

Abstract
Invasive tumors (cancers or malignant lesions) typically develop in the setting in which there is the presence of putative non-invasive lesions and the development of these non-invasive lesions frequently precedes the development of cancers. For some organs, such as the oral cavity, cervix and skin, the respective putative pre-invasive lesions can be observed over time and documented to progress to invasive lesions. However, for less readily observable lesions, such as those of the prostate, the progression of the pre-invasive lesions, e.g., prostatic intraepithelial neoplasia (PIN) and prostatic proliferative inflammatory atrophy (PIA) to prostatic cancer are more difficult to document. Thus, for most organ systems, specific pre-invasive neoplastic lesions have been proposed based upon the apparent observations of one or more of the following: 1) microinvasive disease developing from a pre-invasive neoplastic lesion, 2) the general association of the pre-invasive lesion with invasive lesions, 3) the subsequent development of invasive lesions following diagnosis of the pre-invasive lesion, 4) correlations of the molecular features of the putative pre-invasive lesion with the matching invasive lesions, and 5) reductions in the rate of cancer following removal of the pre-invasive lesion. When there are mixtures of pre-invasive lesions with actual cancers in the same case, some of the above specific associations are more difficult to make. Several terms have been used to describe pre-invasive lesions, many of which are now less useful as our knowledge of these lesions increases. It is now commonly accepted that these lesions are a features of the spectrum of neoplastic development and most are accepted as “neoplastic lesions” with associated molecular features, even though they may be reversible even if they have mutations in suppressor genes (e.g., p53) or are associated with viral etiologies (e.g., cervical intraepithelial neoplasia). The overall term, “pre-invasive neoplasia”, seems to best describe these putative pre-invasive lesions. Thus, terms such as incipient neoplasia should be abandoned. The term “intra-epithelial neoplasia” with an associated grade, which has been developed for pre-invasive neoplastic lesions of the cervix, i.e. cervical intraepithelial neoplasia (CIN), seems to be a terminology that adds consistency across epithelial organs. Thus, adoption of these terms for the additional organ sites of pancreas (PanIN) and prostate (PIN) seems accepted. Less descriptive terms such as the degrees of dysplasia of the oral cavity and bronchopulmonary system and actinic keratosis and Bowen's disease of the skin might be better designated as oral intraepithelial neoplasia (OIN), pulmonary intraepithelial neoplasia (PulIN) and dermal intraepithelial neoplasia (DIN). The etiology of pre-invasive neoplasia is the etiology of the matching cancers. Some obvious initiating factors include exposure to the whole range of ionizing and non-ionizing radiation, tobacco abuse and a broad range of other carcinogens (e.g., benzene). A frequent initiation factor is the setting of long standing continuing damage, inflammation and repair (LOCDIR) which leads to early molecular features associated with neoplasia after about one year. An excellent example of this is ulcerative colitis (UC) in which dysregulation of microsatellite repair enzymes have been documented one year following diagnosis of UC. While the nomenclature, description, diagnosis and etiology of pre-invasive neoplasia has advanced, approaches to therapy of such lesions have not progressed adequately even though it has been identified that, for example, removal of polyps periodically from the colorectum, DCIS from the breast, and high grade CIN from the cervix, results in a reduction in the development of cancers of the colorectum, breast, and cervix, respectively. With the development of more molecularly targeted therapy with fewer side effects, preventive therapies may be more successfully targeted to pre-invasive neoplastic lesions.
Keywords: Intraepithelial neoplasia, pre-invasive neoplasia, prostatic intraepithelial neoplasia, pancreatic intraepithelial neoplasia, cervical intraepithelial neoplasia, adenomatous polyps, ductal carcinoma in situ, lobular carcinoma in situ, inflammation, radiation, viral infections, carcinogens, dysplasia, actinic keratosis, repair, angiogenesis, LOCDIR
1. Introduction
Malignant (invasive neoplastic) lesions are thought to develop from specific “pre-invasive neoplastic lesions” whose histopathologic features have been described for most epithelial organ systems. Sometimes these lesions are referred to as incipient, pre-cancerous, or pre-neoplasia; however, for many epithelial organ systems such lesions are now referred to as intraepithelial neoplasia or, less specifically, e.g., for the oral cavity, as epithelial dysplasia or pre-invasive neoplasia. The characterization of these lesions as “neoplasia” is justified because about 30% to 60% of the most aggressive of these lesions are thought to progress to malignant (invasive) lesions. Also, these lesions have specific molecular changes which separate them when compared to uninvolved epithelial areas [1–3]. In addition, the higher grade intraepithelial lesions usually have molecular changes which are different from lower grade intraepithelial lesions.
For some organ systems such as the breast, even earlier histopathologic lesions thought to lead to pre-invasive neoplasia (i.e., the pre-invasive lesions of ductal carcinoma in situ [DCIS] or lobular carcinoma in situ [LCIS]) have been identified. These have been designated as lobular or ductal atypical hyperplasia, LAH and DAH respectively. Although patients with LAH and DAH as well as even earlier lesions described as hyperplasia or fibrocystic changes carry an increased risk of developing mammary carcinoma [4], these lesions are not classified as neoplastic. In the breast, DCIS and LCIS are divided into low grade and high grade lesions. In other organ systems such as cervix, prostate and pancreas, intraepithelial neoplasia also is graded, i.e., intraepithelial neoplasia 1, 2 and 3, with less aggressive lesions in the 1 category. For example, for the prostate we refer to one type of pre-invasive neoplastic lesions as prostatic intraepithelial neoplasia with grades PIN 1, PIN 2 or PIN 3 with PIN2 and PIN3 referred to as high grade PIN to indicate the lesions most at risk for progressing to prostatic adenocarcinoma [5–7]. Rarely, in addition to the main intraepithelial neoplastic lesions hypothesized to lead to invasive neoplasia, other alternative pathways (lesions) that may lead to invasive cancer also have been identified or proposed. In the case of pancreas, one such lesion, intraductal papillary mucinous neoplasia (IPMN), [8] and in the prostate, proliferative inflammatory atrophy (PIA), have been proposed as alternative pre-invasive neoplastic lesions [9].
Pre-invasive neoplastic lesions typically occur in small areas of an organ; these areas may frequently be multifocal throughout the organ. A good example of this is the skin in which there may be multiple areas of actinic keratosis on the sun-exposed skin (e.g., arms and face). For the cervix, multifocal areas and grades of CIN may arise along the squamocolumnar junction. Each grade of pre-invasive neoplasia usually has a distinct histopathologic microscopic appearance upon which the grade of intraepithelial neoplasia is based.
In some cases, small tumors have been described as adenomas with the view that malignant tumors may or may not develop from these small adenomas. This was once the view, for example, of small tumors of the cortex of the kidney, which if less than 3 cm were once designated as renal adenomas. We now view most such small tumors of the renal cortex as just early forms of renal cortical carcinomas which have not had time to metastasize; thus, because these small adenomas were not associated with metastatic disease, they were initially considered to be benign. However, some tumors such as adrenal cortical adenomas which meet specific histopathologic and biochemical criteria grow very slowly and are still considered to be benign neoplastic lesions [10].
In general, most intraepithelial neoplastic lesions have been identified by their association with invasive or metastatic cancer (guilt by association) or rarely by areas in intraepithelial neoplasia in which there appears to be invasion into stroma from the intraepithelial lesion [11]. In the case of colorectal neoplasia, adenomatous polyps have been identified to be a pre-invasive neoplastic lesion because if all such polyps are removed from the colorectum, patients very rarely develop colorectal adenocarcinomas [12,13]. Similarly effective local treatment of CIN2 and CIN3 of the cervix prevents the development of squamous cell carcinoma (SCC) of the cervix.
2. Spontaneous resolution of pre-invasive neoplastic lesions
The pre-invasiveneoplastic lesion is not always committed to progressing to invasive cancer. Specifically, we have noted that areas of dysplastic leukoplakia may spontaneously move geographically in the oral cavity as well as spontaneously partially or completely resolve [14].
Similarly, it is known that if cervical intraepithelial neoplasia–3 (CIN-3) lesions are not treated over a decade, about 25 to 35% will regress. If the initial lesion is moderate dysplasia (CIN2) a larger proportion, 45 to 65%, will regress to normal and the proportion regressing increases in patients who are over 50 years of age (Reviewed in Koss [15]). Whether or not there is potential spontaneous regression of intraepithelial neoplastic lesions in other organs is unknown; however, by analogy this is our guess.
3. Molecular changes in pre-invasive neoplasia
The identification of molecular changes during the development of intraepithelial neoplasia in some organ systems has been possible via analysis of familially inherited neoplasia. Dr. Vogelstein and colleagues [16] developed a model of molecular changes in inherited colorectal neoplasia based on neoplasia that develops from families with familial adenomatosis polyposis coli (FAP). In fact, the first gene identified as being dysregulated in FAP was designated adenomatous polyposis coli (APC); identification of mutations in APC was followed by an understanding of how APC interacts with β catenin and an identification of subsequent mutations or dysregulation of K-ras, DCC, SMAD4, MCC and p53 [15]. Mutations in microsatellite repair genes such as MSH2 and MLH1 also have been identified as the cause of colorectal neoplasia in hereditary non-polyposis colon cancer (HNPCC) [17]. Figure 1 demonstrates how hereditary tumors may develop based on inheritance of a mutation in a single gene.
Fig. 1.

Describes a general pathway by which a mutation in a single copy of inherited gene such as APC may cause the development of pre-invasive neoplasia of, for example, the colorectum. Mutations in such genes as BRCA1, BRCA2, APC, MSH2 and MLH1 may follow such a pathway in other tissues. Mutations in DNA repair pathways do not typically produce the pattern of large polyps of the colorectum seen in mutations with APC.
In general, some of the same mutations that occur in familial cancers occur in sporadic cancers, but such mutations may not occur in the same temporal order. Nevertheless, understanding the specific dysregulation of genes and the pattern of genetic dysregulation of familial cancers has provided important insights to the development of genetic dysregulation in sporadic cancers; thus the genetic changes in cancers in individuals from families with FAP and HNPCC follow a sequence that is useful in understanding inherited as well as sporadic colorectal cancers.
Probably in most cases, specific molecular changes may precede the development of discernable histopathologic lesions. In mice exposed to UV-B, mutations in p53 precede discernable lesions by several months [18]. Also, in humans up to 4% of sun exposed skin has been estimated to involve cells with mutations in p53 [19, 20]. Obviously, most of these areas of cells with p53 mutations never progress even into actinic keratoses.
4. Stochastic versus stem cell models in the development of intraepithelial neoplasia
There are two major hypotheses of how preneoplasia develops and progresses – i.e., the stem cell hypothesis in which preneoplasia develops, only from mutations in stem cells and/or in pluripotent early progenitor cells versus the stochastic model in which preneoplasia develops from any cells in which mutations of specific genes occur.
A stem cell or early progenitor cell is defined as a cell that continually self-renews even while producinga more differentiated cell during the same mitosis. Such mitoses are hypothesized to be asymmetric as to DNA transfer to child cells. Stem cells are described as a small subpopulation (< 5%) of the cells within a tissue compartment which upon removal and transplantation to an appropriate environment will, regenerate the phenotypic characteristics of the original epithelial compartment. Thus, as a stem cell divides, both new stem cells are created while the non-stem cells created on each mitosis differentiate to form the differentiated characteristics of the cells of the tissue compartment. In some cases, one characteristic stem cell may maintain more than one cellular compartment via its differentiated children, e.g., alveolar, ductal and myoepithelial cells of the breast. Typically, each stem cell controls only a small geographic area (niche) of a tissue compartment.
The difference between a stem cell model for the development of cancers and the stochastic model is represented in Fig. 2. In general, in the stem cell model only stem cells give rise to cancers. In the stochastic model, cancers can arise from any proliferative cell with specific mutations. It is likely that after one clone of specifically mutated cells develops, it rapidly over-grows the remaining cells of the compartment no matter which model is valid.
Fig. 2.
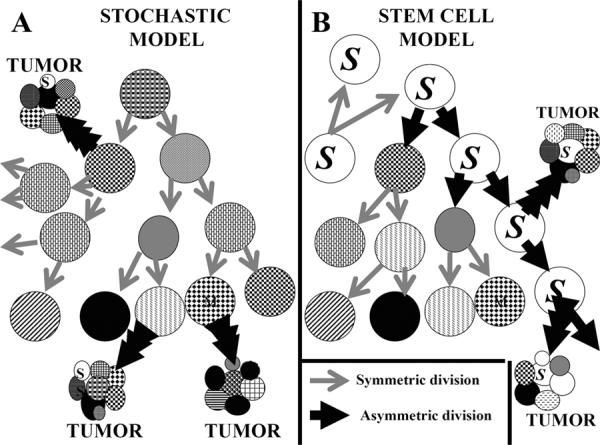
Panel A demonstrates the model of the stochastic development of cancers. In this model, each child cell is somewhat molecularly different as is indicated by the different patterns of the cells. Note that a tumor may develop from any of the child cells. Most divisions until the pattern of cancer develops are considered symmetric. Multiple tumors are demonstrated to arise, but this is unlikely to occur. Once a pre-invasive neoplastic lesion develops, it is likely to overgrow all surrounding pre-invasive neoplastic cells and hence destroy the rest of the group of cells. Panel B demonstrates the model of how cancers develop from stem cells. In this model, cancers may develop only from stem cells (i.e., cells labelled by an S). While these two models for illustration demonstrate multiple tumors developing from a group of cells, this would be unlikely to occur as discussed above. Note that a stem cell generating two stem cells is considered a symmetric division; in contrast, a stem cell generating a more differentiated cell plus a stem cell has undergone an asymmetric division.
The major evidence for the stem cell hypothesis is that normal tissues and cancers develop preferentially from cells expressing stem cell markers and only a few of these cells are necessary to produce a neoplastic lesion in vivo. Specifically, it takes the implantation of 100,000 or more unsorted cells to grow a xenograft tumor, but implantation of less than one hundred stem cells has been demonstrated to be necessary to produce a xenograft tumor [21]. One of the main consequences of the stem cell model is that to treat cancer and other neoplasia successfully, the therapy needs to be directed at the stem cells of the cancer, as well as the typical cells of the cancer; otherwise, the stem cells that are frequently resistant to most therapies will remain after therapy and the lesion will recur [22–24].
An additional aspect of the current model of stem cells is that the stem cells express a characteristic set of biomarkers; however, these molecular features may vary with each specific organ system. The stem cells for neoplasias of the breast are perhaps the best characterized; the stem cells of the breast express aldehyde dehydrogenase activity (ALDH) and the ALDH1 protein. These stem cells also are CD44-v6+ and CD24− and LIN− [23,73].
In the stochastic model, Fig. 2, Panel 1A, the initial parental cell can be any cell, not necessarily a stem cell. This initial parental cell begins dividing and each of the cellular children is similar to, but different from the parent. As with grandchildren, the subsequent generations are even more diverse than the original parent. At any point, any offspring may generate a “bad seed” which develops a molecular change that leads to child cells with characteristics of a neoplastic lesion. The more proliferative child cells are more likely to develop a new mutation, each new mutation may increase proliferation as well as the mutational rate and decrease rate of apoptosis; ultimately, a pre-invasive neoplastic lesion may develop. In contrast, in Fig. 2, Panel 1B, cancers only develop from stem cells, but the “bad seed” pathway is similar.
5. Environmental exposures and the development of neoplasia
Multiple types of environmental exposures increase the risks for developing specific types of cancers. Broad categories of such environmental exposures are ionizing and non-ionizing radiation, infectious microbiota, chemicals and physical agents such as asbestos fibers, wood dust, soot and rock (silica) fragments. Chemicals may range from ions and elements, especially metals, to complex molecules produced by microbiota such as fungi (e.g., aflatoxin B1), manufactured by humans (e.g., diethylstilbestrol) or produced by changes in dietary components on cooking (e.g., heterocyclic amines).
6. Radiation and the development of neoplasia
Radiation is a major cause of specific cancers. For example, although controversial, the gas, radon, which may accumulate in houses and in mines, has been implicated in the development of cancer of the lungs, especially in smokers [25] with a relative risk 1.8 (1.1 to 2.9). Actually, the most common exposure to radiation that leads to the development of neoplasia is solar radiation of the skin.
Frequent molecular changes are noted in squamous cell carcinoma (SCC) and in basal cell carcinoma (BCC) of the skin. Specifically, there are mutations in p53 in about 70 % of SCCs and 50% of BCCs. Of interest, about 4% of the epidermis of normal appearing human skin from individuals with sun exposure but without cancer contains p53 mutated clones of 60 to 3000 cells in size [20] and the risk of developing SCCs and BCCs is correlated with the extent of mutations in p53 [26]. A mutational pattern in p53 is more common in sun exposed skin than in sun shaded skin (×40). These mutations in p53 are usually C→T or CC→TT [27,28]. For clones of smaller size, the clones were conical with their apex frequently at the basal epidermis suggesting an origin from this area where there are basal cells and/or stem cells [20]. Other studies in mouse models have noted UV-B induced clones of p53 are noted months before the development of skin tumors. Also, based on mouse models, by using UV blockers (SPF-15 sun screens) the occur-rence of most p53 mutational clones can be prevented or reversed [18].
In general, primarily UV-B solar radiation and to some very small extent UV-A solar radiations cause p53 mutation in the epidermis. These mutations of suspected stem cells induced by UV-B are thought to be promotional events that lead to clonal expansions of p53 mutated stem cells. These clones then grow, remain static or potentially regress until other molecular changes occur that lead to the development of actinic keratoses and ultimately to SCC.
The major recognized pre-invasive neoplastic lesion of the skin is actinic keratosis which progresses at a low rate to squamous cell carcinoma (SCC) of the skin. Clones of cells with mutated p53 are likely to be the precursors of most actinic keratoses, especially Bowenoid actinic keratosis [28].
7. Chemical effects on the development of neoplasia
A broad range of chemicals can be involved in the development and/or progression of various neoplastic processes. For example, metals such as cadmium, beryllium, arsenic and chromium have carcinogenic potential; however, the methods of action of these metals are not understood. Organic compounds such as cyclic hydrocarbons and cyclic amines have been associated with numerous malignancies. Most have been noted in humans with various environmental exposures; in addition, these chemicals have proved to be potent carcinogens in animals. Some potential carcinogens are listed in Table 1; the associated cancers were primarily determined in animal studies.
Table 1.
Chemical carcinogens[32]
| Chemical agent | Postulated associated malignancy |
|---|---|
| Aflatoxin B1 | Hepatocellular carcinoma |
| Arylamines (e.g., 2 naphthylamines) | Bladder carcinoma |
| Benzene | Acute myeloid leukemia |
| Benzidine | Bladder |
| Diethylstilbestrol | Vagina, cervix |
| Heterocyclic amines | SCC of the lung |
| Metals | |
| – Arsenic | Skin, bronchus, liver |
| – Beryllium | Lung |
| – Cadmium | Lung, prostate, pancreas, kidney |
| – Chromium | Lung |
| – Nickel | Nasal sinus, bronchus |
| N-nitrosamines | Gastrointestinal; adenocarcinoma of the lung |
| Polycyclic aromatic amine | Lung, skin, urinary |
| Vinyl chloride | Liver |
Many chemicals act via the formation of adducts with DNA which following DNA duplication subsequently cause mutations in DNA. For example, mycotoxins typically react with nitrogens of adenine and guanine molecules. In theory, stem cells are usually the sensitive target with some adducts functioning in initiation and frequently different adducts in promotion of neoplastic lesions. Initiation and promotion are probably sufficient to lead to early pre-invasive neoplasia. Subsequently, mutations occur that lead to progression and to eventual malignancy.
In other cases, the actual chemical must be metabolized to reach its optimal carcinogenic potential. Many human enzymes rapidly metabolize carcinogens to noncarcinogens and are hence protective; other human enzymes modify chemicals to produce optimal carcinogens. These enzymes vary in their potency among individuals due to genetic polymorphisms in the enzymes. Polymorphisms can be identified by analysis of SNPs and may either represent positive or negative risk factors for the development of neoplastic lesions. For example, ethanol is associated with a greater risk of developing colorectal cancer in individuals with the E487k polymorphism of aldehyde dehydrogenase 2 (ALDH2) which results in greatly reduced enzymatic activity of ALDH2 [29]. Similarly, polymorphisms in 5, 10- methylenetetrahydrofolate reductase plus low folate levels increase the risk of colorectal cancer associated with ethanol consumption [30,31].
One of the reasons smoking of tobacco may be so strongly associated with the development of pre-invasive neoplasias and cancer of the oral cavity, larynx, lung, urinary tract, and colorectum are the many (> 50) carcinogens in tobacco smoke, as well as the many different types of carcinogens, (e.g., metals, nnitrosamine, polycyclic aromatic hydrocarbons). Thus constant smoking is likely both to initiate and to promote the development of pre-invasive neoplasia in multiple organs exposed to these carcinogens.
Other chemicals may affect cancers once the pre-invasive or advanced stages of cancer have developed. For example, copper has been reported to potentiate the progression of cancers [33] via the involvement of copper in the development of angiogenesis and/or other features that affect the development and progression of neoplasia.
8. Microbiota and infections
After the infectious etiology of neoplasia was first identified in birds, viral transmission of various neoplastic processes was confirmed in multiple animals including rabbits and mice. One of the earliest identified neoplastic processes associated with viral infection in humans is cervical squamous cell neoplasia.
Several examples of human neoplasias that arise secondary to viral infections are listed in Table 2. The most recently identified examples of neoplasias arising from viral etiologies include Merkel cell tumor of the skin [34].
Table 2.
Examples of viral causes of neoplasia
| Virus | Tumor |
|---|---|
| Hepatitis V virus (Hepadnovirus) | Hepatocellular carcinoma |
| Hepatitis C virus (Flavivirus) | |
| Epstein-Barr virus (Herpesvirus) | Burkitt lymphoma |
| Hodgkin lymphoma | |
| Nasopharyngeal carcinoma | |
| Kaposi sarcoma herpesvirus (HSV-8) | Kaposi sarcoma |
| Lymphoma | |
| Genital Human Papilloma Viruses | Anal carcinoma |
| Cervical carcinoma | |
| Human T Cell Leukemia Virus Type-1 | Adult T cell lymphoma |
| (HTLV-1) | |
| Polyomavirus | Merkel cell tumor of skin |
The viral etiology of cervical neoplasia is one of the better understood models of viral interactions in humans to produce neoplastic changes in cells. After infection with multiple HPV serotypes especially 16 or 18, there is a dysregulated production of the E6 and E7 viral genes. E6 binds and inactivates p53 through ubiquination and it also activates hTERT. E7 binds pRb and associated genes p107 and p139 causing increased cellular proliferation. However, these actions of E6 and E7 are not considered sufficient for the production of cervical cancer.
Once cells are infected with HPV and the viral regulatory proteins bind to p53 and pRb, a cascade of several very characteristic changes occur in the cervical epithelium. One of the first observable morphologic changes is koilocytic changes in the mid-epithelium of the cervix. This change results because of the accumulation of material in the cytoplasm that does not stain with eosin of the hematoxylin and eosin (H&E) stain causing a halo appearance in cells. An area of the cervix with only such koilocytic changes is classified as CIN1. Thereafter, more and more cells of the squamous epithelium of the cervix become less differentiated, leading to various grades of cervical intraepithelial neoplasia. CIN3 lesions are thought to be at greatest risk of progression with at least 1/3 of CIN3 lesions progressing to squamous cell carcinoma if not treated [15]. To aid in deciding which cervical lesions require careful follow, analyses can now be performed to identify infections by the thirteen most common high risk serotypes of HPV infections including HPV16, 18, 31, 33, 39 and 45 which can lead to cancer and HPV serotypes 6, 11,42, 43 and 44 which can cause genital warts. Also, young women are now being vaccinated against specific high risk HPV serotypes including 16 and 18 as well as HPV 6 and 11 in order to reduce the risk and costs associated with HPV infections.
Infection of the gastrointestinal system with the bacterium Helicobacter pylori (H. pylori) has been associated with the development of gastric carcinomas and/or gastric lymphomas of MALT type. There also is an association of H. pylori infections in most cases of chronic gastritis and peptic ulcers. Products of H. pylori may also affect tissues downstream of the primary site of infection.
Few other classes of bacteria have been definitely identified or even associated with the development of neoplasia; however, fungal infections that produce toxins such as aflatoxin may cause liver cancer in certain areas of the world. Similarly, infections with Schistosoma haematobium cause areas of localized continuing damage, inflammation and repair in the bladder. Such infections may lead to carcinoma of the bladder, especially squamous cell carcinoma of the bladder.
9. Longstanding continuing damage, inflammation and repair in the etiology of neoplastic processes
Many neoplastic processes/lesions develop in the setting of “long term continuing damage, inflammation and repair (LOCDIR).” We define LOCDIR as severe damage to tissue that continues constantly for more than 1 year and which induces continuing inflammatory responses, severe cellular damage and tissue repair. In this setting recognizable molecular and histopathologic neoplastic lesions (dysplasia) may develop, usually after more than 5 years of LOCDIR. We chose 1 year because dysregulation of microsatellite (MS) repair genes have been noted in continuing ulcerative colitis after 1 year's duration while more acute forms of colitis do not have changes in microsatellites [35]. Examples of lesions classified as LOCDIR include ulcerative colitis, Crohn's disease or non-specific colitis leading to colorectal neoplastic lesions and chronic pancreatitis in which pancreatic cancers may develop [36] and Barrett's esophagus caused by acid reflux from which esophageal adenocarcinomas may develop. Similarly, squamous cell carcinoma may develop along chronic draining sinus tracks or in the bladder secondary to Schistosoma haematobium. As with most categories of LOCDIR, dysplastic and molecular changes may develop in the cells of the affected organ. Even actinic damage in specific sun exposed individuals who work primarily outside may be so constant that the condition could be classified as LOCDIR.
Multiple potential mechanisms have been proposed for the development of neoplastic lesions in the setting of continuing extensive damage to cells and resulting inflammation. Continuing damage may cause the release of inflammatory mediators as well as reactive oxygen species (ROS) or reactive nitrogen species (RNS) which can further damage DNA and cause DNA adduct formation. Similarly, DNA adduct formation may result from local carcinogens associated with the inflammation, damage and repair. Our view is that neoplastic changes occur because the severe cellular damage requires extensive repair of the damage with increased proliferation of cells. Increased proliferation in areas of damaged tissues, frequently may lead to molecular changes in cells due to DNA replication in the setting of genomic damage. In the setting of severe LOCDIR, cellular injury also may result in such extensive damage to cellular DNA that the ability of cells to repair the damage to DNA prior to replication may be exceeded; also ROS and RNS may prevent accurate repiair of the DNA [35]. Thus, during cellular replication, mutations and other changes in DNA may develop. As increased numbers of isolated mutations and other changes in DNA develop, the mutational rates of such damaged cells are likely to increase.
An excellent example of LOCDIR with the development of mutations in normal appearing cells is ulcerative colitis (UC) in which molecular changes in DNA can be identified in normal appearing cells in as little as 1 year after the onset of UC [35]. UC is an example of how years of damage and repair may cause the development of pre-invasive neoplasia (dysplasia) and an increased risk of the development of carcinoma within 7 to 10 years after the onset of UC, depending upon the severeity of the UC [37].
One of the pathways that may be affected by LOCDIR is the microsatellite (MS) repair system. When UC mucosa is evaluated for microsatellite changes, MS instability can be detected in about 15% of randomly collected specimens of UC, but not in ischemic colitis which is a short term inflammatory condition [35–37]. Of interest, some loss or changes in MS were found adjacent to the genes, APC, p53 and DCC [38–40].
When changes in DNA including loss of chromosomal areas occur adjacent to major suppressor genes such as p53 or Rb, the changes in DNA may frequently involve only genes geographically adjacent to the major suppressor genes or oncogenes on the affected areas of the chromosome, but not the major suppressor genes or oncogenes. A new concept is that some genes geographically related on chromosomes to the major suppressor genes frequently have a biological relationship to these major suppressor genes. Although these geographically related genes are usually not considered to be major suppressor genes, in the bladder losses or mutations in such genes have been associated with clonal proliferations. These genes have been designated as “forerunner genes” because their initial involvement usually leads to subsequent involvement of the major suppressor genes in their chromosomal area e.g., p53 or Rb [41,42]. Subsequently, the involvement of major suppressor genes would then lead to the development of pre-invasive neoplasia. Alternatively, defective repair of DNA could lead to haplodeficiency of important regulatory genes such as TGFβR2 or TGFβR1 [43–45] and subsequently to pre-invasive neoplastic lesions. Also, some individuals may be inherently more sensitive to such changes because they have inherited SNPs which cause overall inefficient functioning of specific genes.
10. Immunesurveillance and immune responses to early neoplastic changes
The escape of early neoplasia from immunesurveillance has been postulated for decades and this concept has been the driving force for some approaches to immunotherapy which seek to increase the effectiveness of the immune system in the management/control of cancer. As discussed, pre-invasive neoplasia of the cervix, oral cavity, and skin may spontaneously change and may regress over months to years. Also, some invasive lesions, especially melanomas, have been reported to regress spontaneously. It is unknown if and how the immune system is involved in such spontaneous changes [15,46]; however, the immune system is hypothesized to modulate the development and progression of neoplastic lesions (reviewed in Zhang and Grizzle [46]).
Some examples of the more clear indications that neoplasias are under immune suppression are that cancers that are infiltrated by lymphocytes such as medullary carcinomas of the breast and stomach tend to have better outcomes than similar cancers without lymphocytic infiltration and that upon immunosuppression for organ transplantation, the incidence of non-melanoma skin cancers, thyroid cancer, head and neck cancer, colorectal cancer, as well as bladder, ureteral and renal cancers all increase [47]. Similarly, patients with organ transplants and patients with HIV are more susceptible to neoplastic lesions caused by viral infections (e.g., Kaposi sarcoma, SCC of cervix, vulvar and anus, hepatocellular carcinoma and EBV induced lymphoproliferative neoplasia). Of interest, some molecular changes leading to preinvasive neoplasia may spontaneously resolve if the stimulus that leads to the molecular change is removed. For example, when p53 clones or actinic keratoses that develop in the skin of mice secondary to UV-B radiation are shielded from UV-B radiation, both the size of the p53 clone and lesions of actinic keratosis tend to resolve. However, because the extent of the resolution of UV-B damage does not change in Rag1 knockout mice which cannot develop B, αβT, δγT or natural killer T cells [48], this suggests that parameters other than the immune system are at work in the response of these lesions to withdrawal of the causative stimulus. If this lesion were considered to be a LOCDIR lesion, perhaps the stimulus of constant severe repair is lost and cells with p53 mutations and/or chromosomal losses (MSI changes) are deleted by non-immune mechanisms.
Neoplastic lesions are usually recognized by the immune system and the response results in increased acute phase reactants (APR), stress proteins and antibodies to specific products of neoplastic lesions (tumor specific antibodies). Cells of neoplastic lesions may contain numerous non-self antigens including mutated proteins, splice variants of proteins, oncofetal proteins and atypically modified proteins (e.g., proteins with increased glycosylation). When tumor cells die (a very frequent occurrence) these non-self molecules as well as numerous products of injured and dying cells are released into the interstitium and are picked up by the vascular system. Also, some of these “non-self” molecules may be displayed on the surface of neoplastic cells and create an immune response. Similarly, larger cancers may activate the immune system by signals of tissue damage and hypoxia. The increases in APR and tumor specific antibodies have been used as non-specific targets in the early detection of cancers [47] but their use in early detection has been criticized as lacking specificity. Although the immune system seeks to control (inhibit) neoplastic lesions, such lesions in turn seek to suppress the immunomodulatory mechanisms directed at their control. Specifically, myeloid suppressor cells (MSC, Gr1+ CD11b+) are increased in the spleen and the bone marrow of humans and animals with specific types of neoplastic lesions. Also, the accumulation of MSCs correlates with increased burden of cancers and poor survival (reviewed in [49,50]).
It is hypothesized that suppression of natural killer cells (NK) by MSCs is important in suppression of the immune system [51] as is the production of T regulatory cells which maintain tolerance to self-antigens [52]. However, the infiltration of neoplastic lesions by mast cells and other granulocytes and macrophages results in products (e.g., MCP4, MCP-6, tumor associated osteopontin, cathepsin D, il-6, il-8) being released that are mitogenic for neoplastic cells, affect cancer remodeling of tissues and increase angiogenesis – all changes that may cause the growth and metastasis of cancers [74].
Some of the above protective changes induced by neoplastic lesions occur secondary to the secretion of exosomes – tiny (40 to 100 nm) membrane bound particles produced by reverse budding of endosomes – multivesicular bodies. Exosomes are released from cancer cells and other cells into the extracellular space and ultimately a proportion of exosomes are picked up by the vascular system. Exosomes may carry hundreds of common “waste” proteins as well as specific proteins that are used by exosomes from different tissue sources to provide specific signals when the exosomes fuse with target cells [53–55].
Examples of signals provided by exosomes include TGFβ from exosomes from the thymus that affect the induction of regulatory T cells [52]. Similarly, TGFβ and PGE2 in exosomes from tumors promote bone marrow myeloid cells to become myeloid-derived suppressor cells (MDSCs) which promote tumor progression; for example MDSCs produce il-6 and VEGF [56] which may induce stromal development and angiogenesis.
Studies in animals have demonstrated that exosomes produced by tumor cells cause a decrease in NK cells and the cytotoxic activity of NK cells [51]. Exosomes also react with immature dendritic cells (Gr-1+, CD11b+) and monocytes (CD14+) and inhibit their maturation to mature dendritic cells [57]. Secretion of exosomes also causes the build-up of myeloid suppressor cells in the spleen and in bone marrow as discussed previously [57]. MDSCs can also then suppress NK and killer T cells [50,56].
11. General characteristics of intraepithelial neoplasia leading to adenocarcinoma
Adenocarcinomas develop in multiple organ systems. This includes the breast, prostate, ovary, en dometrium, cervix, lung, pancreas, salivary glands, esophagus, stomach, small bowel and colorectum. Carcinomas develop in the kidney, adrenal, and liver, but these are not usually referred to as adenocarcinomas. Of the adenocarcinomas, only breast, prostate, pancreas, esophagus, stomach, small bowel, colorectum, lung and endometrium have lesions clearly accepted as intra-epithelial or pre-invasive neoplasia (Table 3).
Table 3.
Adenocarcinomas and associated pre-invasive neoplastic lesions
| Organ | Pre-invasive neoplasia leading to adenocarcinomas | Principal features of lesions containing dysplastic epithelial cells |
|---|---|---|
| Breast | Low and high grade: ductal carcinoma in situ (DCIS) and lobular carcinoma in situ (LCIS) | Expanded duct with reduced discontinuous and flattened myoepithelial cells surrounding dysplastic epithelial cells. One type of high grade DCIS has central necrosis “comedo” type |
| Colorectum | Tubular or villous adenoma (adenomatous polyp) with mild, moderate or severe dysplasia | Extent of dysplasia determines the likelihood of conversion to colorectal cancer |
| Endometrium | Endometrial intraepithelial neoplasia (EIN) | Crowded glands with stroma comprising less than 50% of area. |
| Esophagus | Focal low and high grade dysplasia in Barrett's metaplasia | Grade of dysplasia is determined by extent of dysplasia |
| Lung | Bronchoalveolar carcinoma (BAC) is now classified as a pre-invasive neoplastic lesion leading to adenocarcinoma. Atypical adenomatous hyperplasia (AAH) also is postulated to be a pre-invasive neoplastic lesion of lung. | This designation of BAC is questionable due to the aggressive behavior and extent of some bronchoalveolar lesions |
| Pancreas | Low (PanIN1) and high (PanIN2 and PanIN3) grade pancreatic intraepithelial neoplasia; Intraductal papillary mucinous neoplasia (IPMN); other cystic lesions |
Low grade PanIN features mucinous tall columnar cells with basal nuclei which transition to high grade PanIN with dysplastic nuclei and disorganized orientation of cells. IPMN feature large > 1 cm ducts and cystic-like structures lined with dysplastic cells |
| Prostate | Low (PIN1) and high (PIN2 and PIN3) grade prostatic intraepithelial neoplasia; proliferative inflammatory atrophy | PIN – Expanded duct with reduced numbers of flattened basal epithelial cells forming a discontinuous layer surrounding dysplastic luminal-type epithelial cells |
| Small bowel | Tubular, tubulovillous, villous adenomas | Villous adenomas are more likely to progress to malignancy |
| Stomach | Adenomas (sessile or pedunculated polyp) low grade and high grade dysplasia in flat gastric mucosa | Degrees of dysplasia determine low and high grades |
The major characteristics of the different types of intraepithelial neoplasia which ultimately progress to adenocarcinomas are typically both histopathologic and molecular.
Of these, pre-invasive neoplastic lesions of the breast and prostate are similar.
In the case of DCIS and PIN there is an apparent expansion of the duct/ductolobular unit at the site of the lesion caused by the proliferation of neoplastic cells. These neoplastic cells may develop with the molecular characteristics of either luminal or basal cells (Fig. 3). Another is the presence of a single usually discontinuous layer of basal cells and/or myoepithelial cells between the proliferating luminal-like cells and a basement membrane (Fig. 3); stem cells are likely present in this basal cell population. Thus, increased proliferation is a major factor which affects the development of pre-invasive neoplastic lesions. Another feature is the accumulation of molecular changes which have the potential to drive proliferation and/or inhibit apoptosis. Of note, it is the histopathologic lesion that is currently accepted as defining these pre-invasive lesions.
Fig. 3.

Panel A (×600) is an immunohistochemical stain of uninvolved ducts (normal appearing ducts in a patient with ductal cancer) of the breast. Note the myoepithelial cells (black arrows) are arranged in a single basal layer, are flattened to cuboidal and stain strongly with smooth muscle actin (SMA). Panel B (×600) is an area of high grade (comedo) DCIS. Note the myoepithelial cells (black arrows) which stain strongly with SMA are flattened or pyramidal, and do not form a continuous layer as they do in uninvolved ducts. The luminal-like cells of DCIS do not stain with SMA and form a cellular compartment which has many layers of proliferating cells.
12. Ductal carcinoma in situ and lobular carcinoma in situ as pre-invasive neoplastic lesions of the breast
A range of proliferative changes can be noted in breast tissue, but even the mild proliferative changes noted in fibrocystic changes carry an increased risk for the development of cancer ≈ × 1.5. Nevertheless, the separation of atypical ductal hyperplasia (ADH) (a relatively benign change that does not require therapy and has a relative risk of breast cancer 4.4) from low grade DCIS requiring surgical resection with a relative risk 9–11 is the line of separation between benign and a classification of pre-invasive neoplasia [58–60]. The molecular separation of such lesions varies. In general, more aggressive molecular changes are reported to occur less frequently in ADH and in low grade DCIS; also, some cases of atypical hyperplasia with molecular changes have developed as DCIS or invasive cancer has “spread” to uninvolved areas of a duct (Fig. 4).
Fig. 4.

Depicts an area which resembles atypical hyperplasia to low grade DCIS but this photograph probably represents a tumor invading a duct; also there is an adjacent duct, most of which is normal. Panel A (×200) is immunostained for Bcl-2 and both basal and luminal cells of the uninvolved duct stain strongly with Bcl-2. Note that the architecture of a portion of this duct next to the lesion is disrupted by larger cells staining weakly for Bcl-2 (marked by wide gray arrows). Also, the basal-like cells of the area of duct expansion stain moderately to strongly with Bcl-2. Panel B (×200) is immunostained for p53. It demonstrates that the area of disruption of the uninvolved duct contains cells with nuclear accumulation of p53 (i.e., with a p53 mutation or dysregulation). Panels C and D (×600) show the area of duct expansion from Panels A and B. Note the 4 black arrows demark basal-like cells surrounded by larger cells more typical of the cells of ADH/DCIS. These basal cells stain moderately for Bcl-2 (Panel C) and do not demonstrate nuclear accumulation of p53 (Panel D). The mixed cellularity of this lesion suggests a tumor invading a duct. Note the aggressive cells are either spreading to an adjacent uninvolved duct or are inducing changes in the cells of this uninvolved duct.
Ductal or lobular carcinomas in situ of the breast have been identified by their atypical histologic features, their frequent association with mammary carcinoma, and the development of mammary carcinoma after initial surgery for 10 to 15% of cases with DCIS or LCIS. In both DCIS and LCIS, myoepithelial cells are present in an incomplete layer at the base of the lesions (Figs 3 and 4) and whole or partial lobules are sometimes involved by pre-invasive neoplasia (e.g., cancerization of lobules) (Fig. 5). However, in the setting of adjacent carcinoma, there is no reliable method of separating DCIS from cases of carcinoma that have invaded a duct. Low grade DCIS and ADH are difficult to separate even when analyzed by experts. DCIS frequently is defined as involving more than one duct and the lesion has a more uniform population of luminal-like cells. If one considers only pure lesions without a component of invasion, both ADH and low grade DCIS tend to be basal cell negative (CK5/6) as well as negative for HER2 and p53. In contrast, high grade DCIS is frequently positive for HER2 and p53, but negative for ER, PR and Bcl-2. For example, high grade (e.g., comedo) DCIS of the breast may exhibit a strong membrane expression of p185erbB−2 and it may have intranuclear accumulation of p53, consistent with a mutation in p53. Of interest, DCIS lesions that have more aggressive molecular changes are frequently associated with ductal carcinomas that have these same changes.
Fig. 5.

(×400 immunostained Bcl-2) demonstrates partial cancerization of a lobule; of interest, the cells associated with the cancerization are stained strongly with Bcl-2 (gray arrows). Panel B (×400, immunostained Bcl-2) demonstrates a small lobular area with cancerization of the ductolobular unit. Note lymphocytes surrounding the ductolobular units are staining strongly with Bcl-2 as would be expected; however the remaining basal cells staining with Bcl-2 (gray arrows) are reduced to only a partial layer and there is only weak staining of the luminal cells.
13. Pre-invasive neoplastic lesions of the prostate
The primary lesion recognized as a pre-invasive neoplastic lesion of the prostate is high grade prostatic intraepithelial neoplasia (PIN 2 plus PIN 3). As discussed, with many forms of pre-invasive neoplasia, the lesions are classified as pre-invasive neoplasia because they are associated with cancer [5–7,11]. Also, some pathologists have reported that when there is high grade PIN, especially the cribriform pattern of PIN3, there frequently are microinvasive foci associated with the PIN [61].
When PIN lesions are studied by computerized cytomorphometry, the morphologic features were found to be intermediate between benign normal appearing prostate glands and prostate cancer. These morphometric features included nuclear size (increased), nucleolar size (increased), as well as nuclear size variability and nuclear crowding. Also nucleolar variability was increased – number, size and nucleolar eccentricity. There have been numerous studies of the molecular features of PIN and most show that the molecular features of PIN are similar to the same as the molecular features of prostate cancer [2]. Some investigators have focused on how the molecular features of PIN mirror basal cell markers of normal glands of the prostate [2], while others have emphasized how the molecular features are similar to the luminal cells of normal glands of the prostate. Some of our results are demonstrated in Table 4.
Table 4.
Biomarker expression in benign, pre-invasive, neoplastic (PIN) and matching malignant (invasive adenocarcinoma) prostate epithelial cells [2]
| Biomarkers | Benign-normal | PIN | Malignant | |||
|---|---|---|---|---|---|---|
|
|
||||||
| Basal | Luminal | Basal | Luminal | |||
| Growth Factors | TGFα | +/− | +/− | + | + | ++ |
| Growth Factor Receptors | EGFR; P185erbB-2; P180erbB-3 | ++; ++; ++ | +; +; + | ++; ++; ++ | +; ++; ++ | +; ++; ++ |
| Glycosylated Tumor Antigens | TAG72; Lewis Y | −; ++ | −; + | −; ++ | ++; ++ | ++; ++ |
| Tumor Suppressor Gene | p53 | − | − | − | +/− | ++ |
| Anti-metastasis Gene | nm23-H1 | ++ | + | ++ | ++ | ++ |
| Metabolic Enzymes | FASE | +/− | + | +/− | ++ | ++ |
14. Preinvasive neoplasia of the colon
Much of our understanding of the development of colorectal neoplasia (CRN) has originated from the study of familial forms of colorectal neoplasia. The two main conditions of inheritable colorectal cancer are familial adenomatous polyposis (FAP) and hereditary non-polyposis colon cancer (HNPCC).
As demonstrated in Fig. 1, in the typical development of cancer in FAP, one copy of a mutated APC gene is inherited. This heterozygous state for the mutated APC gene results in increased proliferation of colorectal epithelial cells with the mutated APC gene as well as an increased mutational rate in these cells; thus, colorectal epithelial stem cells would be at risk for developing mutations in the other wild type copy of the APC gene. When cells develop mutations in the second native APC gene, interactions of APC with other molecular pathways change (e.g., β catenin accumulates in the cytoplasm and transfers to the nucleus), proliferation and the mutational rates increase further causing stem cells with homozygous mutations in APC to take over a local area of the colorectum. This causes an architectural change with the formation of atypical crypt foci (ACF) as well as the accumulation of additional mutations including mutations in K-ras and SMAD4 and increased expression of COX-2 (Fig. 1). The architectural disruption continues through the atypical crypt state. At some point, an adenomatous polyp forms and begins to grow. Currently, the “transitional lesion” that leads to adenomatous polyps has not been identified and the development of adenomatous polyps from ACF is not understood. Nevertheless, adenomatous polyps develop. Most small polyps are inflammatory polyps; however, these may develop into serrated adenomatous polyps. As adenomatous polyps increase in size to greater than 3 cm, mutations in p53 become more likely and such a mutation may be the forerunner of local invasion. These changes are demonstrated in Fig. 1.
The development of many cases of hereditary non-polyposis colorectal cancer (HNPCC) is similar to adenomatous polyposis coli except the mutated gene that is inherited is frequently one of the DNA microsatellite mismatch repair genes such as MSH2. In HNPCC, ACFs may not develop or be as prominent as they are in FAP or in chemical induction of CRN. Also, as microsatellite instability develops (mutator phenotype), key genes such as TGFβR2 may be inactivated as well as β catenin, bax and caspase 5. The development of HNPCC tumors may be similar molecularly to FAP tumors; however, sometimes inactivation of expression of mismatch repair genes by methylation of their promoters may be involved.
In some cases of HNPCC, neither mutation of mismatch repair genes nor methylation of the promoters of these genes can be identified. There are cases of HNPCC probably secondary to mutations and other changes in multiple genes such as myh which may be occasionally mutated in HNPCC. Alternatively, these cases may be secondary to the inheritance of genes whose polymorphisms may increase the risk of developing cancer. For example, a potential risk factor for the development of HNPCC has been hypothesized to be related to deficiency or haplodeficiency of transforming growth factor β receptor 1 (TGFβR1). The under expression of TGFβR1 has been associated with germline allel-specific expression of several polymorphisms of this gene [43,44].
If an adenomatous polyp of the colorectum is an attached component of an adenocarcinoma, molecular changes noted in the polyp (e.g. mutation in p53) usually are present in the cancer [62]. Thus, in general, molecular changes probably precede histopathologic changes (e.g., mutations in p53 of cells of the colorectum may lead to an invasive tumor with the same dysregulation of p53).
The colorectum is not a homogenous organ, in that the proximal colon (from cecum to proximal 2/3 of transverse colon) develops embryologically from the midget and its vascular system is via the anterior mesenteric system while the distal colon (last 1/3 of transverse colon to rectum) and the rectum develop from the hind-gut and the vascular system is from the posterior mesenteric system. Similarly, the molecular features of the normal proximal colon may vary from those of the distal colorectum [63]. Thus, it is not surprising that tumors of the proximal colon may vary as to their molecular features from those of the distal colorectum. Also, rectal tumors typically are treated differently than tumors of the colon. Thus, our view is that tumors of the colorectum should not be grouped together in evaluations; instead, for example, tumors of the proximal colon should be evaluated separately.
15. General characteristics of intraepithelial neoplasia leading to squamous cell carcinoma
Squamous cell carcinomas (SCC) develop in the skin, oral cavity, larynx, bronchial system, esophagus, cervix, vagina, labia and anus. For the anus of male homosexuals, cervix, vagina and labia, these lesions are usually associated with a viral infection with one of the human papilloma viruses, usually type 16 or 18 as discussed previously. Also, more controversial is the development of subgroups of oral SCC and bronchial SCCs secondary to HPV infections. Most other squamous lesions usually develop as a consequence of noxious exposures. For the skin, this is UV-B exposure, for the oral cavity and esophagus – tobacco and alcohol, for the larynx and bronchial system – cigarette smoke and/or radon exposure. Such exposures usually cause damage and incorporation of adducts into the DNA of the damaged cells. Subsequently, in the resulting repair and replication of DNA and the associated proliferation the adducts increase the likelihood that deleterious, somatic mutations may develop. Continuing noxious exposures causing damage and repair are a subtype of LOCDIR lesions.
The pre-invasive neoplastic lesions leading to SCCs are designated as intraepithelial neoplasia primarily in the cervix as previously discussed. In the skin, these pre-invasive neoplastic lesions are designated as actinic keratoses and in most other sites they are referred to as dysplasia/carcinoma in situ. “Dysplasia” in oral lesions is not in general graded because accurate correlations between the histopathology of these lesions and their likelihood of progressing to cancer have not been made as they have for cervical lesions.
Oral dysplasia usually is a lesion with extensive extracellular keratin on its surface (Fig. 6) which gives the lesions a white-gray appearance on visual examination. This appearance of oral dysplasia is called leukoplakia. Other areas of oral dysplasias may have extensive subepithelial vascularity and these lesions are called erythroplakia. Our experience from studies of longitudinal biopsies of leukoplakia is that squamous dysplasia of the oral cavity is a lesion whose boundaries move over a period of several months [64]. Of interest, is that the boundary is not only a boundary in which a profound difference in histopathologic appearance on H&E staining, but also is a boundary of extensive molecular changes. Typically the boundary of an area of squamous dysplasia is quite sharp and linear. This is demonstrated in Fig. 7, Panel A, in which the boundary on H&E staining is easily identified. Also, the boundary of dysplasia is marked by a matching boundary of sharp molecular changes,including TGFα, EGFr and CD44v6 as shown in Panels B, C and D. In other boundaries of dysplasia, both the nuclear expression of p53 and membrane expression of p185erbB−2 have been shown to increase in such dysplastic cells of the oral mucosa.
Fig. 6.

In Panel A (H&E ×200), the black arrows demonstrate the sharp linear transition between an area of mild epithelial hyperplasia of the oral cavity and dysplastic leukoplakia (in direction of gray arrow). A higher power view (×600) of the basal area of the hyperplasia is shown in Panel B and Panel C shows a high power view of the base of the dysplastic leukoplakia. Note that the nuclei are larger in the dysplastic leukoplakia (fat gray arrow) and that the basal area is disorganized and lacks apparent cellular cohesion as compared with the area of mild hyperplasia (fat white arrow). The dysplastic cells demonstrate nuclear accumulation of p53 in about 30% of the basal cells in the area of dysplasia (not shown) and none in area of hyperplasia (not shown).
Fig. 7.

The panels A–D of Fig. 7 represents a linear transition from mild hyperplasia of the epithelium of the oral cavity to epithelial dysplasia with the separation marked by the gray arrows and the black arrows pointing to the area of dysplasia. Panel A (×200, H&E) represents the histomorphology of this lesion. Panel B (×200, TGFα) demonstrates how increased expression of TGFα follows the exact linear transition of the epithelial boundary as does EGFr in Panel C (×200, EGFr) and CD44v6 in Panel D (×200, CD44v6). Increased proliferation follows this same pattern (data not shown). As will be discussed subsequently, differentiated expression of molecular markers and proliferation follow the boundary of dysplasia almost exactly. The overlap of increased expression of TGFα and EGFr would be characteristic of an area of autocrine interaction.
Areas of transition between CIN and uninvolved cervical epithelium and between oral dysplasia and un-involved epithelium are usually very distinct due to the histomorphological changes as shown in Figs 6–9. These areas of transition also demonstrate distinct molecular changes as well as physiologic changes including increased proliferation (Fig. 8). Of interest, the molecular changes of pre-invasive neoplasia usually mirror the molecular changes observed in the tumors which can develop from these pre-invasive lesions. For example, mutations in p53 as represented by visible nuclear accumulation of p53 does not usually occur in SCC of the cervix or in CIN lesions (Panel A, Fig. 9) while visible nuclear accumulation of p53 is common in both SCCs of dysplastic epithelium of the oral cavity as well as in SCCs of the oral cavity.
Fig. 9.
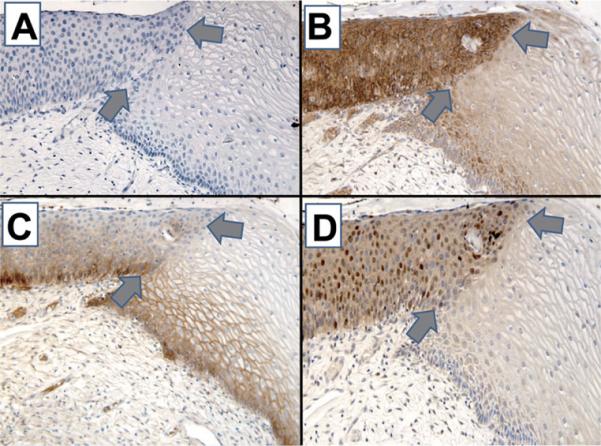
Represents molecular changes occurring between apparent normal appearing uninvolved epithelium of the cervix and CIN. In Panel A (X200, p53 counterstain hematoxylin), there is no staining for p53 as would be expected. Note how clearly the basal cells are arranged in the uninvolved epithelium and how disorganized they appear in the area of CIN. Like the prior cases, the transition between uninvolved epithelium and the epithelium of CIN is linear (gray arrows point to edge of lesion, and top gray arrow to the direction of lesion). Proliferation (not shown) follows this sharp boundary as does increased expression of erbB-2 in Panel B (X200, erbB-2), EGFr in Panel C (X200, EGFr) and TGFα in Panel D (X200, TGFα). The increased expression of TGFα and EGFr in the CIN lesion may suggest an autocrine interaction within CIN.
Fig. 8.

Represents a transition between normal appearing epithelium of the cervix and CIN epithelium. Black arrows mark the transition. The histomorphometric boundary of CIN is relatively linear and is very distinct. As can be seen, there is increased proliferation in the CIN lesion as measured by Ki67/MIB-1 in Panel A (X200, Ki67/MIB-1) and by PCNA in Panel B (X200, PCNA). Proliferation in the uninvolved (normal appearing) cervix is primarily limited to the base of the lesion, but in CIN, is throughout the lesion. In sections not shown of this same area, increased expression of EGFr and erbB-2 follow the exact histopathologic boundary as does the proliferation and as would be expected, there is not a mutation or dysregulation of p53.
Of interest, pre-invasive neoplastic lesions of squamous cell type (e.g., CIN or oral dysplasia) do not appear to gradually develop from the basal cells and do not involve the adjacent epithelium as, for example, spreading laterally; rather, there is a sharp boundary that is almost perpendicular to the basal cells. Note the patterns that we do not see as shown in Fig. 10 are informative; we see only pattern (D).
Fig. 10.

The 4 cartoons demonstrate 4 potential patterns which could mark the spread of a squamous pre-invasive lesion. In general, the patterns (A), (B), and (C) are not usually noted, but pattern (D) is seen in patients with dysplastic lesions of the oral cavity and/or CIN lesions of the cervix, as shown previously.
Why is this boundary so sharp? This may be due to a tendency of neoplastic lesions to “stick” together via various attractions such as those described by Norton et al. [65,66].
15.1. Other pre-invasive lesions
Pre-invasive neoplasia leading to urothelial neoplasia, sarcomas or hematopoietic neoplasia are beyond this section in that they represent a small component of pre-invasive neoplasia or are not currently well defined.
15.2. Treatment of pre-invasive neoplastic lesions
Some better known in situ neoplastic lesions are treated while other in situ lesions are not. The best four examples of organs in which in situ lesions are treated are DCIS and LCIS of the breast and CIN2 – CIN3 of the cervix, actinic keratoses of the skin and leukoplakia of the oral cavity. For example, the whole area of DCIS of the breast is surgically removed and in addition, if complete removal is uncertain, microinvasion is suspected, or the lesions are very high grade, the area of the breast may be subjected to radiation therapy after surgery. Such treatment appears very effective although this is difficult to ascertain because the natural histories of DCIS and LCIS are not known in detail. Nevertheless, in UAB's experience, less than 10% of cases return as either DCIS or invasive cancer. For some CIN2, but all CIN3 of the cervix, the area where CIN typically develops is treated with a laser, cryotherapy and/or is removed surgically.
16. Mouse models of inheritable pre-invasive neoplasia
One of the important aspects of identifying the molecular changes that occur in inheritable cancers is that the pre-invasive neoplastic lesions can be reproduced in some organ systems of transgenic mouse models that carry the same or very similar specific genetic changes that have been identified in inheritable neoplasia in humans. As discussed by Sandgren [67], “Mice are not people, and thus, how do we transfer our understanding of the disease in mice to disease in humans?” He proposes 3 criteria – 1) Morphology – at all levels – gross, microscopic and ultrastructural, i.e., the neoplastic lesions of mice and men should look alike. 2) Molecular – human and murine tumors should display the same molecular alterations and 3) Behavioral – the neoplastic lesions of mice and men should have similar growth, invasiveness, metastases and effects on the species. To this I would add a 4th, Normal Biology – the biology of the cells of the organ of interest should be similar in mouse and man. For example, because the biology of the prostate is different in mice and men (e.g., cellular location of androgen receptors), it is unlikely that excellent mouse models are likely to be developed for the prostate. Thus, transgenic mice do not develop models of useful development of neoplasia for all organ systems usually because the model of disease in rodents does not mirror some aspect of the disease in humans.
16.1. Mouse FAP
A transgenic model of familial adenomatous polyposis (FAP) of the colon is the multiple intestinal neoplasia (MIN) mouse which carries a germ-line mutation in the mouse homologue of the APC gene (ApcMin). Even though this model primarily develops many small adenomas of the small bowel, it has been quite useful in studying pharmacologic agents that may modify intestinal tumor development such as COX-2 inhibitors which markedly inhibit adenoma formation in this animal model. Also, when crossed with a mouse haplodeficient in TGFβR1, large dysplastic adenomas develop in the colon indicating that TGFβR1 may be an important gene in the development of CRN that interacts with ApcMin [44].
16.2. Mouse PanIN
Similarly there are several mouse models of preinvasive pancreatic cancer. One of the more interesting aspects of the embryologic development of the pancreas is that both the exocrine pancreas and the endocrine pancreas are hypothesized to develop from the cells of the ductal epithelium of the pancreas. This can be observed in some transgenic mice models in which ductal-alveolar metaplasia as well as islet metaplasia can be observed frequently in association with pancreatic ducts [68,69]. Very rarely we and others have observed islet metaplasia in a human pancreatic adeno-carcinoma or mixed acinar-endocrine carcinomas [70].
Pre-invasive neoplasia of the pancreas is also a lesion for which a pattern of genetic changes have been identified. Similar to other organ systems, pancreatic intraepithelial neoplasia (PanIN) is designated as PanIN 1, 2 and 3 in which PanIN3 is the most severe lesion and hence the lesion at greatest risk for developing pancreatic cancer. Histopathologic models which mirror the various grades of PanIN as well as the biological changes of PanIN develop in various murine models of pancreatic neoplasia.
The typical ductal adenocarcinoma of the pancreas (PDAC) frequently carries mutations in p16/CDKN2A, p53, SMAD4/DPC4 and K-ras. Because about 90% of PDACs are aneuploid due to early chromosomal instability secondary to defective telomeres, some of these same genes (the suppressor genes, p16, p53 and SMAD4) also are affected by LOH via partial chromosomal loss of 9p, 17p, 18q, respectively. Losses of portions of multiple other chromosomes are also characteristic of pancreatic neoplasia though less common.
Mutations of K-ras-2 begin in PanIN and continue to accumulate in more advanced lesions until approximately over 90% of PDACs have K-ras-2 mutations. Of tumors with native K-ras-2, 1/3 have defects in BRAF downstream of K-ras-2. BRAF is present in about 7% of PDACs, but only in those without mutations in K-ras-2. Of interest, mutations in BRAF occur much more frequently in mismatch repair deficient cases of cancer. Mutations or homozygous deletions of p16 also begin in early stage pancreatic neoplasia until about 80% of tumors are involved. Almost all the other advanced PDACs have hypermethylated p16 or dysregulation of Rb or Cyclin E. In contrast, p53 which is mutated and inactivated in 50–70% of PDACs is a late event in the development of pancreatic neoplasia.
The early mouse models of pancreatic cancer demonstrated several interesting features. In some of the first models, the elastase gene regulatory elements (Ela) which are found specifically in pancreatic acinar cells were combined with one of several molecules associated with pancreatic cancer. These included TGFαwhich when induced in transgenic mice produced a severe diffuse pancreatic fibrosis and acinar to ductal metaplasia. When the TGFα transgenic mice were combined with p53 null mutations, exocrine carcinomas developed in all mice within 4 months; these lesions were complex, but did not metastasize. Of interest, 50% of p53 heterozygous crossed animals developed methylation of the promotor of CDKN2A, DNA gains affecting EGFr, Rel and c-myc and DNA losses of Rb pathway. Subsequently, the Ela promoter was used with mutant (activated K-ras) to produce mice, but activating acinar to ductal metaplasia and PanIN and IPMN lesions. When K-ras mutations were combined with p53 null mice, acinar carcinomas that could metastasize developed [67].
Subsequently, mice with activating K-ras mutations were combined with mice with deletions of Ink4a/Arf (p16/p19) tumor suppressor alleles; this produced early development of PanIN lesions which rapidly developed into highly invasive and a metastatic pancreatic cancers that caused deaths of all such animals by 11 weeks; however, SMAD4 protein was not lost and p53 was native. Also, EGFr and HER2/neu were overex-pressed [68].
A recent study of mice with activating K-ras mutations and loss of SMAD4 found that these mice developed PanIN and IPMN-like lesions as well as locally invasive pancreatic exocrine lesions. Ductal-alveolar metaplasia also was noted. Thus, many of the molecular changes observed in pancreatic ductal adenocarcinoma and the pre-invasive lesions of this tumor (PanIN and IPMN) are features of mouse models of pancreatic cancer [69–72].
17. Summary
In summary, there are multiple causes of pre-invasive neoplasia ranging from radiation, chemicals, infections, and long-term continuing damage, inflammation and repair. Pre-invasive neoplasia may progress to invasion, may remain static for many years or may regress. Some types such as DCIS/LCIS, oral dysplasia, adenomatous polyps and cervical dysplasia are well described and are treated. Other forms such as high grade PIN and PanIN remain as warning signs of an at risk state.
Acknowledgement
Supported in part by the Early Detection Research Network (EDRN) (5U24 CA86359), Department of Defense, “Biomarkers in the Detection of Prostate Cancer in African-Americans” (PC093309), the Breast (5P50CA089019) and Pancreatic (2P50CA101955) SPORES at UAB, the Susan G. Komen Breast Cancer Foundation (BCTR0600484), the Skin Disease Research Center at UAB (5P30AR50948) to William E. Grizzle, and (POP138306) to Upender Manne.
References
- [1].Grizzle WE, Myers RB, Manne U. The use of biomarker expression to characterize neoplastic processes. Biotech Histochem. 1997;72(2):96–104. doi: 10.3109/10520299709082218. [DOI] [PubMed] [Google Scholar]
- [2].Myers RB, Grizzle WE. Biomarker Expression in Prostatic Intraepithelial Neoplasia. Eur Urol. 1996;30(2):153–166. doi: 10.1159/000474165. PMID: 8875196. [DOI] [PubMed] [Google Scholar]
- [3].Grizzle WE, Myers RB, Arnold MA, Srivastava S. Evaluation of biomarkers in breast, and prostate cancer. J Cell Biochem. 1994;19:259–266. [PubMed] [Google Scholar]
- [4].Dupont WD, Page DL. Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med. 1985;312:146–151. doi: 10.1056/NEJM198501173120303. [DOI] [PubMed] [Google Scholar]
- [5].McNeal JE, Bostwick DG. Intraductal dysplasia; a pre-malignant lesion of the prostate. Hum Pathol. 1986;17:64–71. doi: 10.1016/s0046-8177(86)80156-3. [DOI] [PubMed] [Google Scholar]
- [6].Bostwick DG, Amin MB, Dundore P. Architectural patterns of high-grade prostatic intraepithelial neoplasia. Hum Pathol. 1993;42:298–310. doi: 10.1016/0046-8177(93)90041-e. [DOI] [PubMed] [Google Scholar]
- [7].Bostwick DC, Brawer MK. Prostate intra-epithelial neoplasia, and early invasion in prostate cancer. Cancer. 1987;59:788–794. doi: 10.1002/1097-0142(19870215)59:4<788::aid-cncr2820590421>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- [8].Adsay NV. Pathological classification of cystic neoplasms of the pancreas in pancreatic cancer. In: Von Hoff DD, Evans DB, Hruban RH, editors. Pancreatic Cancer. 2005. pp. 716–735. [Google Scholar]
- [9].De Marzo AM, Marchi VL, Epstein JI, Nelson WG. Proliferative inflammatory atrophy of the prostate: Implications for prostatic carcinogenesis. Am J Pathol. 1999;155:1985–1199. doi: 10.1016/S0002-9440(10)65517-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gandour MJ, Grizzle WE. A Small Adrenocortical Carcinoma with Aggressive Behavior. An evaluation of criteria for malignancy. Arch Pathol Lab Med. 1986;110(11):l076–l079. [PubMed] [Google Scholar]
- [11].McNeal JE, Villers A, Redwine EA, Freiha FS, Stamey TA. Microcarcinoma in the prostate: its association with duct-acinar dysplasia. Human Pathol. 1991;22:644–652. doi: 10.1016/0046-8177(91)90286-x. [DOI] [PubMed] [Google Scholar]
- [12].Winawer SJ, Zauber AG, Ho MN, O'Brien MJ, Gottlieb LS, Sternberg SS, Waye JD, Shapiro M, Bond JH, Panish JF, Ackroyd F, Shike M, Kurtz RC, Hornsby-Lewis L, Gerdes H, Stewart ET. The National Polyp Study Workgroup, Prevention of colorectal cancer by colonoscopic polypectomy. New Engl J Med. 1993;329(27):1977–1981. doi: 10.1056/NEJM199312303292701. [DOI] [PubMed] [Google Scholar]
- [13].Rex DK. Colonoscopy, A review of its yield for cancers, and adenomas by indication. Am J Gastroenterol. 1995;90(3):353–365. [PubMed] [Google Scholar]
- [14].Beenken S, Sellers M, Huang P, Peters G, Krontiras H, Dixon P, Stockard C, Listinsky C, Grizzle WE. Transforming growth factor α (TGFα) expression in dysplastic oral leukoplakia: modulation by 13-cis retinoic acid. Head Neck. 1999;21(6):566–573. doi: 10.1002/(sici)1097-0347(199909)21:6<566::aid-hed11>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- [15].Koss LG. LG Koss's Diagnostic cytology, and its histopathologic bases. 3rd edition J.B. Lippincott Company; Philadelphia: 1979. Epidermoid carcinoma of the uterus, cervix, and related precancerous lesions; pp. 285–375. [Google Scholar]
- [16].Fearon ER, Vogelstein BA. A genetic model for colorectal carcinogenesis. Cell. 1990;61:579–567. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- [17].Ionov Y, Peinado MA, Malkhosyan S, Shibata S, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:5658–5561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- [18].Honnavara N, Ananthaswamy SM, Loughlin PC, Evans RL, Ullrich SE, Kripke ML. Sunlight, and skin cancer: inhibition of p53 mutations in UV-irradiated mouse skin by sunscreens. Nature Medicine. 1997;3(5):510–526. doi: 10.1038/nm0597-510. [DOI] [PubMed] [Google Scholar]
- [19].Ling G, Persson A, Berne B, Uhlén M, Lundeberg J, Ponten F. Persistent p53 mutations in single cells from normal human skin. Am J Pathol. 2001;159:1247–1253. doi: 10.1016/S0002-9440(10)62511-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jonason AS, Kunala S, Price GJ, Restifo RJ, Spinelli HM, Persing JA, Leffell DJ, Tarone RE, Brash DE. Frequent clones of p53-mutated keratinocytes in normal human skin. PNAS. 1996;93(24):14025–14029. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal, and malignant human mammary stem cells, and a predictor of poor clinical outcomes. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kaharala M, Wicha MS. Implications of the cancer stem-cell hypothesis for breast cancer prevention, and therapy. J Clin Oncol. 2008;26:2813–2820. doi: 10.1200/JCO.2008.16.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Charafe-Jauffret E, Monville F, Ginestier C, Dontu G, Birnbaum D, Wicha MS. Cancer stem cells in breast: Current opinion, and future challenges. Pathobiology. 2008;75:75–84. doi: 10.1159/000123845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur M-H, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, Birnbaum D, Wicha MS. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity, and a distinct molecular signature. Cancer Res. 2009;69(4):1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pershagen G, Akerblom G, Axelson O, Clavensjo B, Damber L, Desai G, Enflo A, Lagarde F, Mellander H, Svartengren M, Swedjemark GA. Residential radon exposure, and lung cancer in Sweden. NEJM. 1994;330:159–164. doi: 10.1056/NEJM199401203300302. [DOI] [PubMed] [Google Scholar]
- [26].Ouhtit A, Nakazawa H, Armstrong BK, ricker A, Tan E, Yamasaki H, English DR. UV-radiation-specific p53 mutation frequently in normal skin as a predictor of risk of basal cell carcinoma. J Natl Cancer Inst. 1998;90:523–531. doi: 10.1093/jnci/90.7.523. [DOI] [PubMed] [Google Scholar]
- [27].Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Pontén J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Campbell C, Quinn AG, Ro Y-S, Angus B, Rees JL. P53 mutations are common, and early events that precede tumor invasion in squamous cell neoplasia of the skin. J Invest Dermatol. 1993;100:746–748. doi: 10.1111/1523-1747.ep12475717. [DOI] [PubMed] [Google Scholar]
- [29].Murata M, Tagawa M, Watanabe S, Kimura H, Takeshita T, Morimoto K. Genotype difference of aldehyde-dehydrogenase 2 gene in alcohol drinkers influences the incidence of Japanese colorectal cancer patients. Japanese Journal of Cancer Research. 1999;90:711–719. doi: 10.1111/j.1349-7006.1999.tb00805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pereira M, Goldberg GM, Bar-Ziv J, Danovitch G. Loss of heterozygosity of methylenetetrahydrofolate reductase in colon carcinomas. Oncology Reports. 1999;6:597–599. doi: 10.3892/or.6.3.597. [DOI] [PubMed] [Google Scholar]
- [31].Shannon B, Gnanasampanthan S, Beilby J, Iacopetta B. A polymorphism in the methylenetetrahydrofolate reductase gene predisposes to colorectal cancers with microsatellite instability. Gut. 2002;50:520–524. doi: 10.1136/gut.50.4.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Abel EL, DiGiovanni J. Environmental carcinogenesis. In: Craig B, Thompson MD, Gray JW, Mendelsohn J, editors. The Molecular Basis of Cancer. 3rd ed. 2008. pp. 91–113. [Google Scholar]
- [33].Daniel KG, Chen D, Oriu S, Cui QC, Miller FR, Dou QP. Clioquinol, and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors, and apoptosis inducers in human breast cancer cells. Breast Cancer Research. 2005;7:R897–R908. doi: 10.1186/bcr1322. Doi:16.1186/bcr1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Brentnall TA, Crispin DA, Bronner MP, Cherian SP, Hueffed M, Rabinovitch PS, Rubin CE, Haggitt RC, Boland CR. Microsatellite instability in nonneoplastic mucosa from patients with chronic ulcerative colitis. Cancer Res. 1996;56:1237–1240. [PubMed] [Google Scholar]
- [36].Brentnall TA, Chen R, Lee JG, Kimmey MB, Bronner MP, Haggitt RC, Kowdley KV, Hecker LM, Byrd Dr. Microsatellite instability, and k-ras mutations associated with pancreatic adenocarcinoma, and pancreatitis. Cancer Res. 1995;55:4264–4267. [PubMed] [Google Scholar]
- [37].Willenbucher RF, Aust DE, Chang CG, Zelman SJ, Ferrell LD, Moore DHII, Waldman FM. Genomic instability is an early event during the progression pathway of ulcerative-colitis-related neoplasia. Am J Pathol. 1999;154:1825–1830. doi: 10.1016/S0002-9440(10)65438-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Loeb LA. A mutator phenotype in cancer. Cancer Res. 2001;61:3230–3239. [PubMed] [Google Scholar]
- [39].Loeb KR, Loeb LA. Genetic instability, and the mutator phenotype. Studies in ulcerative colitis. American Journal of Pathology. 1999;154:1621–1626. doi: 10.1016/S0002-9440(10)65415-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Heinen CD, Noffsinger AE, Belli J, Straughen J, Fischer J, Groden J, Fenoglio-Preiser CM. Regenerative lesions in ulcerative colitis are characterized by microsatellite mutation. Genes Chromosomes, and Cancer. 1997;19:170–175. [PubMed] [Google Scholar]
- [41].Lee S, Jeong J, Majewski T, Scherer SE, Kim M-S, Tuziak T, Tang KS, Baggerly K, Grossman HB, Zhou J-H, Shen L, Bondaruk J, Ahmed SS, Samanta S, Spiess P, Wu X, Filipek S, McConkey D, Bar-Eli M, Issa J-P, Benedict WF, Czerniak B. Forerunner genes contiguous to RB1 contribute to the development of in situ neoplasia. PNAS. 2007;104(34):13732–13737. doi: 10.1073/pnas.0701771104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kim M-S, Jeong J, Majewski T, Kram A, Yoon D-S, Zhang R-D, Li J-Z, Ptaszynski K, Kuang TC, Zhou J-H, Sathyanarayana UG, Tuziak T, Johnston DA, Grossman HB, Gazdar AF, Scherer SE, Benedict WF, Czerniak B. Evidence for alternative candidate genes near RB1 involved in clonal expansion of in situ urothelial neoplasia. Laboratory Investigation. 2006;86:175–190. doi: 10.1038/labinvest.3700378. [DOI] [PubMed] [Google Scholar]
- [43].Valle L, Serena-Acedo T, Liyanarachi S, Hampel H, Com-eras I, Li Z, Zeng Q, Zhang H-T, Pennison MJ, Sadim M, Pasche B, Tanner SM, de la Chapelle A. Germline allele-specific expression of TGFBR1 confers an increased risk of colorectal cancer. Science. 2008;321:1361–1365. doi: 10.1126/science.1159397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zeng Q, Phukan S, Xu Y, Sadim M, Rosman DS, Pennison M, Liao J, Yang G-Y, Huang C-C, Valle L, Di Cristofano A, de la Chappelle A, Pasche A. Tgfbr1 haploinsufficiency is a potent modifier of colorectal cancer development. Cancer Res. 2009;69(2):678–686. doi: 10.1158/0008-5472.CAN-08-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kaklamani VG, Wisinski KB, Sadim M, Gulden C, Do A, Offit K, Baron JA, Ahsan H, Mantzoros C, Pasche B. Variants of the adiponectin (ADIPOQ), and adiponectin receptor 1 (ADIPOR1) genes, and colorectal cancer risk. JAMA. 2008;300(13):1523–1131. doi: 10.1001/jama.300.13.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang HG, Grizzle WE. Aging, immunity,, and tumor susceptibility. Immunol Allergy Clin N Am. 2003;23:83–102. doi: 10.1016/s0889-8561(02)00085-1. [DOI] [PubMed] [Google Scholar]
- [47].Grizzle WE, Semmes OJ, Bigbee W, Zhu L, Malik G, Oelschlager DK, Manne B, Manne U. The need for the review, and understanding of SELDI/MALDI mass spectroscopy data prior to analysis. Cancer Informatics. 2005;1(1):86–97. [PMC free article] [PubMed] [Google Scholar]
- [48].Remenyik É , Wikonkál NM, Zhang W, Pailwal V, Brash DE. Antigen-specific immunity does not mediate acute regression of UVB-induced p53-mutant clones. Oncogene. 2003;22:6369–6376. doi: 10.1038/sj.onc.1206657. [DOI] [PubMed] [Google Scholar]
- [49].Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, Zhang H-G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood. 2007;109:4336–4342. doi: 10.1182/blood-2006-09-046201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Liu C, Yu S, Zinn K, Wang J, Zhang L, Jia Y, Kappes JC, Barnes S, Kimberly RP, Grizzle WE, Zhang HG. Murine mammary carcinoma exosomes promote tumor growth by suppression of NK cell function. J Immunol. 2006;176:1375–1385. doi: 10.4049/jimmunol.176.3.1375. [DOI] [PubMed] [Google Scholar]
- [52].Wang G-J, Liu Y, Qin A, Shah SV, Den Z-B, Xiang X, Chang Z, Liu C, Wang J, Zhang L, Grizzle WE, Zhang H-G. Thymus exosomes-like particles induce regulatory T cells. J Immunol. 2008;181:5242–5248. doi: 10.4049/jimmunol.181.8.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Clayton A, Court J, Navabi H, Adams M, Mason MD, Hobot JA, Newman GR, Jasani B. Analysis of antigen presenting cell derived exosomes, based on immuno-magnetic isolation, and flow cytometry. J Immunol Methods. 2001;247:163–174. doi: 10.1016/s0022-1759(00)00321-5. [DOI] [PubMed] [Google Scholar]
- [54].Blanc L, Barres C, Bette-Bobillo P, Vida M. Reticulocyte-secreted exosomes bind natural IgM antibodies: involvement of a ROS-activatable endosomal phospholipase iPLA2. Blood. 2007;110:3407–3416. doi: 10.1182/blood-2007-04-085845. [DOI] [PubMed] [Google Scholar]
- [55].Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis, and function. Nat Rvw Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- [56].Xiang X, Poliakov A, Liu C, Liu Y, Deng Z-B, Wang J, Cheng Z, Shah SV, Wang G-J, Zhang L, Grizzle WE, Mobley J, Zhang H-G. Induction of myeloid-derived suppressor cells by tumor exosomes. In J Cancer. 2009;124:2621–2623. doi: 10.1002/ijc.24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yu S, Liu C, Su K, Wang J, Liu Y, Zhang L, Li C, Cong Y, Kimberly R, Grizzle WE, Falkson C, Zhang H-G. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J Immunol. 2007;178:6867–6875. doi: 10.4049/jimmunol.178.11.6867. [DOI] [PubMed] [Google Scholar]
- [58].Dupont WD, Page DL. Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med. 1985;312:146–151. doi: 10.1056/NEJM198501173120303. [DOI] [PubMed] [Google Scholar]
- [59].Page DL, Dupont WD, Rogers LW, Landenberger M. Intraductal carcinoma of the breast: follow-up after biopsy only. Cancer. 1982;49:751–758. doi: 10.1002/1097-0142(19820215)49:4<751::aid-cncr2820490426>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- [60].Page DL, Dupont WD, Rogers LW, Jensen RA, Schuyler PA. Continued local recurrence of carcinoma 15–25 years after a diagnosis of low grade ductal carcinoma in situ of the breast treated only by biopsy. Cancer. 1995;76:1197–1200. doi: 10.1002/1097-0142(19951001)76:7<1197::aid-cncr2820760715>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- [61].Epstein JI. Pathology in prostate cancer, principles, and practice. In: Kantoff PW, Carroll PR, D'Amico AV, editors. Philadelphia: Lippincott William, and Wilkins. 2002. pp. 232–253. [Google Scholar]
- [62].Shanmugam C, Katkoori VR, Jhala NC, Grizzle WE, Siegal GP, Manne U. p53 Nuclear Accumulation, and Bcl-2 Expression in Contiguous Adenomatous Components of Colorectal Adenocarcinomas Predict Aggressive Tumor Behavior. J Histochem Cytochem. 2008;56(3):305–312. doi: 10.1369/jhc.7A7362.2007. PMCID: PMC2324183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Grizzle WE, Shibata D, Manne U, Myers RB, Frost AR. Molecular, and Histopathologic Changes in the Development of Colorectal Neoplasia. In: Srivastava S, Henson DE, Gazdar A, editors. Molecular Pathology of Early Cancer. Chapter 10. IOS Press; Amsterdam, Netherlands: 1999. pp. 135–170. [Google Scholar]
- [64].Beenken S, Hockett R, Jr., Grizzle W, Weiss HL, Pickens A, erloff M, Malone WF, Bland KI. Transforming growth factor α (TGF-α): A surrogate endpoint biomarker? J Am Coll Surg. 2002;195(2):149–158. doi: 10.1016/s1072-7515(02)01219-x. [DOI] [PubMed] [Google Scholar]
- [65].Norton L, Massagué J. Is cancer a disease of self-spreading? Nature Medicine. 2006;12(8):875–878. doi: 10.1038/nm0806-875. [DOI] [PubMed] [Google Scholar]
- [66].Norton L. Cancer stem cells, self-seeding,, and decremented exponential growth: theoretical, and clinical implications. Breast Dis. 2008;29:27–36. doi: 10.3233/bd-2008-29104. PubMed PMID: 19029622. [DOI] [PubMed] [Google Scholar]
- [67].Sandgren EP. Mouse models of exocrine pancreatic cancer. In: Hoff DD, Evans DB, Hruban RH, editors. Pancreatic Cancer. Chapter 6. Jones, and Bartlett Publishers; Sudbury, Massachusetts: 2005. pp. 87–100. [Google Scholar]
- [68].Aguirre AJ, Bardessy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras, and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes, and Development. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bell RH, Jr., Memoli VA, Longneker DS. Hyperplasia, and tumors of the islets of Langerhans in mice bearing an elastase I-SV40 T-antigen fusion gene. Carcinogenesis. 1990;11:1393–1398. doi: 10.1093/carcin/11.8.1393. [DOI] [PubMed] [Google Scholar]
- [70].Klimstra DS, Rosai J, Heffess CS. Mixed acinarendocrine carcinomas of the pancreas. Am J Surg Pathol. 1994;18:765–778. doi: 10.1097/00000478-199408000-00002. [DOI] [PubMed] [Google Scholar]
- [71].Kojima K, Vickers SM, Adsay V, Jhala NC, Kim H-G, Schoeb TR, Grizzle WE, Klug CA. Inactivation of Smad4 accelerates KrasG12D-mediated pancreatic neoplasia. Cancer Res. 2007;67(17):8121–8130. doi: 10.1158/0008-5472.CAN-06-4167. [DOI] [PubMed] [Google Scholar]
- [72].Winslow MM, Jacks T. Genetic mouse models of cancer. In: Mendelsohn J, Howley PM, Israel MA, Gray JW, Thompson CB, editors. The Molecular Basis of Cancer. 3rd edition Chapter 9. Saunders Elsevier; 2008. pp. 129–138. [Google Scholar]
- [73].Cole K, Tabernero M, Anderson KS. Biologic characteristics of premalignant breast disease. Cancer Biomark. 2011;9:177–192. doi: 10.3233/CBM-2011-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Srivastava S, Grizzle WE. Biomarkers, and the genetics of early neoplastic lesions. Cancer Biomark. 2011;9:41–64. doi: 10.3233/CBM-2011-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]