 8,200 × g for 10 min and resuspended (3 ml/g of cell [wet weight]) in 25 mM Tris-HCl (pH 8) (buffer A). Cells were lysed using an Emulsiflex C5 high-pressure homogenizer (Avestin) and centrifuged for 15 min at 12,857 × g. Supernatant was incubated with 1% (wt/vol) streptomycin on ice for 15 min and then centrifuged for 15 min at 12,857 × g. The supernatant was loaded using a fast-protein liquid chromatography (FPLC) system (AktaPurifier; GE Healthcare) onto a Q-Sepharose column (GE Healthcare) preequilibrated with buffer A. The column was washed with buffer A, and the sample was eluted at a flow rate of 2 ml/min with a 0 to 70% gradient of 25 mM Tris-HCl (pH 8)–1 M NaCl (buffer B) over 20 column volumes. The collected elutant was analyzed by SDS-PAGE, and the fractions eluting with 10 to 15% buffer B, containing the desired protein of 41.4 kDa, were pooled. Each pooled sample was treated with 40 and 50% saturating concentrations of ammonium sulfate with incubation on ice for 15 min, followed by centrifugation at 31,209 × g for 15 min after each step. The 50% ammonium sulfate pellet containing the desired protein was resuspended in 10 ml of 25 mM Tris-HCl (pH 8)–300 mM NaCl (buffer C) and loaded using an FPLC onto a Sephacryl S-200 column (2.5 by 60 cm; GE Healthcare) preequilibrated with buffer C at a flow rate of 1 ml/min. The elutant was analyzed by SDS-PAGE, and the desired fractions were pooled and pelleted in liquid nitrogen for storage at −80°C.
B. subtilis ThiI protein was expressed by transforming pDS108 into E. coli Arctic Express competent cells (Stratagene). Single colonies were inoculated in 3 liters of LB medium with ampicillin and grown to an OD600 of 0.5 at 25°C. Cell cultures were induced with l-arabinose (0.2%), grown overnight (16 h) at 15°C, and then harvested by centrifugation at 8,200 × g for 10 min. Cell pellets were resuspended (5 ml/g cells [wet weight]) in 25 mM Tris-HCl (pH 8)–20% glycerol (buffer D) and lysed using an Emulsiflex C5 high-pressure homogenizer (Avestin). Cell lysate was centrifuged for 15 min at 12,857 × g, and then the supernatant was treated with 1% (wt/vol) streptomycin on ice for 15 min, followed by centrifugation for 15 min at 12,857 × g. The soluble protein was treated with 50 and 70% saturating concentrations of ammonium sulfate and incubation on ice for 15 min, followed by centrifugation for 15 min at 31,209 × g after each step. The desired protein of 45.4 kDa was contained in the 70% ammonium sulfate pellet, which was resuspended in 10 ml of 25 mM Tris-HCl (pH 8)–300 mM NaCl–20% glycerol (buffer E) and loaded onto a Sephacryl S-200 column (2.5 by 60 cm; GE Healthcare) preequilibrated with buffer E. Fractions containing the desired protein were pooled and pelleted in liquid nitrogen for storage at −80°C.
Azotobacter vinelandii IscS was produced from E. coli BL21(DE3) cells containing pDB943 and isolated as previously described (33). E. coli ThiI was produced from E. coli BL21(DE3) and isolated through Ni-NTA chromatography as previously reported (18). All of the protein purifications were monitored by SDS-PAGE (9), and the protein concentrations were determined by the method of Bradford et al. (3) using a Bio-Rad protein assay kit and bovine serum albumin as the standard. Pyridoxal-5′-phosphate bound to NifZ and IscS was quantified as described previously (27).
Cysteine desulfurase activity.
Cysteine desulfurase activity was determined through formation of S2− and alanine as previously described (23). Assays were performed with fixed concentrations of NifZ (0.01 mg, 0.3 μM), varying the concentrations of l-cysteine (0.0125 to 0.5 mM), and in the presence or absence of 2 mM DTT and/or 6 μM ThiI.
35S-sulfur transfer assays.
Assay reactions (40 μl) contained 4.26 μM NifZ or IscS and 3.89 μM ThiI–1 mM ATP–5 mM MgCl2 in 50 mM Tris-HCl (pH 7.5) with 50 μM l-cysteine and 10 μCi of [35S]cysteine (Perkin-Elmer). Reactions were incubated at 37°C for 1 h and quenched with 0.4 mM N-ethylmaleimide for 5 min. Reactions were analyzed by nonreducing SDS-PAGE. Protein size and migration were observed by Coomassie brilliant blue staining and 35S-persulfurated proteins were visualized using a phosphorimager (Bio-Rad).
32P-adenylation assays.
B. subtilis ThiI and E. coli ThiI (10 μM) were incubated with 100 μM synthetic E. coli tRNAMet in 30 μl 50 mM Tris-HCl (pH 8)–1 mM MgCl2. The reaction was started with 10 μCi of [α-32P]ATP (Perkin-Elmer), followed by incubation at 37°C for 2 h. Reaction components were separated by 12% acrylamide–8 M urea gel electrophoresis in 1× TBE buffer. The migration pattern of tRNA was observed by UV after ethidium bromide straining, while the α-32P-adenylated tRNA intermediate was visualized using a phosphorimager (Bio-Rad).
RESULTS
Gene inactivation of nifZ and thiI in Bacillus subtilis.
The chromosomal location of thiI coding sequence is 4 bases downstream of the nifZ gene encoding for a cysteine desulfurase and 94 bases upstream of the sspB gene encoding for the small, acid-soluble spore protein (16). Transcript sequence analysis in Bacillus anthracis indicated that, along with nifZ and sspB, thiI is part of a polycistronic operon (13). Therefore, in order to avoid any potential polar effect on the downstream genes, pMutin integrational plasmid containing fragments of ′nifZ′ or ′nifZ were used in the generation of knockout strains. For the construction of B. subtilis strain DD14, pDS54, an internal ′nifZ′ fragment cloned into pMutin4 (25), was inserted into the chromosome, simultaneously disrupting nifZ expression and placing thiI under the control of pSPAC, an IPTG-inducible promoter. A similar strategy was used for construction of B. subtilis strain DD5; pMutin containing a ′nifZ fragment (pDS19) was inserted into B. subtilis chromosome placing thiI and sspB under the control of the pSAPC promoter.
In order to confirm that the strain DD14 was able to express thiI upon the addition of IPTG, RT-PCR analysis was performed on purified total RNA samples of strains PS832 and DD14. Amplification of cDNA generated from the RNA samples was used to determine the presence of thiI transcripts in the wild type and the DD14 samples in the presence or absence of IPTG (Fig. 1). The sufC gene amplification was used as an internal positive control (Fig. 1, lanes 2, 4, and 6), while suf promoter region amplification was used as negative control of any potential DNA contamination in purified total RNA samples (data not shown). As shown in the gels from RT-PCR experiments (Fig. 1), B. subtilis strain DD14 only expresses ThiI when cultured with 0.5 mM IPTG.
Fig 1.
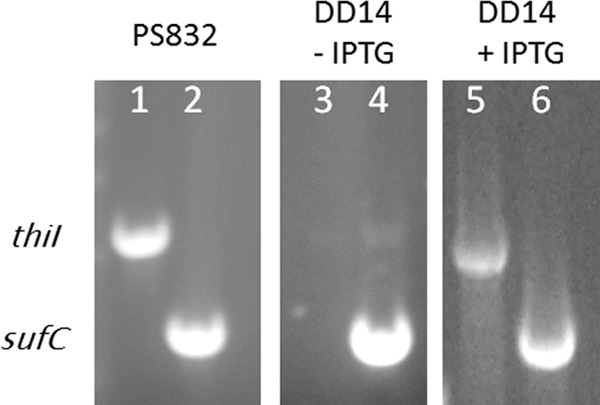
IPTG-dependent expression of ThiI in the DD14 strain. B. subtilis PS832 wild-type (lanes 1 and 2) and DD14 (lanes 3 to 6) were cultured in LB in the absence (lanes 1 to 4) or presence (lanes 5 and 6) of IPTG. Total RNA was extracted and used in RT-PCR analysis of thiI (lanes 1, 3, and 5) and control sufC (lanes 2, 4, and 6) transcripts.
B. subtilis strains lacking nifZ and/or thiI display a specific disruption in the s4U8 synthesis.
The analysis of modified nucleosides from isolated tRNA samples was performed using HPLC-MS and/or UV detection. B. subtilis strains PS832, DD5, and DD14 were cultured in the absence or in the presence of 0.5 mM IPTG. The samples were analyzed for the presence of s4U by monitoring the A330. The peak associated with s4U in these samples was verified by comparison to s4U standard and by the observance of the 259 M/Z−, corresponding to the mass of s4U-H (see Fig. S1 in the supplemental material). For every sample, the intensity of s4U signal was normalized to the intensity of the pseudouridine (ψ) peak, which was used as an internal control. Cells lacking nifZ and/or thiI displayed dramatically lower levels of s4U (Table 1; see Fig. S1 in the supplemental material), thus indicating the participation of both proteins in this thiolation pathway.
Table 1.
tRNA modification levels in B. subtilis strains
| Strain | Medium additive | Relevant genotype | s4U/ψ (SD)a | Relative level (%) |
|---|---|---|---|---|
| PS832 | None | nifZ+ thiI+ | 0.117 (0.017) | |
| DD5 | None | nifZ+ ΔthiI | 0.008 (0.002) | 7 |
| DD5 | IPTG | nifZ+ thiI+ | 0.113 (0.012) | 96 |
| DD14 | None | ΔnifZ ΔthiI | 0.012 (0.007) | 10 |
| DD14 | IPTG | ΔnifZ thiI+ | 0.014 (0.001) | 11 |
That is, the absorbance ratio of the s4U peak area from the A330 chromatogram to the ψ peak area from A260.
In B. anthracis and B. subtilis, genetic defects influencing the stringent response pathway can be manifested by defects in sporulation (26). Because of the association between UV-dependent stringent response and s4U8 tRNA in E. coli (18), we hypothesized that the lack of s4U8 could lead to defects on spore formation or spore resistance to UV radiation. Interestingly, the sspB gene located downstream of thiI is involved in spore resistance to heat and UV radiation (16). The spore count was estimated on cultures exposed to UV radiation, iron, and H2O2 challenges. Under these conditions, the pattern of sporulation of B. subtilis strain DD14 was affected but not significantly different than the wild-type (data not shown). We have also not detected any differences on spores isolated from cells lacking NifZ and/or ThiI to survive upon exposure to UV radiation or heat.
In addition to the s4U8 synthesis in E. coli, IscS and ThiI also participate in sulfur mobilization during the formation of the thiazole moiety in the biosynthesis of thiamine (6). Strains lacking IscS or ThiI do not synthesize s4U8, and they are thiamine auxotrophs (12, 18, 29). In B. subtilis, however, strain DD14 showed no growth defects when cultured in minimal medium in the absence of thiamine (see Fig. S2 in the supplemental material). Although this experimental approach does not completely rule out the involvement of NifZ and ThiI in sulfur transfer reactions for the synthesis of thiazole, it does provide an indication that these steps can be bypassed or substituted by other cellular components in vivo. For example, B. subtilis genome encodes for NifZ and three additional cysteine desulfurases (YrvO, NifS, and SufS) which could act as alternative sulfur donors in strains lacking NifZ, thus providing functional replacement in the initial sulfur transfer step in thiazole biosynthesis. The lack of a specific sulfur donor in thiazole biosynthesis is also manifested under certain growth conditions in S. enterica; the thiamine auxotrophy of a thiI mutant strain is suppressed in cysteine-enriched cultures (14).
ThiI is a sulfur acceptor of NifZ cysteine desulfurase.
The purified form of NifZ showed identical catalytic activity in the standard cysteine desulfurase assay for sulfide (Fig. 2) and alanine formation (see Fig. 4, left). The affinity of the enzyme for cysteine (KmCys = 29.7 ± 8.7 μM), was similar to the values determined for other cysteine desulfurases (15, 23, 30). The pH dependence activity profile (Fig. 2, inset) was suggestive that, at lower pH values, the rate-limiting step is controlled by the deprotonation of the active-site cysteine residue that performs the nucleophilic attack onto the substrate-activated thiol. The calculated pKa of 7.19 ± 0.09 for NifZ was slightly lower for a cysteine residue, but within the expected range of thiol-dependent enzymes.
Fig 2.

Activity profile of NifZ cysteine desulfurase. Substrate saturation curve of NifZ activity determined through quantification of sulfide production. Assays were performed with a fixed concentration of NifZ (0.01 mg, 0.3 μM) and various concentrations of l-cysteine in the presence of 2 mM DTT. Catalytic rates were calculated by the slopes of three-time point reactions. The saturation curve was a best fit for the Michaelis-Menten equation, determining the kinetic constants Km = 29.7 ± 8.7 μM and Vmax of 350 ± 26 nmol/min/mg. (Inset) The pH profile of NifZ activity demonstrates the pH dependence of cysteine desulfurase activity. Assays detecting sulfide were conducted with fixed concentrations of NifZ (0.01 mg, 0.3 μM), l-cysteine (0.5 mM), and DTT (2 mM) over a range of pHs. The line is a best fit for the Henderson-Hasselbalch equation resulting in a pKa of 7.19 ± 0.09.
Fig 4.

NifZ activity is enhanced by sulfur acceptor ThiI in nonreducing conditions. Alanine formation monitored over time upon incubation with NifZ (0.01 mg, 0.3 μM) in the absence (▲) or presence of 6 μM ThiI (■). (Left) The linear rate of alanine formation under reducing (2 mM DTT) conditions shows activity levels of 354 ± 12 nmol/min/mg (solid line, NifZ) and 262 ± 24 nmol/min/mg (dashed line, NifZ + ThiI). (Right) NifZ activity under nonreducing conditions shows a linear rate of alanine formation (80 ± 1.4 nmol/min/mg), while in the presence of ThiI, displays a biphasic reaction profile for alanine production. The reaction velocity associated with the first phase of the reaction (250 ± 9.0 nmol/min/mg) was faster than the rate calculated for the second phase of the reaction (84 ± 5.5 nmol/min/mg). The graphs show representative curves from individual experiments. The velocities were calculated from a linear fit of alanine formation over time for each reaction or distinct phase within each reaction. The specific activities indicated above are the average of at least three independent experiments.
SDS-PAGE analysis showed that in the presence of [35S]cysteine, NifZ becomes covalently modified with 35S. In addition, NifZ was able to covalently modify ThiI under nonreducing conditions (Fig. 3). These results provide further support for involvement of the NifZ active-site cysteine in forming a persulfide intermediate during NifZ's catalytic cycle and the occurrence of a persulfide sulfur transfer step from NifZ to ThiI during the biosynthesis of s4U8.
Fig 3.
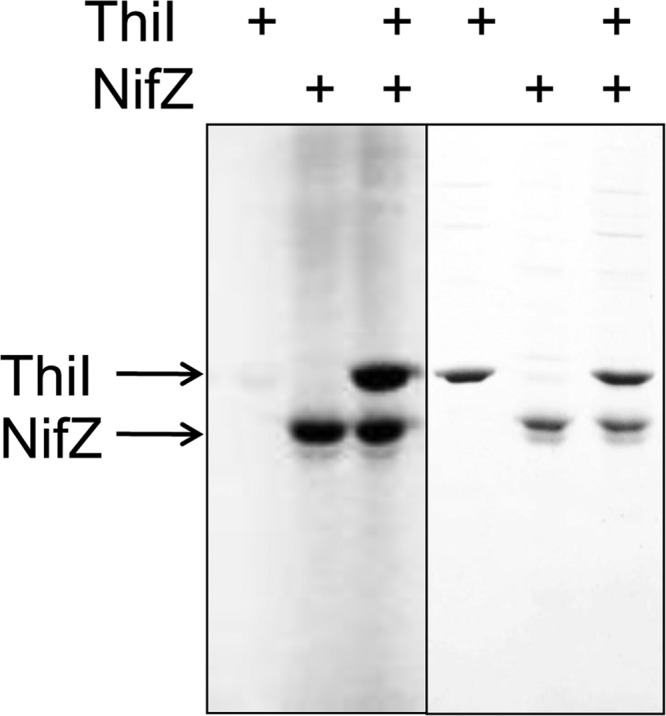
In vitro sulfur transfer between NifZ and ThiI. NifZ and/or ThiI (4 μM each) were incubated with 35S-labeled l-cysteine (10 μCi), l-cysteine (50 μM), ATP (1 mM), and MgCl2 (5 mM) and quenched with N-ethylmaleimide (0.4 mM). Nonreducing SDS-PAGE gels were stained with Coomassie brilliant blue (right) and exposed using a phosphorimager (left). Labeling of NifZ and ThiI shows the 35S covalent modification in the absence of DTT.
Recently, we have described the kinetic mechanism of the bisubstrate cysteine-SufU sulfurtransferase reaction catalyzed by the B. subtilis SufS enzyme (23). The presence of the sulfur acceptor SufU increases the catalytic rate of the SufS reaction by >100-fold. In addition, E. coli ThiI was able to elicit a 2-fold enhancement of IscS cysteine desulfurase activity under reducing conditions (our unpublished results and see reference 7). The B. subtilis ThiI, however, was not able to enhance NifZ activity of sulfide or alanine formation (Fig. 4, left). It is possible that in some representatives of class I cysteine desulfurases, such as NifZ, the active-site cysteine, contained in a longer structural loop, is easily susceptible to cleavage by reducing agents (i.e., DTT could compete with ThiI for the enzyme's persulfide-sulfur). Therefore, we determined the activity profile of NifZ in the presence or absence of ThiI under nonreducing conditions (without DTT). Figure 4 (right panel) shows the linear steady-state rate of alanine formation catalyzed by NifZ in the absence of DTT (80 ± 1.4 nmol of Ala/min/mg). The activity is lower than that observed in the standard assay with DTT (354 ± 12 nmol of Ala/min/mg), which is in agreement with the slower catalytic formation of polysulfide by cysteine desulfurases under nonreducing conditions (33).
In contrast, under nonreducing conditions, the NifZ reaction in the presence of ThiI showed a biphasic profile of alanine formation (Fig. 4, right). The activity associated with the initial reaction rate was calculated to be 250 ± 9.0 nmol of Ala/min/mg. Interestingly, the activity in the first phase in a reaction containing 20 molar equivalent of ThiI (4.8 nmol) generated nearly stoichiometric amount of alanine (∼4 nmol, 16 turnovers). The fast consumption of ThiI led to the second phase of the reaction (84 ± 5.5 nmol of Ala/min/mg), which displayed a rate similar to the one observed for polysulfide formation (i.e., activity of NifZ without DTT and ThiI).
NifZ and ThiI are sufficient and capable of synthesizing s4U8 in vitro.
The proposed catalytic cycle involving the s4U8 formation can be divided in three discrete steps: sulfur transfer from the cysteine desulfurase to ThiI, the adenylation of tRNA by ThiI, and the final transfer of the persulfide sulfur to the activated C-4 of the uridine 8 of tRNA to form s4U8 (17). We have investigated these three steps by probing: (i) the persulfide formation on ThiI by NifZ (as described above), (ii) the [α-32P]ATP ThiI-dependent adenylation of tRNA (Fig. 5, left), and (iii) the overall synthesis of s4U8 in unmodified tRNA transcripts (Fig. 5, right). The in vitro reaction required the presence of DTT (i.e., no s4U8 was formed under nonreducing conditions), suggesting the participation of an additional, as-yet-unidentified molecule serving as a reducing agent in this pathway.
Fig 5.

In vitro synthesis of s4U8 by B. subtilis ThiI and NifZ. (Left) Formation of adenylated-tRNA was performed by incubation of synthetic tRNA transcripts (100 μM) with [α-32P]ATP (10 μCi) and MgCl2 (1 mM). Reactions containing 10 μM E. coli or B. subtilis ThiI (+) were compared to reactions in the absence of ThiI (−). Samples were analyzed by urea-PAGE and visualized in a phosphorimager. The lower-molecular-weight 32P signal is attributed to degradation of tRNA-adenylated during PAGE analysis. (Right) In vitro generation of s4U8 was performed in synthetic tRNA transcripts in the presence of NifZ and ThiI as described in Materials and Methods. The A330 chromatographic profiles of tRNA-digested nucleoside resulting from reactions in the presence of separately expressed and purified NifZ and ThiI (dashed line, short dashes), and coexpressed and copurified NifZ-ThiI (dotted line) were compared to the chromatographic profile of the s4U standard (solid line), and digested unmodified tRNAMet transcript (dashed line, long dashes).
Specific partnership between NifZ and ThiI.
The occurrence of analogous steps in the biosynthesis of s4U8 in B. subtilis and E. coli led us to investigate if the B. subtilis NifZ and/or ThiI were capable of replacing E. coli IscS and/or ThiI in vivo. Initial complementation studies were performed for individual components: (i) E. coli cells lacking IscS and expressing NifZ (E. coli CL100 strain transformed with pDS21 and cultured in the presence of arabinose) and (ii) E. coli cells lacking ThiI and expressing B. subtilis ThiI (E. coli JLD26501 strain transformed with pDS108 and cultured in the presence of arabinose). In both cases, analysis of nucleosides isolated from tRNA samples showed no detectable s4U (Fig. 6). However, in vivo s4U8 formation was observed when these strains coexpressed both B. subtilis proteins NifZ and ThiI (E. coli JLD26501 or CL100 strains transformed with pDS115 and cultured in the presence of arabinose) (Fig. 6). The ability of B. subtilis NifZ and ThiI to heterologously synthesize s4U8 provides additional in vivo evidence for specific interactions promoted during sulfur transfer and rules out the potential involvement of an additional B. subtilis specific component participating in sulfur trafficking in this pathway.
Fig 6.

In vivo synthesis of s4U8 by B. subtilis ThiI and NifZ in E. coli. Complementation of E. coli CL100, the iscS deletion strain (dashed line, long dashes), or E. coli JLD26501, the thiI deletion strain (dotted line), was performed with plasmids expressing B. subtilis NifZ and/or B. subtilis ThiI. HPLC A330 chromatograms of tRNA-digested nucleosides show the distinct presence of s4U in cells coexpressing both B. subtilis nifZ and thiI (pDS115, dashed and dotted black line). Chromatograms from individual complementation experiments along with negative controls (both strains transformed with pAra13) displayed absorbances near detection limit (gray). The peak corresponding to s4U from complementation studies was compared to the s4U standard (inset) and to s4U from E. coli MG1655 (solid line). The levels of s4U, from at least three independent experiments, were determined from the ratio of absorbances from assigned peak areas, A330 (s4U)/A260 (ψ). The calculated average absorbance ratios of s4U/ψ were as follows: E. coli MG1655 (wild type), 0.712 ± 0.279; E. coli JLD26501(pDS115), 0.361 ± 0.246; E. coli JLD26501(pDS108), 0.007 ± 0.0004; E. coli JLD26501(pAra13), 0.001 ± 0.0007; E. coli CL100(pDS115), 0.464 ± 0.234; E. coli CL100(pDS21), 0.007 ± 0.007; and E. coli CL100(pAra13), 0.007 ± 0.004.
Despite general similarities between enzymes performing analogous steps in this pathway for E. coli and B. subtilis, the mechanism of sulfur transfer from the cysteine desulfurase to ThiI likely follows a different route. This observation is evident by the lack of individual-protein complementation with the E. coli system, which could be justified by the absence of the sulfur-receiving Rhd domain in the B. subtilis ThiI.
DISCUSSION
We have demonstrated the specific involvement of ThiI and NifZ in the biosynthesis of s4U8 in B. subtilis. Despite similarities of the general scheme involving s4U8 formation, the present study provides the first experimental evidence that, in some species, tRNA modification involving ThiI dispenses the need for a sulfur-acceptor Rhd domain. This observation seems to be linked with the direct involvement of a “devoted” cysteine desulfurase that provides specific partnership during sulfur transfer. A sequence search on the microbial database of fully sequenced genomes indicated the presence of 229 genes encoding proteins similar to ThiI, 148 of which were sequences lacking the Rhd domain. In species representing the Firmicutes (87 genomes), ThiI sequences always lacked the Rhd domain, and the thiI genomic location was always adjacent to a cysteine desulfurase gene similar to nifZ; the only two exceptions were Enterococcus faecalis and Lactococcus lactis.
Interestingly, none of the thiI genes encoding a ThiI-Rhd fusion protein were located next to a cysteine desulfurase gene. One possible function of the sulfur-acceptor Rhd-handle of ThiI is to interact more easily with general cysteine desulfurases. In E. coli and S. enterica, IscS is a general cysteine desulfurase that interacts with a suite of different sulfur acceptors in the biosynthesis of most, if not all, thio-cofactors (17). Therefore, the activity of this enzyme has to be partitioned among the various physiological partners including ThiI. In these cases, the Rhd-sulfurtransferase domain seems to be more suitable for competing with other sulfur-accepting biomolecules. Conversely, ThiI sequences lacking this handle do not compete well in a one-donor/multi-acceptor environment. In B. subtilis, for example, this biological problem is circumvented with the presence of a devoted cysteine desulfurase.
The genomic synteny observed in the Firmicutes provides a good indicator for the partnership between a cysteine desulfurase and ThiI. Several lines of experimental evidence presented here not only support this proposal but also provide additional insight into specific steps of sulfur trafficking. First, NifZ and ThiI are required for the biosynthesis of s4U8 in B. subtilis resulting from the phenotypes observed upon gene inactivation. Second, the in vitro synthesis of s4U8 is achieved in the presence of purified forms of NifZ and ThiI. Third, complementation studies in E. coli indicated that the sulfur transfer step from NifZ to B. subtilis ThiI are mutually specific: (i) NifZ is not capable of persulfide sulfur transfer to E. coli ThiI (NifZ does not complement IscS in vivo) and (ii) B. subtilis ThiI is cannot accept a persulfide sulfur from IscS (B. subtilis ThiI does not complement E. coli ThiI in vivo). Collectively, these results indicate that B. subtlis NifZ and ThiI take an alternate route for sulfur acquisition and delivery compared to studied systems involving IscS and ThiI-Rhd enzymes.
The requirement of a reducing agent, such as DTT, for the in vitro synthesis of s4U8 by B. subtilis NifZ and ThiI indicates the participation of another as-yet-unidentified reducing agent in the catalytic cycle. Unlike the B. subtilis ThiI, in the absence of reducing agent the E. coli ThiI is able to undergo one turnover reaction thus generating one equivalent of s4U8. The proposed mechanism for this reaction involves the regeneration of E. coli ThiI through reduction of the Cys344-Cys456 disulfide bond at the end of each catalytic cycle (17, 19). It is possible that in the NifZ-ThiI reaction, a reducing agent could also be involved in the reduction of the persulfide sulfur on the cysteine desulfurase or on ThiI to release S2−. Although the identity of the physiological reductant, as well as the biosynthetic step(s) affected by such a molecule, are still undefined, results from complementation studies rule out the participation of a species-specific molecule acting as the reducing agent.
It has been proposed that short ThiI sequences similar to the one from B. subtilis would lack sulfurtransferase activity (i.e., the ability to accept a sulfur from the cysteine desulfurase and transfer it to the adenylated-tRNA intermediate), and this role would be fulfilled by a separate polypeptide (28). However, the results presented here do not support this proposal. Despite the lack of an Rhd domain, B. subtilis ThiI is able to directly interact with NifZ as these proteins copurify when isolated from cells coexpressing NifZ and ThiI. However, we do not believe that a stable complex is mandatory for the functionality of this pathway since separately purified enzymes are equally competent of performing in vitro s4U8 synthesis.
Kinetic analysis of cysteine desulfurase under nonreducing conditions revealed the role of ThiI as a direct sulfur acceptor NifZ. The high catalytic rate of the Cys-ThiI sulfurtransferase reaction, displayed in the first phase of the reaction, showed stoichiometric dependence of ThiI (i.e., reactions containing 4.8 nmol of ThiI generated ∼4 nmol of alanine in the first phase). The involvement of B. subtilis ThiI in persulfide sulfur transfer from NifZ cysteine desulfurase, shown in 35S-labeling experiments and kinetic analysis of NifZ under nonreducing conditions, suggests the formation of a catalytic intermediate on ThiI.
Nevertheless, the lack of the sulfur-acceptor Rhd-domain indicates that this protein possesses an alternate transient site for sulfur modification. The B. subtilis ThiI Cys344 residue holds an equivalent position to the E. coli ThiI Cys344, which is involved in the final step of sulfur transfer through formation of a disulfide bond with Cys456 located in the Rhd domain (19, 20). While the B. subtilis ThiI Cys344 residue likely displays a function analogous to the Cys344 of E. coli ThiI, the identity of the residue carrying the ThiI sulfurtransferase activity remains unidentified. The lack of an obvious Cys456 corresponding residue suggests the participation of another cysteine residue in ThiI fulfilling this role. The B. subtilis ThiI sequence contains three additional cysteine residues (Cys81, Cys229, and Cys345). Among them, only Cys344 is completely conserved in all ThiI sequences. Interesting to note is the presence of the adjacent Cys345 residue, which is conserved only in ThiI sequences from Gram-positive species. Either the participation of an additional cysteine residue of ThiI or the occurrence of an alternate sulfur transfer step involving the direct participation of NifZ are plausible mechanisms for the synthesis of s4U8 tRNA. These two hypotheses are currently being explored.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. J. Larson for providing E. coli strains used in this work, as well as for helpful feedback, and R. W. Alexander for assisting with the synthesis of tRNA.
This project was supported by the National Science Foundation (MCB-1054623).
Footnotes
Published ahead of print 6 July 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Agris PF, Vendeix FA, Graham WD. 2007. tRNA's wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 366:1–13 [DOI] [PubMed] [Google Scholar]
- 2. Anagnostopoulos C, Spizizen J. 1961. Requirements for transformation in Bacillus subtilis. J. Bacteriol. 81:741–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 4. Caldeira de Araujo A, Favre A. 1985. Induction of size reduction in Escherichia coli by near-ultraviolet light. Eur. J. Biochem. 146:605–610 [DOI] [PubMed] [Google Scholar]
- 5. Iwata-Reuyl D. 2008. An embarrassment of riches: the enzymology of RNA modification. Curr. Opin. Chem. Biol. 12:126–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jurgenson CT, Begley TP, Ealick SE. 2009. The structural and biochemical foundations of thiamin biosynthesis. Annu. Rev. Biochem. 78:569–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kambampati R, Lauhon CT. 1999. IscS is a sulfurtransferase for the in vitro biosynthesis of 4-thiouridine in Escherichia coli tRNA. Biochemistry 38:16561–16568 [DOI] [PubMed] [Google Scholar]
- 8. Kessler D. 2006. Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol. Rev. 30:825–840 [DOI] [PubMed] [Google Scholar]
- 9. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 10. Lauhon CT. 2006. Orchestrating sulfur incorporation into RNA. Nat. Chem. Biol. 2:182–183 [DOI] [PubMed] [Google Scholar]
- 11. Lauhon CT, Erwin WM, Ton GN. 2004. Substrate specificity for 4-thiouridine modification in Escherichia coli. J. Biol. Chem. 279:23022–23029 [DOI] [PubMed] [Google Scholar]
- 12. Lauhon CT, Kambampati R. 2000. The iscS gene in Escherichia coli is required for the biosynthesis of 4-thiouridine, thiamin, and NAD. J. Biol. Chem. 275:20096–20103 [DOI] [PubMed] [Google Scholar]
- 13. Martin J, Zhu W, Passalacqua KD, Bergman N, Borodovsky M. 2010. Bacillus anthracis genome organization in light of whole transcriptome sequencing. BMC Bioinform. 11(Suppl 3): S10 doi:10.1186/1471-2105-11-S3-S10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martinez-Gomez NC, Palmer LD, Vivas E, Roach PL, Downs DM. 2011. The rhodanese domain of ThiI is both necessary and sufficient for synthesis of the thiazole moiety of thiamine in Salmonella enterica. J. Bacteriol. 193:4582–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mihara H, Esaki N. 2002. Bacterial cysteine desulfurases: their function and mechanisms. Appl. Microbiol. Biotechnol. 60:12–23 [DOI] [PubMed] [Google Scholar]
- 16. Moeller R, Setlow P, Reitz G, Nicholson WL. 2009. Roles of small, acid-soluble spore proteins and core water content in survival of Bacillus subtilis spores exposed to environmental solar UV radiation. Appl. Environ. Microbiol. 75:5202–5208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mueller EG. 2006. Trafficking in persulfides: delivering sulfur in biosynthetic pathways. Nat. Chem. Biol. 2:185–194 [DOI] [PubMed] [Google Scholar]
- 18. Mueller EG, Buck CJ, Palenchar PM, Barnhart LE, Paulson JL. 1998. Identification of a gene involved in the generation of 4-thiouridine in tRNA. Nucleic Acids Res. 26:2606–26010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mueller EG, Palenchar PM, Buck CJ. 2001. The role of the cysteine residues of ThiI in the generation of 4-thiouridine in tRNA. J. Biol. Chem. 276:33588–33595 [DOI] [PubMed] [Google Scholar]
- 20. Palenchar PM, Buck CJ, Cheng H, Larson TJ, Mueller EG. 2000. Evidence that ThiI, an enzyme shared between thiamin and 4-thiouridine biosynthesis, may be a sulfurtransferase that proceeds through a persulfide intermediate. J. Biol. Chem. 275:8283–8286 [DOI] [PubMed] [Google Scholar]
- 21. Phizicky EM, Alfonzo JD. Do all modifications benefit all tRNAs? FEBS Lett. 584:265–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rozenski J, Crain PF, McCloskey JA. 1999. The RNA Modification Database: 1999 update. Nucleic Acids Res. 27:196–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Selbach B, Earles E, Dos Santos PC. 2010. Kinetic analysis of the bisubstrate cysteine desulfurase SufS from Bacillus subtilis. Biochemistry 49:8794–8802 [DOI] [PubMed] [Google Scholar]
- 24. Sherlin LD, et al. 2001. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA 7:1671–1678 [PMC free article] [PubMed] [Google Scholar]
- 25. Vagner V, Dervyn E, Ehrlich SD. 1998. A vector for systematic gene inactivation in Bacillus subtilis. Microbiology 144(Pt 11): 3097–3104 [DOI] [PubMed] [Google Scholar]
- 26. van Schaik W, Prigent J, Fouet A. 2007. The stringent response of Bacillus anthracis contributes to sporulation but not to virulence. Microbiology 153:4234–4239 [DOI] [PubMed] [Google Scholar]
- 27. Wada H, Snell EE. 1962. Enzymatic transamination of pyridoxamine. I. With oxaloacetate and alpha-ketoglutarate. J. Biol. Chem. 237:127–132 [PubMed] [Google Scholar]
- 28. Waterman DG, Ortiz-Lombardia M, Fogg MJ, Koonin EV, Antson AA. 2006. Crystal structure of Bacillus anthracis ThiI, a tRNA-modifying enzyme containing the predicted RNA-binding THUMP domain. J. Mol. Biol. 356:97–110 [DOI] [PubMed] [Google Scholar]
- 29. Webb E, Claas K, Downs DM. 1997. Characterization of thiI, a new gene involved in thiazole biosynthesis in Salmonella typhimurium. J. Bacteriol. 179:4399–4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ye H, Abdel-Ghany SE, Anderson TD, Pilon-Smits EA, Pilon M. 2006. CpSufE activates the cysteine desulfurase CpNifS for chloroplastic Fe-S cluster formation. J. Biol. Chem. 281:8958–8969 [DOI] [PubMed] [Google Scholar]
- 31. Yi C, Pan T. Cellular dynamics of RNA modification. Acc. Chem. Res. 44:1380–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. You D, Xu T, Yao F, Zhou X, Deng Z. 2008. Direct evidence that ThiI is an ATP pyrophosphatase for the adenylation of uridine in 4-thiouridine biosynthesis. Chembiochem 9:1879–1882 [DOI] [PubMed] [Google Scholar]
- 33. Zheng L, Cash VL, Flint DH, Dean DR. 1998. Assembly of iron-sulfur clusters. Identification of an iscSUA-hscBA-fdx gene cluster from Azotobacter vinelandii. J. Biol. Chem. 273:13264–13272 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.