

Abstract
The nucleotide messenger cyclic di-GMP (c-di-GMP) plays a central role in the regulation of motility, virulence, and biofilm formation in many pathogenic bacteria. EAL domain-containing phosphodiesterases are the major signaling proteins responsible for the degradation of c-di-GMP and maintenance of its cellular level. We determined the crystal structure of a single mutant (R286W) of the response regulator RocR from Pseudomonas aeruginosa to show that RocR exhibits a highly unusual tetrameric structure arranged around a single dyad, with the four subunits adopting two distinctly different conformations. Subunits A and B adopt a conformation with the REC domain located above the c-di-GMP binding pocket, whereas subunits C and D adopt an open conformation with the REC domain swung to the side of the EAL domain. Remarkably, the access to the substrate-binding pockets of the EAL domains of the open subunits C and D are blocked in trans by the REC domains of subunits A and B, indicating that only two of the four active sites are engaged in the degradation of c-di-GMP. In conjunction with biochemical and biophysical data, we propose that the structural changes within the REC domains triggered by the phosphorylation are transmitted to the EAL domain active sites through a pathway that traverses the dimerization interfaces composed of a conserved regulatory loop and the neighboring motifs. This exquisite mechanism reinforces the crucial role of the regulatory loop and suggests that similar regulatory mechanisms may be operational in many EAL domain proteins, considering the preservation of the dimerization interface and the spatial arrangement of the regulatory domains.
INTRODUCTION
First discovered as an allosteric regulator of cellulose synthase in Glucoacetobacter xylinus, cyclic-di-GMP (c-di-GMP) mediates a wide variety of bacterial cellular functions mainly associated with the transition between a planktonic and a community-based biofilm lifestyle (7, 17, 35, 36). Dissection of the signaling networks that regulate cellular c-di-GMP levels is expected to unveil the molecular mechanisms underlying biofilm formation, a major contributor to the persistent infections caused by many pathogenic bacteria. Cellular concentrations of c-di-GMP are controlled by two types of enzymes with opposing activities: GGDEF domain proteins with diguanylate cyclase activity and proteins containing either an EAL or a HD-GYP domain, which have c-di-GMP phosphodiesterase (PDE) activity (38–41). While GGDEF domains catalyze the synthesis of c-di-GMP, EAL and HD-GYP domains hydrolyze c-di-GMP to generate either linear 5′-pGpG or GMP. Many bacterial genomes contain multiple copies of genes encoding GGDEF, EAL and HD-GYP domains, an observation consistent with their prominent roles in c-di-GMP signaling. GGDEF, EAL, or HD-GYP domains usually do not function as stand-alone proteins but in association with various regulatory domains that modulate their enzymatic activities in response to external stimuli (4, 14, 29). A wide variety of regulatory domains that sense the environmental signals from the surroundings are found fused to the three enzymatically active domains. Elucidating how the regulatory domains modulate the enzymatic activities of c-di-GMP metabolizing proteins is crucial for a better understanding of the molecular mechanism of c-di-GMP signaling. Recent structural studies on proteins containing GGDEF domain, including the response regulators PleD and WspR, suggested that enzymatic regulation is mainly achieved through the control of their oligomerization state (9, 50). The crystal structure of the protein YkuI from Bacillus subtilis that contains an enzymatically inactive EAL domain and a putative regulatory PAS domain revealed the β/α barrel structure of the EAL domain (26). Based on site-directed mutagenesis studies, a regulatory mechanism was proposed for the EAL domain-containing protein RocR from Pseudomonas aeruginosa (31). In this mechanism, a loop of RocR [named “loop 6” to be consistent with other (α/β)8 barrel proteins, where the corresponding loop acts as a lid for substrate binding and product release] plays an important role for binding the catalytic metal ion and the substrate. More recently, Barends et al. proposed a regulatory mechanism for the BLUF photoreceptor-regulated EAL domain in the BlrP1 protein from Klebsiella pneumoniae, based on the crystal structures of the protein crystallized at different pH values. The mechanism proposed for BlrP1 involves the subtle repositioning of two catalytic metal-ions through conformational changes in the β5-α5 loop (equivalent to loop 6 in RocR), as well as in other structural motifs (2).
The response regulator RocR from the opportunistic pathogen P. aeruginosa contains an N-terminal phosphoreceiver (REC) domain and a C-terminal EAL domain that possesses c-di-GMP specific PDE activity (23, 24, 32). Based on sequence homology and in vivo studies, the REC domain of RocR is predicted to accept a phosphate group from the cognate membrane-bound histidine kinase sensor RocS1. Phosphorylation of RocR putatively modulates the enzymatic activity of its EAL domain and hence of the local level of c-di-GMP. To understand how structural changes originating from the regulatory REC domain of RocR are transmitted to its EAL domain and hence regulate its catalytic activity, we set out to determine the crystal structure of RocR. Although the wild-type RocR could not be crystallized, a 2.5-Å crystal structure of the R286W mutant that exhibits lower catalytic activity was determined. The crystal structure reveals a highly unusual tetrameric arrangement featuring two markedly different conformations for the four subunits of RocR, with only two active sites accessible for substrate binding. The overall quaternary structures of the wild-type RocR protein and of two single mutants (R286W and D56N) are indistinguishable in solution, as assessed by small-angle X-ray scattering (SAXS) experiments. In conjunction with previously acquired biochemical and biophysical data, we propose a regulatory mechanism that involves the propagation of structural changes from the REC domain to the active site of the EAL domain through a dimerization interface and an important functional loop motif.
MATERIALS AND METHODS
Cloning, expression, purification, and crystallization.
The cloning, expression, and purification of RocR was described previously (20). In brief, the RocR protein was crystallized at 18°C via the sitting-drop vapor-diffusion method by mixing 1 μl of protein with an equal volume of a solution containing 15 to 20% (wt/vol) PEG 3350 (Hampton Research), 0.1 M Na HEPES (pH 7.5), 0.2 M sodium tartrate, and 5% glycerol. Rod-shaped crystals measuring up to 0.3 mm in length grow over the course of 10 to 14 days. DNA sequencing revealed a mutation introduced by the PCR at position 286 of the amino acid sequence (R286W). The wild-type RocR protein fails to crystallize in the same condition as the RocR mutant, possibly because of a crystal contact established between side chains of Trp286 (subunit A) and Arg378 from a neighboring molecule. C-di-GMP used for cocrystallization or soaking was synthesized enzymatically using a thermophilic DGC protein (30).
Expression, purification, and crystallization of the seleniated RocR protein.
The RocR-pET26b plasmid was transformed into Escherichia coli B834 (DE3) (Novagen). Upon reaching an optical density at 600 nm (OD600) of 0.4, cells grown at 310 K in Luria-Bertani medium supplemented with 50 μg of kanamycin/ml were harvested by centrifugation and resuspended in selenomethionine base medium (Molecular Dimensions). The washing step was repeated, and the cells were inoculated into 2 liters of preaerated, prewarmed selemethionine expression medium (Molecular Dimensions) supplemented with 40 mg of l-selenomethionine/liter and 50 μg of kanamycin/ml. Upon reaching an OD600 of 0.6, the culture was cooled to 301 K, and protein expression was induced with IPTG (isopropyl-β-d-thiogalactopyranoside) at a final concentration of 0.1 mM. Protein purification and crystallization was carried out as described for the native protein (20), except the concentration of PEG 3350 was 10 to 15% (wt/vol).
Enzymatic assay.
The phosphodiesterase activity of the wild-type RocR and mutant R286W was assessed by monitoring the formation of the product 5′-pGpG from the hydrolysis of c-di-GMP. The reaction assay was performed by incubating the enzymes (1 μM) and c-di-GMP (20 μM) at various temperatures (25, 40, 50, 60, and 70°C) in 100 mM Tris-HCl (pH 8.0), 20 mM KCl, and 25 mM MgCl2 for 5 min. Reactions were stopped by adding 1/10 volume of 1 M CaCl2, and the progress of c-di-GMP hydrolysis was monitored by using the Agilent LC1200 system (mobile phase of 20 mM triethylammonium bicarbonate [pH 7.0] and 5% methanol, at a rate 1 ml/min) with a XDB-C18 column.
X-ray data collection and structure determination.
Data collection statistics for crystals of the native protein, for the selenomethionyl protein (SeMet) and the tetrachloroplatinate(II) derivative are given in Table 1. Heavy-metal derivative crystals were prepared using the JBScreen Heavy kit (Jena Biosciences). Native RocR crystals were soaked with 10 mM K2PtCl4 in a modified reservoir solution devoid of sodium tartrate for 10 min. The data sets collected at the Swiss Light Source (Paul Scherrer Institut, Switzerland, beamline PXIII) for the Pt derivative at the LIII edge, and at the National Synchrotron Radiation Research Center (Taiwan) for the selenomethionyl protein and native protein, were processed with the CCP4 program suite (6) or with the program XDS (18). Using the program SHELXD (43), four Pt sites were found. Electron density maps using either SIRAS or SAD phases calculated with the program SHARP (49) were not interpretable. However, the phases derived from the Pt derivative allowed the identification of 38 Se positions (out of a total of 40 Se), using anomalous differences collected at the selenium absorption edge. Using the program PROFESS (6), a 2-fold NCS could be located that mapped one set of Se positions onto the other. An initial map using SAD phases calculated using program SHARP could thus be averaged using 2-fold NCS and the program DM (6), yielding a partially interpretable map. Further density averaging was performed using a mask covering a protein dimer. Program BUCANEER (8) was then used to build a partial model that contained 1414 residues. Several rounds of manual model building were then performed using program COOT (11), interspersed with refinement with program REFMAC (Table 2) (6). Interface areas and solvation energies were calculated by the protein interfaces, surfaces, and assemblies service PISA (22). Movie morphs were created by the Database of Macromolecular Movements (12), and figures were generated with the program PyMOL (Warren DeLano Scientific). Structure-based alignment of RocR against homologs was generated with ESPript (16). Interface areas buried during tetramer formation are listed in Table S1 and Fig. S1B in the supplemental material.
Table 1.
Data collection statistics
| Data set (synchrotron) | Native (NSRRC) | SeMet (NSRRC) | K2PtCl4 (SLS PXIII) |
|---|---|---|---|
| Wavelength (Å) | 1.00 | 0.97910 | 1.07216 |
| Resolution (Å)a | 30–2.50 (2.59–2.50) | 30–3.95 (4.09–3.95) | 20.00–4.29 (4.55–4.29) |
| Space group | P6122 | P6122 | P6122 |
| Cell parameters a/b/c (Å) | 118.8/118.8/495.1 | 119.1/119.1/449.5 | 120.3/120.3/491.1 |
| α/β/γ (°) | 90/90/120 | 90/90/120 | 90/90/120 |
| No. of unique reflections | 60,479 | 19,193 | 26,870 |
| Redundancy | 11.9 (8.8) | 20.1 (19.7) | 2.43 |
| I/σ | 61.9 (2.9) | 28.7 (7.4) | 10.81 (2.23) |
| Completeness (%) | 84.1 (75.0) | 99.7 (98.4) | 98.5 (97.0) |
| Rmergeb | 0.046 (0.495) | 0.161 (0.427) | 0.094 (0.579) |
Values in parentheses indicate values in the highest resolution shell.
Rmerge = ∑|Ij − 〈I〉 |/∑Ij, where Ij is the intensity of an individual reflection, and 〈I〉 is the average intensity of that reflection.
Table 2.
Refinement statistics
| Parametera | Observation |
|---|---|
| Data range (Å) | 20.00–2.50 (2.57–2.50) |
| No. of used reflections | 57,265 (1,983) |
| Nonhydrogen atoms | 11,886 |
| Rwork | 0.216 (0.310) |
| Rfree | 0.285 (0.353) |
| RMSD bond length (Å) | 0.015 |
| RMSD bond angles (°) | 1.635 |
| Ramachandran plot (no. of residues)b | |
| Allowed | 1,320 (99.5) |
| Generously allowed | 5 (0.4) |
| Disallowed | 1 (0.1) |
Rwork = ∑‖Fo| − |Fc‖/∑|Fc|, where Fo denotes the observed structure factor amplitude, and Fc is the structure factor amplitude calculated from the model. Rfree is the same as for Rwork but calculated with 5% (3,044) of the randomly chosen reflections omitted from the refinement. RMSD, root mean square deviation.
The percentage is given in parentheses.
Small angle X-ray scattering.
X-ray scattering data from the wild-type RocR and its mutants were collected according to standard procedures on the X33 beamline (34) of the EMBL Hamburg at the Deutsches Elektronen Synchrotron using a robotic sample changer (37) and a Pilatus 1 M detector (Dectris, Villigen, Switzerland). The scattering patterns were measured using a sample detector distance of 2.7 m and a wavelength of λ = 1.5 Å, covering the range of momentum transfer 0.01 < s < 0.5 Å−1. To monitor for radiation damage, eight 15-s exposures were collected for each protein sample, and no radiation effect was observed. All of the constructs were measured for at least three solute concentrations each, in the range from 1 to 8 mg/ml. The data were normalized to the intensity of the transmitted beam, and the scattering of the buffer was subtracted. The difference curves were scaled for concentration, the data was extrapolated to infinite dilution using PRIMUS (19) and the radius of gyration Rg. The pair distribution functions were computed by GNOM (44), providing also the maximum particle size Dmax, and the low-resolution shape of the wild-type RocR was determined ab initio by DAMMIF (13). The scattering from the high-resolution models was computed by CRYSOL (45).
RESULTS
Overall structure of the R286W mutant of RocR.
The RocR monomer is composed of an N-terminal regulatory REC domain connected by a linker of ∼13 residues to a C-terminal EAL domain (Fig. 1A). Despite extensive screening of crystallization conditions, the wild-type RocR could not be crystallized. Instead, a mutant protein (R286W) was found to be more amenable to crystallization. Enzymatic assays show that at ambient temperatures, the R286W mutant is active with lower enzymatic activity than the wild-type protein (Fig. 1D). However, the mutant protein shares great thermostability with the wild-type RocR, recovering robust enzymatic activity at elevated temperatures. The crystal structure of the R286W mutant was determined at a resolution of 2.5 Å using SAD phasing at the Se absorption edge (see Materials and Methods and Tables 1 and 2). The asymmetric unit of the present crystal form contains one tetramer assembled around a single noncrystallographic dyad (Fig. 1B), a finding consistent with the observation that RocR forms tetramers in solution (31, 32). Unexpectedly, the four RocR subunits in the crystal structure adopt two distinct conformations (Fig. 1C and 2). Two subunits adopt a closed conformation (thereafter labeled “A” and “B”, with a root mean square deviation [RMSD] of 1.01 Å) with their REC domain situated on top of the (β/α)8 barrel of their EAL domain (Fig. 2A), while the other two subunits adopt an open conformation (“C” and “D”, RMSD = 1.06 Å) with their REC domains shifted to the side of their EAL domains (Fig. 2B). Having brought the EAL domains of a closed and an open subunit in coincidence, a 67° rotation and a translation of 29 Å is required to superimpose their REC domains (Fig. 2C). The tetrameric structure of RocR is rather unique because most EAL domain proteins form dimers through a conserved EAL-EAL dimerization interface (2, 26, 46). A dimeric form of RocR was observed in solution for a few RocR mutants with compromised enzymatic activity, suggesting that the RocR tetramer could be formed by two EAL dimers (31, 32). The crystal structure reveals a set of interactions between residues projecting from helices α8 and α10 and from loop 6 that stabilize the EAL-EAL interface between subunits A and C (or their equivalents B and D, by the dyad) (Fig. 1C and see Fig. S1 in the supplemental material). This mode of interaction is also found in the dimeric EAL proteins TBD1265 from Thiobacillus denitrificans (46), YkuI from Bacillus subtilis (26), and BlrP1 from Klebsiella pneumoniae (2) with the same structural elements involved in stabilizing the EAL-EAL interface (see Fig. S1A in the supplemental material). However, in contrast to these proteins, the unusual tetrameric structure of RocR arises from additional intra- and intersubunit interactions between EAL and REC domains (see Fig. S1B in the supplemental material), which result in a total buried surface area of 13,820 Å2. The most extensive interactions are observed between the open subunits C and D, which lead to a head-to-tail association between their respective REC and EAL domains (see Fig. S1B in the supplemental material). The RECA and RECB domains from the closed subunits occupy the central core of the tetramer with the putative phosphorylation sites Asp56 pointing inside the tetramer and largely inaccessible from the solvent. Further stabilization of the tetramer is provided by interactions between RECB and EALC (or their equivalent by the dyad RECA with EALD). Importantly, the latter interactions give rise to the inhibition of the EAL domains in trans as described in details below.
Fig 1.

Structure of the RocR R286W mutant protein. (A) Domain organization of RocR. (B) The RocR tetramer is shown from three angles separated by 90°. Its subunits are colored green (subunit A), cyan (subunit B), magenta (subunit C), and yellow (subunit D). The position of the single dyad that runs through the crystallographic tetramer is indicated in each panel. The region involved in forming intermolecular EAL-EAL contacts is colored in white, and a magnified view of this interaction is shown in panel C. (C) Schematic depiction of the RocR tetramer highlighting the open conformation of its C and D subunits, which form a saddle-like structure into which the REC domains of the closed subunits A and B are inserted, forming the core of the tetramer. The EAL active sites are represented as three black triangles. EAL active sites of subunits C and D are concealed (crossed circle) in trans by the REC domains located at the center of the structure. The interdomain linker is depicted as a line (dashed line if it is partially disordered in the crystal structure). (D) Enzymatic activity of the wild-type and R286W mutant RocR. (Left panel) HPLC analysis of c-di-GMP degradation in the presence of no enzyme and R286W mutant at 25 and 60°C. (Center and right panels) HPLC analysis of c-di-GMP degradation for wild type and R286W mutant across temperatures. The mutant exhibits diminished activity that is recovered at elevated temperatures. Reaction conditions are described in the experimental section.
Fig 2.

(A and B) Closed (subunits A and B, in cyan, in panel A) and open conformations (subunits C and D, in magenta, in panel B) that constitute the RocR tetramer. Residues from the REC and EAL active sites are represented as yellow sticks. (C) Superposition of the two conformers based on their EAL domains. The residual rotation (angle needed to bring their respective REC domains into coincidence) is indicated.
Quaternary structures of wild-type and mutant RocR in solution.
The observation of two markedly distinct conformations for the RocR subunits raises the possibility of the existence of a partially or fully open tetrameric structure in solution, with domains RECA and RECB swung outward and the catalytic sites of domains EALC and/or EALD accessible. A hypothetical fully open conformation of RocR featuring a tetramer with a 222 symmetry is shown in Fig. S2A in the supplemental material. We used the technique of SAXS to address the important question of whether the wild-type and R286W mutant proteins adopt closed structures in solution, similar to the mutant structure revealed by X-ray crystallography. The processed experimental scattering from the wild-type RocR displayed in Fig. 3A (curve 1), and the overall parameters computed from the scattering data clearly indicate that the protein is tetrameric in solution. The experimental radius of gyration Rg = 37 ± 1 Å and the maximum particle size Dmax = 110 × 10 Å fully agree with the values computed from the crystal structure of RocR (Rg = 36.5Å, Dmax = 116 Å) but not with a partially or fully open tetrameric structure (see Fig. S2A in the supplemental material). Moreover, the low-resolution molecular shape determined from the experimental data neatly matches the crystallographic model (Fig. 3B). The scattering pattern computed from the closed crystal structure fits the experimental data very well with discrepancy χ = 1.08 (Fig. 3A, curve 2). In contrast, the scattering patterns computed from the putative half-open and open models (Fig. 3A, curves 3 and 4, respectively) display substantial systematic deviations from the experimental data and yield very high discrepancy values of χ = 4.7 and 11.3, respectively. These findings suggest that, despite the altered catalytic activity exhibited by the R286W mutant, the overall closed conformation observed in the crystal structure is likely to be adopted by both the wild-type RocR and the R286W mutant in solution. Note that the residue Trp286 is from the helix α7 of the EAL domain and situated at the RECC-EALD and RECD-EALC interfaces that may play a role in the regulatory mechanism, as we will discuss later. It was previously shown that the D56N RocR mutant exhibits catalytic properties different from the wild-type RocR. Comparative SAXS experiments showed that the scattering patterns from the mutant D56N coincides with the scattering from the wild-type RocR within the experimental error (see Fig. S2B in the supplemental material), indicating that the effect of the D56N mutation on catalytic property is also likely to be exerted through local structural changes rather than large alterations to the overall quaternary structure.
Fig 3.

SAXS analysis of the conformation of RocR in solution. (A) Experimental scattering from the wild-type protein (1, black dots), calculated scattering from the closed tetramer observed in the crystal (2, blue line); calculated scattering from the putative half-open and open RocR tetramers (3 and 4, red and green dashed lines). The logarithm of the scattering intensity is plotted as a function of momentum transfer s = 4π sin(θ)/λ, where θ is the scattering angle and λ is the X-ray wavelength. (B) ab initio low-resolution shape reconstructed from the wild-type RocR data (gray mesh) superimposed on the structure of the crystallographic tetramer using SUPCOMB (21).
REC domain and interdomain linker.
The overall structure of the REC domain of RocR resembles the chemotaxis protein CheY (48). A central sheet of five parallel β-strands is flanked by helices α2′, α3′, and α4′ on one side and α1′ and α5′ on the other, resulting in a (β/α)5 topology (Fig. 4A and see Fig. S3A in the supplemental material). Canonical REC domains contain five essential residues: situated at the end of strand β1′, Glu10 and Asp11 are needed to coordinate a divalent metal ion that is absent here (42), whereas Asp56 at the end of β3′ is the putative residue that receives a phosphate group from the cognate histidine kinase RocS1. Residues Ser83 and Lys111 are predicted to stabilize the phospho-aspartyl adduct and to relay conformational changes through the protein (3). In the current structure, no electron density that could account for a phosphate group is visible next to Asp56 and RocR appears to be nonphosphorylated. Residues projecting from the face contributed by α4′-β5′-α5′ are usually involved in forming specific protein-protein interactions (1, 25, 33). Residue Phe105, located near its center, adopts a single rotamer conformation, with its phenyl ring buried inside the hydrophobic core. Two structural differences (overall RMSD of 2 Å) are visible between the REC domains of the open and closed conformers (Fig. 4A). Local structural remodeling is seen in the β4′-α4′ loop, which undergoes conformational changes upon phosphorylation in other REC domain-containing proteins (3, 15). Moreover, the β3′-α3′ loop adjacent to Asp56 shows significant differences between the two conformers with a RMSD of 4.38 Å. Previous NMR studies on the prototypical CheY protein revealed that even unphosphorylated REC domains can sample an ensemble of conformations ranging from inactivated to fully activated (5, 15). Given the decreased accessibility of the phosphorylation site and the surface properties of the α4′-β5′-α5′ face, the conformation adopted by RECA and RECB (closed subunits) might resemble the phosphorylated state.
Fig 4.

Observed conformations in the individual domains of RocR. (A) Superposition of RECB (cyan, side chains shown as white sticks) with RECC (yellow, side chains shown as black sticks). Phe105, the “switch residue” located on strand β5′, adopts the same rotamer in a buried conformation. The β3′-α3′ and β4′-α4′ loop regions display different conformations between the open (subunit C) and closed conformers (subunit B). (B) Close-up view of the EAL domain of RocR using a diagram representation (helices shown as cyan ribbons and β-strands as magenta arrows). Secondary structure elements of the additional lobe that protrudes over the TIM barrel are labeled. EAL active-site residues and loop 6 are represented as sticks, and the Mg2+ ion is represented as a green sphere. A surface representation of the EAL domain is overlaid. (C) Magnified view of the EAL active-site region with electron density displayed at a level of 3 σ calculated using Fourier coefficients Fo-Fc, where the metal and one bound water were omitted from the phase calculation. (D) Close-up view of the entrance to EAL active sites in the open (yellow) and closed (cyan) conformers. The catalytically important Loop 6 displays two distinct conformations, with Glu296 in two rotameric conformations and Tyr301 flipped toward opposite directions.
The interdomain linker (Q130DLPRQIEVAELP142) is visible in subunit B, where it adopts an extended conformation stabilized by interactions with Ser74, Gly75, and His78 of the RECB domain (see Fig. S3C in the supplemental material). The linker is only partially visible in subunits A, C, and D. The disordered residues from the linker are residues 135 to 138 in subunit A, residues 131 to 140 in subunit C, and residues 130 to 135 in subunit D. The intrinsic flexibility for the linker region is in agreement with the elevated deuteration levels observed by using the method of hydrogen-deuterium (H/D) exchange-coupled mass spectrometry (31).
Phosphodiesterase EAL domain.
The EAL domains of RocR adopt a (β/α)8 barrel-like fold, similar to other reported EAL domain structures (Fig. 4B and see Fig. S3B in the supplemental material) (2, 26–28, 46). The structure is a variant of the TIM barrel fold with the β1 strand running antiparallel to the other β-strands and also by the presence of additional α-helices α1, α2, and α3 that, together with loop β2-α2, form an additional lobe that protrudes above the barrel, near the putative c-di-GMP binding pocket. The catalytic residues are located at the C-terminal end of the barrel, including residues that form the metal ion-binding site and the evolutionarily conserved residues of loop 6: D296FGAGYSS303 (see Fig. S3B in the supplemental material). A single Mg2+ ion is present in each EAL active site of RocR (Fig. 4C). Together with Asn233, Glu265, and Asp295 and a water molecule, residue Glu175 from the signature EVL motif completes the octahedral metal ion coordination shell. As predicted by our previous model (32), a water molecule is coordinated by the Mg2+ ion and at hydrogen bonding distance from Glu352 and Glu175. In the crystal structures of the enzymatically active BlrP1 and TBD1265, the EAL domains were seen to bind one or two metal ions when high concentration of Mn2+ was used for cocrystallization. For RocR, extensive cocrystallization and soaking experiments with Mg2+, Mn2+, Ca2+, or Zn2+ in the presence or absence c-di-GMP did not produce a RocR structure with two metal ions bound.
c-di-GMP is expected to bind to RocR in a manner similar to BlrP1 and TBD1265, considering that the key residues that bind c-di-GMP are conserved among these three proteins. When we modeled c-di-GMP in the EAL active sites of RocR using the crystal structure of BlrP1 as a guide, EALA and EALB from the closed subunits highly resemble the EAL domain of BlrP1 and appear in the correct conformation for binding c-di-GMP. However, despite repeated attempts, cocrystallization and soaking experiments with c-di-GMP did not yield an enzyme-substrate complex. One structural element known to affect the catalytic properties of EAL domains is loop 6 (2, 31). Interestingly, loop 6 in the four EAL domains adopts two different conformations, as evidenced by the repositioning of residue Asp296 and a large rotation of the phenyl ring of Tyr301 (Fig. 4D). The observations confirm our previous proposal that loop 6 can readily undergo conformational changes and that structural changes in the REC domain could be coupled with conformational changes in loop 6. Another conserved function of loop 6 in EAL domains is to contribute to the dimerization interface along with helix α10. Despite the different conformation adopted by loop 6 in EALA and EALC (or EALB and EALD), the loops and the two α10 helices form a nonsymmetric dimer interface that is stabilized by numerous polar interactions (Fig. 1C and see Fig. S1 in the supplemental material).
Inhibition of c-di-GMP binding in EALC/D domains in trans by REC domain.
One of the most striking features of the RocR crystal structure is that the quaternary structure adopted by the tetramer places the REC domains of subunits A and B against the top of the β/α barrel of the EAL domains from the subunits C and D, thus occluding their active site from the solvent (Fig. 1C and 5). The interaction between the EAL and REC domains is mainly mediated through the interaction of helices α4′ and α5′ from the REC domain. A motif consisting of three nonpolar residues (Pro88, Ile89, and Leu90) located at the end of α4′ helix of the RECA or RECB domain is packed into a concave hydrophobic region in the EALC or EALD domain (Fig. 5C). The neighboring Gln92 residue is also seen to form hydrogen bonds with two main chain groups (Pro87 and Ile89) from the EALC/D domain. In addition, the residue Tyr195 from the lobe region is tightly sandwiched by several residues that project from helix α5′ of the REC domain (Fig. 5D). Together, these interactions lock RECA and RECB at the center of the tetrameric structure. As a result, superimposition of subunits C and D with c-di-GMP bound EAL domains of BlrP1 and TBD1265 reveals insufficient space to accommodate c-di-GMP in either EALC or EALD. It should be noted, however, that this mode of inhibition is only possible for two of the four subunits because the space at the center of the tetrameric RocR can only accommodate up to two REC domains at the same time (Fig. 1C). Importantly, when the RECA and RECB domains are located at the central core of the tetramer, the Asp56 phosphorylation sites are shielded from the solvent and thus inaccessible to the cognate histidine kinase RocS1. Hence, the only Asp56 residues that can be phosphorylated are located in the RECC and RECD domains.
Fig 5.

Inhibition of c-di-GMP binding in trans by REC domains. (A) In the observed crystal structure, binding of c-di-GMP to the open RocR conformer (subunit C, magenta) is prevented due to physical occlusion by the REC domain of a closed conformer (subunit B, blue). (B) The in trans REC-EAL interaction is mediated mainly through the interactions between hydrophobic residues preceding REC helix α4′ and an EAL hydrophobic pocket (C) and interactions between Y195 of the EAL lobe and residues projecting from helix α5′ (D).
DISCUSSION
Given the large number of two-component signaling systems, response regulators comprise a major family of signaling proteins in prokaryotes with the REC domain fused to a wide variety of DNA-binding or enzymatic domains. RocR represents a subfamily of response regulators that contain a c-di-GMP specific phosphodiesterase domain, with RocR homologues readily identified in various bacterial species (see Fig. S4 in the supplemental material). The crystal structure and SAXS results reported here revealed that RocR adopts a highly unusual quaternary structure in solution with its four subunits adopting two distinct conformations. Surprisingly, two of the four substrate-binding pockets of RocR (C and D) are not accessible to c-di-GMP and thus likely to be constitutively enzymatically inhibited. Inhibition of the enzymatic activity of the subunits C and D is achieved in trans by using the REC domains from the other two subunits to physically block the access of the c-di-GMP substrate. The EAL domains from the subunits A and B are likely to contain the functional active sites with open c-di-GMP binding pockets. A catalytic mechanism utilizing a single Mg2+ or Mn2+ ion was proposed for RocR previously, while a slightly different mechanism based on two metal ions was proposed for the EAL domain proteins BrlP1 and TBD1265 (2, 46). Both mechanisms share a same set of residues critical for catalysis, with the exception of Asp296, which is proposed to bind a second metal ion in the two-metal-ion mechanism but with a minor role in the one-metal-ion mechanism. Residue Glu352 was proposed to be essential in both mechanisms, albeit with different catalytic roles. Despite our repeated attempts to cocrystallize the protein with c-di-GMP and high concentration of Mg2+, Mn2+, or Ca2+ ion, all of the crystal structures we obtained only contain a single metal ion per protein subunit. Because the second metal ion may only bind in the presence of c-di-GMP, the current structure does not allow us to discriminate between the one- and two-metal mechanisms for RocR.
Sequence comparisons of the REC domain with other canonical REC domains suggest that the key residues for phosphorylation are fully conserved. Hence, the REC domain is likely to be phosphorylated at position Asp56 by the cognate histidine kinase RocS1, and the catalytic activity of the EAL domain could be modulated by the phosphorylation-induced structural changes. Our efforts to prepare phosphorylated forms of RocR using several small phosphate donors were unfortunately futile, as indicated by the negligible effect on enzymatic activity. The inability of the small phosphate donors to phosphorylate RocR may be due to the strong interactions between the REC and EAL domains, similar to some of the response regulators characterized by Barbieri et al. (1). Our repeated effort to crystallize the phosphate mimic BeF3− complexed protein for structural studies was also unsuccessful. Here we deduce a regulatory mechanism based on the current crystal structure and the results of previous biochemical and biophysical studies. Notably, the regulatory mechanism shares some similarity with the mechanism proposed for the blue-light receptor-regulated BlrP1 (2).
Both the crystal structure and the SAXS results for the wild type and R286W and D56N mutants suggest that two REC domains are locked at the center of the tetrameric protein by a large network of specific interactions (Fig. 1 and 5). Even though the D56N mutation significantly alters the catalytic properties of RocR (31), the D56N mutant appears to adopt the same quaternary structure as the wild type and the R286W mutant, according to the SAXS experiment. Thus, given the extensive set of interactions that maintain two REC domains at the center of the RocR tetramer, the regulatory mechanism controlling the activity of the EAL domain is likely to involve local conformational changes rather than large quaternary conformational alterations such as a swinging of the centrally located REC domains to the outside of the molecule, leading to a 222 symmetric RocR tetramer (see Fig. S2A in the supplemental material). In addition, site-directed mutagenesis studies on RocR showed that the enzymatic activity of the EAL domain is very sensitive to conformational changes in a functional loop (loop 6). As revealed by the hydrogen/deuterium (H/D) exchange experiments with the wild type and D56N mutant, the D56N mutation causes significant structural changes in loop 6 in correlation with changes in the kinetic parameters kcat and Km. Based on all of the information, we propose a regulatory mechanism, as depicted in Fig. 6. For clarity, only the relevant domains of one half of the symmetric protein are shown and discussed here. Phosphorylation of the accessible Asp56 in RECD (or RECC) triggers significant conformational changes within the REC domain. Based on the H/D exchange results and structural studies on other activated CheY-like proteins (10, 47), the greatest structural changes occur in the region that encompasses the α4′ and α5′ helices. These structural changes are propagated to the neighboring EALC domain through the direct contact between the terminal residues Ile89 and Leu90 of the α5′ helix of RECD (Fig. 5C) and residues Phe310 and Pro311 that follow loop 6 (amino acids 295 to 305) of the EALC domain (Fig. 6) Because loop 6 (EALC) forms multiple interactions with the adjacent loop 6 (EALA) at the EAL/EAL dimer interface, the structural changes incurred by one loop are likely to be transmitted to the other. Conformational changes in loop 6 would affect the catalytic efficiency of the EAL domains by modulating binding of the c-di-GMP substrate or of the Mg2+ ion or through desolvation of the active site required for effective catalysis. The signal transmission pathway (Fig. 6), which is initiated at the Asp56 position of the RECD domain and ends at the Mg2+ ion in the EALA domain, spans an approximate distance of 48 Å and traverses the RECD/EALC and EALC/EALA interfaces.
Fig 6.
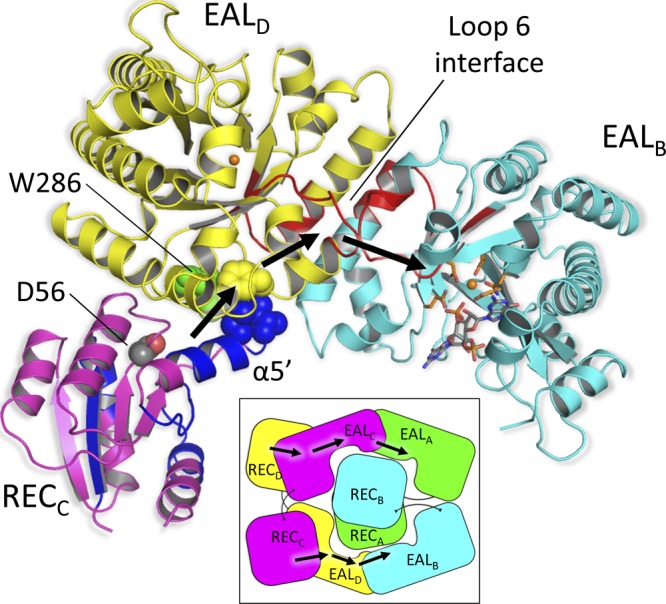
Proposed regulatory mechanism. Only the relevant domains from one half of the symmetric protein are shown for clarity. Phosphorylation of Asp56 of the RECC/D domain induces local conformational changes in the REC domains (see the text). These structural changes are transmitted to the adjacent EAL domain through the direct contact that exist between the terminal residues of helix α5′ (Ile89 and Leu90, depicted as blue spheres) and residues Phe310 and Pro311 (depicted as yellow spheres) situated immediately downstream of loop 6 of EALD/C. The signal is further transmitted down to the active site of EALB/A through loop 6 that constitute EALD/C-EALB/A dimer interface. Based on the H/D exchange-coupled mass spectrometry results reported previously (31), the peptides of RocR that exhibit significant conformational changes upon D56N mutation are colored in blue (REC) and red (EAL), respectively. W286 (green spheres) is located at the REC-EAL interface and may reduce the R286W mutant's activity by disrupting the signal propagation. See also Fig. S6 in the supplemental material. (Inset) Schematic view of the signal transmission pathway across the RocR tetramer. The same color code is adopted.
In addition to the crystal structure, several lines of evidence support the proposed regulatory mechanism and the central role of the highly conserved loop 6 (D296FGAGYSS302) in the mechanism. First, alteration of the conserved residues in the loop 6 region has a profound impact on catalysis. For example, the F297A mutation resulted in a 33-fold decrease in kcat and a 5-fold decrease in Km, whereas the S302A mutation gave rise to strong substrate inhibition. Replacement of residue Glu268 by Gln stabilizes the conformation of loop 6 and led to a reduced kcat by 447-fold and Km by 10-fold (2). These experimental observations suggest that perturbations in the conformation of loop 6 are a very effective way to regulate the catalytic efficiency of the EAL domain. Second, our previous H/D exchange studies are consistent with the proposed structural changes. Comparison of the H/D exchange patterns between RocR and the D56N mutant (the phosphorylation site) suggested that the replacement of Asp56 by Asn induces significant structural changes in both REC and EAL domains (31). The most significant changes occur in the regions that encompass the α4′ and α5′ helices of the REC domain, loop 6, and part of the long helix α10 at the dimer interface. Thus, the regions undergoing structural or conformational changes as determined by H/D exchange experiments, coincide with the proposed signal propagation pathway (Fig. 6). Third, a signal transmission pathway that also involves loop 6 and other structural motifs was proposed for the EAL domain-containing protein BlrP1 (2). A superposition of the BlrP1 structure with the RocR structure shows that the regulatory BLUF and REC domains occupy similar positions close to the bottom of the α/β barrel near the α5/α7 helices (see Fig. S5 in the supplemental material), suggesting a conserved mode for signal transmission between the two proteins. Nonetheless, while the regulatory domain of BlrP1 only regulates the activity of the neighboring EAL domain, RocR needs to further propagate the structural changes to the second EAL domain across the EAL-EAL interface (Fig. 6 and see Fig. S5 in the supplemental material). And lastly, the regulatory mechanism can explain why the R286W mutant exhibits altered enzymatic activity. The Trp286 is seen at the interface of the RECC-EALD and RECD–EALC interfaces in the crystal structure and is part of the proposed signal transmission pathway (Fig. 6 and see Fig. S6 in the supplemental material). Hence, the Arg286 residue in the wild-type protein could play an important role in propagating the conformational change originated from the D56 site. The mutation R286W not only disrupts the pathway but also exerts an effect on the active site of the EAL domain through allosteric regulation, which accounts for the altered enzymatic activity of the R286W mutant.
Many bacterial genomes encode large numbers of EAL domains that are putatively regulated by a wide range of sensory domains for perceiving environmental signals. It is not known whether the EAL domain proteins share similar regulatory mechanisms. The crystal structures of BlrP1 and RocR point to an overall conservation of the mechanism controlling the enzymatic activity of their EAL domain despite the use of different regulatory domains and variation of input signals. In essence, this exquisite regulatory mechanism involves the structural remodeling of the functional loop at the EAL domain dimer interface triggered by a signal originating from a strategically positioned regulatory domain located near the α5/α7 helices of the EAL domain. The structural changes in the loop and its vicinity region modulate the catalytic efficiency of the PDE domain by influencing the binding of the metal ion and c-di-GMP. It will be of great interest to see whether this mechanism is generally preserved in other EAL domain proteins, including the prevalent proteins that contain the GGDEF-EAL didomain unit.
Supplementary Material
ACKNOWLEDGMENTS
We thank the scientists and staff on the BL13B1 (National Synchrotron Radiation Research Center, Taiwan), PXIII (Paul Scherrer Institut, Switzerland), and X33 (DESY, Hamburg, Germany) beamlines for their expert assistance.
The laboratory of J.L. was supported by a CRP-2008 grant, a BMRC grant (08/1/22/19/589), and an ATIP grant from the CNRS. The laboratory of Z.-X.L is supported by an ARC Tier II grant from MOE (Singapore).
Footnotes
Published ahead of print 29 June 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Barbieri CM, Mack TR, Robinson VL, Miller MT, Stock AM. 2010. Regulation of response regulator autophosphorylation through interdomain contacts. J. Biol. Chem. 285:32325–32335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barends TRM, et al. 2009. Structure and mechanism of a bacterial light-regulated cyclic nucleotide phosphodiesterase. Nature 459:1015–1018 [DOI] [PubMed] [Google Scholar]
- 3. Bourret RB. 2010. Receiver domain structure and function in response regulator proteins. Curr. Opin. Microbiol. 13:142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang AL, et al. 2001. Phosphodiesterase A1, a regulator of cellulose synthesis in Acetobacter xylinum, is a heme-based sensor. Biochemistry 40:3420–3426 [DOI] [PubMed] [Google Scholar]
- 5. Cho HS, et al. 2000. NMR structure of activated CheY. J. Mol. Biol. 297:543–551 [DOI] [PubMed] [Google Scholar]
- 6. Collaborative 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. Sect. D 50:760–763 [DOI] [PubMed] [Google Scholar]
- 7. Cotter PA, Stibitz S. 2007. c-di-GMP-mediated regulation of virulence and biofilm formation. Curr. Opin. Microbiol. 10:17–23 [DOI] [PubMed] [Google Scholar]
- 8. Cowtan K. 2006. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. Sect. D Biol. Crystallogr. 62:1002–1011 [DOI] [PubMed] [Google Scholar]
- 9. De N. 2008. Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biol. 6:e67 doi:10.1371/journal.pbio.0060067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Djordjevic S, Goudreau PN, Xu Q, Stock AM, West AH. 1998. Structural basis for methylesterase CheB regulation by a phosphorylation-activated domain. Proc. Natl. Acad. Sci. 95:1381–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 12. Flores S, et al. The Database of Macromolecular Motions: new features added at the decade mark. Nucleic Acids Res. 34:D296–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Franke D, Svergun DI. 2009. DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42:342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Galperin MY, Nikolskaya AN, Koonin EV. 2001. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol. Lett. 203:11–21 [DOI] [PubMed] [Google Scholar]
- 15. Gardino AK, et al. 2009. Transient non-native hydrogen bonds promote activation of a signaling protein. Cell 139:1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gouet P, Courcelle E, Stuart DI, Métoz FM. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308 [DOI] [PubMed] [Google Scholar]
- 17. Hengge R. 2009. Principles of c-di-GMP signaling in bacteria. Nat. Rev. Microbiol. 7:263–273 [DOI] [PubMed] [Google Scholar]
- 18. Kabsch W. 2010. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D 66:133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36:1277–1282 [Google Scholar]
- 20. Kotaka M, et al. 2009. Expression, purification and preliminary crystallographic analysis of Pseudomonas aeruginosa RocR protein. Acta Crystallogr. Sect. F 65:1035–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kozin MB, Svergun DI. 2001. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34:33–41 [Google Scholar]
- 22. Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372:774–797 [DOI] [PubMed] [Google Scholar]
- 23. Kuchma SL, Connolly JP, O'Toole GA. 2005. A three-component regulatory system regulates biofilm maturation and type III secretion in Pseudomonas aeruginosa. J. Bacteriol. 187:1441–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kulasekara HD, et al. 2005. A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol. Microbiol. 55:368–380 [DOI] [PubMed] [Google Scholar]
- 25. Lee SY, et al. 2001. Crystal structure of an activated response regulator bound to its target. Nat. Struct. Biol. 8:52–56 [DOI] [PubMed] [Google Scholar]
- 26. Minasov G, et al. 2009. Crystal structures of YkuI and its complex with second messenger c-di-GMP suggests catalytic mechanism of phosphodiester bond cleavage by EAL domains. J. Biol. Chem. 284:13174–13184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Navarro MVAS, Bae DNN, Wang Q, Sondermann H. 2009. Structural analysis of the GGDEF-EAL domain-containing c-di-GMP receptor FimX. Structure 17:1104–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Navarro MVAS, et al. 2011. Structural basis for c-di-GMP-mediated inside-out signaling controlling periplasmic proteolysis. PLoS Biol. 9:e1000588 doi:10.1371/journal.pbio.1000588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qi Y, Rao F, Luo Z, Liang Z-X. 2009. A flavin cofactor-binding PAS domain regulates c-di-GMP synthesis in AxDGC2 from Acetobacter xylinum. Biochemistry 48:10275–10285 [DOI] [PubMed] [Google Scholar]
- 30. Rao F, et al. 2009. Enzymatic synthesis of c-di-GMP using a thermophilic diguanylate cyclase. Anal. Biochem. 389:138–142 [DOI] [PubMed] [Google Scholar]
- 31. Rao F, et al. 2009. The functional role of a conserved loop in EAL domain-based c-di-GMP specific phosphodiesterase. J. Bacteriol. 191:4722–4731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rao F, Yang Y, Qi Y, Liang ZX. 2008. Catalytic mechanism of c-di-GMP specific phosphodiesterase: a study of the EAL domain-containing RocR from Pseudomonas aeruginosa. J. Bacteriol. 190:3622–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robinson VL, Wu T, Stock AM. 2003. Structural analysis of the domain interface in DrrB, a response regulator of the OmpR/PhoB subfamily. J. Bacteriol. 185:4186–4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roessle MW, et al. 2007. Upgrade of the small-angle X-ray scattering beamline X33 at the European Molecular Biology Laboratory, Hamburg. J. Appl. Crystallogr. 40:s190–s194 [Google Scholar]
- 35. Romling U, Gomelsky M, Galperin MY. 2005. C-di-GMP: the dawning of a novel bacterial signaling system. Mol. Microbiol. 57:629–639 [DOI] [PubMed] [Google Scholar]
- 36. Ross P. 1987. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylate. Nature 325:279–281 [DOI] [PubMed] [Google Scholar]
- 37. Round AR, et al. 2008. Automated sample-changing robot for solution scattering experiments at the EMBL Hamburg SAXS station X33. J. Appl. Crystallogr. 41:913–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ryan RP, et al. 2006. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. U. S. A. 103:6712–6717 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39. Ryjenkov DA, Tarutina M, Moskvin OV, Gomelsky M. 2005. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J. Bacteriol. 187:1792–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schirmer T, Jenal U. 2009. Structural and mechanistic determinants of c-di-GMP signaling. Nat. Rev. Microbiol. 7:724–735 [DOI] [PubMed] [Google Scholar]
- 41. Schmidt AJ, Ryjenkov DA, Gomelsky M. 2005. The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J. Bacteriol. 187:4774–4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schuster M, Silversmith RE, Bourret RB. 2001. Conformational coupling in the chemotaxis response regulator CheY. Proc. Natl. Acad. Sci. U. S. A. 98:6003–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sheldrick G. 2010. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. Sect. D 66:479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Svergun D. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25:495–503 [Google Scholar]
- 45. Svergun D, Barberato C, Koch MHJ. 1995. CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28:768–773 [Google Scholar]
- 46. Tchigvintsev A, et al. 2010. Structural insight into the mechanism of c-di-GMP hydrolysis by EAL domain phosphodiesterases. J. Mol. Biol. 402:524–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Toro-Roman A, Mack TR, Stock AM. 2005. Structural analysis and solution studies of the activated regulatory domain of the response regulator ArcA: a symmetric dimer mediated by the α4-β5-α5 face. J. Mol. Biol. 349:11–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Volz K. 1993. Structural conservation in the CheY superfamily. J. Biol. Chem. 32:11741–11753 [DOI] [PubMed] [Google Scholar]
- 49. Vonrhein C, Roversi BEP, Bricogne G. 2007. Automated structure solution with autoSHARP. Methods Mol. Biol. 364:215–230 [DOI] [PubMed] [Google Scholar]
- 50. Wassmann P, et al. 2007. Structure of BeF3—modified response regulator PleD: implications for diguanylate cyclase activation, catalysis, and feedback inhibition. Structure 15:915–927 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.