

Abstract
Cervical cancer (CC) is the second most common cancer in women. Currently no tractable molecular based therapeutic targets exist for patients with invasive CC and no predictive markers of risk assessment for progression of precancerous lesions are identified. New molecular insights into CC pathogenesis are urgently needed. Towards this goal, we first determined the copy number alterations of chromosome 4 and then examined the role of PCDH10 mapped to 4q28 as a candidate tumor suppressor gene. We identified monosomy 4 in 47% of 81 invasive CC studied by SNP array and found that 91% of 130 invasive CC harboring methylation in the promoter region of the PCDH10 gene. We then showed that aberrant promoter hypermethylation of PCDH10 is associated with down-regulation of gene expression and cell lines exposed to demethylating agent resulted in profound reactivated gene expression. We also showed that the promoter methylation in the PCDH10 gene occurs at an earliest identifiable stage of low-grade squamous intraepithelial lesion (LSIL). Our studies demonstrate that inactivation of PCDH10 may be a critical event in CC progression and form a potentially useful therapeutic target for CC.
INTRODUCTION
Chromosome 4 frequently undergoes nonrandom loss in cervical cancer (CC) as documented by loss of heterozygosity (LOH) and chromosomal comparative genomic hybridization (cCGH) analyses (Mitra et al., 1994; Hampton et al., 1996; Rao et al., 2004). Furthermore, introduction of chromosome 4 in HeLa cells suppresses telomerase activity, induces senescent-like phenotype, and the distal region on the long arm of chromosome 4 has been shown to carry putative senescence genes (Backsch et al., 2001, 2005). Additionally, 4q deletions have been reported to occur at early stages in CC progression (Backsch et al., 2005; Singh et al., 2007). These findings suggest the loss of function of one or more proliferation-regulating genes on chromosome 4 and their involvement in malignant progression of cervical epithelium. However, no known tumor suppressor genes on chromosome 4 have been implicated so far in CC. Chromosome 4 carries at least three protocadherin family genes, PCDH7, PCDH10, and PCDH18. The protocadherins are a subfamily of the cadherin superfamily genes that encodes cadherin-related neuronal receptors that play a role in the establishment and function of specific cell-cell connections (Yagi, 2008). Recently a number of PCDH genes have been implicated as tumor suppressor genes (Waha et al., 2005; Imoto et al., 2006; Yu et al., 2008). Previously, it has been shown that PCDH10 is hypermethylated and functions as a tumor suppressor in multiple human cancer types (Ying et al., 2006, 2007; Yu et al., 2009).
Despite the successful use of pap-smear screening programs in early detection and treatment, CC remains a major cause of cancer deaths in women world-wide (Waggoner, 2003). CC progresses by distinct morphological changes from normal epithelium to carcinoma through low-grade squamous intraepithelial lesions (LSIL) and high-grade SILs (HSIL). Although infection of high-risk human papillomavirus (HPV) is recognized as an important initiating event in cervical tumorigenesis, HPV alone is not sufficient for the progression to invasive cancer (zur Hausen, 2002). Even though the prophylactic HPV vaccine can prevent onset of precancerous cervical lesions, no biological or genetic markers are available to predict which precancerous lesions progress to invasive CC. The molecular genetic aspects of progression of precursor SILs to invasive cancer remain poorly understood. Therefore identification of critical “genetic hits” in CC is important in understanding the natural history and biology.
In the present study, we examined the role of PCDH10 in CC tumorigenesis and identified promoter hypermethylation as a major mechanism of inactivation of this gene. We also have shown that the promoter hypermethylation of PCDH10 occurs very early in the progression suggesting a role for PCDH10 in the progression of CC.
MATERIALS AND METHODS
Patients, Tumor Tissues, and Cell Lines
A total of 398 samples of DNA representing various stages of cervical cancer progression were utilized in the present study. These include 130 invasive CCs (nine cell lines and 121 cases of primary tumors), 268 cytologic pap smears from normal and various stages of precancerous lesions. The cell lines (HT-3, ME-180, CaSki, MS751, C-4I, C-33A, SW756, HeLa, and SiHa) were obtained from American Type Culture Collection (ATCC, Manassas, VA) and grown in tissue culture as per the supplier's specifications. All specimens were obtained from Columbia University Medical Center (New York), Instituto Nacional de Cancerología (Santa Fe de Bogota, Colombia), the Department of Gynecology of Campus Benjamin Franklin, Charité-Universitätsmedizin Berlin (Germany) and Department of Clinical Oncology, The Chinese University of Hong Kong with appropriate informed consent and approval of protocols by institutional review boards. All primary tumors were diagnosed as squamous cell carcinoma except seven that were diagnosed as adenocarcinoma. Clinical information such as age, stage and size of the tumor, follow-up data after initial diagnosis and treatment was obtained for the majority of tumors from the review of institutional medical records. Tissues were frozen at -80°C immediately after resection and were embedded with tissue freeze medium (OTC) before microdissection. All primary tumor specimens were determined to contain at least 60% tumor by examination of hematoxylin and eosin (H&E) staining of adjacent sections. Cytologic specimens were collected after visualization of the cervical os, the ectocervix was sampled with a spatula and endocervical cells obtained with a brush rotated three hundred sixty degrees. Exfoliated cells were immediately placed in PreservCyt Solution (Cytc Corporation, Marlborough, MA) for routine processing by a cytopathologist. Cells were collected from normal and precancerous lesions simultaneously from the same spatula for both cytology and DNA. Cells collected in phosphate buffered saline were stored at -80°C for DNA isolation. A total of 63 pap smears were diagnosed as normal, 35 as atypical squamous cells of undetermined significance (ASC-US), 107 as LSIL, and 63 as HSIL. The diagnosis of all HSILs was also confirmed by a biopsy. High-molecular-weight DNA from frozen tumor tissues, cell lines, and cell pellets from pap smears was isolated by standard methods. Human papillomavirus types were identified as described earlier (Narayan et al., 2003).
SNP Array Analysis
The Affymetrix 250K NspI SNP chip with an average coverage of 11.5 kb of the genome was utilized for copy number analysis as per the manufacturer's protocol. We performed SNP array on 81 CC cases (9 cell lines and 72 primary tumors enriched for tumor cells by microdissection) selected based on high tumor content and 7 microdissected normal cervical squamous epithelial samples as controls to serve as the reference for copy number analysis. Acquisition of SNP data and analysis of copy number data for chromosome 4, representing 7.1% SNPs on 250K array, using CytoBand information files from the dChip website (http://biosun1.harvard.edu/complab/dchip/chromosome.htm#refgene) was performed as previously described (Scotto et al., 2008a, b). Copy numbers <1.5 were considered as deletion, 2.5–4.0 as gain, and >4.1 as amplification in the raw copy number view.
Methylation Specific PCR (MSP) and Sequencing
Genomic DNA was treated with sodium bisulphite as described (Narayan et al., 2003). Placental DNA treated in vitro with SssI methyltransferase (New England BioLabs, Beverly, MA) and normal lymphocyte DNA converted with sodium bisulphite was used as methylated and unmethylated controls, respectively. Following three sets of primers for amplification of methylated (M) DNA and one set of primers for unmethylated (U) DNA spanning the CpG Island of cDNA clone NM_032961 were designed:
PCDH10-MF-1 5’-GATTTTCGTCGCGTATTGGT-3’, and
PCDH10-MR-1 5’-CACCGCTTCCTTCACGATAC-3’ spanning exon 1 (+905 to +1101 bp),
PCDH10-MF-2 5’-GCGTTTTTATTAGGGCGTAAC-3’, and
PCDH10-MR-2 5’-CGATACTATAAATAAACCGAACGTT-3’ spanning exon 1 (+1726 to +1862 bp)
PCDH10-MF3 5’-ATTGTAGTGGTTCGGGATCG-3’, and
PCDH10-MR3 5’-CGCCAAACACGTTATTTTCA-3’ spanning exon 1 (+1294 to 1438)
PCDH10-UF 5’-TGTTTTGGGTGTGGGAGTTT-3’ and
PCDH10-UR 5’-CCCTCACTCACCACTTCCTT-3’ spanning exon 1(+881 to +1090 bp).
The following additional primer set spanning 40 CpG sites between +1121 to +1618 bp and the sequence common to both methylated and unmethylated templates was used for cloning and sequencing.
PCDH10-cl-F 5’-GTATTGTGGTGGTTTTTTTTAG-3’, and
PCDH10-cl-R 5’-TAAAATCCTTCAACTACTCATAATC-3’.
MSP of a proportion of precancerous lesions obtained from Hong Kong was performed as previously described (Ying et al., 2006). PCR was performed by using standard conditions for 30 cycles on primary invasive cancer and 35 cycles for precancerous lesions with annealing temperatures varying between 56-62°C.
PCR products were run on 2% agarose gels and visualized after ethidium bromide staining. All MSP experiments were performed in triplicate and the promoter hypermethylation was considered positive when present in at least one of the regions. MSP products were either directly sequenced or sub-cloned into pCR2.1-TOPO (Invitrogen) followed by sequencing of multiple clones using M13 primers.
Drug Treatment
Cells in culture were treated with 5 or 10 μM 5-Aza-2’deoxycytidine (5-aza-CdR) for 5 days as described (Narayan et al., 2003).
Real Time RT-PCR Analysis
Total RNA isolated from normal cervical squamous epithelium (three samples obtained from different commercial sources and five samples from hysterectomy specimens), tumor tissues, and cervical cancer cell lines was reverse transcribed as described (Narayan et al., 2003). Relative quantitation of expression of PCDH10 (Assay ID Hs00263133) and Human GAPDH as endogenous control (FAM / MGB Probe) genes was performed in triplicate experiments using TaqMan Gene Expression Assay using the Applied Biosystems 7500 Fast Real-Time PCR system (Foster City, CA, USA). Briefly, PCR was performed using standard thermal cycle conditions of 2 min 50°C, 10 min 95°C, 15 sec 95°C and 1 min 60°C for 40 or 45 cycles with input cDNA ranging from 50-100 ng. The CT and comparative CT (ΔΔCT) values were calculated using Applied Biosystems real-time PCR system software package setting at auto CT and baseline.
RESULTS AND DISCUSSION
Monosomy of Chromosome 4 is a Frequent Genomic Alteration in Invasive CC
Affymetrix 250K NspI SNP array analysis was performed on a panel of 81 CC cases (72 primary tumors and 9 cell lines) to identify genome-wide copy number alterations (CNA) (unpublished data). Analysis of the dataset of chromosome 4 CNA showed copy number losses on chromosome 4 in 38 (47%) CC cases. Of these, 34 (89.5%) cases showed loss of the entire chromosome 4 (monosomy) (Fig. 1)(Supplementary Table). Of note, no detectable amplifications were found on chromosome 4, while only rare non-recurrent copy number gains of smaller regions on chromosome 4 were observed in 4 of 81 cases (4.9%). These data demonstrate that chromosome 4 is frequently lost in CC. To identify the clinical significance, we evaluated the association of chromosome 4 losses with pathologic features such as histology, age, stage and size of the tumor, treatment outcome, and HPV type by univariate analyses and found no significant associations. This finding thus suggests that chromosome 4 loss is a common genomic alteration resulting in dysregulation of important genes that play a role in the development of CC. Cytogenetic studies have identified frequent loss of chromosome 4 in a wide-variety of hematologic malignancies such as diffuse large B-cell lymphoma, follicular lymphoma, multiple myeloma, acute lymphoblastic leukemia/lymphoma, acute and chronic myelogenous leukemia, and solid tumors such as carcinoma of breast, large intestine, kidney, ovary, pancreas, brain, testicular germ cell tumor, and Wilms tumor (http://cgap.nci.nih.gov/Chromosomes/RecurrentAberrations). These data, therefore, suggest that chromosome 4 losses may play an important role in multiple tumor types implicating for the presence of one or more tumor suppressor genes on this chromosome.
Figure 1.
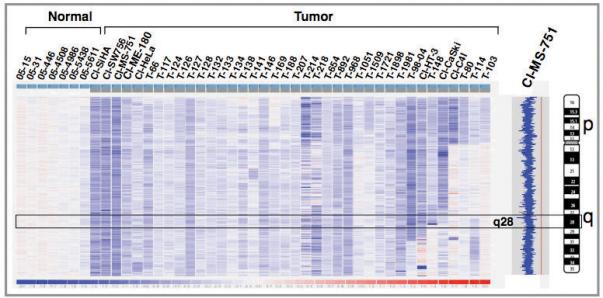
Identification of chromosome 4 copy number alterations in invasive CC by 250K NspI SNP array. Each vertical column represents a sample with genomic regions representing from pter (top) to qter (bottom). Prefix “T” indicates primary tumor; “Cl” indicates cell line. The blue-red scale bar (-2 to +2) at the bottom represents the copy number changes relative to mean across the samples. The intensities of blue and red indicate relative decrease and increase in copy numbers, respectively. G-banded ideogram of chromosome 4 is shown on the extreme right. All tumors that exhibited chromosome 4 losses are shown from largest to smallest region of losses. Inferred copy number view of MS-751 cell line showing 4p monosomy from normal (2N) (red line) is shown on right.
Previously, we have shown that PCDH10, a gene mapped to 4q28, is hypermethylated in CC cell lines and may act as tumor suppressor (Ying et al., 2006, 2007). Here, we have examined the role of PCDH10 promoter methylation in primary tumors from CC and its role in progression in detail.
Epigenetic Inactivation of PCDH10 is a Frequent Phenomenon in Invasive CC
Methylation status of PCDH10 was assessed using three sets of MSP primers spanning the CpG island (CGI) to assess qualitatively hypermethylation within the promoter region and exon 1 of the gene in invasive CC. We identified CGI hypermethylation in 118 (90.8%) of the 130 tumor DNA samples (9 cell lines and 121 primary tumors) examined from CC. Both cell lines (88.9%) and primary tumors (90.9%) showed a similar frequency of promoter hypermethylation (Table 1). Similar analysis on DNA isolated from 63 normal cervical epithelia did not reveal hypermethylation in the CGI of PCDH10. AHRR was the only other gene shown to be methylated in such a high frequency in CC (Zudaire et al., 2008). This high frequency of promoter methylation of PCDH10 in CC suggests that the inactivation of this gene plays an important role in its development.
TABLE 1.
Frequency of Promoter Hypermethylation of the PCDH10 Gene in Cervical Cancer Progression
| Tissue type | No. Cases studied | Methylated (%)* |
|---|---|---|
| Normal cervix | 63 | 0 |
| ASC-US | 35 | 2 (5.7%) |
| LSIL | 107 | 14 (13.1%) |
| HSIL | 63 | 29 (46.0%) |
| Primary invasive cancer | 121 | 110 (90.9%) |
| Cervical cancer cell lines | 9 | 8 (88.9%) |
ASC-US, atypical squamous cells of undetermined significance; LSIL, low-grade squamous intra-epithelial lesion; HSIL, high-grade squamous intra-epithelial lesion.
All MSP experiments were performed thrice and the promoter methylation was considered positive only when confirmed twice in at least one of the primer sets.
Among the cell lines studied, 8 of 9 were MSP positive and no unmethylated allele was found in the methylated cell lines. These data combined with SNP array data (Fig. 1) suggest deletion of one copy of PCDH10 due to monosomy 4 and methylation of the 2nd PCDH10 allele in all cell lines carrying monosomy 4 results in loss of function of the gene (Fig. 2A)(Table 2). Therefore, we provide evidence that PCDH10 is one of the targets on chromosome 4. The cell line C-33A that was negative by MSP showed no evidence of copy number alterations of chromosome 4 by SNP array. Thus, these data suggest that the PCDH10 inactivation follow the Knudson's “two-hit” hypothesis (Knudson and Strong, 1972). Similarly, 23 primary tumors showed simultaneous loss of 4q and PCDH10 gene methylation. However, 41 of the primary tumors representing the large majority showed PCDH10 methylation but no evidence of loss of 4q by SNP array (Table 2). Because a tumor suppressor gene can be inactivated by methylation of both copies and mutation in the 2nd copy, additional mechanisms of inactivation of the gene cannot be excluded.
Figure 2.
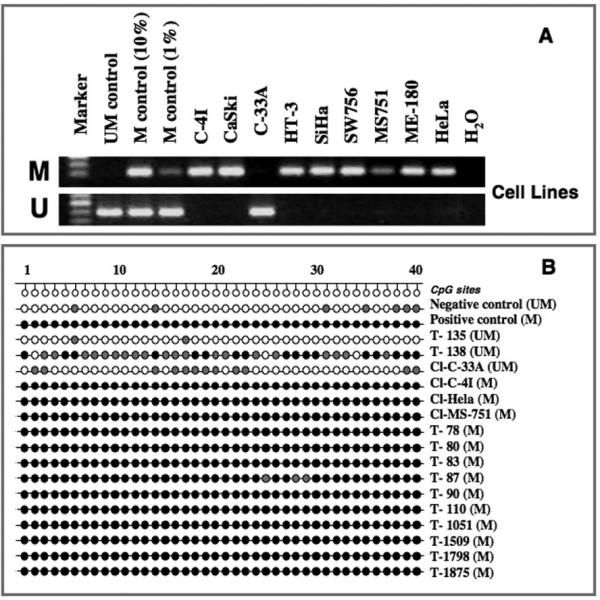
Analysis of PCDH10 methylation in cervical cancer cell lines and primary tumors. A. MSP analysis in cervical cancer cell lines. U, unmethylated; M, methylated. B. Bisulphite MSP cloning and sequencing of the PCDH10 gene. T, tumor; Cl, cell line. CpG sites examined are numbered sequentially as shown above. Filled circles indicate methylated CpG sites, semi-filled circles indicate partial/heterogeneous methylation, and empty circles indicate unmethylated CpGs. Methylation status by MSP is shown on the right (in bracket), M, methylated; U, unmethylated.
TABLE 2.
Status of 4q Loss by SNP array, PCDH0 Methylation by MSP, and Expression by Real Time PCR in Cervical Cancer Cell Lines and Primary Tumors
| Tumor | Status of 4q Loss | PCDH10 MSP status | PCDH 10 Expression ∇∇CT value* |
|---|---|---|---|
| C4I | 4q31.3-q32.1 | M | +4.48 |
| Ca Ski | 4q | M | +9.3 |
| C33-A | 0 | UM | +0.63 |
| HT-3 | 4q11-q31.3, 4q34.3 | M | +8.48 |
| SiHa | 4q | M | +9.7 |
| SW-756 | 4q | M | +10.38 |
| MS751 | 4q | M | +11.18 |
| ME180 | 4q | M | +9.61 |
| HeLa | 4q | M | +9.43 |
| T-24 | 0 | M | ND |
| T-55 | 0 | M | ND |
| T-57 | 0 | M | ND |
| T-66 | 4q | M | ND |
| T-75 | 0 | UM | ND |
| T-78 | 0 | M | ND |
| T-79 | 0 | M | ND |
| T-80 | 0 | M | ND |
| T-86 | 0 | M | ND |
| T-87 | 0 | M | ND |
| T-92 | 0 | M | ND |
| T-98 | 0 | M | ND |
| T-103 | 0 | M | ND |
| T-106 | 0 | M | ND |
| T-107 | 0 | M | ND |
| T-108 | 0 | M | ND |
| T-114 | 4q28.3-qter | UM | ND |
| T-116 | 0 | M | ND |
| T-117 | 4q | M | ND |
| T-118 | 0 | M | ND |
| T-124 | 4q | M | ND |
| T-126 | 4q | M | ND |
| T-127 | 4q | M | ND |
| T-128 | 4q | M | ND |
| T-130 | 0 | M | ND |
| T-132 | 4q | M | ND |
| T-133 | 4q | M | ND |
| T-134 | 4q | M | ND |
| T-135 | 0 | UM | ND |
| T-137 | 0 | M | ND |
| T-138 | 4q | UM | ND |
| T-140 | 0 | M | +2.73 |
| T-141 | 4q13-q21.1 | M | UD |
| T-146 | 4q | M | ND |
| T-148 | 4qcen-28.3 | M | ND |
| T-160 | 0 | M | ND |
| T-161 | 0 | M | ND |
| T-166 | 0 | M | ND |
| T-167 | 0 | M | ND |
| T-169 | 4q12-qter | UM | ND |
| T-188 | 4q | M | ND |
| T-190 | 0 | M | ND |
| T-194 | 0 | M | ND |
| T-205 | 0 | M | ND |
| T-207 | 4q | M | ND |
| T-214 | 0 | M | ND |
| T-218 | 4q21.1-q22.1; 4q26-qter | M | ND |
| T-222 | 0 | M | ND |
| T-224 | 0 | M | UD |
| T-654 | 4q | M | +2.5 |
| T-841 | 0 | M | +1.7 |
| T-869 | 0 | M | ND |
| T-892 | 4q | M | ND |
| T-939 | 0 | UM | ND |
| T-940 | 0 | M | ND |
| T-954 | 0 | M | ND |
| T-966 | 0 | M | ND |
| T-968 | 4q | M | ND |
| T-984 | 0 | M | ND |
| T-1051 | 4q28.3-qter | M | ND |
| T-1068 | 0 | UM | ND |
| T-1509 | 0 | M | UD |
| T-1721 | 4q | UM | ND |
| T-1875 | 0 | M | ND |
| T-1898 | 4q | M | ND |
| T-1900 | 0 | M | UD |
| T-1907 | 0 | M | ND |
| T-1981 | 4q | M | ND |
| T-2035 | 0 | M | ND |
| T-20-04 | 0 | M | +2.48 |
| T-56-04 | 0 | UM | ND |
| T-98-04 | 4qcen-q32.1 | M | UD |
| T-197 | ND | M | UD |
| T-1434 | ND | M | +3.6 |
| T-103-04 | ND | M | UD |
| T-54 | ND | M | +1.05 |
0, no loss of 4q by SNP analysis; MSP, methylation specific PCR; M, methylated; UM, unmethyalted; ND, not done; UD, undetectable
∇∇CT value is a comparative CT between the target and endogenous control. Positive values indicate decreased target (PCDH10) RNA compared to the control (GAPDH).
To validate the MSP data and to assess the extent of methylation of CpG sites, direct bisulphite sequencing of a region covering 40 CpGs within the CGI was performed in one negative and one positive control, and three unmethylated and 13 methylated tumors chosen randomly to represent each of these classes (Fig. 2B). We confirmed the MSP results in all positive tumors that showed methylation in almost all CpGs in the tested region (Fig. 2B). The unmethylated cell line C-33A and one of the unmethylated primary tumor (T-135) showed heterogeneous methylation of a few CpG residues, while one (T-138) of the two unmethylated tumors showed methylation of the majority of CpGs and a heterogeneous methylation in the remaining CpGs. A possible explanation for MSP negativity of the later tumor was that the MSP targeted region might not be completely methylated in all CpG sites. These data further suggest that the frequency of promoter methylation assessed by MSP is an underestimate as this method can only interrogate a limited number of CpGs.
To examine the prognostic role of PCDH10 hypermethylation, we performed a correlative analysis of methylation with clinico-pathologic features such as age, tumor stage, and size of the tumor, clinical outcome, and HPV type in primary tumors and found no significant associations (data not shown). These data therefore suggest that promoter hypermethylation and inactivation of PCDH10 play an important role in the development of CC and may be an early event in progression.
Since aberrant promoter hypermethylation of tumor suppressor genes is generally associated with transcriptional inactivation through loss of expression, we performed real-time quantitative PCR analysis of PCDH10 expression on a panel of 6 normal cervical epithelia, 9 cervical cancer cell lines, and 13 primary tumor specimens. Real-time qPCR data were subjected to statistical analysis to generate ΔCT , ΔΔCT values and fold-changes using replicate samples. All normal cervical epithelia showed lower levels of expression of the PCDH10 gene (mean CT value of 32.12±2.85) compared to GAPDH (mean CT value of 21.5±1.46). Most primary tumor samples analyzed showed either no detectable levels of expression or very high CT values of PCDH10 (mean/SD 35.55±2.52) compared to the control gene (mean/SD 19.39±3.54) suggesting a complete loss or a down regulated expression. Of the 13 primary tumors, all of which were methylated, 7 did not show any detectable levels of PCDH10 expression, while GAPDH showed high-levels of expression (Fig. 3A). Among the cell lines examined, all 8 methylated cell lines showed 80-99-fold decrease in relative expression of PCDH10. Whereas the unmethylated cell line C-33A showed 2.7-fold higher expression relative to normal cervix (Fig. 2B)(Table 2). The data on PCDH10 hypermethylation and gene expression together suggest that epigenetic inactivation is a frequent phenomenon in CC and may play a role in its development.
Figure 3.
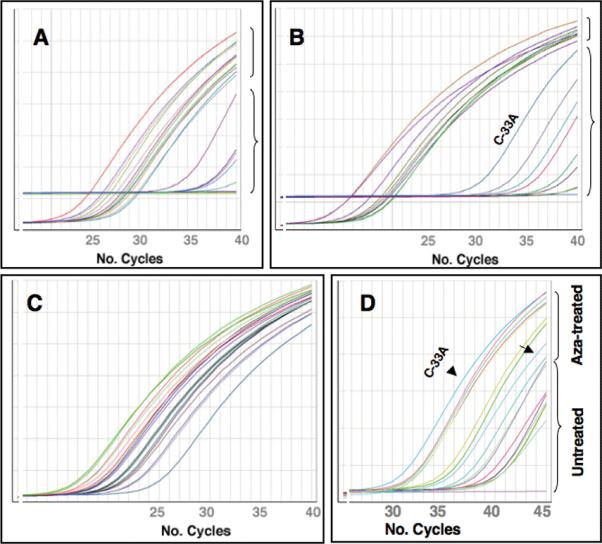
Real-time quantitative PCR analysis of PCDH10 gene expression compared to control gene. A. Expression plot showing PCDH10 (brace) and GAPDH (bracket) gene expression in primary invasive cervical cancer. B. Expression plot showing PCDH10 (brace) and GAPDH (bracket) gene expression in cervical cancer cell lines. Note higher expression of PCDH10 in unmethylated cell line C-33A. C and D. Expression plots showing levels of expression of control gene GAPDH (C) and PCDH10 (D) in cell lines after exposure to azacytidine. Note no difference in control gene in untreated and azacytidine-treated cell lines, while PCDH10 gene expression is reactivated in treated cell lines. Arrow and arrowhead indicate high basal as well as after Azacytidine-exposure, respectively, of PCDH10 expression levels in an unmethylated cell line C-33A.
Since DNA hypermethylation-mediated gene silencing is closely associated with histone modifications and the DNA demethylating agent 5-aza-2’-deoxycytidine (5-aza-CdR) and the HDAC inhibitor trichostatin (TSA) reactivates expression of epigenetically silenced genes, we examined whether the promoter hypermethylation-mediated down modulated gene expression can be reversed after 5-aza-CdR treatment of CC cell lines. Of the 9 cell lines studied, 8 exhibited complete methylation of the CGI and the remaining cell line (C-33A) was unmethylated by MSP (Fig. 2A). All methylated cell lines showed reactivation of the PCDH10 gene after 5-aza-CdR at 7- (SiHa) to 471-fold (SW756) (Fig. 3D). Albeit low, the unmethylated cell line C-33A also showed about 2.2-fold increased expression of PCDH10 gene after treatment. A possible explanation for this low-level induction in C-33A after 5-aza-CdR treatment is due to sparse and mosaic pattern of methylation seen in bisulphite-converted DNA where only 28% CpG sites were methylated compared to nearly all CpGs methylated in MSP positive cell lines (Fig. 2B). A similar increased expression was observed after treatment with TSA and combination of TSA and 5-aza-CdR (data not shown). Of note, no change in the levels of expression of GAPDH gene was found against treatment (Fig. 3C). Thus, these data indicate that the demethylation of PCDH10 promoter effectively reactivates gene expression by reversing the methylation affect.
PCDH10 Promoter Hypermethylation is an Early Event in Tumor Progression
To identify the role of promoter hypermethylation of PCDH10 gene in CC progression, we studied methylation status of PCDH10 CGI in DNA obtained from 268 cytological smears diagnosed as normal (N=63), ASC-US (N=35), low-grade squamous intraepithelial lesion (LSIL) (N=107), and high-grade SIL (HSIL) (N=63). We found no evidence of promoter hypermethylation in DNA isolated from cytologic smears diagnosed as normal. In contrast, 2 (5.7%) of 35 cases diagnosed as ASC-US, 14 (13.1%) of 107 LSILs and 29 (46.0%) of 63 HSILs showed positivity by MSP analysis (P=0.0001)(Table 1, Fig. 4A). To validate the MSP data on precancerous lesions, we performed bisulphite sequencing after cloning of the same region of CGI that was analyzed in invasive tumors in one MSP negative and five MSP positive precancerous lesions (Fig. 4B). We found no evidence of methylation of the region in the MSP negative HSIL, while all MSP-positive SILs showed the presence of heterogeneous and low frequency of methylated CpGs in small fraction of clones in the tested region (Fig. 4B). We noticed substantial differences in the number of CpG sites methylated between invasive cancer and precancerous lesions where the MSP-positive invasive cancers exhibit virtually 100% methylated CpGs and the MSP-positive precancerous lesions showed only 7.2% in LSIL and 13.3-24.1% in HSILs. Thus, these data provide evidence that promoter hypermethylation of PCDH10 initiate in early developmental stage and proceed gradually during CC tumorigenesis.
Figure 4.

Analysis of PCDH10 methylation in cervical precancerous lesions. A. MSP analysis. B. Bisulphite MSP cloning and sequencing of the PCDH10 gene. U, unmethylated; M, methylated. LSIL, low-grade squamous intraepithelial lesions; HSIL, high-grade squamous intraepithelial lesions. CpG sites examined are numbered sequentially as shown above in panel B. Filled circles indicate methylated CpG sites and empty circles indicate unmethylated CpGs. Pap smear numbers indicating methylation status by MSP are shown on right.
The natural history of cervical precancerous lesions varies where approximately 1% of low-grade and 15% of high-grade cervical intraepithelial neoplastic (CIN) lesions progress to invasive cancer (Syrjanen, 1996; McCredie et al., 2008; Schiffman and Rodriguez, 2008). Furthermore, precancerous lesions harboring high-risk HPV infection predict higher risk for progression to invasive cancer, particularly HPV 16 possess greater risk than other HPV types (Syrjanen, 1996; zur Hausen, 2002). In order to examine the relation of HPV infection with PCDH10 methylation status in precancerous lesions, we were able to obtain HPV data in 8 of 14 cases of LSIL that showed PCDH10 methylation by MSP. Seven of 8 (87.5%) LSILs showed infection with high-risk HPV types. Therefore, the combined data on PCDH10 methylation documented here and high proportion of cases with infection of high-risk HPV suggests that epigenetic changes and HPV plays a synergistic role in transformation of dysplastic cells. These data suggest that PCDH10 methylation is initiated very early in CC development and its inactivation may form a potential signature in defining risk of precancerous lesions to progress to invasive cancer.
A cytological diagnosis of ASC-US is reported in around 5% of women undergoing pap screening but the clinical relevance of ASC-US is largely unclear. Follow up studies of patients with this unequivocal diagnosis show histologically high-grade disease (CIN) lesions or even invasive cancer in a proportion of cases [The ASCUS-LSIL Triage Study (ALTS) Group, (2003)]. However, no optimal strategy for ASC-US triage to identify high-grade disease that requires follow-up and treatment is available in limiting the number of women who receive unnecessary procedures. Since we found PCDH10 methylation in 2 (5.7%) of 35 patients with the diagnosis of ASCUS, whether these patients represent already underlying high-grade disease remains to be examined.
In this study, we showed that PCDH10 is epigenetically silenced in a large majority of CC. The consequence of this hypermethylation of PCDH10 in CC biology remains to be understood for its mechanistic role, and implications in predicting the behavior of CIN. It is important to evaluate the consequence of PCDH10 promoter methylation since we identified gradual increase in methylation density and rates in CC progression. The protocadherin gene family members contain 6 extra cellular cadherin domains, a transmembrane domain and a cytoplasmic tail differing from those of the classical cadherins. The PCDH10 encodes a cadherin-related neuronal receptor thought to play a role in the establishment and function of specific cell-cell connections in the brain (Frank and Kemler, 2002). Although the exact functions of protocadherins are not well understood, they are believed to play a role in signal transduction and growth control. It has been shown that cadherins play an important role in tumor progression by functioning as suppressors of invasion and metastasis (Jeanes et al., 2008). Recently, a number of protocadherins, however, have been implicated as tumor suppressors, including PCDH10 (Waha et al., 2005; Imoto et al., 2006; Ying et al., 2006, 2007; Yu et al., 2008, 2009).
Although the role of PCDH10 in cancer has not been fully elucidated, the epigenetic silencing may contribute to tumorigenesis and tumor progression (Ying et al., 2006). Most recently, PCDH10 was reported to be methylated in the early stage of gastric carcinogenesis and its methylation was associated with poor prognosis of gastric cancer patients (Yu et al., 2009). Our present work suggests translational implications of PCDH10 methylation in CC progression since this gene was a target of epigenetic silencing in invasive tumors compared to their respective normal epithelium, together with the evidence for its occurrence at initial tumorigenic steps. In addition, PCDH10 methylation was significantly associated with increasing tumor progression. Therefore, we hypothesize that PCDH10 represents a potential therapeutic target for demethylating drugs to achieve its reactivation. Assessing PCDH10 methylation in cytology specimens obtained from precancerous lesions may represent a potential alternative adjunct procedure for the early detection and follow-up of these patients. In summary, our study identifies the novel methylation of PCDH10 in CC and provides evidence for its role in progression from early precancerous lesions to invasive cancer.
Acknowledgments
Supported by the grant CA095647 from National Institutes of Health
REFERENCES
- Backsch C, Rudolph B, Kuhne-Heid R, Kalscheuer V, Bartsch O, Jansen L, Beer K, Meyer B, Schneider A, Durst M. A region on human chromosome 4 (q35.1-->qter) induces senescence in cell hybrids and is involved in cervical carcinogenesis. Genes Chromosomes Cancer. 2005;43:260–272. doi: 10.1002/gcc.20192. [DOI] [PubMed] [Google Scholar]
- Backsch C, Wagenbach N, Nonn M, Leistritz S, Stanbridge E, Schneider A, Durst M. Microcell-mediated transfer of chromosome 4 into HeLa cells suppresses telomerase activity. Genes Chromosomes Cancer. 2001;31:196–198. doi: 10.1002/gcc.1134. [DOI] [PubMed] [Google Scholar]
- Frank M, Kemler R. Protocadherins. Curr Opin Cell Biol. 2002;14:557–562. doi: 10.1016/s0955-0674(02)00365-4. [DOI] [PubMed] [Google Scholar]
- Hampton GM, Larson AA, Baergen RN, Sommers RL, Kern S, Cavenee WK. Simultaneous assessment of loss of heterozygosity at multiple microsatellite loci using semi-automated fluorescence-based detection: subregional mapping of chromosome 4 in cervical carcinoma. Proc Natl Acad Sci U S A. 1996;93:6704–6709. doi: 10.1073/pnas.93.13.6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoto I, Izumi H, Yokoi S, Hosoda H, Shibata T, Hosoda F, Ohki M, Hirohashi S, Inazawa J. Frequent silencing of the candidate tumor suppressor PCDH20 by epigenetic mechanism in non-small-cell lung cancers. Cancer Res. 2006;66:4617–4626. doi: 10.1158/0008-5472.CAN-05-4437. [DOI] [PubMed] [Google Scholar]
- Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008;27:6920–6929. doi: 10.1038/onc.2008.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson AG, Jr., Strong LC. Mutation and cancer: neuroblastoma and pheochromocytoma. Am J Hum Genet. 1972;24:514–532. [PMC free article] [PubMed] [Google Scholar]
- McCredie MR, Sharples KJ, Paul C, Baranyai J, Medley G, Jones RW, Skegg DC. Natural history of cervical neoplasia and risk of invasive cancer in women with cervical intraepithelial neoplasia 3: a retrospective cohort study. Lancet Oncol. 2008;9:425–434. doi: 10.1016/S1470-2045(08)70103-7. [DOI] [PubMed] [Google Scholar]
- Mitra AB, Murty VV, Li RG, Pratap M, Luthra UK, Chaganti RS. Allelotype analysis of cervical carcinoma. Cancer Res. 1994;54:4481–4487. [PubMed] [Google Scholar]
- Narayan G, Arias-Pulido H, Koul S, Vargas H, Zhang FF, Villella J, Schneider A, Terry MB, Mansukhani M, Murty VV. Frequent promoter methylation of CDH1, DAPK, RARB, and HIC1 genes in carcinoma of cervix uteri: Its relationship to clinical outcome. Mol Cancer. 2003;2:24. doi: 10.1186/1476-4598-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PH, Arias-Pulido H, Lu XY, Harris CP, Vargas H, Zhang FF, Narayan G, Schneider A, Terry MB, Murty VV. Chromosomal amplifications, 3q gain and deletions of 2q33-q37 are the frequent genetic changes in cervical carcinoma. BMC Cancer. 2004;4:5. doi: 10.1186/1471-2407-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman M, Rodriguez AC. Heterogeneity in CIN3 diagnosis. Lancet Oncol. 2008;9:404–406. doi: 10.1016/S1470-2045(08)70110-4. [DOI] [PubMed] [Google Scholar]
- Scotto L, Narayan G, Nandula SV, Arias-Pulido H, Subramaniyam S, Schneider A, Kaufmann AM, Wright JD, Pothuri B, Mansukhani M, Murty VV. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer. 2008a;47:755–765. doi: 10.1002/gcc.20577. [DOI] [PubMed] [Google Scholar]
- Scotto L, Narayan G, Nandula SV, Subramaniyam S, Kaufmann AM, Wright JD, Pothuri B, Mansukhani M, Schneider A, Arias-Pulido H, Murty VV. Integrative genomics analysis of chromosome 5p gain in cervical cancer reveals target over-expressed genes, including Drosha. Mol Cancer. 2008b;7:58. doi: 10.1186/1476-4598-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Indra D, Mitra S, Mondal RK, Basu PS, Roy A, Roychowdhury S, Panda CK. Deletions in chromosome 4 differentially associated with the development of cervical cancer: evidence of slit2 as a candidate tumor suppressor gene. Hum Genet. 2007;122:71–81. doi: 10.1007/s00439-007-0375-6. [DOI] [PubMed] [Google Scholar]
- Syrjanen KJ. Spontaneous evolution of intraepithelial lesions according to the grade and type of the implicated human papillomavirus (HPV). Eur J Obstet Gynecol Reprod Biol. 1996;65:45–53. doi: 10.1016/0028-2243(95)02303-a. [DOI] [PubMed] [Google Scholar]
- The ASCUS-LSIL Triage Study (ALTS) Group Results of a randomized trial on the management of cytology interpretations of atypical squamous cells of undetermined significance. Am J Obstet Gynecol. 2003;188:1383–1392. doi: 10.1067/mob.2003.457. [DOI] [PubMed] [Google Scholar]
- Waggoner SE. Cervical cancer. Lancet. 2003;361:2217–2225. doi: 10.1016/S0140-6736(03)13778-6. [DOI] [PubMed] [Google Scholar]
- Waha A, Guntner S, Huang TH, Yan PS, Arslan B, Pietsch T, Wiestler OD. Epigenetic silencing of the protocadherin family member PCDH-gamma-A11 in astrocytomas. Neoplasia. 2005;7:193–199. doi: 10.1593/neo.04490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T. Clustered protocadherin family. Dev Growth Differ. 2008;50(Suppl 1):S131–40. doi: 10.1111/j.1440-169X.2008.00991.x. [DOI] [PubMed] [Google Scholar]
- Ying J, Gao Z, Li H, Srivastava G, Murray PG, Goh HK, Lim CY, Wang Y, Marafioti T, Mason DY, Ambinder RF, Chan AT, Tao Q. Frequent epigenetic silencing of protocadherin 10 by methylation in multiple haematologic malignancies. Br J Haematol. 2007;136:829–832. doi: 10.1111/j.1365-2141.2007.06512.x. [DOI] [PubMed] [Google Scholar]
- Ying J, Li H, Seng TJ, Langford C, Srivastava G, Tsao SW, Putti T, Murray P, Chan AT, Tao Q. Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene. 2006;25:1070–80. doi: 10.1038/sj.onc.1209154. [DOI] [PubMed] [Google Scholar]
- Yu J, Cheng YY, Tao Q, Cheung KF, Lam CN, Geng H, Tian LW, Wong YP, Tong JH, Ying JM, Jin H, To KF, Chan FK, Sung JJ. Methylation of protocadherin 10, a novel tumor suppressor, is associated with poor prognosis in patients with gastric cancer. Gastroenterology. 2009;136:640–51. e1. doi: 10.1053/j.gastro.2008.10.050. [DOI] [PubMed] [Google Scholar]
- Yu JS, Koujak S, Nagase S, Li CM, Su T, Wang X, Keniry M, Memeo L, Rojtman A, Mansukhani M, Hibshoosh H, Tycko B, Parsons R. PCDH8, the human homolog of PAPC, is a candidate tumor suppressor of breast cancer. Oncogene. 2008;27:4657–4665. doi: 10.1038/onc.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zudaire E, Cuesta N, Murty V, Woodson K, Adams L, Gonzalez N, Martinez A, Narayan G, Kirsch I, Franklin W, Hirsch F, Birrer M, Cuttitta F. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 2008;118:640–650. doi: 10.1172/JCI30024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]