

Abstract
Anti-NMDA receptor (NMDAR) encephalitis is a newly identified autoimmune disorder that targets NMDARs, causing severe neurological symptoms including hallucinations, psychosis, and seizures, and may result in death (Dalmau et al., 2008). However, the exact epitope to which these antibodies bind is unknown. A clearly defined antigenic region could provide more precise testing, allow for comparison of immunogenicity between patients to explore potential clinically relevant variations, elucidate the functional effects of antibodies, and make patients' antibodies a more effective tool with which to study NMDAR function. Here, we use human CSF to explore the antigenic region of the NMDAR. We created a series of mutants within the amino terminal domain of GluN1 that change patient antibody binding in transfected cells in stereotyped ways. These mutants demonstrate that the N368/G369 region of GluN1 is crucial for the creation of immunoreactivity. Mass spectrometry experiments show that N368 is glycosylated in transfected cells and rat brain regions; however, this glycosylation is not directly required for epitope formation. Mutations of residues N368/G369 change the closed time of the receptor in single channel recordings; more frequent channel openings correlates with the degree of antibody staining, and acute antibody exposure prolongs open time of the receptor. The staining pattern of mutant receptors is similar across subgroups of patients, indicating consistent immunogenicity, although we have identified one region that has a variable role in epitope formation. These findings provide tools for detailed comparison of antibodies across patients and suggest an interaction between antibody binding and channel function.
Introduction
N-methyl-d-aspartate receptors are ionotropic glutamate receptors comprised of two GluN1 subunits and two GluN2/3 subunits (Dingledine et al., 1999; Cull-Candy and Leszkiewicz, 2004). There are eight alternatively spliced GluN1 isoforms and two GluN3 subunits (A, B) that bind glycine and four GluN2 subunits (A–D) that bind glutamate. GluN subunits contain two large extracellular domains, the amino terminal domain (ATD) and S1 and S2 domains, which form the ligand binding domain; three membrane-spanning domains (TM1, 3, 4); a membrane loop (TM2); and an intracellular C-terminal domain that links to scaffolding proteins and messenger systems. The ATD is further subdivided into two lobes (Karakas et al., 2009). NMDA receptors (NMDARs) are critical in learning and memory, and hypofunction of the receptor has been implicated in schizophrenia (Coyle, 2006).
Recently, we described a new autoimmune disorder designated anti-NMDAR encephalitis (Dalmau et al., 2007) in which patients produce autoantibodies to the GluN1 subunit of the NMDAR (Dalmau et al., 2008). The frequency of the disorder may be underappreciated; a number of cases of encephalitis of unknown etiology have been retroactively diagnosed as anti-NMDAR encephalitis (Prüss et al., 2010). These antibodies crosslink the receptor, causing it to be internalized and destroyed (Hughes et al., 2010), and antibody application to hippocampal slices attenuates LTP (Zhang et al., 2012). Patients present with psychiatric symptoms, including memory loss, hallucinations, and paranoia. The disorder progresses to neurological dysfunction, including seizures, dyskinesias, and autonomic instability, which frequently requires mechanical ventilation (Dalmau et al., 2008). The degree of recovery depends on speed of diagnosis (Dalmau et al., 2008; Finke et al., 2012; Kashyape et al., 2012), making accurate and rapid diagnosis critical.
The initial patients diagnosed with anti-NMDAR encephalitis were mainly adult women in whom the presence of NMDAR antibodies coincided with an ovarian teratoma or cyst that expressed NMDA receptors (Dalmau et al., 2007), which led to the hypothesis that the tumor is the precipitating autoimmune event in this subgroup (Dalmau et al., 2008). Although the clinical symptoms are similar, the patient population now includes more men and children without tumors, making the immunological trigger more obtuse (Florance et al., 2009; Irani et al., 2010; Kashyape et al., 2012). In contrast, other disorders associated with NMDAR antibodies, such as systemic lupus erythematosis (SLE), are clinically distinct, and the role of NMDAR antibodies in disease pathogenesis is less clear (Lauvsnes and Omdal, 2012). Understanding the biochemical and physiological requirements of antibody binding is a crucial component in understanding the pathophysiology of autoimmune disorders, from myasthenia gravis to the rapidly evolving field of synaptic autoimmune encephalitides (Kayser and Dalmau, 2011), of which anti-NMDAR encephalitis is among the most common.
In the present work, we explore the components of GluN1 that are necessary for patient antibody binding and the possible involvement of post-translational modifications and binding partners, identify a region of the receptor that influences both antibody binding and receptor function, investigate the effect of acute antibody application on channel function, and find a receptor subdomain that has a variable effect on antibody binding.
Materials and Methods
HEK293 transfection.
HEK293 cells were cultured and transiently transfected as described previously (Wu et al., 2007). Briefly, cells were plated on poly-l-lysine-coated dishes (immunocytochemistry and electrophysiology: 0.5 μg/ml on glass coverslips; immunoprecipitations and cell-surface biotinylations: 5 μg/ml on plastic dishes) in minimum essential medium containing 7.5% fetal bovine serum, 2.5% horse serum, 1% penicillin/streptomycin, and 1% l-glutamine, maintained at 37°C/5% CO2. Cells were transfected 1 d later using the calcium phosphate method. For most experiments, the transfection solution contained 2 μg of total DNA per milliliter of medium, divided equally between the DNA constructs used; for single-channel recordings and cell-surface biotinylations, the total amount of DNA was decreased to 1.33 μg of DNA per ml in medium, with a ratio of 1:3:3 GluN1:GluN2:GFP (Gielen et al., 2009). For GluN1/GluN2 cotransfections, medium was supplemented with 500 μm ketamine (Sigma) to prevent cell death. In experiments blocking N-linked glycosylation, medium was supplemented with 2 μg/ml tunicamycin (Sigma). In most experiments the GluN1a splice variant was used; therefore, residue numbering reflects GluN1a numbering.
Patient material.
CSF was collected in accordance with the University of Pennsylvania Institutional Review Board guidelines and stored at −80°C. Unless otherwise noted, patient material used is CSF from adult female patients, the majority of whom had ovarian cysts or teratomas (i.e., the originally defined patient population). In immunocytochemistry experiments, patient CSF was used at a dilution of 1:10 to 1:100, depending on patient titer; control CSF was used at 1:10 to 1:20.
Immunocytochemistry.
Transfected HEK293 cells were stained as described previously (Dalmau et al., 2008). Briefly, 16–24 h after transfection cells were fixed in cold 4% paraformaldehyde in PBS for 10 min at room temperature, washed once with PBS, permeabilized in 0.3% Triton X-100 in PBS for 10 min at room temperature, washed once with PBS, blocked with 10% bovine serum albumin (BSA) in PBS for 1 h at 37°C, and washed twice with PBS. Cells were then incubated in commercial NMDAR antibody and CSF (range 1:10 – 1:100) in 1% BSA in PBS overnight, 4°C. Each experiment included CSF from at least one individual without anti-NMDAR encephalitis as well; none of these showed CSF staining of NMDARs. For most experiments, we used a commercial anti-GluN1 antibody against the TM3–4 loop (BD Biosciences 556308, 1:1000). For experiments using the ATD-TM4 deletion mutant, we used a C-terminal GluN1 commercial antibody (Millipore. catalog no. AB9864; 1:200); for those experiments investigating the role of GluN2B domains, we used a C-terminal GluN2A/2B antibody (Millipore, catalog no. AB1548; 1:200). GluN3A expression was verified by Western blot (Tocris Bioscience, catalog no. 2059; 1:1000). (GluN3A plasmid kindly provided by Dr. Stuart Lipton, Sanford-Burnham Medical Research Institute, La Jolla, CA).
The next day, coverslips were washed twice with PBS and put into secondary antibody (1:1000 Alexa Fluor 488 goat anti-human, 1:1000 Alexa 568 goat anti-mouse or goat anti-rabbit, 1% BSA in PBS) for 2 h at 37°C. Cells were washed once with PBS and once with dH2O and mounted on glass slides in mounting medium with DAPI (Vectashield), and stored at 4°C until imaging. Cells were imaged on a Leica DMR microscope. Staining intensity was quantified using ImageJ software. Overall background was subtracted from each image using a sliding paraboloid with a set radius for each experiment. Mean pixel intensity of both the entire field and untransfected cells within the field was measured for both the 488 channel (human antibody) and 568 (commercial antibody). The mean intensity of the untransfected cells was subtracted from the overall mean intensity, yielding the mean intensity of transfected cells only. The 488 staining intensity was then normalized to 568 intensity to control for the level of GluN expression. Each 488/568 set of images was then further normalized to the average intensity of its matched wild-type GluN1 images, which were transfected, stained, and imaged identically to the mutant images, yielding a relative value of patient antibody staining of mutant versus wild-type receptor.
Site-directed mutagenesis and large-scale deletion mutagenesis.
Point mutations were made using the Stratagene QuikChange Mutagenesis kit according to manufacturer's instructions. Larger deletions of GluN1 were made using a PCR-based method with the QuikChange kit (Makarova et al., 2000). Briefly, primers were designed with two halves: the upstream half primes to the region immediately preceding the intended deletion, while the downstream half primes to the region immediately following the intended deletion. GluN1-ATD deletion (delete residues 26–382): Forward (Fwd), 5′-cgc gcc gcc tgc gac ccc aag gga gga gag aca gag aaa cct cg; Reverse (Rev), 5′-gg ttt ctc tgt ctc tcc tcc ctt ggg gtc gca ggc. GluN1-TM4 deletion (delete residues 401–792): Fwd, 5′-cc acc aga cta aag ata tgg tt cgg tat cag gaa tgc; Rev, 5′-gca ttc ctg ata ccg aac cca tat ctt tag tct ggt gga c. GluN1–144-156 deletion: Fwd, 5′-cgc acg gtg ccg ccc gtc tac aac tgg aac cac atc; Rev, 5′- gtg gtt cca gtt gta gac ggg cgg cac cgt gcg aag g. GluN1-ATD top lobe deletion 1 (delete residues 26–140), Fwd: 5′-cgc gcc gcc tgc gac ccc aag gtg ccg ccc tac tcc cac cag; Rev, 5′- gct gga ctg gtg gga gta ggg cgg cac ctt ggg gtc gca ggc. GluN1-ATD top lobe deletion 2 (delete residues 275–349), Fwd: 5′-gga ctt cag ctc atc aat ggc aac tat agt atc atg aac c; Rev, 5′- ctg cag gtt cat gat act ata gtt gcc att gat gag ctg aag.
Large-scale deletions of GluN2B were made by inserting restriction enzyme sites into wild-type receptor. The GluN2B-ATD deletion was formed by inserting BglII sites at residues 27 (Fwd, 5′-cgt atc ggg cag caa agc tag atc tca aaa gag cgc ccc; Rev, 5′-ggg gcg ctc ttt tga gat cta gct ttg ctg ccc gat acg) and 382 (Fwd, 5′-ggg tgg gaa aat gga aag aca gat ctc tgc aga tga aat act acg; Rev, 5′-cgt agt att tca tct gca gag atc tgt ctt tcc att ttc cca ccc). The GluN2B-S2 deletion was formed by inserting MluI sites at residues 703 (Fwd, 5′-gca gaa atg cac gcg tac atg gga aag ttc aac caa agg gg; Rev, 5′-ccc ctt tgg ttg aac ttt ccc atg tac gcg tgc att tct gc) and 793 (Fwd, 5′-gga gat gga aga act gga cgc gtt gtg gct cac tgg cat ttg c; Rev: 5′-gca aat gcc agt gag cca caa cgc gtc cag ttc ttc cat ctc c). The resulting DNA was cleaved with BglII or MluI respectively and run on a 1% agarose gel; the larger fragment was extracted and ligated overnight with T4 ligase (Invitrogen) according to the manufacturer's instructions. All mutations were confirmed by sequencing.
To verify mutant expression levels, HEK293 cells were transfected with wild-type or mutant DNA, lysed in 1.5× stop buffer (75 mm Tris pH 6.8, 3.75 mm EDTA, 15% glycerol, 3% SDS), boiled, and run on Western blots. GluN2B expression levels were probed with a C-terminal GluN2A/2B antibody (Millipore, catalog no. AB1548; 1:100). GluN1 levels were probed with a TMIII-IV loop antibody (BD Biosciences, catalog no. 556308; 1:1000), with the exception of the GluN1-TM4 deletion mutant, which was probed with an ATD-specific antibody (Millipore, catalog no. 07-362; 1:1000). Blots were quantified using ImageJ and normalized to GFP levels (Sigma, catalog no. G1544; 1:3000) to control for transfection efficiency. Two mutants expressed at very low levels (N239Q and N276Q), so other mutation strategies were employed; no other mutants expressed at levels that were significantly different from wild type (one-way ANOVA, p > 0.05). Levels of mutants not reported in Results, percent compared to wild-type ± SEM, n = 5–6: GluN1-ATD deletion, 214.5 ± 28.9; GluN1-TM4 deletion, 221.3 ± 48.1; GluN1-ATD top lobe deletion, 114.2 ± 15.7; GluN1–144-156 deletion, 71.9 ± 13.6; GluN1-N61Q, 148.2 ± 88.7; GluN1-N203Q, 85.3 ± 36.1; GluN1-N239A, 57.29 ± 15.6; GluN1-N276A, 118.6 ± 37.8; GluN1-N300Q, 72.9 ± 14.5; GluN1-N350Q, 198.3 ± 59.6; GluN1-N368Q, 154.1 ± 74.9; GluN1-T370A, 99.6 ± 34.1; GluN2B-ATD deletion, 129.9 ± 36.6; GluN2B-S2 deletion, 78.1 ± 19.8.
Immunoprecipitation.
For immunoprecipitations (IPs) of heterologously expressed GluN1, HEK293 cells were transfected with GluN1 and GluN2B expression plasmids and lysed 16–24 h later. Cells were washed twice with PBS with 0.1 mm CaCl and 1 mm MgCl2 and lysed in 1 ml of lysis buffer per 100 mm plate. Lysis buffer contained 150 mm NaCl, 1 mm EDTA, 100 mm Tris-HCl, 1% Triton X-100, and 1% sodium deoxycholate, pH 6.5, supplemented the day of use with 1:500 EDTA-free protease inhibitor cocktail III (Calbiochem) and 1% N-octyl-β-d-glucopyranoside (Sigma). Cells were lysed for 1 h at 4°C and collected. Debris was cleared by centrifugation at 16100 × g for 20 min at 4°C. Supernatants were stored at −80°C until use.
For immunoprecipitations from rat brain regions, adult male CD rats were sacrificed by CO2 inhalation in accordance with Children's Hospital of Philadelphia Institutional Animal Care and Use Committee guidelines. The brain was immediately removed and dissected on ice. Cortex, hippocampus, and cerebellum were immediately transferred to dry ice. Regions were homogenized in 20 ml lysis buffer per 1 g wet weight, and lysed for 1 h at 4°C. Debris was cleared by centrifugation at 39,000 × g for 1 h at 4°C. Supernatants were stored at −80°C until use.
Immunoprecipitations were performed using the Pierce kit for crosslink IPs (Thermo Scientific), according to the manufacturer's instructions. To immunoprecipitate all GluN1 variants, a C-terminal commercial antibody was used (Millipore, catalog no. 05-432; 10 μg/column). CSF IPs were performed with 20–50 μl of CSF from patients or control subjects. All columns were incubated with lysate for 60–90 min at 4°C and reused up to 10 times. Eluates were collected in tubes containing 5 μl of 1 m Tris, pH 8.0. For mass spectrometry, eluates were pooled and concentrated with 10 kDa molecular weight cutoff centrifugal concentrators (Pall). To minimize protein adsorption into the concentrator membrane, concentrators were spun in 5 min intervals at 5000 × g at 4°C, and the reservoir was mixed between spins. Concentrated eluate was run on a 10% Bio-Rad Ready Gel fixed in 25% ethanol/7% acetic acid for 20 min and stained with G250 until bands appeared. For Western blot, eluates were run on 10% Bio-Rad Ready Gel, transferred to nitrocellulose membranes, and probed with a different commercial GluN1 antibody (BD Biosciences, catalog no. 556308; 1:1000).
Mass spectrometry analysis.
For in-gel digestion, Coomassie-stained gels were excised and cut into 1 mm3 cubes. Briefly, gels were destained with 50% methanol/1.25% acetic acid, reduced with 5 mm DTT (Thermo Scientific), and alkylated with 40 mm iodoacetamide (Sigma). Gels were then washed with 20 mm ammonium bicarbonate (Sigma). Gel pieces were dehydrated with acetonitrile. Trypsin (Promega) (10 ng/ml in 20 mm ammonium bicarbonate) was added to the gel pieces. Proteolysis was allowed to proceed for 4 h at 37°C (Tang et al., 2005). Peptides were then extracted with 0.3% trifluoroacetic acid. Diisopropyl fluorophosphate (DFP) (Sigma) (Jansen et al., 1949) was added to a final concentration of 5 mm and incubated overnight at 37°C. Supernatant was pulled off followed by a second extraction in 50% acetonitrile/5 mm DFP at room temperature for 4 h. Supernatants were combined and lyophilized. One microliter (500,000 U) of PNGase F (New England Biolabs) was lyophilized in 20 μl of 50 mm ammonium phosphate (Sigma), reconstituted in 20 μl of H218O (Cambridge Isotopes), and then added to lyophilized extracts. Extracts were incubated at 37°C for 4 h, frozen, and lyophilized.
Tryptic digests were analyzed on a hybrid LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) coupled with a NanoLC pump (Eksigent) and autosampler. Tryptic peptides were separated by reverse phase (RP)-HPLC on a nanocapillary column, 75 μm inner diameter × 20 cm ProteoPep2 (New Objective). Mobile phase A consisted of 1% methanol/0.1% formic acid, and mobile phase B consisted of 1% methanol/0.1% formic acid/80% acetonitrile. Peptides were eluted into the mass spectrometer at 300 nl/min, with each RP-LC run comprising a 15 min sample load at 3% B and a 90 min linear gradient from 5 to 45% B. The mass spectrometer was set to repetitively scan m/z from 300 to 1800 (r = 100,000 for LTQ-Orbitrap) followed by data-dependent tandem mass spectrometry (MS/MS) scans on the 10 most abundant ions, with a minimum signal of 1500, dynamic exclusion with a repeat count of 2, repeat duration of 15 s, exclusion size of 500, and duration of 60 s, isolation width of 2.0, normalized collision energy of 28, and waveform injection and dynamic exclusion enabled. Fourier transform (FT) mass spectrometry (FTMS) full scan automatic gain control (AGC) target value was 1e6, while multiple spectrometry (MS2) AGC was 5e3, respectively. FTMS full scan maximum fill time was 500 ms, while ion trap MSn fill time was 50 ms; microscans were set at 1. FT preview mode, charge state screening, and monoisotopic precursor selection were all enabled with rejection of unassigned and 1+ charge states.
All MS/MS data were analyzed using Mascot (version 2.3.02; Matrix Science). Mascot was set up to search a subset of the UniProt-SwissProt/Tremble protein sequence database containing human, mouse, rat, rabbit, chicken, and guinea pig sequences (version 14.5; 171,404 entries) or the Escherichia coli database appended with mutant sequences of the NMDA receptor (version 2011_05; 24,290 entries), assuming the digestion enzyme trypsin. Mascot was searched with a fragment ion mass tolerance of 0.80 Da and a parent ion tolerance of 50 ppm. Iodoacetamide derivative of cysteine was specified in Mascot as a fixed modification. Deamidation of asparagine and glutamine, 18O label of the C terminus, deamidation in the presence of 18O of asparagine and glutamine, 18O label at both C-terminal oxygens, and oxidation of methionine were specified in Mascot as variable modifications. Scaffold (version Scaffold_3_00_08; Proteome Software) was used to validate MS/MS-based peptide and protein identifications using the Peptide Prophet algorithm (Keller et al., 2002; Nesvizhskii et al., 2003) to establish a 1% false discovery rate.
Outside-out single channel recordings.
HEK293 cells were transfected with a 1:3:3 ratio of GluN1:GluN2B:GFP (Gielen et al., 2009) for 16–18 h in the presence of ketamine. Medium was changed and cells were maintained in fresh medium with ketamine for 2–14 h. Transfected cells were identified by GFP fluorescence. Cells were voltage clamped at −60 mV using borosilicate glass pipettes (World Precision Instruments) with resistances of 6–9 MΩ. Intrapipette solution contained 150 mm potassium gluconate, 10 mm HEPES, 10 mm EGTA, 2 mm MgCl2, 1.4 mm CaCl2, and 2 mm Mg-ATP, pH 7.35, 310 mOsm. Currents were recorded at room temperature in Mg2+-free extracellular solution containing 155 mm NaCl, 3 mm KCl, 0.5 mm CaCl2, and 10 mm HEPES, pH 7.35, 310–320 mOsm. Treatment applications were performed using a ValveBank 8.2 perfusion system with Lee valves (Automate Scientific); all traces analyzed were taken after at least 30 s of continuous application of 100 nm glutamate/1 μm glycine.
To examine the effects of acute patient antibody application, outside-out patches were exposed to 100 nm glutamate/1 μm glycine (agonist) for a minimum of 2 min followed by at least 7 min of exposure to agonist alone, agonist plus 1:100 CSF from patients without anti-NMDAR encephalitis, or agonist plus 1:100 CSF from anti-NMDAR encephalitis patients. CSF was dialyzed against extracellular solution to remove endogenous glutamate. CSF from two control individuals and three patients was used, with at least two patches per individual. One minute of trace before antibody application was analyzed and compared to 1 min of trace from 6 to 7 min of antibody application.
In all cases, signals were amplified using an Axopatch-1D amplifier (Molecular Devices/Molecular Devices Corporation), acquired at 20 kHz, filtered at 2 kHz, and saved using pClamp10 software for off-line analysis. A minimum of 1 min of traces were filtered at 1 kHz Bessel and analyzed using Clampfit 10. Most patches contained at least one double opening; these were excluded from analysis by suppression (Stocca and Vicini, 1998). Closed dwell times were graphed in log histograms (Erreger et al., 2005) and fitted with six exponential components (Wyllie et al., 2006). The two longest components of the exponential fit were used to estimate critical shut time (Tcrit) values for the wild-type GluN1 patches and each mutant (Colquhoun and Sakmann, 1985), such that equal numbers of events from each component of the distribution will be misclassified. Tcrit values represent the amount of time within which multiple openings can be assumed to originate from a single channel; therefore, a burst analysis of channel closed dwell times within the channel's Tcrit allows an assessment of channel closed time regardless of the number of channels within the patch. Individual Tcrit values were then used for burst analysis (Clampfit 10).
Cell surface biotinylations.
Cell-surface biotinylations were performed as described previously (Hughes et al., 2010) after 16–20 h transfections. Briefly, cells were washed twice with PBS containing 0.1 mm CaCl2 and 1 mm MgCl2 (rinsing solution) and incubated with gentle rocking for 30 min at 4°C in rinsing solution with 1 mg/ml Sulfo-NHS-Biotin (Thermo Scientific). Cells were washed twice with rinsing solution plus 100 mm glycine (quenching solution) and incubated with gentle rocking for 20–30 min at 4°C in quenching solution. Cells were washed twice with rinsing solution and lysed in RIPA buffer containing 150 mm NaCl, 1 mm EDTA, 100 mm Tris HCl, 1% Triton-X, 1% sodium deoxycholate, 0.1% SDS, and 1:500 protease inhibitor cocktail for 1 h at 4°C. Cells were collected and cleared by centrifugation at 12,400 × g for 20 min. One aliquot of supernatant was taken for lysate fraction and stored at −20°C until use. A second aliquot was incubated with washed avidin-linked agarose beads overnight at 4°C while shaking. Beads were spun down at 12,400 × g for 15 min, and the supernatant (intracellular fraction) was removed and stored at −20°C until use. The beads were rinsed four times with (1) RIPA buffer, (2) high-salt wash buffer, (3) high-salt wash buffer (4), and no-salt wash buffer and stored at −20°C until use. Surface fraction proteins were eluted from the beads with equal volume 2× sample buffer, incubated for 10 min at room temperature and 30 min at 37°C, and spun down. Lysate fraction protein concentrations were determined by BCA protein assay and equal amounts of protein were loaded; surface fraction samples were loaded in the same volume as their lysate counterparts. Gels were probed with anti-GluN1 antibody (BD Biosciences, catalog no. 556308; 1:1000). Lysate GFP levels were measured to control for transfection efficiency (Sigma, catalog no. G1544; 1:3000); surface transferrin receptor levels were measured as a surface fraction loading control (Zymed/Invitrogen, catalog no. 13-6800; 1:2000). Surface actin levels were also measured to verify clean fractionation (Sigma, catalog no. A2103; 1:3000).
Statistical analysis.
Quantifications of immunocytochemistry images and cell surface biotinylation Western blots are expressed as mean ± standard error of the mean (SEM). Statistical comparisons were made by Student's t test and one-way or two-way ANOVA plus Tukey's post hoc testing or Kruskal–Wallis test plus Dunn's post hoc testing, as indicated in figure legends. Correlation of antibody staining with model peptide half-lives (Robinson et al., 2004) was analyzed with nonlinear regression and one phase decay. Single-channel closed and open times were measured with cumulative fit histograms. For single channel burst analysis, the number of events per burst was evaluated with histograms, which were then fit with one-phase decay exponentials; all r2 > 0.97. The tau values of those exponentials were then correlated with antibody staining using linear regression. All statistical analyses were performed with GraphPad Prism (GraphPad Software), and values of p < 0.05 were considered significant.
Results
We previously showed that the ATD of GluN1 is required for binding of anti-NMDAR patients' antibodies (Dalmau et al., 2008), a region that is found in NMDARs across the CNS. However, patients' antibodies do not appear to stain all regions equally; immunohistochemistry experiments show robust hippocampal staining, some cortical staining, and very little cerebellar staining. Similarly, the symptomatology of patients suggests hippocampal and cortical involvement, with less evidence of cerebellar dysfunction (Dalmau et al., 2008). These regions express different levels of GluN1 splice variants (Laurie et al., 1995), and one alternatively spliced cassette, exon 5, is found in the ATD (Dingledine et al., 1999). Therefore, we expressed GluN1 splice variants with and without exon 5 in HEK293 cells and stained them with patients' antibodies. Some GluN1 isoforms do not reach the cell surface when expressed without a GluN2/3 (McIlhinney et al., 1998); therefore, by using permeabilized cells, we can evaluate the role of GluN1 alone. The presence or absence of exon 5 did not affect staining by patients' antibodies (Fig. 1A). The adult hippocampus and cortex also express much higher levels of GluN2B than the cerebellum (Watanabe et al., 1992; Akazawa et al., 1994); although antibody binding only requires GluN1, it is possible that the presence of GluN2B modulates staining. We expressed GluN1 alone, with GluN2B, and with GluN2B mutants missing either the ATD or the S2 domain, either of which could potentially alter GluN1-ATD conformation and therefore staining. None of the GluN2B variants impacted antibody staining (Fig. 1B), confirming the importance of GluN1 in antibody recognition.
Figure 1.
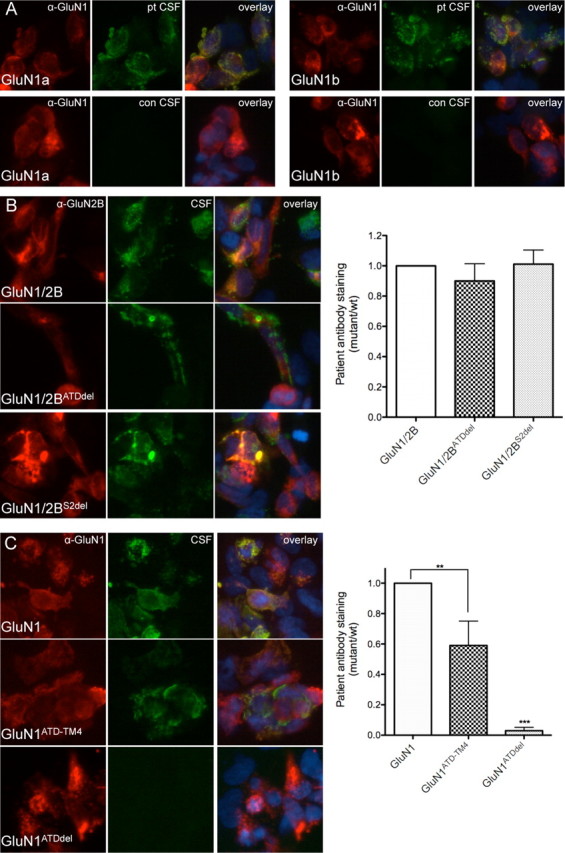
Patients' antibodies recognize the ATD of GluN1 and are not dependent on splice variant or GluN2 domains. A, Patients' antibodies (pt CSF) recognize both splice variants that lack (GluN1a) and include (GluN1b) exon 5 (GluN1a = 0.482 ± 0.088 AU; GluN1b = 0.586 ± 0.073 AU (where AU is arbitrary units); n = 5 patients, p > 0.05, t test with Welch's correction). CSF from individuals without anti-NMDAR encephalitis (con CSF) does not stain GluN1-transfected HEK cells. B, Antibody staining is not affected by the absence of domains within GluN2B (n = 7 patients, p > 0.05, one-way ANOVA plus Tukey's post hoc testing). C, The ATD of GluN1 is both necessary and sufficient for staining by patients' antibodies, although the lack of other extracellular and transmembrane domains does decrease antibody staining (n = 7–10 patients, **p < 0.01, ***p < 0.001, one-way ANOVA plus Tukey's post hoc testing).
We next sought to determine whether the ATD is also sufficient for antibody recognition. Therefore, we created a GluN1 mutant that lacks S1, S2, and several transmembrane domains, linking the end of the ATD directly to TM4 (ATD-TM4). This mutant expressed well and stained robustly with patients' antibodies (Fig. 1C). In contrast, GluN1 lacking the ATD did not stain at all, confirming both the necessity and sufficiency of the ATD for epitope creation.
Potential role of post-translational modifications in immunogenicity
These data indicate that while a specific region in GluN1 is necessary and sufficient for immunoreactivity, it is not associated with known variability in primary sequence in different regions of the brain. Therefore, we considered whether post-translational modifications might alter immunoreactivity. Post-translational modifications (PTMs) can occur differentially in different parts of the CNS (Grigoriadis and De Souza, 1989). They can also alter immunogenicity of proteins, which can trigger autoimmune responses; the presence of an epitope that is not expressed in the thymus appears to make it easier to escape self-tolerance (Doyle and Mamula, 2001). Among PTMs that are found on extracellular domains, N-linked glycosylation is one of the most common. The ATD of GluN1 includes seven N-linked glycosylation consensus sites. When we blocked N-linked glycosylation by growing cells in the presence of tunicamycin, an inhibitor of N-acetylglucosamine transferases, patients' antibodies no longer stained cells transfected with GluN1, indicating that N-linked glycosylation may be necessary for creation of the epitope (Fig. 2A).
Figure 2.
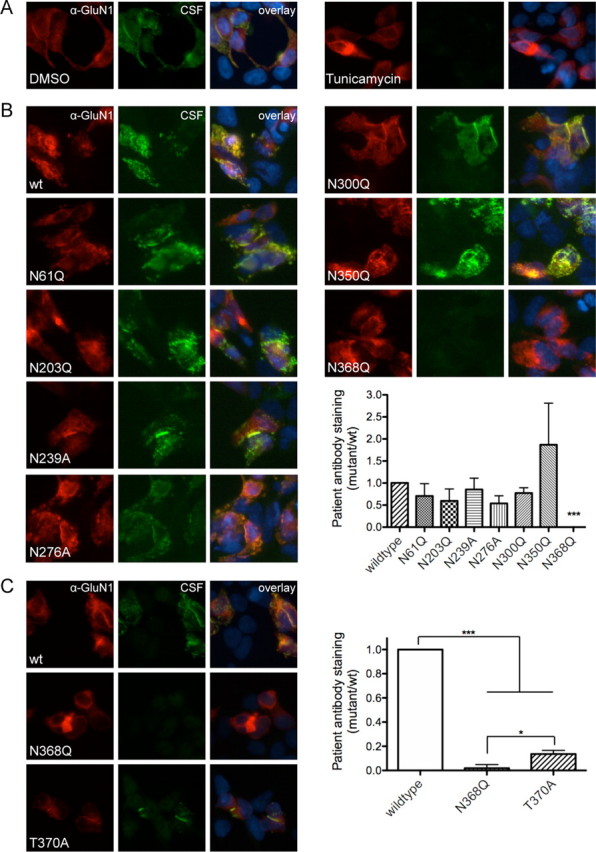
GluN1–N368 affects epitope formation, independent of glycosylation state. A, Blocking N-linked glycosylation with tunicamycin (2 μg/ml) blocks antibody staining (DMSO = 0.605 ± 0.117 AU, tunicamycin = −0.016 ± 0.020 AU (where AU is arbitrary units); n = 10 patients, p < 0.001, t test with Welch's correction). B, Mutation of six of the seven N-linked glycosylation sites within the ATD of GluN1 does not affect staining by patients' antibodies, while mutation of N368 to Q blocks antibody staining (n = 6–9 patients; wild-type vs N368Q, p < 0.001; wild-type vs all others, p > 0.05; Kruskal–Wallis test). C, Blocking N-linked glycosylation of N368 by mutating T370 to A, however, decreases but does not abolish antibody staining (n = 11 patients; *p < 0.05, one-way ANOVA plus Tukey's post hoc testing, ***p < 0.001).
However, NMDA receptor expression and formation can be altered by tunicamycin (Chazot et al., 1995; Everts et al., 1997), and secondary effects on receptor structure could explain the lack of staining in tunicamycin-treated cultures. Therefore, to confirm the role of N-linked glycosylation in epitope formation, we created a series of receptors with single point mutations. The canonical N-linked glycosylation consensus sequence is N-X-S/T, where X can be anything except proline (Kornfeld and Kornfeld, 1985). Each of the seven ATD glycosylation sites was mutated from N to Q; N239Q and N276Q did not express sufficiently (data not shown), so the additional mutants N239A and N276A were made. While six of the N-linked mutants did not affect staining with patients' antibodies, N368Q abolished antibody staining (Fig. 2B), indicating that glycosylation of residue N368 may be a necessary component of the epitope.
To verify the role of N368 glycosylation, we made an additional mutation of the glycosylation consensus site, T370A. Unlike N368Q, T370A decreased antibody staining but did not fully abolish it (Fig. 2C), indicating that glycosylation state is not the only determining factor in this region's role in epitope formation. In addition to being a consensus site for glycosylation, GluN1-N368 may undergo deamidation, a nonenzymatic PTM in which asparagine or aspartate residues are spontaneously converted first to a succinimidyl intermediate before resolving into either aspartate or isoaspartate (IsoAsp) (Robinson and Robinson, 2004). The process is determined by the flexibility of the region; in model peptides, NG sequences deamidate with a half-life of roughly one day, while more constrained NI sequences deamidate with a half-life closer to a year (Robinson et al., 2004). Deamidation is also more common in long-lived proteins and occurs in unfolded portions of the protein. Like glycosylation, deamidation has also been associated with autoimmunity and escape from self-tolerance (Molberg et al., 1998; Moss et al., 2005). The NMDA receptor tends to be long-lived (Huh and Wenthold, 1999), GluN1-N368 appears to be in an unstructured region (Farina et al., 2011; Karakas et al., 2011), and the C-terminal residue is a glycine, all of which make it possible that GluN1-N368 may natively deamidate and that this event may play a role in epitope formation.
We therefore made a series of mutations to residue G369 that would affect deamidation of N368: G369A, G369I, G369L, G369S, and G369T, which should slow deamidation to different degrees. These mutants were expressed in HEK293 cells and stained with patients' antibodies. As expected for a deamidation-based epitope, the G369 mutants stained to different extents (Fig. 3A) and correlated with deamidation rates of model peptides (Robinson et al., 2004) (Fig. 3B). Additionally, we made an N368D mutant, which should allow deamidation, and an N368D/G369I double mutant, which would allow formation of Asp but block the formation of IsoAsp. N368D maintained antibody staining, although less robustly than wild-type, while N368D/G369I was not stained by patients' antibodies (Fig. 3C), potentially suggesting a role for isoaspartate rather than aspartate in the creation of the immunoreactive region.
Figure 3.

Patient antibody staining has the same structural components as deamidation. A, Mutating residue G369 decreases staining by patients' antibodies to different extents (n = 8–9 patients, ***p < 0.001, one-way ANOVA plus Tukey's post hoc testing). B, Mutation of G369 to residues that increasingly slow deamidation in model peptides correlate with decreased patient antibody staining (r2 = 0.87, nonlinear correlation, one phase decay; where wt is wild type). C, Mutation of N368 to D maintains antibody staining at a decreased level while dual mutation of N368D/G369I blocks staining (n = 8–9 patients, ***p < 0.001, one-way ANOVA plus Tukey's post hoc testing).
To determine whether N368 is deamidated in culture systems and in vivo, GluN1 was immunoprecipitated from HEK293 cells expressing wild-type GluN1 or different N368/G369 mutants, as well as native GluN1 from rat brain cortex, hippocampus, or cerebellum, and analyzed by mass spectrometry. GluN1 was digested with trypsin followed by PNGase F in H218O. PNGase F deglycosylates N-glycosylated peptides, converting Asn to Asp; in the presence of H218O, these Asp residues are marked with 18O (Gonzalez et al., 1992). This allows quantification of amidated or unmodified GluN1 as well as natively deamidated GluN1 (+1 Da) and GluN1 deglycosylated by PNGase F (+3 Da). GluN1-N368D serves as a positive control for “deamidated” GluN1. PNGase F-treated wild-type GluN1 showed exclusively a +3 Da shift in expected mass or a +2 Da shift when compared to N368D (Fig. 4A,B), indicating that GluN1 is fully glycosylated under normal circumstances. This was also seen across G369 mutants as well as GluN1-N368 isolated from different brain regions (Fig. 4C).
Figure 4.

GluN1–N368 is fully glycosylated in heterologous systems and rat brain regions. A, Peptide ion isotope abundance profiles. GluN1–N368D has a one Da increase in peptide mass due to the presence of D instead of N (top), while deglycosylating hippocampal GluN1 with PNGase F in 18O water leads to a further two Da increase in peptide size due to transfer of 18O during deglycosylation (bottom) and conversion of N to D. B, Example MS-MS spectra of GluN1–368-containing peptides from N368D (top) or deglycosylated hippocampal GluN1 (bottom). All hippocampal fragments that include deglycosylated N368 show a 2 Da increase in mass due to 18O incorporation. C, Observed unmodified, deamidated (+1 Da), or deglycosylated (+3 Da) GluN1–368 peptides from N368/G369 mutants and rat brain regions, immunoprecipitated with commercial GluN1 antibody, and rat hippocampus, immunoprecipitated with CSF from two patients. “Unmodified” refers to peptides of the expected mass; N368D and N368D/G369I show no evidence of post-translational modification but do show a 1 Da increase due to the presence of Asp residues. D, Western blot (WB) of GluN1 immunoprecipitations (IP) using commercial antibody or CSF from two different patients. Br, Whole brain; Ctx, cortex; Hp, hippocampus; Cb, cerebellum; all rat.
Immunoprecipitation with patients' antibodies
Having shown that GluN1-N368 is glycosylated in different brain regions, we next sought to determine the exact PTM profile of GluN1 molecules that are recognized by patient antibody. Therefore, we immunoprecipitated GluN1 with CSF from a high-titer patient and analyzed it by mass spectrometry. GluN1 was positively identified, including N368 deglycosylated by PNGase F treatment (Fig. 4C). Thus, patients' antibodies recognize glycosylated N368. Together, these results suggest that although deamidation of this residue is not the key event leading to immunogenicity, the structural determinants for creation of the epitope match those for deamidation of N368.
The results of the N368-related experiments suggest that glycosylation and deamidation of N368 are neither necessary nor sufficient for epitope formation, but more likely contribute by creating favorable conformations of this small region. Therefore, we sought to identify conformational components of epitope formation in other paradigms. Having determined conditions under which patients' CSF can immunoprecipitate GluN1, we further explored the issue of regional specificity of antibody with immunoprecipitation rather than immunohistochemistry. CSF from both a high titer patient and a lower titer patient immunoprecipitated GluN1 from whole rat brain lysate as well as cortex, hippocampus, and, in contrast with immunohistochemistry experiments, cerebellum (Fig. 4D). These immunoprecipitations show that patients' CSF is capable of binding cerebellar GluN1 when presented in the correct context, and further demonstrate the presence of conformational features of the epitope. This conformational dependency of antibody binding is also probably responsible for inability of patients' antibodies to recognize NMDAR on Western blots (data not shown).
Lack of role of GluN2/3 subunits
Recent crystal structures show that the NMDA receptor ATD exists as a dimer of dimers, with a number of points of contact between the top lobes of dimerized GluN1 and GluN2 subunits, but with little contact between the bottom lobes due to a conformational rotation (Karakas et al., 2009; Stroebel et al., 2011). GluN1 residues N368 and G369 are found in the bottom lobe of the domain; therefore, we suspected that their effects on antibody binding would be independent of GluN2 or GluN3 variant. Because we have created GluN1 mutants that show a wide spectrum of patient antibody binding, cotransfection with this panel of mutants allows us to identify even a minor impact of GluN2/3 subunit on antibody binding. Antibody staining patterns of N368/G369 mutants did not vary with GluN2 subunit or GluN3A (Fig. 5), showing that patients' antibody staining is controlled by GluN1 alone.
Figure 5.

GluN1–N368/G369 modulates patient antibody staining with no contribution from GluN2/3 subunits. A, Example overlay pictures of wild-type GluN1, G369S, and G369L subunits coexpressed with GluN2 subunits, GluN3A, or vector. Red, Commercial GluN1 antibody; green, patient CSF. B, Quantification of coexpression studies. GluN1 mutant accounts for most of the total variance (n = 3 patients; GluN1 = 69.2%, p < 0.0001, two-way ANOVA), while GluN2 subunit effects are nonsignificant (2.52%, p = 0.14). GluN3A also does not impact patient antibody staining (n = 3 patients; GluN1 = 86.4%, p < 0.0001; GluN3A = 0.2%, p = 0.52, two-way ANOVA).
Electrophysiological effects of N368/G369 mutations
Residues N368 and G369 are located near the top of the bottom lobe of the ATD, close to the hinge between the two lobes, a region that is postulated to control receptor physiology (Gielen et al., 2009; Yuan et al., 2009; Hansen et al., 2010). Therefore, mutations in this area could induce conformational changes in the GluN1 ATD, which in turn affect the single channel properties of the receptor. To explore this possibility, we performed single channel recordings on outside-out patches from HEK293 cells cotransfected with different GluN1 mutants and GluN2B (Fig. 6A). While there is no consistent relationship between the open time of the receptor and patient antibody staining (Fig. 6B), the relationship between antibody staining and the closed duration of the receptor is fairly unambiguous; the GluN1 variants that stain most robustly are closed the shortest time. This does not reflect differential ability to reach the cell surface, as all N368/G369 mutants reach the surface to similar levels as wild type by cell-surface biotinylation (Fig. 6C).
Figure 6.
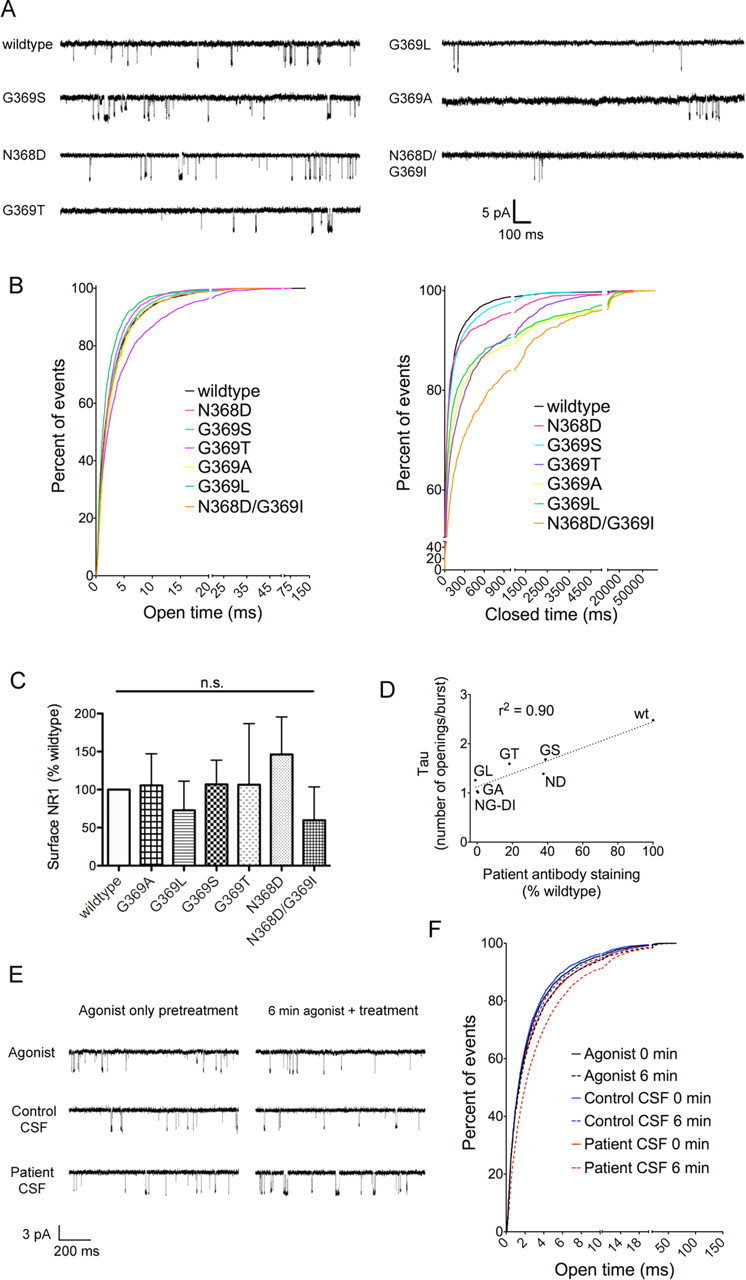
GluN1–N368/G369 mutations affect receptor closed duration but not surface expression, and antibody application prolongs open time. A, Example single-channel outside-out traces from GluN1 mutants, coexpressed with GluN2B in HEK293 cells. All traces were recorded with coapplication of 100 nm glutamate and 1 μm glycine. B, Cumulative frequency histograms of open duration and closed duration of single-channel traces (n = 4–5 patches per mutant). C, Cell-surface biotinylations show no difference in surface expression of GluN1, normalized to transfection efficiency (lysate GFP levels) and a surface loading control (transferrin receptor) (n = 3, p > 0.05, one-way ANOVA). D, The number of events in a burst correlates with patient antibody staining of N368/G369 mutants, as measured by the tau of the exponential fit of event number histogram (r2 = 0.90, linear regression; n (single channel burst analysis) = 4–5 patches, n (staining) = 8–11 patients). E, Example single-channel outside-out traces from GluN1/GluN2B-transfected HEK293 cells exposed to a minimum of 2 min 100 nm glutamate/1 μm glycine (agonist only pretreatment) followed by a minimum of 7 min agonist only, agonist with control CSF (1:100), or agonist with patient CSF (1:100) (6 min agonist plus treatment). F, Cumulative frequency histograms of open duration (n = 7–8 patches per condition). 0 min = open duration during 1 min of recordings in agonist immediately preceding treatment; 6 min = open duration during 1 min of recordings in agonist only, agonist + control CSF (1:100, 2 individuals), or agonist + patient CSF (1:100, 3 individuals), measured after 6–7 min of treatment.
However, closed durations alone may reflect differences in the numbers of channels in a given patch; therefore, we performed a burst analysis of closed durations (see Materials and Methods). This revealed increases in the percentage of single opening events per burst among mutant receptors. Because single openings are excluded from a burst analysis-based calculation of Popen (Plested et al., 2007), we instead quantified the role of N368/G369 mutation on open probability by fitting single decay exponential curves to histograms of the numbers of events per burst. The tau of those exponential fits correlates strongly with patient antibody staining, suggesting that patients' antibodies stain GluN1 ATD forms that are prone to opening more frequently.
If antibodies are more likely to bind to an ATD conformation that promotes short closed durations, antibodies themselves may have an effect on receptor function. While prolonged (>24 h) incubation leads to a decrease in NMDAR levels (Hughes et al., 2010), the acute effects of binding patients' antibodies are unknown. Therefore, we performed single-channel recordings on outside-out patches from GluN1/GluN2B-expressing HEK293 cells and applied agonist alone for at least 2 min (100 nm glutamate/1 μm glycine), followed by agonist alone or with patient or control CSF (1:100) for an additional 7 min. Agonist alone or agonist with control CSF did not have a sizeable effect on open duration of the receptor (Fig. 6E,F). Agonist with patient CSF, however, prolonged the open duration of the receptor (Fig. 6E,F). None of the treatments had a consistent effect on receptor closed time (data not shown), indicating that patient antibody does not itself promote opening. Rather, these data suggest that patient antibody binds to a receptor that is more prone to opening and stabilizes the open state.
Immunogenic profile of adult male and pediatric patients
Although patients with anti-NMDA receptor encephalitis generally have similar symptoms and all show GluN1 reactivity (Dalmau et al., 2008; Florance et al., 2009), different patient subgroups, based on demographic or etiological factors, could generate different immune responses. Having created a series of point mutations that alter patient antibody staining in stereotyped ways, we tested whether antibodies from the more recently identified patient populations recognize N368/G369 mutants similarly. Both adult male and male and female pediatric patients (Fig. 7A,B) showed the same staining patterns, indicating that all anti-NMDAR encephalitis patients, regardless of age, sex, presence of tumor, or variability in presentation, have similar epitopic determinants and share similar immunological processes.
Figure 7.

Adult male and pediatric patients show similar reactivity to GluN1 mutants as the original adult female patient population. A, Example staining patterns of a patient from the originally identified patient population (female with ovarian teratoma) and a more recently identified patient (pediatric male). B, Quantification of staining patterns in patients that match the original cohort (n = 9–11 patients) and those that have been identified more recently (males and pediatric patients without tumors, n = 7 patients). The patient population does not contribute to the variability within the samples (0.01%, p = 0.82, two-way ANOVA); most variability is accounted for by the GluN1 mutant (76.71%, p < 0.0001). Most of the significant differences between GluN1 mutants are identical between the two cohorts: wild-type versus all mutants, p < 0.001 to p < 0.0001; N368Q and G369I versus G369S and N368D, p < 0.05 to p < 0.0001. However, the differences between T370A and N368D are significant in the male/pediatric cohort (p < 0.05) but not the original (p > 0.05). *p < 0.05, **p < 0.01, ***p < 0.001.
Deletion of additional ATD subdomains
The recently published GluN1 ATD crystal structures (Farina et al., 2011; Karakas et al., 2011) show that residues N368 and G369 are located in the bottom lobe of the ATD, near the hinge of the two lobes. To determine whether the N368/G369 mutations cause a conformational change that affects a distant epitope or are themselves near the epitope, we created additional deletion mutants. The crystal structures reveal an α-helix spanning residues 144–156 in close proximity to N368/G369, although far removed in primary sequence. Therefore, we deleted these residues and stained with patients' antibodies. Deletion of residues 144–156 abolishes antibody staining (Fig. 8A, D), providing further evidence that this region of the bottom lobe is a necessary component of the epitope. Because of the proximity of N368/G369 to the top lobe, we also created a top lobe deletion mutant. This mutant, unlike the other mutants we have made and tested, shows a wide range of staining with patients' antibodies (Fig. 8E). While some antibodies maintain staining with this mutant (Fig. 8A), others lose staining entirely (Fig. 8B), and still others show an enhanced response to the deletion of the top lobe (Fig. 8C). These data indicate that the antibody binding site on GluN1, while universally involving residues N368/G369, shows more variability than the N368/G369 staining patterns suggest.
Figure 8.

Additional deletions of GluN1-ATD subdomains support an epitope located near the hinge of the top and bottom lobes and reveal a degree of antibody heterogeneity. A–C, Binding of patients' antibodies is blocked by deletion of an α-helix spanning residues 144–156, which is near N368/G369. Deletion of the top lobe (top del) of the ATD, however, has variable effects. It can preserve antibody staining at levels close to that of wild-type (A), destroy antibody staining (B), or increase antibody staining (C). D, Deletion of residues 144–156 uniformly destroys antibody binding (n = 4 patients, ****p < 0.0001 vs wild-type, Student's t test), while deletion of the top lobe has different effects on different patients (n = 15 patients, p > 0.05 vs wild-type, Student's t test). E, Individual staining intensities of ATD top lobe deletion versus wild-type. F, Model of ATD with proposed antibody binding site and effect of G369 mutations. Open-cleft conformation of the ATD both allows antibody binding and leads to channel opening, and the nearby α-helix spanning residues 144–156 also seems to play a role. Antibody binding to this open conformation then stabilizes the conformation and prolongs open time. The proximity of the top lobe of the ATD makes it likely that a small shift in epitope location could result in the variable staining patterns of the ATD top lobe deletion mutants. G, Substitution of larger residues at position 369 block antibody binding and promote a closed-cleft conformation that keeps the channel closed.
Discussion
This study has shown that the ATD of GluN1 is necessary and sufficient for recognition by antibodies from patients with anti-NMDAR encephalitis. Within the ATD, we have identified a small region that is crucial for antibody recognition. The importance of this region is similar across multiple demographic and etiological subgroups of the disorder, and specific structural features of antibody recognition are linked to specific physiological properties. Furthermore, acute antibody application prolongs the open time of the receptor. We have also identified a region that impacts antibody staining in variable ways. Together, these studies identify biochemical features common across the rapidly expanding field of anti-NMDAR encephalitis patients, with implications for receptor physiology in the presence and absence of antibody, as well as a less prominent variation that may ultimately explain some of the differences between patients. This is an important step toward a more complete understanding of the pathophysiology behind this recently discovered and increasingly prominent disorder.
While the distribution of patient antibody staining resembles that of GluN2B (Watanabe et al., 1992; Akazawa et al., 1994) and/or GluN1a splice variants (Laurie et al., 1995), antibody staining is unaffected by GluN2 and instead appears to be controlled by the conformation of the GluN1 ATD. Patients' CSF can immunoprecipitate cerebellar GluN1, suggesting that there is no inherent structural difference between cerebellar GluN1 and hippocampal/cortical GluN1 that abolishes immunoreactivity when assayed in solution. The different regional pattern of antibody staining could instead reflect conformational differences in GluN1 in receptors as presented in fixed slices versus receptors assayed in solution. Cerebellar NMDARs are conformationally distinct from cortical/hippocampal NMDARs as measured by pharmacological approaches (Murphy et al., 1987; Sanchez-Perez et al., 2005). Such differences may be less prominent in more flexible detergent-solubilized receptors, allowing antibody binding.
The single channel studies of N368/G369 mutants link recognition by patients' antibodies with specific physiological properties. The ATD has a number of well defined roles in NMDAR physiology (Paoletti, 2011; Furukawa, 2012). It is necessary for receptor assembly (Meddows et al., 2001; Hansen et al., 2010) and contains the binding site for a number of allosteric modulators (for review, see Paoletti, 2011). The ATD consists of a top and bottom lobe, creating a clamshell-like structure (Jin et al., 2009; Karakas et al., 2009; Sobolevsky et al., 2009). The conformation of this clamshell may reflect interactions with other receptor subunits as well as binding of modulators (Karakas et al., 2011; Furukawa, 2012), which then alter channel opening and closing (Gielen et al., 2009; Paoletti, 2011). N368/G369 are adjacent to the hinge of the clamshell; changing the size of these residues could readily change the open/closed clamshell dynamics.
The mutations to residues N368/G369 could affect the closed time of the receptor by changing the clamshell conformation, which then impacts antibody staining. Incrementally increasing the size of the G369 residue causes a corresponding decrease in patient antibody staining; this indicates that G369 may act as a hinge, inducing progressively greater conformational change that increasingly distorts the epitope and prevents antibody binding while also affecting receptor open probability (Fig. 8F,G). We propose that acute antibody application to wild-type receptors stabilizes the open conformation, leading to the observed increase in open duration. Alternatively, mutant-induced conformational changes could alter the relative exposure of an antibody binding site located elsewhere in the ATD. Changing the glycosylation state of N368 could have a similar effect, inducing variable conformational changes depending on the mutation introduced.
While our data suggest that the clamshell conformation of the GluN1 ATD affects patient antibody binding, how this relates to the differences in antibody binding across brain regions in immunohistochemical experiments is unclear. Differences in regional reactivity are not directly modulated by the GluN2/3 subunit or post-translational modifications of N368/G369. Other interactions such as regional binding partners or region-specific release of modulators could play a role in antibody recognition. The expression of physiological NMDAR modulators is not usually restricted to one or two brain regions, as large pools of presynaptic zinc, an NMDAR modulator, are found in most brain regions (Palmiter et al., 1996; Wang et al., 2002); polyamines, which can modulate NMDARs through multiple mechanisms (for review, see Yamakura and Shimoji, 1999), are released throughout the brain (Paschen et al., 1992); and EphB receptors, which can bind NMDARs (Dalva et al., 2000), are expressed throughout the nervous system (Willson et al., 2006). Therefore, while several candidate modulators of ATD conformation could confer specificity on antibody recognition, none have obvious regional restrictions. Further work remains to be done to determine the causes of differential regional antibody binding in tissue and, based on symptomatology, in vivo.
In addition to the potential importance of the N368/G369 region on channel function, the current study provides tools that will be useful in refining the diagnosis and evaluation of subjects with potential anti-NMDAR encephalitis. Here, distinct patient groups all have similar antibody reactivity when evaluated by point mutations of N368/G369. This work can be expanded to include more patients to test for variability within the population. These mutants also allow more selective evaluation of anti-NMDAR encephalitis patients compared with other patient populations having NMDAR antibodies, such as lupus (DeGiorgio et al., 2001) and those with predominant psychiatric symptoms or abnormal movements (Chapman and Vause, 2011; Rubio-Agusti et al., 2011). The detailed antibody response in such patients might correlate with a wider range of symptoms displayed. We found that deletion of the top lobe of the ATD can maintain, enhance, or abolish antibody staining. Because of the proximity of N368/G369 to the top lobe, it seems reasonable that a small shift in the exact epitope between patients could result in variable responses to the presence of the ATD top lobe, while maintaining the N368/G369 region as a primary component of the immunogen. This would be similar to the known epitopic region of the nicotinic acetylcholine receptor in myasthenia gravis, in which many antibodies are directed against a “main immunogenic region,” which is fairly small but not a single epitope (Tzartos et al., 1998).
Anti-NMDAR encephalitis patients may also make additional GluN1 antibodies. A number of autoimmune disorders have diverse antibody responses, including SLE, in which only one-third of patients have anti-NMDAR antibodies (Lauvsnes and Omdal, 2012), and myasthenia gravis, in which roughly half of the antibody response is directed against the main immunogenic region (Lindstrom, 2002). We have previously shown that patients' CSF and serum IgG have similar effects on NMDARs in cultured neurons (Hughes et al., 2010). However, it is possible that those effects are all mediated by a specific antibody population, namely the conformation-dependent ATD antibodies described here. While those appear to be the only NMDAR antibody population in the CSF, it is possible that the serum antibody response is more heterogeneous, as has been described for other disorders (Raju et al., 2005). These mutants provide a means for testing the heterogeneity of the serum antibody population, which may be helpful for monitoring disease progression. Additionally, IgA antibodies to the NMDAR were recently described in association with cognitive decline (Prüss et al., 2012); it would be interesting to test whether these antibodies are directed against the same region of GluN1.
The region around the glycosylation site GluN1-N368 controls recognition by patients' antibodies, but glycosylation itself is not necessary or sufficient for antibody staining. Still, it is becoming increasingly clear that glycosylation affects channels in a variety of ways: GluN2B glycosylation was recently identified as a determinant of synaptic integration (Storey et al., 2011), and ASIC1a trafficking is regulated by N-linked glycosylation (Jing et al., 2012). The regulation of glycosylation is still largely unknown; however, one potential mechanism of regulation is the restricted expression of different glycosyltransferases (Yamamoto et al., 2003). As the detailed investigation of specific glycopeptides becomes more commonplace, the mechanisms underlying glycosylation regulation should become more clear.
While the present work has made substantial progress in determining which components of the ATD are necessary for antibody binding, developing tools that can be used to both study the role of ATD conformation on channel function and make a thorough examination of the patient population, considerable work remains. Based on the present data, N368 PTMs are not a tenable explanation for the relative lack of cerebellar antibody staining and symptomatology, but the reason for the sparing of the cerebellum is unknown. It is unclear how ATD modulatory factors interact with patient antibody binding and what effect that might have on disease progression. Also, while the present work provides strong evidence that the epitope is the same across patients with tumors and those without, how the autoimmune response is generated in the latter group is unclear. There remain a large number of potential triggers for the initiation of the immune process; as this disorder becomes more widely identified and studied, there should be significant advances in this area. Because recovery rates are much greater when the tumor is found and removed (Dalmau et al., 2008), determining the immunological trigger must be a priority in future work. The tools developed here should help in that process.
Footnotes
This work was supported by NIH Grants NS045986, NS067270, and P30 HD026979 (D.R.L.); NS077851, MH094741, and CA89054 (J.D.); and MH083395 (A.J.G.). J.D. receives additional funding from Euroimmun, Fundació la Marató de TV3, and the McKnight Foundation. We thank Stefano Vicini for helpful discussions and technical aid on single-channel recordings, and Rita Balice-Gordon for useful discussions.
D.R.L and J.D. receive royalties from the sale of testing for NMDAR autoantibodies. J.D. receives additional royalties from the editorial board of UpToDate and from a patent for the use of Ma2 as an autoantibody test.
References
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential expression of five N-methyl-d-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol. 1994;347:150–160. doi: 10.1002/cne.903470112. [DOI] [PubMed] [Google Scholar]
- Chapman MR, Vause HE. Anti-NMDA receptor encephalitis: diagnosis, psychiatric presentation, and treatment. Am J Psychiatry. 2011;168:245–251. doi: 10.1176/appi.ajp.2010.10020181. [DOI] [PubMed] [Google Scholar]
- Chazot PL, Cik M, Stephenson FA. An investigation into the role of N-glycosylation in the functional expression of a recombinant heteromeric NMDA receptor. Mol Membr Biol. 1995;12:331–337. doi: 10.3109/09687689509072435. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol. 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. 2006;26:365–384. doi: 10.1007/s10571-006-9062-8. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE, Voloschin A, Baehring JM, Shimazaki H, Koide R, King D, Mason W, Sansing LH, Dichter MA, Rosenfeld MR, Lynch DR. Paraneoplastic anti-N-methyl-d-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36. doi: 10.1002/ana.21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, Dessain SK, Rosenfeld MR, Balice-Gordon R, Lynch DR. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091–1098. doi: 10.1016/S1474-4422(08)70224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103:945–956. doi: 10.1016/s0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7:1189–1193. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Doyle HA, Mamula MJ. Post-translational protein modifications in antigen recognition and autoimmunity. Trends Immunol. 2001;22:443–449. doi: 10.1016/s1471-4906(01)01976-7. [DOI] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 2005;563:345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts I, Villmann C, Hollmann M. N-Glycosylation is not a prerequisite for glutamate receptor function but is essential for lectin modulation. Mol Pharmacol. 1997;52:861–873. doi: 10.1124/mol.52.5.861. [DOI] [PubMed] [Google Scholar]
- Farina AN, Blain KY, Maruo T, Kwiatkowski W, Choe S, Nakagawa T. Separation of domain contacts is required for heterotetrameric assembly of functional NMDA receptors. J Neurosci. 2011;31:3565–3579. doi: 10.1523/JNEUROSCI.6041-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke C, Kopp UA, Prüss H, Dalmau J, Wandinger KP, Ploner CJ. Cognitive deficits following anti-NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry. 2012;83:195–198. doi: 10.1136/jnnp-2011-300411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florance NR, Davis RL, Lam C, Szperka C, Zhou L, Ahmad S, Campen CJ, Moss H, Peter N, Gleichman AJ, Glaser CA, Lynch DR, Rosenfeld MR, Dalmau J. Anti-N-methyl-d-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol. 2009;66:11–18. doi: 10.1002/ana.21756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H. Structure and function of glutamate receptor amino terminal domains. J Physiol. 2012;590:63–72. doi: 10.1113/jphysiol.2011.213850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielen M, Siegler Retchless B, Mony L, Johnson JW, Paoletti P. Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature. 2009;459:703–707. doi: 10.1038/nature07993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez J, Takao T, Hori H, Besada V, Rodriguez R, Padron G, Shimonishi Y. A method for determination of N-glycosylation sites in glycoproteins by collision-induced dissociation analysis in fast atom bombardment mass spectrometry: identification of the positions of carbohydrate-linked asparagine in recombinant alpha-amylase by treatment with peptide-N-glycosidase F in 18O-labeled water. Anal Biochem. 1992;205:151–158. doi: 10.1016/0003-2697(92)90592-u. [DOI] [PubMed] [Google Scholar]
- Grigoriadis DE, De Souza EB. Heterogeneity between brain and pituitary corticotropin-releasing factor receptors is due to differential glycosylation. Endocrinology. 1989;125:1877–1888. doi: 10.1210/endo-125-4-1877. [DOI] [PubMed] [Google Scholar]
- Hansen KB, Furukawa H, Traynelis SF. Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol Pharmacol. 2010;78:535–549. doi: 10.1124/mol.110.067157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, Parsons TD, Lynch DR, Dalmau J, Balice-Gordon RJ. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. 2010;30:5866–5875. doi: 10.1523/JNEUROSCI.0167-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh KH, Wenthold RJ. Turnover analysis of glutamate receptors identifies a rapidly degraded pool of the N-methyl-d-aspartate receptor subunit, NR1, in cultured cerebellar granule cells. J Biol Chem. 1999;274:151–157. doi: 10.1074/jbc.274.1.151. [DOI] [PubMed] [Google Scholar]
- Irani SR, Bera K, Waters P, Zuliani L, Maxwell S, Zandi MS, Friese MA, Galea I, Kullmann DM, Beeson D, Lang B, Bien CG, Vincent A. N-methyl-d-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain. 2010;133:1655–1667. doi: 10.1093/brain/awq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen EF, Nutting MDF, Jang R, Balls AK. Inhibition of the proteinase and esterase activities of trypsin and chymotrypsin by diisopropyl fluorophosphate; crystallization of inhibited chymotrypsin. J Biol Chem. 1949;179:189–199. [PubMed] [Google Scholar]
- Jin R, Singh SK, Gu S, Furukawa H, Sobolevsky AI, Zhou J, Jin Y, Gouaux E. Crystal structure and association behaviour of the GluR2 amino-terminal domain. EMBO J. 2009;28:1812–1823. doi: 10.1038/emboj.2009.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L, Chu XP, Jiang YQ, Collier DM, Wang B, Jiang Q, Snyder PM, Zha XM. N-glycosylation of acid-sensing ion channel 1a regulates its trafficking and acidosis-induced spine remodeling. J Neurosci. 2012;32:4080–4091. doi: 10.1523/JNEUROSCI.5021-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, Furukawa H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009;28:3910–3920. doi: 10.1038/emboj.2009.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, Furukawa H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature. 2011;475:249–253. doi: 10.1038/nature10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyape P, Taylor E, Ng J, Krishnakumar D, Kirkham F, Whitney A. Successful treatment of two paediatric cases of anti-NMDA receptor encephalitis with Cyclophosphamide: the need for early aggressive immunotherapy in tumour negative paediatric patients. Eur J Paediatr Neurol. 2012;16:74–78. doi: 10.1016/j.ejpn.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Kayser MS, Dalmau J. The emerging link between autoimmune disorders and neuropsychiatric disease. J Neuropsychiatry Clin Neurosci. 2011;23:90–97. doi: 10.1176/appi.neuropsych.23.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Putzke J, Zieglgansberger W, Seeburg PH, Tolle TR. The distribution of splice variants of the NMDAR1 subunit mRNA in adult rat brain. Brain Res Mol Brain Res. 1995;32:94–108. doi: 10.1016/0169-328x(95)00067-3. [DOI] [PubMed] [Google Scholar]
- Lauvsnes MB, Omdal R. Systemic lupus erythematosus, the brain, and anti-NR2 antibodies. J Neurol. 2012;259:622–629. doi: 10.1007/s00415-011-6232-5. [DOI] [PubMed] [Google Scholar]
- Lindstrom J. Autoimmune diseases involving nicotinic receptors. J Neurobiol. 2002;53:656–665. doi: 10.1002/neu.10106. [DOI] [PubMed] [Google Scholar]
- Makarova O, Kamberov E, Margolis B. Generation of deletion and point mutations with one primer in a single cloning step. Biotechniques. 2000;29:970–972. doi: 10.2144/00295bm08. [DOI] [PubMed] [Google Scholar]
- McIlhinney RA, Le Bourdelles B, Molnar E, Tricaud N, Streit P, Whiting PJ. Assembly intracellular targeting and cell surface expression of the human N-methyl-d-aspartate receptor subunits NR1a and NR2A in transfected cells. Neuropharmacology. 1998;37:1355–1367. doi: 10.1016/s0028-3908(98)00121-x. [DOI] [PubMed] [Google Scholar]
- Meddows E, Le Bourdelles B, Grimwood S, Wafford K, Sandhu S, Whiting P, McIlhinney RA. Identification of molecular determinants that are important in the assembly of N-methyl-d-aspartate receptors. J Biol Chem. 2001;276:18795–18803. doi: 10.1074/jbc.M101382200. [DOI] [PubMed] [Google Scholar]
- Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, Fugger L, Scott H, Noren O, Roepstorff P, Lundin KE, Sjostrom H, Sollid LM. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4:713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- Moss CX, Matthews SP, Lamont DJ, Watts C. Asparagine deamidation perturbs antigen presentation on class II major histocompatibility complex molecules. J Biol Chem. 2005;280:18498–18503. doi: 10.1074/jbc.M501241200. [DOI] [PubMed] [Google Scholar]
- Murphy DE, Schneider J, Boehm C, Lehmann J, Williams M. Binding of [3H]3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid to rat brain membranes: a selective, high-affinity ligand for N-methyl-d-aspartate receptors. J Pharmacol Exp Ther. 1987;240:778–784. [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Cole TB, Quaife CJ, Findley SD. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc Natl Acad Sci U S A. 1996;93:14934–14939. doi: 10.1073/pnas.93.25.14934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P. Molecular basis of NMDA receptor functional diversity. Eur J Neurosci. 2011;33:1351–1365. doi: 10.1111/j.1460-9568.2011.07628.x. [DOI] [PubMed] [Google Scholar]
- Paschen W, Widmann R, Weber C. Changes in regional polyamine profiles in rat brains after transient cerebral ischemia (single versus repetitive ischemia): evidence for release of polyamines from injured neurons. Neurosci Lett. 1992;135:121–124. doi: 10.1016/0304-3940(92)90150-6. [DOI] [PubMed] [Google Scholar]
- Plested AJ, Groot-Kormelink PJ, Colquhoun D, Sivilotti LG. Single-channel study of the spasmodic mutation alpha1A52S in recombinant rat glycine receptors. J Physiol. 2007;581:51–73. doi: 10.1113/jphysiol.2006.126920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüss H, Dalmau J, Harms L, Holtje M, Ahnert-Hilger G, Borowski K, Stoecker W, Wandinger KP. Retrospective analysis of NMDA receptor antibodies in encephalitis of unknown origin. Neurology. 2010;75:1735–1739. doi: 10.1212/WNL.0b013e3181fc2a06. [DOI] [PubMed] [Google Scholar]
- Prüss H, Holtje M, Maier N, Gomez A, Buchert R, Harms L, Ahnert-Hilger G, Schmitz D, Terborg C, Kopp U, Klingbeil C, Probst C, Kohler S, Schwab JM, Stoecker W, Dalmau J, Wandinger KP. IgA NMDA receptor antibodies are markers of synaptic immunity in slow cognitive impairment. Neurology. 2012;78:1743–1753. doi: 10.1212/WNL.0b013e318258300d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju R, Foote J, Banga JP, Hall TR, Padoa CJ, Dalakas MC, Ortqvist E, Hampe CS. Analysis of GAD65 autoantibodies in Stiff-Person syndrome patients. J Immunol. 2005;175:7755–7762. doi: 10.4049/jimmunol.175.11.7755. [DOI] [PubMed] [Google Scholar]
- Robinson NE, Robinson A. Molecular clocks: deamidation of asparaginyl and glutaminyl residues in peptides and proteins. Cave Junction, OR: Althouse; 2004. [Google Scholar]
- Robinson NE, Robinson ZW, Robinson BR, Robinson AL, Robinson JA, Robinson ML, Robinson AB. Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J Pept Res. 2004;63:426–436. doi: 10.1111/j.1399-3011.2004.00151.x. [DOI] [PubMed] [Google Scholar]
- Rubio-Agusti I, Dalmau J, Sevilla T, Burgal M, Beltran E, Bataller L. Isolated hemidystonia associated with NMDA receptor antibodies. Mov Disord. 2011;26:351–352. doi: 10.1002/mds.23315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Perez A, Llansola M, Cauli O, Felipo V. Modulation of NMDA receptors in the cerebellum. II. Signaling pathways and physiological modulators regulating NMDA receptor function. Cerebellum. 2005;4:162–170. doi: 10.1080/14734220510008003. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey GP, Opitz-Araya X, Barria A. Molecular determinants controlling NMDA receptor synaptic incorporation. J Neurosci. 2011;31:6311–6316. doi: 10.1523/JNEUROSCI.5553-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroebel D, Carvalho S, Paoletti P. Functional evidence for a twisted conformation of the NMDA receptor GluN2A subunit N-terminal domain. Neuropharmacology. 2011;60:151–158. doi: 10.1016/j.neuropharm.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Tang HY, Ali-Khan N, Echan LA, Levenkova N, Rux JJ, Speicher DW. A novel four-dimensional strategy combining protein and peptide separation methods enables detection of low-abundance proteins in human plasma and serum proteomes. Proteomics. 2005;5:3329–3342. doi: 10.1002/pmic.200401275. [DOI] [PubMed] [Google Scholar]
- Tzartos SJ, Barkas T, Cung MT, Mamalaki A, Marraud M, Orlewski P, Papanastasiou D, Sakarellos C, Sakarellos-Daitsiotis M, Tsantili P, Tsikaris V. Anatomy of the antigenic structure of a large membrane autoantigen, the muscle-type nicotinic acetylcholine receptor. Immunol Rev. 1998;163:89–120. doi: 10.1111/j.1600-065x.1998.tb01190.x. [DOI] [PubMed] [Google Scholar]
- Wang Z, Danscher G, Kim YK, Dahlstrom A, Mook Jo S. Inhibitory zinc-enriched terminals in the mouse cerebellum: double-immunohistochemistry for zinc transporter 3 and glutamate decarboxylase. Neurosci Lett. 2002;321:37–40. doi: 10.1016/s0304-3940(01)02560-5. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K, Mishina M. Developmental changes in distribution of NMDA receptor channel subunit mRNAs. Neuroreport. 1992;3:1138–1140. doi: 10.1097/00001756-199212000-00027. [DOI] [PubMed] [Google Scholar]
- Willson CA, Foster RD, Onifer SM, Whittemore SR, Miranda JD. EphB3 receptor and ligand expression in the adult rat brain. J Mol Histol. 2006;37:369–380. doi: 10.1007/s10735-006-9067-0. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hsu FC, Gleichman AJ, Baconguis I, Coulter DA, Lynch DR. Fyn-mediated phosphorylation of NR2B Tyr-1336 controls calpain-mediated NR2B cleavage in neurons and heterologous systems. J Biol Chem. 2007;282:20075–20087. doi: 10.1074/jbc.M700624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie DJ, Johnston AR, Lipscombe D, Chen PE. Single-channel analysis of a point mutation of a conserved serine residue in the S2 ligand-binding domain of the NR2A NMDA receptor subunit. J Physiol. 2006;574:477–489. doi: 10.1113/jphysiol.2006.112193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakura T, Shimoji K. Subunit- and site-specific pharmacology of the NMDA receptor channel. Prog Neurobiol. 1999;59:279–298. doi: 10.1016/s0301-0082(99)00007-6. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Yamamoto F, Luong TT, Williams T, Kominato Y. Expression profiling of 68 glycosyltransferase genes in 27 different human tissues by the systematic multiplex reverse transcription-polymerase chain reaction method revealed clustering of sexually related tissues in hierarchical clustering algorithm analysis. Electrophoresis. 2003;24:2295–2307. doi: 10.1002/elps.200305459. [DOI] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J Neurosci. 2009;29:12045–12058. doi: 10.1523/JNEUROSCI.1365-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Tanaka K, Sun P, Nakata M, Yamamoto R, Sakimura K, Matsui M, Kato N. Suppression of synaptic plasticity by cerebrospinal fluid from anti-NMDA receptor encephalitis patients. Neurobiol Dis. 2012;45:610–615. doi: 10.1016/j.nbd.2011.09.019. [DOI] [PubMed] [Google Scholar]