

Summary
The genome of Leishmania major, the causative agent of cutaneous Leishmaniasis, contains three nearly identical genes encoding putative glutathione peroxidases, which only differ at their N- and C-termini. Since the gene homologues are essential in trypanosomes, they may also represent potential drug targets in leishmania. Recombinant protein for the shortest of these showed negligible peroxidase activity with glutathione as electron donor indicating that it is not a bone fide glutathione peroxidase. In contrast, high peroxidase activity was obtained with tryparedoxin indicating that these proteins belong to a new class of monomeric tryparedoxin-dependent peroxidases (TDPX) distinct from the classical decameric 2-Cys peroxiredoxins (TryP). Mass spectrometry studies revealed that oxidation of TDPX1 with peroxides results in formation of an intramolecular disulphide bridge between Cys35 and Cys83. Site-directed mutagenesis and kinetic studies showed that Cys35 is essential for peroxidase activity whereas Cys83 is essential for reduction by tryparedoxin. Detailed kinetic studies comparing TDPX1 and TryP1 showed that both enzymes obey saturation ping pong kinetics with respect to tryparedoxin and peroxide. Both enzymes show high affinity for tryparedoxin and broad substrate specificity for hydroperoxides. TDPX1 shows higher affinity towards hydrogen peroxide and cumene hydroperoxide than to t-butyl hydroperoxide whereas no specific substrate preference could be detected for TryP1. TDPX1 exhibits rate constants up to 8 × 104 M−1s−1 whereas TryP1 exhibits higher rate constants ~106 M−1s−1. All three TDPX proteins together constitute about 0.05 % of the Leishmania major promastigote protein content whereas the TryPs are ~40 times more abundant. Possible specific functions of TDPXs are discussed.
Keywords: Glutathione peroxidase, tryparedoxin peroxidase, peroxiredoxin, trypanothione, Leishmania
Introduction
Leishmaniasis is a disease complex caused by over 18 species of Leishmania infecting 12 million people (World Health Organisation). Dependent on the species these eukaryotic parasites affect a wide range of clinical symptoms: from cutaneous (self-healing skin ulcers) (e.g. L. major) to mucocutaneous (e.g. L. braziliensis) to visceral forms (e.g. L. donovani, L. infantum). The latter form is invariably fatal if left untreated. Current treatments are unsatisfactory and better drugs are urgently required.
Most parasites, including Leishmania spp., are more susceptible to reactive oxygen species than their hosts [1;2]. Mammalian cells have a battery of enzymatic systems for metabolising hydroperoxides: catalase; selenium- and sulphur-dependent glutathione peroxidases (GPXs); glutathione-dependent 1-cys peroxiredoxins; and thioredoxin-dependent 2-cys peroxiredoxins. With the exception of catalase, reducing equivalents for the reduction of hydroperoxides are derived from NADPH either via glutathione reductase or thioredoxin reductase. In contrast, leishmania lack catalase, selenium-dependent peroxidases, glutathione reductase and thioredoxin reductase. Instead, the entire antioxidant defence system (either haem-dependent ascorbate peroxidases [3;4] or thiol-dependent peroxidases) is mediated via the unique dithiol trypanothione (N1,N8-bis(glutathionyl)spermidine) together with NADPH-dependent trypanothione reductase (TryR), an essential enzyme in Leishmania spp. [5-7].
The first class of thiol-dependent peroxidases belongs to the classical 2-Cys peroxiredoxins. This comprises: NADPH; TryR; trypanothione; tryparedoxin (TryX), an 18 kDa protein with similar functions to thioredoxin; and tryparedoxin peroxidase type I (TryP), a 2-Cys peroxiredoxin [8]. TryPs were originally identified and characterised in C. fasciculata [8-12] and these have been subsequently identified and studied in a variety of trypanosomatids [13-18]. Additional roles for TryP have been proposed such as metastasis in L. guyanensis [19] and arsenite-resistance in L. amazonensis [20]. L. major TryP is also a putative vaccine candidate [21].
The second class of thiol-dependent peroxidases are GPX-like with closest similarity to the mammalian selenoprotein GPX4. On the basis of their thiol substrate specificity these can be subdivided into two types. The first type, exemplified by T. cruzi GPXII apparently shows specific, but low activity with glutathione and none at all with tryparedoxin [22]. This enzyme is specific for linoleic hydroperoxide and shows no activity towards hydrogen peroxide or short chain hydroperoxides. The second type, exemplified by T. cruzi GPXI [23] and T. brucei Px III [24], are actually tryparedoxin-dependent peroxidases with low, non-physiological activity with glutathione [24;25]. Both enzymes will use cumene hydroperoxide as substrate, whereas T. cruzi GPXI is inactive with hydrogen peroxide. RNA interference studies in T. brucei demonstrated that Px III as well as TryP are essential for parasite survival [26;27]. This suggests that these enzymes may represent much needed novel drug targets. However, their unique roles in trypanosome metabolism still need to be identified. Since the glutathione peroxidase-like proteins do not contain selenocysteine and show negligible activity with glutathione we subsequently refer to type II tryparedoxin-dependent peroxidases as TDPXs to distinguish them from the structurally unrelated decameric type I tryparedoxin peroxidases (TryP). Despite the fact that Leishmania spp. are obligate intracellular parasites of macrophages, and therefore live in a potentially hostile oxidising environment in the mammalian stage of their life cycle, none of these TDPX proteins have been characterised in any Leishmania spp.. The cytosolic L. major TryP has been shown to have tryparedoxin-dependent peroxidase activity but no kinetic analysis has been performed [13]. Comparative studies on TryP and TDPX tryparedoxin peroxidases have not been reported.
Using the recently published genome of L. major [28] we have identified three GPX-like proteins encoded in a tandem array on chromosome 26. These proteins merely differ in their N- and C-terminal sequences, suggesting a common reaction mechanism, but different subcellular localizations. In this paper we analyse the physicochemical, mechanistic and kinetic properties of the putative cytosolic GPX-like protein (TDPX1) and compare it to the classical tryparedoxin peroxidase (TryP1) from the L. major Friedlin genome strain.
Results
Recently, glutathione peroxidase-like proteins from T. brucei and T. cruzi have been shown to be tryparedoxin-dependent peroxidases [25;27]. The aim of this study was to analyse the homologous proteins in L. major and compare them to classical TryP, a 2-Cys peroxiredoxin-like peroxidase. The genome of the L. major Friedlin strain revealed three genes (TDPX1, 2 and 3; Figure 1) arranged in an array on chromosome 26 encoding proteins with homology to mammalian glutathione peroxidases. A selenocysteine, a tryptophan and a glutamine residue form a catalytic triad in the active site in mammalian GPX4 and are essential for peroxidase activity [29]. Selenocysteine is replaced by a cysteine in all three L. major glutathione peroxidase-like proteins, but the tryptophan and glutamine residues are conserved (Cys35, Gln71 and Trp125, LmTDPX1 numbering; Figure 1). The three L. major sequences differ only in their N- and C-terminal sequences whereas the core proteins are identical from residues 2 to 161. The corresponding nucleotide sequences encoding this region are also identical. TDPX2 and TDPX3 have an additional extension at the N-terminus which is a putative mitochondrial targeting sequence. TDPX3 also has a putative glycosomal targeting sequence (SKI) at the shorter C-terminus [30]. TDPX1 lacks these putative signals and is therefore likely to be a cytosolic protein. Thus the three different genes encode an almost identical protein possibly targeted to different subcellular localizations. TDPX1 from L. major protein has 64 and 65% identity with the homologous proteins in T. cruzi and T. brucei, respectively, and only 35% with human GPX4, the most similar among the mammalian GPXs. Interestingly, the L. major glutathione peroxidase-like proteins have six Cys residues, whereas only three Cys residues are conserved in most other organisms including T. brucei and T. cruzi (Figure 1).
Figure 1. Multiple sequence alignment of experimentally characterized and predicted glutathione peroxidase-like proteins.

Parasite proteins are encoded by following open reading frames in GeneDB: Leishmania major: LmTDPX1 (LmjF26.0820), LmTDPX2 (LmjF26.0810), LmTDPX3 (LmjF26.0800), Trypanosoma brucei: TbTDPX1 (Tb927.7.1120), TbTDPX2 (Tb927.7.1130), TbTDPX3 (Tb927.7.1140), Trypanosoma cruzi: TcTDPX1 (Tc00.1047053503899.110), TcTDPX2 (Tc00.1047053503899.119), TcTDPX3 (Tc00.1047053503899.130). The other proteins have the following ExPASy Swiss-Prot accession numbers: Arabidopsis thaliana AtGPX6 (O48646), Saccharomyces cerevisiae ScGPX2 (P38143), Homo sapiens HsGPX4 (P36969). Conserved (black background) and similar residues (grey background) are indicated by asterisks and dots respectively. The cysteine residues are coloured in yellow and the three conserved amino acids involved in peroxidase activity are marked with an inverted triangle. The cysteine shown in this paper to be involved in disulphide bridge formation with the active site cysteine is marked with a square. The differences in the three L. major GPX-like proteins are marked in red. Percent identities to LmTDPX1 indicated at the end of the alignments.
The full length open reading frame of LmjF26.0820, which encodes the putative cytosolic protein TDPX1, was cloned into pET-15b and expressed in BL21 (DE3) pLysS with an N-terminal His-tag. The protein was purified by Ni-NTA chromatography with a yield of about 20 mg l−1 of E. coli culture (Figure 2A). After removal of the 6xHis-tag and further purification, the protein was analysed by size exclusion chromatography and found to elute under reducing conditions as single peak with an apparent molecular weight of 16.6 kDa (Figure 2B) indicating that TDPX1 is monomeric (Mr 19.6 kDa). At higher protein concentrations (> 2 mg ml−1) a second less abundant peak corresponding to a dimer was observed (data not shown). Analysis of both peaks by SDS-PAGE under non-reducing conditions showed both peaks ran as monomers. Thus the native monomeric protein can form non-covalent dimers at high protein concentrations (data not shown). In addition, prolonged storage under non-reducing conditions with exposure to air can promote the formation of TDPX1 aggregates linked by disulphide-bridges at high protein concentration (data not shown).
Figure 2. Purification of recombinant TDPX1 from E. coli.
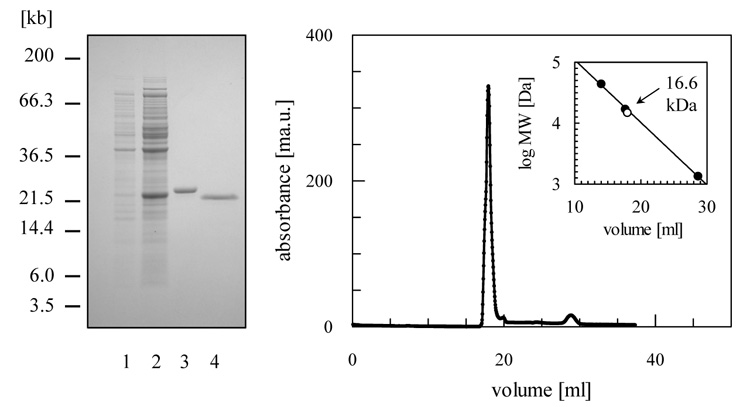
(A) SDS-PAGE analysis: lane 1, un-induced fraction of BL21 Star (DE3) pLysS (pET-15b – LmjF26.0820); lane 2, 4 h after induction with isopropyl-β-D-thiogalactopyranoside; lane 3, 2 μg of (His)6--tagged protein after chromatography on a nickel-chelating Sepharose column; lane 4, 2 μg of LmTDPX1 after removal of (His)6-tag with thrombin. (B) Gel filtration profile of LmTDPX1. The inset shows a plot of elution volume versus log molecular mass of a standard protein mixture (closed circles; ovalbumin, 44 kDa; myoglobin, 17 kDa; Vitamin B-12, 1.35 kDa). The open circle represents the elution volume of LmTDPX1.
The gene sequence of the published L. major TryP [13] differs slightly to those in the L. major genome. Thus, for comparative purposes, we re-cloned and expressed cytosolic TryP1 (LmjF15.1120) as well as the putative cytosolic tryparedoxin (TryX, LmjF29.1160) from the genome strain. TryP1 and TryX are both highly expressed proteins and could be purified in a single step as his-tagged proteins (15-20 mg l−1 bacterial culture).
Peroxidase activity
To analyse the peroxidase activity of the putative glutathione peroxidase-like protein an assay was established containing NADPH, glutathione reductase, GSH as reducing agent and hydrogen peroxide (Figure 3A). With the L major peroxidase there was a negligible difference (0.00145 ± 0.00023 s−1) in the decrease of absorption due to NADPH consumption with or without peroxidase in the assay (Figure 3B), which is much less than the rate of the direct reduction of hydrogen peroxide by GSH alone. In contrast, when selenocysteine-dependent bovine GPX was used as a positive control, GSH-dependent peroxidase activity could be readily detected (Figure 3C).
Figure 3. Peroxidase activity of TDPX1.

(A) Scheme for glutathione-dependent peroxidase assay. (B) Reaction traces plus 5 μM LmTDPX1 or (C) plus bovine GSH peroxidase. (D) Scheme for tryparedoxin-dependent peroxidase assay. (E) Reaction traces plus 5 μM LmTDPX1 or (F) bovine GSH peroxidase. All reactions were started with the addition of 300 μM H2O2 (arrow). Symbols: without enzyme (open circles); plus enzyme (closed circles). For further details see “experimental procedures”.
Replacing GSH and glutathione reductase in the assay with the tryparedoxin system (T. cruzi TryR, trypanothione and L. major TryX, Figure 3D) efficient peroxidase activity of TDPX1 could be detected (Figure 3E). Thus the L. major glutathione peroxidase-like protein is a tryparedoxin-dependent peroxidase (TDPX1). In contrast, bovine GPX could not be reduced by the tryparedoxin system indicating major differences in substrate specificity between mammalian GPX and parasite TDPX1 (Figure 3F).
Kinetic mechanism
The peroxidase activity of TDPX1 towards different hydroperoxides was analysed in the tryparedoxin-dependent assay. TryX was held constant at several fixed concentrations while the hydroperoxide concentration was varied. Assay conditions were checked to ensure that neither TryR nor trypanothione were limiting in the assays at the highest concentration of TryX.
The individual data sets obey simple Michaelis-Menten kinetics and the double-reciprocal transformation yields parallel lines (Figure 4A) consistent with a ping pong mechanism. In a secondary plot (Figure 4B) the reciprocal TryX concentrations are plotted against the intercepts from the primary plot (Figure 4A). The intercept of the second plot is not zero and represents the reciprocal value of the maximal velocity (kcat). Thus the protein shows saturation kinetics. Values for kcat and Km were determined using a global fit of the data sets to equation 1 for TryX and each of the hydroperoxide substrates and are summarised in Table 2.
Figure 4. Kinetic analysis of TDPX1.

Representative data are shown for cumene hydroperoxide and kinetic parameters for this and other substrates are reported in Table 2. TryX was fixed at 0.5 μM (open circle), 1 μM (filled circle), 2 μM (open square), 3 μM (filled square) or 5 μM (open triangle) and cumene hydroperoxide concentrations were varied (50-1000 μM). Initial velocities were determined and globally fitted by non-linear regression to an equation describing a ping-pong mechanism (see “experimental procedures for further details). (A) Double reciprocal transformation of primary data showing the best fit. (B) Secondary plot of the intercepts of the primary plot A versus the reciprocal TryX concentrations.
Table 2.
Kinetic properties of TDPX1 and TryP1 with TryX as reducing agent with different hydroperoxides
| peroxide substrate |
k1 (ROOH) [M-1 s-1] × 105 |
k2 (TryX) [M-1 s-1] × 106 |
k
cat
[s−1] |
Km (ROOH) [μM] |
Km (TryX ) [μM] |
|---|---|---|---|---|---|
| TDPX1 | |||||
| hydrogen peroxidea |
0.80 ± 0.18 | 6.9 ± 1.5 | 15.4 ± 1.4 | 193 ± 27 | 2.2 ± 0.3 |
| hydrogen peroxideb |
1.0 ± 0.06 | 6.2 ± 0.6 | 21.4 ± 7.4 | 211 ± 74 | 3.5 ± 1.2 |
|
t-butyl hydroperoxidea |
0.068 ± 0.0019 | 5.2 ± 1.6 | 15.2 ± 2.0 | 2244 ± 353 | 2.9 ± 0.5 |
| cumene hydroperoxidea |
0.79 ± 0.09 | 6.2 ± 0.7 | 16.2 ± 0.7 | 207 ± 14 | 2.6 ± 0.2 |
| TryP1 | |||||
| hydrogen peroxideb |
13 ± 2 | 1.7 ± 0.1 | 8.8 ± 1.0 | 6.3 ± 0.8 | 4.9 ± 0.6 |
|
t-butyl hydroperoxideb |
8.9 ± 0.8 | 1.8 ± 0.1 | 7.8 ± 0.8 | 10.5 ± 1.4 | 4.3 ± 0.5 |
| cumene hydroperoxideb |
11 ± 1.5 | 3.0 ± 0.2 | 8.6 ± 0.5 | 8.0 ± 0.7 | 2.8 ± 0.2 |
The initial velocities of 30 individual assays with different TryX and hydroperoxide concentrations were globally fitted to the equation describing a ping-pong mechanism (see “experimental procedures”). Values are the means and standard errors obtained by non-linear regression.
The data were calculated using the integrated Dalziel rate equation (see “experimental procedures”). Values are the weighted means and standard deviations of two independent experiments obtained by linear regression.
LmTDPX1 exhibited highest affinity towards hydrogen peroxide (Km 193 ± 27 μM) and cumene hydroperoxide (Km 207 ± 14 μM), but lowest affinity to t-butyl hydroperoxide (Km 2.24 ± 0.35 mM) (Table 2). The affinity towards TryX was independent of the hydroperoxide substrate with a mean Km of 2.5 ± 0.2 μM. Likewise kcat (mean: 16 ± 0.8 s−1) was not significantly different with the three peroxide substrates, yielding an overall rate constant (k2 = kcat/K ) of 6.4 × 106 M−1 m s−1. Similar values were obtained with hydrogen peroxide as substrate using the integrated Dalziel rate equation 2 for a bi-substrate mechanism (see experimental procedures and Table 2). However, analysis with varying the hydroperoxide concentrations yields more accurate kinetic parameters.
Under the same conditions the kinetic properties of TryP1 were analysed to compare them with TDPX1. However, high hydroperoxide concentrations inactivate TryP1 in a time and concentration dependent manner (Figure 5A). This is similar to other peroxiredoxin-like peroxidases, where a sulphinic acid (-SO2H) is formed due to oxidation of the sulphenic acid (-SOH) intermediate in the reaction cycle [31;32]. Sulphinic acids cannot be reduced directly by thioredoxins or tryparedoxins and consequently inactivation of the peroxidases occurs. Thus, the classical analytical method cannot be used and single curve progression analysis was performed instead using the integrated rate equation 2 with different concentrations of TryX and a fixed, non-inhibitory concentration of hydroperoxide [8;33]. Representative plots are shown in Figure 5B and 5C with cumene hydroperoxide as substrate. In the primary plot (Figure 5B) the integrated reciprocal initial velocity multiplied by the enzyme concentration was plotted against the integrated reciprocal hydroperoxide concentrations. The reciprocal slope corresponds to the rate constant k1 for the reduction of hydroperoxides. In a secondary plot (Figure 5C) the ordinate intercepts of the first plot are re-plotted against the reciprocal TryX concentrations. The reciprocal intercept gives the value for the maximum velocity (kcat) and the reciprocal slope corresponds to the rate constant k2 for TryX reduction. The Km values can be obtained by dividing kcat by the rate constants k1 or k2 (Table 2). An average limiting kcat of about 8 to 10 s−1 could be observed for all three hydroperoxides tested. Also the rate constants for the reduction of the hydroperoxides are all in a similar range from about 0.9 to 1.3 (x 106) M−1 s−1. The rate constants for TryX (k2 = kcat/Km) are in the range 1.7-3 (x 106) M−1 s−1 and only slightly higher than k1. The Km values towards the different hydroperoxides are also quite similar ranging from 6.3 to 10.5 μM. Thus TryP1 shows good activity with all three substrates with no specific preference.
Figure 5. Kinetic analysis of TryP1 and inactivation by cumene hydroperoxide.

(A) Initial rates as a function of cumene hydroperoxide concentration. Assays were performed with 5 μM TryX and varying amounts of cumene hydroperoxide (50-1000 μM). Reactions were started with the addition of either TDPX1 (0.2 μM) or TryP1 (0.2 μM) and initial rates determined. TDPX1 (open circles) follows Michaelis-Menten kinetics, whereas TryP1 (closed circles) is inactivated with increasing hydroperoxide concentration. (B) Linear plot of the integrated Dalziel rate equation for a two substrate reaction. Activity of TryP1 was determined with 50 μM cumene hydroperoxide and varying concentrations of TryX (2 μM, open circles; 3 μM, filled circles; 5 μM, filled squares; 10 μM, open squares) as described in the “experimental procedures”. (C) Secondary Dalziel plot. The slope corresponds to φ2 (Km/kcat) for TryX and the ordinate intercept to φ0 (1/kcat). Details of other results are shown in Table 2.
Expression of TDPX, TryP and TryX in L. major promastigotes
Western blot analysis was used to estimate the concentration of TDPX, TryP and TryX in L. major promastigotes using different amounts of non-tagged recombinant protein as calibration standards (Figure 6A). L. major protein extracts were prepared from exponentially growing and stationary phase cells. The same amount of protein extract was loaded in each lane and verified by Coomassie-staining (Figure 6B). Representative Western-blots are shown in Figure 6A. The antisera were highly specific and only a single band was detected in L. major protein extracts at the expected size of each individual recombinant non-tagged protein, (data not shown). No major differences in the expression levels of TDPX, TryP and TryX could be observed between the exponentially growing and stationary phase. A protein content of 5.8 ± 0.7 μg [106 parasites]−1 and a mean cell volume of 37.4 ± 0.3 nl [106 parasites]−1 was obtained in logarithmic or stationary phase of growth. By densitometric analysis TDPX is estimated to represent 0.02-0.08 % of the total protein content. Likewise TryP and TryX represent 1-4% and 0.1-0.3% of total protein. With the calculated molecular mass of TryX (16.5 kDa), TDPX1 (19.3 kDa), and TryP1 (22.1 kDa) the concentrations in L. major promastigotes can be estimated to be 9.4-28.2 μM, 1.6-6.4 μM, and 70-280 μM, respectively. TDPX1, TDPX2 and TDPX3 and the different TryP proteins cannot be separated by SDS-PAGE and are not distinguished by Western blot analysis so that these values represent overall estimations of the relative abundance.
Figure 6. Estimation of TDPX, TryP and TryX concentrations in L. major promastigotes.

Proteins and parasite extracts were separated by SDS-PAGE under reducing conditions and analysed by western blotting as described under experimental procedures. Equal amounts of L. major promastigotes from mid-log (L) and stationary phase (S) of growth were analysed: 3.0 × 106 cells for TDPX or 1.5 × 106 cells for TryP and TryX. Recombinant non-tagged proteins were used as calibration standards (TDPX1: 4-20 ng, TryP1: 250-1000 ng, TryX: 4-20 ng). Band intensity was proportional to the amount of recombinant protein added. At least two independent experiments were performed. The right hand panel is stained with Coomassie to show equal loading for extracts prepared from either phase of growth.
TDPX1 forms an intramolecular disulphide bridge
Most 2-Cys peroxiredoxins form two intermolecular disulphide-bridges upon oxidation resulting in a homo-dimer as smallest functional subunit. Consistent with this, TryP1 is detected as a monomer under reducing SDS-PAGE and as a dimer following oxidation with peroxide and separation under non-reducing conditions (Figure 7). In contrast, reduced and oxidized TDPX1 show only slightly different mobility and thus covalent dimer-formation clearly does not occur following oxidation by peroxide (Figure 7). However, this minor change in mobility could be due to formation of an intramolecular disulphide-bridge. To test this hypothesis, the thiol content of reduced and peroxide oxidised protein was analysed using DTNB. After reduction by DTT and separation by size exclusion chromatography native TDPX1 was found to contain 5.2 ± 0.3 thiol groups per monomer in good agreement with the six predicted from the gene sequence (Figure 1). Addition of SDS (2% final concentration) did not alter this result indicating that all cysteine residues are accessible to the thiol reagent. After oxidation with a 5-fold excess of hydrogen peroxide and removal of residual peroxide using a desalting column, the thiol content decreased to 3.5 ± 0.1 thiol groups per monomer. The difference of 1.7 ± 0.3 thiol groups between the two preparations is thus consistent with formation of an intramolecular disulphide-bridge following oxidation by hydrogen peroxide.
Figure 7. SDS-PAGE analysis of reduced and oxidized TDPX1, TDPX1 mutants and TryP1.

Proteins were first reduced with DTT or oxidized with H2O2 and then residual sulphydryl groups were alkylated with iodoacetamide as described in the experimental procedures. Aliquots (2 μg per lane) were separated by SDS-PAGE and stained with Coomassie: lanes 1 and 2, TDPX1 wild type; lanes 3 and 4, TDPX1 Cys35Ala; lanes 5and 6, TDPX1 Cys83Ala; lanes 7 and 8 TryP1 wild type. Odd numbered lanes are reduced with DTT and even numbered lanes oxidized with H2O2. The schematics show the predicted disulphide bond arrangement for TDPX1 and TryP1.
To determine the nature of the disulphide bridge formed, reduced and oxidized TDPX1 were digested with trypsin and the peptides analysed by mass spectrometry (Figure 8A, B). In the spectrum of the oxidized protein one additional peak is apparent which cannot be found in the spectrum of the reduced protein. The mass of this peak can be assigned to the sum of two peptides containing two cysteine residues (−2 H +1), namely those containing the Cys35 and the Cys83 residues (see Figure 1). An additional cysteine corresponding to Cys64 is conserved in all TDPXs. Although no peak with the corresponding mass could be assigned to a peptide containing Cys64, it is possible that we were not able to detect this under our experimental conditions. To eliminate this possibility, a second sample was digested with chymotrypsin and analysed as above (Figure 8C, D). Again one additional peak was detected in the oxidized spectrum which was absent in the reduced one and again the mass fitted to the sum of the two peptides (−2H +1) containing the same cysteine residues, Cys35 and Cys83. Additionally in the spectra of the oxidized and reduced protein the peptides containing the conserved Cys64 residue was detected. Thus, these results suggest specific disulphide bridge formation between Cys35 and Cys83, not involving Cys64.
Figure 8. Disulphide-bond analysis by mass spectrometry.

Reduced and oxidized TDPX1 wild-type was separated by SDS-PAGE and stained by Coomassie (see Figure 7). The proteins were excised from the gel and digested by trypsin or chymotrypsin. The resulting peptides were analysed by mass-spectrometry. Peptides derived from digestion by trypsin (A, B) or chymotrypsin (C, D) from reduced protein (A, C) or oxidized protein (B, D), respectively. Only the relevant part of the spectrum which shows differences is shown.
Site-directed mutagenesis
Site-directed mutagenesis of Cys35, Cys64 and Cys83 to Ala were performed to extend the findings of the mass spectrometry analysis. The Cys35Ala and Cys83Ala mutants were expressed and purified as before. However, the Cys64Ala mutant was less soluble than the wild-type protein, did not bind specifically to the Ni-NTA column and precipitated during concentration. The Cys35Ala and Cys83Ala mutants showed partial mobility shifts under reducing and oxidizing conditions by SDS-PAGE with some higher aggregate formation evident following peroxide treatment (Figure 7). Abrogation of the mobility shift is more pronounced in the Cys35Ala mutant. Extending the alkylation reaction to 3 h with the addition of 2% SDS part way through the incubation in the presence of increased (300 mM) iodoacetamide did not change the protein pattern, suggesting that incomplete alkylation is not responsible for the observed partial mobility shifts of the mutants. Dimer formation is most evident in Cys83Ala, with lesser amounts in the Cys35Ala mutant and none in the wild-type, which only shows aggregation at high protein concentration (data not shown). In contrast to the wild-type TDPX, no specific disulphide-bridge formation could be detected by mass spectrometry analysis of either oxidized mutant proteins (data not shown). The Cys35Ala mutant was completely devoid of peroxidase activity in the TryX-dependent assay and the Cys83Ala mutant showed only around 1% residual peroxidase activity in comparison to the wild-type protein (Table 3). However, the Cys83Ala mutant displayed 25-fold greater peroxidase activity with DTT as reducing agent than the wild-type protein, equivalent to 64% of the wild-type activity in the TryX-dependent assay. In contrast, GSH did not show this effect. The Cys35Ala mutant exhibited no peroxidase activity at all with DTT or GSH. These results demonstrate that Cys35 is the essential catalytic residue and suggest Cys83 is important for regeneration of Cys35 by TryX.
Table 3.
Peroxidase activity of TDPX1 wild-type and cysteine mutants. Enzymatic activity was determined using 300 μM H2O2 and TryX (5 μM), GSH (3 mM) or DTT (10 mM) as reducing agent. Activity is expressed as a percentage of the wild-type TDPX1 assayed with TryX (6.89 ± 0.06 s−1). See “experimental procedures” for further details. The data are the means ± standard error, n = 3.
| relative activity, % | |||
|---|---|---|---|
|
|
|||
| TryX | GSH | DTT | |
| WILD-TYPE | 100 | 0.022 ± 0.035 | 2.65 ± 0.22 |
| TDPX1 Cys35Ala | 0 ± 0.033 | −0.045 ± 0.015 | 0.25 ± 0.16 |
| TDPX1 Cys83Ala | 1.27 ±1.05 | 0.048 ± 0.036 | 64.5 ± 7.5 |
Intrinsic tryptophan fluorescence
Classical 2-Cys peroxiredoxins are well known for their conformational changes dependent on their redox state [34;35]. As TDPX1 has only one tryptophan residue (see Figure 1) this can be utilized to analyse whether a conformational change occurs during the reaction cycle of the enzyme. The emission spectrum of the indole group of tryptophan is highly dependent on the nature of its environment. The emission maximum of free indole is near 340 nm, whereas it is blue-shifted when it is in a hydrophobic environment, for instance when it is buried within a native protein [36]. Wild-type TDPX1 and the mutants Cys35Ala and Cys83Ala were reduced with 10 mM DTT or oxidized with two equivalents of hydrogen peroxide, respectively. DTT, trace amounts of oxidized DTT or hydrogen peroxide did not influence the spectra (data not shown). The emission maximum in the spectrum of the oxidized wild-type protein is 341.5 nm (Figure 9), suggesting the tryptophan residue is located in a hydrophilic environment likely at the protein surface. Reduction with DTT mediates a blue-shift of the emission maximum to 332 nm indicating a movement of the tryptophan into a more hydrophobic environment, probably into the interior of the protein. The reduced and oxidized spectra of the C83A mutant look similar to the corresponding wild-type spectra. Therefore the Cys83 residue and the disulphide-bridge formation are not essential for the redox-dependent change in fluorescence emission. The spectrum of the oxidised Cys35Ala mutant has an emission maximum of 340 nm, similar to the wild-type oxidised protein. The spectrum of the reduced protein showed no blue-shift of the emission maximum. This suggests that oxidation of the active site cysteine residue triggers a conformational change in TDPX1. In the wild-type spectrum of the reduced protein another effect can be observed: the overall fluorescence is largely quenched. Thus two major effects can be observed upon reduction of TDPX1 wild-type: Firstly, blue-shift of the emission maximum and secondly, quenching of the fluorescence. In all, it can be concluded that the tryptophan environment is different in the two redox stages and thus it can be speculated that a conformational change has to take place during the reaction cycle.
Figure 9. Emission spectra of TDPX1 and cysteine mutants Cys35Ala and Cys83Ala.

The proteins (20 μM) were measured under reduced (10 mM DTT) and oxidized (40 μM H2O2) conditions, respectively. The excitation wavelength was 280 nm.
Discussion
The results presented here represent the first comprehensive comparison of the TDPX and TryP classes of tryparedoxin peroxidases in Leishmania spp.. Despite significant sequence similarity to mammalian GPX4, LmTDPX1 is a bone fide tryparedoxin peroxidase with no physiologically relevant activity with GSH as electron donor. This agrees with previous reports on the orthologues TbTDPX3 [24] and TcTDPX2 [25].
Kinetic analysis of LmTDPX1 with LmTryX as reducing agent shows saturation kinetics obeying a Bi Bi ping pong mechanism. This kinetic behaviour matches that for TcTDPX2 [37], but is in contrast with TbTDPX3 which has been reported to follow an unsaturated ping pong mechanism with infinite Km and kcat with TryX [24]. The reason for this discrepancy is not clear – all three species contain additional tryparedoxin-like proteins containing a WCPPC motif, but the LmTryX used here shows greatest similarity (58% identity) to TbTryX used in earlier studies. Differences in assay conditions or the presence of a histidine tag on the longer N-terminus of TbTDPX3 might also be contributing factors. Nonetheless, in terms of substrate specificity towards hydroperoxides, LmTDPX1 closely resembles TbTDPX3 rather than TcTDPX2, which is apparently inactive with H2O2 as substrate. Thus substrate specificity and mechanism can not be deduced simply on the basis of sequence similarity alone.
Our biochemical studies on LmTDPX1 reveal that the enzyme is functional as a monomer with the loss of 2 thiols per mole of enzyme following oxidation with H2O2. Mass spectrometry and mutagenesis studies indicate formation of a specific disulphide bridge between Cys35 and Cys83. Cys35 is the equivalent residue to the active site selenocysteine in mammalian GPXs and the Cys35Ala mutant is devoid of enzyme activity, indicating that Cys35 is involved in catalysis. The Cys83Ala mutant shows only 1% of wild-type activity with TryX indicating that formation of an intramolecular disulphide is important for interaction with TryX. In contrast, this mutant displayed significant activity with DTT (65% of wild type) suggesting that the putative Cys35 sulphenic acid intermediate is readily accessible to DTT, but much less so to GSH or TryX. Attempts to trap the putative intermediate with 4-chloro-7-nitrobenz-2-oxa-1,3-diazole (NBD chloride) were unsuccessful. The role of the highly conserved Cys64 is less clear, but appears to contribute to the stability of the native conformation of the protein, since we were unable to isolate this mutant. Although it could be involved in disulphide bond formation in the absence of Cys83, it is less likely to be involved in the reaction mechanism because the equivalent mutation (Cys76Ser) in T. brucei TDPX3 had no effect on enzyme activity [38]. Non-specific intramolecular and intermolecular disulphide formation cannot be ruled out based on our current findings.
During completion of this study, Schlecker et al. reported that, for TbTDPX3, Cys-47 is essential for catalytic activity and that oxidation promotes formation of an intramolecular disulphide bridge between Cys47 and Cys95 [38]. These residues in the trypanosome enzyme are the equivalent of Cys35 and Cys83 in the leishmania enzyme. Thus our results support and complement each other in a distantly related parasite. Using molecular modelling, Schlecker et al. suggested that a large conformational change in TbTDPX3 would be necessary to bring the Cys95 region into proximity with the postulated Cys47 sulphenate intermediate for disulphide bond formation. Our studies on intrinsic tryptophan fluorescence could support this hypothesis. In this model this conformational change would cause a shift of the tryptophan into a more polar environment [39]. Clearly, formation of a disulphide-bridge is not an absolute requirement since this effect is observed with the Cys83Ala mutant. However, these spectral changes are only observed when a free Cys35 thiol is present (i.e. wild type and Cys83Ala, reduced forms). Thus, the observed fluorescence quenching and spectral shift could also be due to a charge interaction between the cysteine-35 thiolate and the tryptophan pyrrole ring [40]. This interpretation lends support to the alternative hypothesis that the structure of the Cys95 containing region is significantly different from that of mammalian GPXs. Structural studies are required to resolve these two alternative proposals.
Comparison between LmTDPX1 and LmTryP1 revealed some interesting similarities and differences. Both enzymes obey saturable ping pong kinetics with similar kcat (~15 and ~9 s−1) and Km (~3 and ~ 5 μM) values for LmTryX, irrespective of hydroperoxide substrate. The intracellular concentration of LmTryX (9-28 μM) indicates that TryX is a physiologically relevant substrate in vivo. Both peroxidases have an N-terminal peroxidative cysteine and a C-terminal resolving cysteine, except in monomeric LmTDPX1 these form an intra-molecular disulphide, whereas in LmTryP1 these form reciprocal intermolecular disulphides between active sites in adjacent monomers forming dimers that oligomerise into decamers. LmTDPX1 is 10-fold less active with t-butyl hydroperoxide than either H2O2 or cumene hydroperoxide, whereas LmTryP1 shows no marked substrate preference. The affinity for hydroperoxides is also at least one order of magnitude less for LmTDPX1 (Km > 200 μM) than LmTryP1 (Km ≤ 11 μM). Like many 2-Cys peroxiredoxins [32] LmTryP1 is sensitive to inactivation by over-oxidation, an important regulatory mechanism in mammalian cells [31]. In contrast, LmTDPX1 did not show any sign of inactivation by hydroperoxides. Finally, TryPs constitute 1-4% of the total cellular protein and are at least 40-fold more abundant than TDPXs on a molar basis such that TDPXs contribute less than 1% to the overall peroxidative capacity in L. major. Thus, although TryPs are susceptible to inactivation by hydroperoxides, it seems unlikely that TDPXs could form a significant second line of defence against oxidant stress.
Despite the apparent redundancy of function in detoxification of peroxides, both TDPX and TryP are essential in T. brucei indicating that they must have additional unique functions [26;27]. Mammalian mitochondrial GPX4 protects against oxidant-stress induced apoptosis, whereas cytosolic GPX4 suppresses the activation of lipoxygenases and cycloxygenases involved in inflammation [41] and the S.cerevisiae homologue (GPx3) is implicated in redox signalling [42]. Further investigations could reveal similar functions for TDPXs in leishmania. The pronounced differences in substrate specificity and mechanism between parasite TDPXs and mammalian GPXs suggest they may be potential drug targets. Gene knockout studies are required to determine whether this also applies in L. major.
Experimental procedures
All chemicals were of the highest grade available from Sigma, BDH and Molecular Probes. Restriction enzymes and DNA-modifying enzymes were from Promega.
Cloning and site directed mutagenesis
The gene sequences for TDPX1, TDPX2 and TDPX3, TRYP1 and TRYX were identified using the genome database GeneDB (http:www.genedb.org/). The complete open reading frame of LmjF26.0820 (TDPX1) was amplified by PCR from genomic DNA of L. major Friedlin strain using forward primer containing an NdeI site and reverse primer containing a BamHI site (see table 1). The 525 bp PCR-product was digested with NdeI and BamHI and cloned directly into the NdeI/BamHI site of pET-15b (Novagen) to generate plasmid pET15b-TDPX1. In a similar fashion the open reading frames for TRYX (LmjF29.1160) and TRYP1 (LmjF15.1120) were amplified by PCR and cloned into pET-15b (for primers see table 1). Site directed mutagenesis of TDPX1 was performed using the QuikChange® Site-Directed Mutagenesis Kit (Stratagene) (for primers see table 1). All DNA sequences were verified by the Sequencing Service (School of Life Sciences, University of Dundee, Scotland, www.dnaseq.co.uk).
Table 1.
Primers used for cloning of TDPX1, TryX and TryP1 and site-directed mutagenesis of TDPX1. The initiator and terminator codons are in bold and the restriction sites are underlined. The codons of the mutated amino acids are in bold.
| cloned protein or mutation | primer |
|---|---|
| F- TDPX1 | 5′- TATAT CAT ATG TCT ATC TAC GAC TTC AAG GTC-3′ |
| R- TDPX1 | 5′- ATATA GGA TCC TCA CGA TTG AGT GCT TGG -3′ |
| F- TryX | 5′-ATATATCATATGTCCGGTGTCGCAAAG- 3′ |
| R- TryX | 5′-ATATAGGATCCTTACTCGTCTCTCCACGG- 3′ |
| F- TryP1 | 5′- ATATAT CATATG TCCTGCGGTAACGCC-3′ |
| R- TryP1 | 5′-ATATA GGA TCC TTA CTG CTT GCT GAA GTA TC- 3′ |
| F- TDPX1 Cys35Ala | 5′- CAACGTAGCCAGCAAGGCCGGCTTCACCAAGGGCG-3′ |
| R- TDPX1 Cys35Ala | 5′- CGCCCTTGGTGAAGCCGGCCTTGCTGGCTACGTTG-3′ |
| F- TDPX1 Cys64Ala | 5′-GGTACTGGCGTTCCCGGCCAACCAGTTCGCCGGTC-3′ |
| R- TDPX1 Cys64Ala | 5′-GACCGGCGAACTGGTTGGCCGGGAACGCCAGTACC-3′ |
| F- TDPX1 Cys83Ala | 5′- AGGTGAAAAGTTTCGCCGCCACGCGTTTCAAGGCTGAG-3′ |
| R- TDPX1 Cys83Ala | 5′-CTCAGCCTTGAAACGCGTGGCGGCGAAACTTTTCACCT-3′ |
Expression and purification of L. major wild type TDPX1 and mutants, TryX and TryP1
Competent BL21 (DE3) pLysS (Merck Bioscience) were transformed with the plasmid pET-15b-TDPX1. At an optical density of ~ 0.5-0.7 the bacteria were induced with 0.4 mM isopropyl-β-D-thiogal-actopyranoside and harvested by centrifugation 4-6 h later. The pellet from 1 L bacteria culture was stored at −80 °C.
The pellet was thawed and resuspended in 50 ml buffer A (50 mM Tris-HCl pH 8.0, 250 mM NaCl, 5 mM imidazole) supplemented with one Complete Protease Inhibitor tablet (Roche)). The solution was mixed with 25 μg ml−1 DNAse and shaken for 30 min on ice to lyse the cells. After sonication (6 × 30 s on ice) the broken cells were centrifuged for 45 min at 50 000 × g at 4°C. The supernatant was filtered sterilized (Steriflip®, Millipore) and loaded on a 1 ml HisTrap column (Amersham) previously equilibrated with buffer A. The column was washed with 50 ml buffer B (buffer A + 20 mM imidazole), 25 ml buffer C (buffer A + 20 mM imidazole, 20% glycerol) and protein eluted with buffer E (buffer A + 250 mM imidazole).
The hexahistidine-tag was removed by incubating pooled fractions with thrombin (1U per 100 μg of TDPX1 at room temperature for 10 h) and dialyzed against 50 mM Tris-HCl, 20 mM NaCl. Thrombin was removed by incubation with benzamidine beads (Amersham) and any residual His-tagged protein with Ni2+-nitrilotriacetate (Ni-NTA) beads (Quiagen). A further purification step was performed using a 5 ml HiTrap Q HP column (Amersham) and 50 mM Tris-HCl, pH 8.0, 20 mM NaCl as equilibration buffer. The protein was found in the flow through and in the first wash fractions with equilibration buffer. Size exclusion chromatography was performed using a Superdex 75 HR 10/30 column (Amersham) and 50 mM Hepes-NaOH pH 7.4. Gel Filtration Standard (Bio-Rad) was used as calibration standards.
TryP1 and TryX were expressed and purified on a Ni-NTA matrix column in a similar manner as TDPX1. The only difference was that TryX was expressed at 25°C after induction overnight. His-tagged proteins were dialyzed against 50 mM Hepes buffer pH 7.4 and stored at −80°C. His-tags from TryP1 and TryX were removed in a similar manner than from TDPX1. A tenfold amount of thrombin (10 U per 100 μg) was necessary for the complete His-tag removal from TryP1.
Protein concentrations of all the purified proteins were determined at different dilutions from the absorbance at 280 nm using the theoretical extinction coefficients calculated from ExPASy (http://us.expasy.org/tools/protparam.html) assuming all cysteine residues were in the reduced state.
Analysis of TDPX1, TryP1 and TryX concentrations in L. major promastigotes
L. major promastigotes (Friedlin strain; WHO designation: MHOM/JL/81/Friedlin) were grown in M199 medium (Caisson Laboratories) with supplements as described earlier [43]. Parasites (2 × 108) were pelleted at 2000 × g for 10 min, washed with 5 ml PBS buffer and centrifuged again under the same conditions. Finally, parasites were resuspended in 0.4 ml of 50 mM Tris-HCl pH 8.0, 9 M urea, and 0.1% Triton X-100 for determination of protein concentration. For Western-blot analysis the parasites were resuspended in 0.4 ml 2x loading buffer. The parasite mixtures were heated for 10 min at 95°C. The crude urea lysates were centrifuged (16000 × g, 15 min) and protein content in the supernatants determined by the Bradford protein assay (BioRad) using BSA as standard. L. major protein extracts in loading buffer (3 × 106 parasites for TDPX1 and 1.5 × 106 parasites for TryX and TryP1) were analysed by SDS-PAGE (12%-NuPAGE-gel, Invitrogen) with varying amounts of non-tagged recombinant TDPX1 (4-20 ng), TryX (4-20 ng) or TryP1 (250-1000 ng) protein included as calibration standards. Cell volumes were determined using a Schärfe CASY cell counter.
Proteins were analysed by Western blotting, using a 1:500 dilution of the TDPX1 antibody, 1:2000 for TryX and 1:5000 for TryP1. Polyclonal antisera against recombinant non-tagged TDPX1, TryX and TryP (LmjF15.1060) were raised in adult male Wistar rats as described elsewhere [43]. Polyclonal rabbit–anti rat immunoglobulin/HR (DAKO A/S) in a 1: 5000 dilution was used as secondary antibody. Finally the proteins were detected using the ECL Plus Western Blotting Detection System (Amersham). The intensity of the protein bands were quantified as absolute integrated optical density using LabWorks Imaging and Analysis Software (UVP, UK). The resulting data were plotted against the protein concentration and linear regression analysis performed using the software GraFit. Results are the means of at least two independent experiments.
Enzyme Assays
Peroxidase activity of TDPX1 was determined using TryX, glutathione or DTT as reducing agents.
Tryparedoxin-dependent assays were performed in a volume of 250 μl containing 50 mM HEPES-NaOH pH 7.4, 1 mM EDTA, 3 U ml−1 T. cruzi TryR [44], 100 μM trypanothione disulphide (T[S]2 Bachem), 250 μM NADPH, 0.5-5 μM TryX, 0.2 μM TDPX1 and either 50-500 μM H2O2, 50 – 2000 μM t-butyl hydroperoxide or 50 – 1000 μM cumene hydroperoxide, respectively. After 5 min of incubation at 27°C to allow complete reduction of T[S]2 to T[SH]2 the background was measured for 2 minutes by addition of the peroxide to the assay lacking TDPX1. Finally, the reaction was started by addition of TDPX1 and the consumption of NADPH due to a decrease of absorbance at 340 nm was measured with a UV-VIS spectrophotometer (Shimadzu, UV-2401 PC). The combined data were fitted by non-linear least squares regression using the software GraFit to the following equation describing a Bi Bi ping-pong mechanism (where A = ROOH and B = TryX):
| 1 |
Glutathione-dependent assays were performed similar to the tryparedoxin-dependent assay in a volume of 250 μl at 27 °C containing 50 mM HEPES, pH 7.4, 1 mM EDTA, 150 μM NADPH, 3 mM GSH, 0.2 U ml−1 yeast glutathione reductase (Sigma), 300 μM H2O2 and 5 μM TDPX1 wild-type or mutants. Bovine GPX (0.05 U ml−1, Sigma) was used as positive control. Assays were corrected for non-enzymatic activity.
DTT-dependent assays were performed in a volume of 150 μl at 27 °C containing 50 mM HEPES, pH 7.4, 10 mM DTT and 5 μM wild-type TDPX1, 5 μM C35A-TDPX1 or 0.5 μM C83A-TDPX1, respectively. The reaction was started with 300 μM H2O2. At different time points 20 μl samples were added to 1 ml Peroxoquant reagent (Perbio Science) and residual H2O2 quantified colourimetrically at 550 nm with a UV-VIS spectrophotometer (Shimadzu, UV-2401 PC). A calibration curve was performed by adding different amounts of hydrogen peroxide in a 20 μl sample volume directly to 1 ml Peroxoquant reagent. Assays were corrected for non-enzymatic activity.
The kinetic properties of TryP1 were determined using the same conditions as in the TryX-dependent TDPX1 assay. The only differences were that the assays were started with the addition of 50 μM hydroperoxide and TryX concentrations were varied between 2 and 10 μM. The consumption of NADPH in the presence of all components except peroxidase was measured to be ≤ 1% of the enzymatic rate and was thus neglected. The data were analysed using the integrated Dalziel rate equation for a two substrate enzymatic system:
| 2 |
where,
The Quantitative Analysis of Sulphydryl Groups
Free sulphydryl groups in TDPX1 were determined using 5, 5′-dithio-bis(2-nitrobenzoic acid) (DTNB) [45]. Reduced and oxidized protein was obtained by treatment with 50 mM DTT or a fivefold excess of hydrogen peroxide, respectively, followed by size exclusion chromatography (S75 HR 10/30) to remove DTT or a PD10 Desalting column (GE healthcare) for the removal of hydrogen peroxide. TDPX1 (10 μM) was added to DTNB (2 mM) in 50 mM Tris-HCl, pH 8.0 and the absorbance at 412 nm was measured against a 2 mM DTNB solution as reference. The amounts of reactive sulphydryl groups were determined using ε412 = 13,600 M–1·cm–1 [46]. Two independent experiments were performed on triplicate samples.
Mobility shift of reduced and oxidized wild-type and mutant TDPX1 and Mass Spectrometry
TryP1, TDPX1 wild-type and Cys mutants (20 μM each) were incubated with 20 mM DTT or 40 μM H2O2 for 10 min. The redox state was fixed by alkylation of any remaining sulphydryl groups by incubation with 100 mM iodoacetamide in the dark for 30 min. Protein samples (2 μg per lane) were separated by SDS-PAGE in a 12% acrylamide gel and stained with Coomassie. Bands of reduced and oxidized TDPX1 wild-type were excised and digested with trypsin or chymotrypsin and peptides analysed by the Mass Fingerprinting Service (Wellcome Trust Biocentre, University of Dundee).
Intrinsic tryptophan fluorescence
Wild-type and Cys mutants of TDPX1 (20 μM) were incubated with either 10 mM DTT or 40 μM H2O2 for 5 min. An excitation wavelength of 280 nm was used and the fluorescence emission spectrum was recorded from 300-500 nm with a Varian Carey Eclipse fluorescence spectrophotometer. An overlay of 15 spectra is shown.
Acknowledgements
This work was funded by the Wellcome Trust. We would like to thank Hongtu Ye for preliminary studies, Susan Wyllie and Neil Greig for producing antiserum to TDPX1, TryX and TryP and Ahilan Saravanamuthu and Anna S.F. Marques for providing recombinant T. cruzi TryR.
Abbreviations
- GPX
glutathione peroxidase
- TryX
tryparedoxin
- TryP
tryparedoxin peroxidase type I
- TDPX
tryparedoxin peroxidase type II
- TryR
trypanothione reductase
- DTNB
5, 5′-dithio-bis(2-nitrobenzoic acid
- ROOH
hydroperoxide
- ROH
alcohol
- T[SH]2 and T[S]2
trypanothione and trypanothione disulphide, respectively.
References
- [1].Schirmer RH, Schollhammer T, Eisenbrand G, Krauth-Siegel RL. Oxidative stress as a defense mechanism against parasitic infections. Free Radic. Res. Commun. 1987;3:3–12. doi: 10.3109/10715768709069763. [DOI] [PubMed] [Google Scholar]
- [2].Croft SL, Sundar S, Fairlamb AH. Drug Resistance in Leishmaniasis. Clin. Microbiol. Rev. 2006;19:111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Adak S, Datta AK. Leishmania major encodes an unusual peroxidase that is a close homologue of plant ascorbate peroxidase: a novel role of the transmembrane domain. Biochem. J. 2005;390:465–474. doi: 10.1042/BJ20050311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wilkinson SR, Obado SO, Mauricio IL, Kelly JM. Trypanosoma cruzi expresses a plant-like ascorbate-dependent hemoperoxidase localized to the endoplasmic reticulum. Proc. Natl. Acad. Sci. U. S. A. 2002;99:13453–13458. doi: 10.1073/pnas.202422899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dumas C, Ouellette M, Tovar J, Cunningham ML, Fairlamb AH, Tamar S, Olivier M, Papadopoulou B. Disruption of the trypanothione reductase gene of Leishmania decreases its ability to survive oxidative stress in macrophages. EMBO J. 1997;16:2590–2598. doi: 10.1093/emboj/16.10.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tovar J, Wilkinson S, Mottram JC, Fairlamb AH. Evidence that trypanothione reductase is an essential enzyme in Leishmania by targeted replacement of the tryA gene locus. Mol. Microbiol. 1998;29:653–660. doi: 10.1046/j.1365-2958.1998.00968.x. [DOI] [PubMed] [Google Scholar]
- [7].Tovar J, Cunningham ML, Smith AC, Croft SL, Fairlamb AH. Down-regulation of Leishmania donovani trypanothione reductase by heterologous expression of a trans-dominant mutant homologue: effect on parasite intracellular survival. Proc. Natl. Acad. Sci. USA. 1998;95:5311–5316. doi: 10.1073/pnas.95.9.5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nogoceke E, Gommel DU, Kiess M, Kalisz HM, Flohé L. A unique cascade of oxidoreductases catalyses trypanothione-mediated peroxide metabolism i. Crithidia fasciculata. Biol. Chem. 1997;378:827–836. doi: 10.1515/bchm.1997.378.8.827. [DOI] [PubMed] [Google Scholar]
- [9].Gommel DU, Nogoceke E, Morr M, Kiess M, Kalisz HM, Flohé L. Catalytic characteristics of tryparedoxin. Eur. J. Biochem. 1997;248:913–918. doi: 10.1111/j.1432-1033.1997.t01-1-00913.x. [DOI] [PubMed] [Google Scholar]
- [10].Tetaud E, Fairlamb AH. Cloning, expression and reconstitution of the trypanothione-dependent peroxidase system of Crithidia fasciculata. Mol. Biochem. Parasitol. 1998;96:111–123. doi: 10.1016/s0166-6851(98)00120-0. [DOI] [PubMed] [Google Scholar]
- [11].Montemartini M, Kalisz HM, Hecht HJ, Steinert P, Flohé L. Activation of active-site cysteine residues in the peroxiredoxin-type tryparedoxin peroxidase of Crithidia fasciculata. Eur. J. Biochem. 1999;264:516–524. doi: 10.1046/j.1432-1327.1999.00656.x. [DOI] [PubMed] [Google Scholar]
- [12].Alphey MS, Bond CS, Tetaud E, Fairlamb AH, Hunter WN. The structure of reduced tryparedoxin peroxidase reveals a decamer and insight into reactivity of 2Cys-peroxiredoxins. J. Mol. Biol. 2000;300:903–916. doi: 10.1006/jmbi.2000.3881. [DOI] [PubMed] [Google Scholar]
- [13].Levick MP, Tetaud E, Fairlamb AH, Blackwell JM. Identification and characterisation of a functional peroxidoxin from Leishmania major. Mol. Biochem. Parasitol. 1998;96:125–137. doi: 10.1016/s0166-6851(98)00122-4. [DOI] [PubMed] [Google Scholar]
- [14].Castro H, Budde H, Flohé L, Hofmann B, Lunsdorf H, Wissing J, Tomás AM. Specificity and kinetics of a mitochondrial peroxiredoxin of Leishmania infantum. Free Radic. Biol. Med. 2002;33:1563–1574. doi: 10.1016/s0891-5849(02)01088-2. [DOI] [PubMed] [Google Scholar]
- [15].Castro H, Sousa C, Santos M, Cordeiro-Da-Silva A, Flohe L, Tomas AM. Complementary antioxidant defense by cytoplasmic and mitochondrial peroxiredoxins in Leishmania infantum. Free Radic. Biol. Med. 2002;33:1552–1562. doi: 10.1016/s0891-5849(02)01089-4. [DOI] [PubMed] [Google Scholar]
- [16].Barr SD, Gedamu L. Cloning and characterization of three differentially expressed peroxidoxin genes from Leishmania chagasi - Evidence for an enzymatic detoxification of hydroxyl radicals. J. Biol. Chem. 2001;276:34279–34287. doi: 10.1074/jbc.M104406200. [DOI] [PubMed] [Google Scholar]
- [17].Jirata D, Kuru T, Genetu A, Barr S, Hailu A, Aseffa A, Gedamu L. Identification, sequencing and expression of peroxidoxin genes from Leishmania aethiopica. Acta Trop. 2006;99:88–96. doi: 10.1016/j.actatropica.2006.08.001. [DOI] [PubMed] [Google Scholar]
- [18].Flohé L, Budde H, Bruns K, Castro H, Clos J, Hofmann B, Kansal-Kalavar S, Krumme D, Menge U, Plank-Schumacher K, Sztajer H, Wissing J, Wylegalla C, Hecht HJ. Tryparedoxin peroxidase of Leishmania donovani: Molecular cloning, heterologous expression, specificity, and catalytic mechanism. Arch. Biochem. Biophys. 2002;397:324–335. doi: 10.1006/abbi.2001.2688. [DOI] [PubMed] [Google Scholar]
- [19].Walker J, Acestor N, Gongora R, Quadroni M, Segura I, Fasel N, Saravia NG. Comparative protein profiling identifies elongation factor-1 beta and tryparedoxin peroxidase as factors associated with metastasis in Leishmania guyanensis. Mol. Biochem. Parasitol. 2006;145:254–264. doi: 10.1016/j.molbiopara.2005.10.008. [DOI] [PubMed] [Google Scholar]
- [20].Lin YC, Hsu JY, Chiang SC, Lee ST. Distinct overexpression of cytosolic and mitochondrial tryparedoxin peroxidases results in preferential detoxification of different oxidants in arsenite-resistant Leishmania amazonensis with and without DNA amplification. Mol. Biochem. Parasitol. 2005;142:66–75. doi: 10.1016/j.molbiopara.2005.03.009. [DOI] [PubMed] [Google Scholar]
- [21].Stober CB, Lange UG, Roberts MTM, Alcami A, Blackwell JM. Heterologous priming-boosting with DNA and modified vaccinia virus Ankara expressing tryparedoxin peroxidase promotes long-term memory against Leishmania major in susceptible BALB/c mice. Infect. Immun. 2007;75:852–860. doi: 10.1128/IAI.01490-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wilkinson SR, Taylor MC, Touitha S, Mauricio IL, Meyer DJ, Kelly JM. TcGPXII, a glutathione-dependent Trypanosoma cruzi peroxidase with substrate specificity restricted to fatty acid and phospholipid hydroperoxides, is localized to the endoplasmic reticulum. Biochem. J. 2002;364:787–794. doi: 10.1042/BJ20020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wilkinson SR, Meyer DJ, Kelly JM. Biochemical characterization of a trypanosome enzyme with glutathione-dependent peroxidase activity. Biochem. J. 2000;352:755–761. [PMC free article] [PubMed] [Google Scholar]
- [24].Hillebrand H, Schmidt A, Krauth-Siegel RL. A second class of peroxidases linked to the trypanothione metabolism. J. Biol. Chem. 2003;278:6809–6815. doi: 10.1074/jbc.M210392200. [DOI] [PubMed] [Google Scholar]
- [25].Wilkinson SR, Meyer DJ, Taylor MC, Bromley EV, Miles MA, Kelly JM. The Trypanosoma cruzi enzyme TcGPXI is a glycosomal peroxidase and can be linked to trypanothione reduction by glutathione or tryparedoxin. J. Biol. Chem. 2002;277:17062–17071. doi: 10.1074/jbc.M111126200. [DOI] [PubMed] [Google Scholar]
- [26].Wilkinson SR, Horn D, Prathalingam SR, Kelly JM. RNA interference identifies two hydroperoxide metabolizing enzymes that are essential to the bloodstream form of the African trypanosome. J. Biol. Chem. 2003;278:31640–31646. doi: 10.1074/jbc.M303035200. [DOI] [PubMed] [Google Scholar]
- [27].Schlecker T, Schmidt A, Dirdjaja N, Voncken F, Clayton C, Krauth-Siegel RL. Substrate specificity, localization, and essential role of the glutathione peroxidase-type tryparedoxin peroxidases in Trypanosoma brucei. J. Biol. Chem. 2005;280:14385–14394. doi: 10.1074/jbc.M413338200. [DOI] [PubMed] [Google Scholar]
- [28].Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clayton C, Coulson RM, Cronin A, Cruz AK, Davies RM, De Gaudenzi J, Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser A, Fuchs M, Gabel C, Goble A, Goffeau A, Harris D, Hertz-Fowler C, Hilbert H, Horn D, Huang Y, Klages S, Knights A, Kube M, Larke N, Litvin L, Lord A, Louie T, Marra M, Masuy D, Matthews K, Michaeli S, Mottram JC, Muller-Auer S, Munden H, Nelson S, Norbertczak H, Oliver K, O’neil S, Pentony M, Pohl TM, Price C, Purnelle B, Quail MA, Rabbinowitsch E, Reinhardt R, Rieger M, Rinta J, Robben J, Robertson L, Ruiz JC, Rutter S, Saunders D, Schafer M, Schein J, Schwartz DC, Seeger K, Seyler A, Sharp S, Shin H, Sivam D, Squares R, Squares S, Tosato V, Vogt C, Volckaert G, Wambutt R, Warren T, Wedler H, Woodward J, Zhou S, Zimmermann W, Smith DF, Blackwell JM, Stuart KD, Barrell B, Myler PJ. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Epp O, Ladenstein R, Wendel A. The refined structure of the selenoenzyme glutathione peroxidase at 0.2-nm resolution. Eur. J. Biochem. 1983;133:51–69. doi: 10.1111/j.1432-1033.1983.tb07429.x. [DOI] [PubMed] [Google Scholar]
- [30].Opperdoes FR, Szikora JP. In silico prediction of the glycosomal enzymes of Leishmania major and trypanosomes. Mol. Biochem. Parasitol. 2006;147:193–206. doi: 10.1016/j.molbiopara.2006.02.010. [DOI] [PubMed] [Google Scholar]
- [31].Wood ZA, Schroder E, Harris JR, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- [32].Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- [33].Hofmann B, Hecht HJ, Flohe L. Peroxiredoxins. Biol. Chem. 2002;383:347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- [34].Konig J, Lotte K, Plessow R, Brockhinke A, Baier M, Dietz KJ. Reaction mechanism of plant 2-Cys peroxiredoxin - Role of the C terminus and the quaternary structure. J. Biol. Chem. 2003;278:24409–24420. doi: 10.1074/jbc.M301145200. [DOI] [PubMed] [Google Scholar]
- [35].Wood ZA, Poole LB, Hantgan RR, Karplus PA. Dimers to doughnuts: Redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- [36].Szabo AG. Fluorescence principles and measurement. In: Gore MG, editor. Spectrophotometry & Spectrofluorimetry. Oxford University Press; Oxford: 2000. pp. 33–67. [Google Scholar]
- [37].Wilkinson SR, Kelly JM. The role of glutathione peroxidases in trypanosomatids. Biol. Chem. 2003;384:517–525. doi: 10.1515/BC.2003.060. [DOI] [PubMed] [Google Scholar]
- [38].Schlecker T, Comini MA, Melchers J, Ruppert T, Krauth-Siegel RL. Catalytic mechanism of the glutathione peroxidase-type tryparedoxin peroxidase of Trypanosoma brucei. Biochem. J. 2007 doi: 10.1042/BJ20070259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Reshetnyak YK, Koshevnik Y, Burstein EA. Decomposition of protein tryptophan fluorescence spectra into log-normal components. III. Correlation between fluorescence and microenvironment parameters of individual tryptophan residues. Biophys. J. 2001;81:1735–1758. doi: 10.1016/S0006-3495(01)75825-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Vivian JT, Callis PR. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 2001;80:2093–2109. doi: 10.1016/S0006-3495(01)76183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 2003;34:145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- [42].Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell. 2002;111:471–481. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- [43].Oza SL, Shaw MP, Wyllie S, Fairlamb AH. Trypanothione biosynthesis in Leishmania major. Mol. Biochem. Parasitol. 2005;139:107–116. doi: 10.1016/j.molbiopara.2004.10.004. [DOI] [PubMed] [Google Scholar]
- [44].Borges A, Cunningham ML, Tovar J, Fairlamb AH. Site-directed mutagenesis of the redox-active cysteines of Trypanosoma cruzi trypanothione reductase. Eur. J. Biochem. 1995;228:745–752. doi: 10.1111/j.1432-1033.1995.tb20319.x. [DOI] [PubMed] [Google Scholar]
- [45].Ellman GL. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- [46].Gething MJH, Davidson BE. Molar absorption-coefficient of reduced Ellmans reagent - 3-Carboxylato-4-nitro-thiophenolate. Eur. J. Biochem. 1972;30:352. [Google Scholar]