

Abstract
Objective:
To describe the phenotype of patients with C9FTD/ALS (C9ORF72) hexanucleotide repeat expansion.
Methods:
A total of 648 patients with frontotemporal dementia (FTD)–related clinical diagnoses and Alzheimer disease (AD) dementia were tested for C9ORF72 expansion and 31 carried expanded repeats (C9+). Clinical and neuroimaging data were compared between C9+ (15 behavioral variant FTD [bvFTD], 11 FTD–motor neuron disease [MND], 5 amyotrophic lateral sclerosis [ALS]) and sporadic noncarriers (48 bvFTD, 19 FTD-MND, 6 ALS).
Results:
All C9+ patients displayed clinical syndromes of bvFTD, ALS, or FTD-MND. At first evaluation, C9+ bvFTD patients had more delusions and greater impairment of working memory, but milder eating dysregulation compared to bvFTD noncarriers. C9+FTD-MND patients had a trend for longer survival and had an earlier age at onset than FTD-MND noncarriers. Voxel-based morphometry demonstrated more thalamic atrophy in FTD and FTD-MND carriers than in noncarriers.
Conclusions:
Patients with the C9ORF72 hexanucleotide repeat expansion develop bvFTD, ALS, or FTD-MND with similar clinical and imaging features to sporadic cases. Other FTD spectrum diagnoses and AD dementia appear rare or absent among C9+ individuals. Longer survival in C9+ FTD-MND suggests slower disease progression and thalamic atrophy represents a novel and unexpected feature.
Frontotemporal dementia (FTD) is a common dementia syndrome among patients presenting before 65 years of age with prevalence equal to Alzheimer disease (AD) dementia.1,2 FTD often overlaps with amyotrophic lateral sclerosis (ALS), with symptoms of FTD occurring in 15%–41% of patients with ALS and features of ALS occurring in 15% of FTD.3,4 Many patients with FTD and ALS exhibit autosomal dominant family histories (FTD 10%5; ALS 5%–10%6; FTD–motor neuron disease [MND] 37%7) and a number of large familial cohorts have been linked to a chromosome 9p region.7–12 Recently, a noncoding expanded hexanucleotide repeat in chromosome 9 open reading frame 72 (C9ORF72) was identified as the cause of chromosome 9p–associated FTD and ALS.13,14 This mutation is the most common genetic cause of familial and sporadic behavioral variant FTD (bvFTD) and ALS.
In one study, the C9ORF72 (C9FTD/ALS) expansion accounted for 11.7% of familial FTD, 22.5% of familial ALS, and 4% of sporadic ALS.13 Previous family studies of chromosome 9p–linked families7,8,10 suggested that some features may distinguish this mutation from sporadic bvFTD and ALS, including ataxia, parkinsonism, visuospatial impairment, psychosis,10 and greater posterior cortical atrophy.7 Here, we describe clinical and neuroimaging features seen in symptomatic C9FTD/ALS carriers at the Memory and Aging Center at the University of California, San Francisco (UCSF).
METHODS
Standard protocol approvals, registrations, and patient consents.
All aspects of the study were approved by an institutional review board. All patients and surrogates (as per UCSF institutional review board protocol) provided written informed consent.
Participants.
Patients.
A total of 648 patients with FTD-spectrum clinical diagnosis (bvFTD [n = 123, 19%], progressive supranuclear palsy [PSP; n = 50, 7.7%], corticobasal syndrome [CBS; n = 53, 8.2%], semantic variant primary progressive aphasia [svPPA; n = 72, 11.1%], nonfluent variant primary progressive aphasia [nfvPPA; n = 21, 3.2%], FTD-MND [n = 35, 5.4%]), logopenic variant primary progressive aphasia (lvPPA; n = 18, 2.8%), posterior cortical atrophy (n = 8, 1.2%), mild cognitive impairment (n = 72, 11.1%), ALS (n = 20, 3.1%), AD dementia (n = 171, 26.4%), and dementia with Lewy bodies with AD dementia (n = 5, 0.8%) evaluated at UCSF were tested for the C9FTD/ALS expansion13 (table e-1 on the Neurology® Web site at www.neurology.org, figure 1). We included patients with AD dementia because previous reports identified AD dementia phenotype associated with the C9ORF72 mutation.9,12 Thirty-one individuals (4.7%) representing 26 different families carried the C9ORF72 repeat expansion (C9+), of which there were 3 pairs of relatives and 1 trio of relatives.
Figure 1. Subjects included in clinical analyses.

(A) Subjects in gray bar were included in demographic and family history analyses shown in table 1. The patients in the dashed boxes inside the gray bar were used for motor symptoms assessment. (B) Subjects in the gray boxes in the bottom row were included in neuropsychiatric, neuropsychological, and survival analyses shown in table 2 and table e-1. aRight temporal variant in 20 patients with semantic variant primary progressive aphasia (svPPA). bBehavioral variant frontotemporal dementia (bvFTD) groups were matched for disease severity (Clinical Dementia Rating sum of boxes) by removing 3 noncarriers. AD = Alzheimer disease dementia; ALS = amyotrophic lateral sclerosis; CBS = corticobasal syndrome; DLB + AD = dementia with Lewy bodies with Alzheimer disease; FHx+ = family history with autosomal dominant pattern of inheritance but no known frontotemporal dementia–associated mutation; FTD-MND = frontotemporal dementia–motor neuron disease; lvPPA = logopenic variant of primary progressive aphasia; MCI = mild cognitive impairment; nfvPPA = nonfluent variant of primary progressive aphasia; PCA = posterior cortical atrophy; PiB+ = positive PET scan with β-amyloid ligand [11C]–Pittsburgh compound B; PSP = progressive supranuclear palsy. Individuals with known MAPT or GRN mutations were not screened for C9ORF72 mutations.
Consecutive patients matched for age, gender, and diagnosis were selected from the remaining 617 patients who did not carry repeat expansions (figure 1). Patients included were required to have complete clinical history, neuropsychological testing data, and a structural MRI. Since our goal was to compare C9+ patients with sporadic FTD cases, noncarriers with an autosomal dominant family history, including those with GRN or MAPT mutations, were excluded (GRN and MAPT mutation carriers were not screened for the expansion). Comparison patients met criteria for bvFTD (international bvFTD research criteria [FTDC]15; n = 48), ALS (clinically definite or probable El Escorial Criteria16; n = 6), or FTD-MND (n = 19). Both C9+ patients and noncarriers were designated as FTD-MND if they met 1) both FTDC and ALS criteria; 2) FTDC and had evidence of an ALS spectrum disease (primary lateral sclerosis or lower motor neuron signs) in bulbar musculature or more than 1 spinal level; or 3) ALS criteria and at least 2 of the core FTDC. Because there were no patients with AD dementia, lvPPA, PCA, mixed DLB and AD, PSP, CBS, nfvPPA, or svPPA (left or right temporal variant) in the C9+ group, noncarriers with these syndromes were not included in the analysis. Likewise, patients who demonstrated amyloid uptake on PET with the β-amyloid ligand 11C-Pittsburgh compound B (noncarriers n = 2, bvFTD; C9+ n = 1, bvFTD) were excluded because the presence of concurrent, underlying AD pathology could confound clinical analyses (appendix e-1).
Healthy controls.
Healthy older adult controls (mean age = 62.9 ± 1.2 years) were recruited from the San Francisco community as part of ongoing longitudinal studies of aging. Controls had a Clinical Dementia Rating Scale sum of boxes (CDR-SB)17 score = 0, a normal neurologic examination, and no cognitive complaints.
Procedures.
Blinded chart review.
Clinical visit summaries from each subject's first UCSF evaluation were reviewed by 1 of 2 neurologists (S.J.S., L.T.T.) who were blinded to mutation carrier status. The following data were recorded: age at symptom onset, time (years) from first symptom to first UCSF visit, first symptom (behavioral, cognitive, or motor), first neuropsychiatric symptom, and results of neurologic examination and neuropsychological testing, including Neuropsychiatric Inventory (NPI)18 scores. Each diagnostic criterion for bvFTD (FTDC), ALS (El Escorial), and combined FTD-MND was ascertained for each patient. Subsequent visit summaries were reviewed for neurologic examination data and diagnostic status, and mortality records were obtained from the UCSF research database.
Determination of family history.
To quantify the number of patients with a positive family history of neurodegenerative disease, psychiatric disorders, or developmental problems, a separate blinded review of family history was performed by a genetic counselor (J.C.F.) of all unrelated patients (n = 99). A family history was considered positive if review of 3-generation pedigrees revealed at least 1 first-degree or second-degree relative with disease (appendix e-1, table 1).
Table 1.
Demographic profile, motor signs, and family history across groupsa

Abbreviations: ALS = amyotrophic lateral sclerosis; bvFTD = behavioral variant of frontotemporal dementia; C9+ = C9ORF72 mutation carriers; FHx = family history; FTD-MND = frontotemporal dementia–motor neuron disease.
Demographic information, family history, and motor signs at first evaluation. Years displayed as least squares means ± standard error (range).
p ≤ 0.01.
Neuropsychological data.
Neuropsychological performance was recorded from the first clinic visit and included standard bedside tests described previously.19 Three more severe noncarriers were removed to best match the carrier groups for disease severity with CDR-SB (p = 0.9 bvFTD and p = 0.8 FTD-MND).
Genotyping.
The presence of expanded GGGGCC hexanucleotide repeats in C9ORF72 was detected using a 2-step protocol. First, in all samples, the hexanucleotide repeat was PCR amplified using 1 fluorescently labeled primer followed by fragment length analysis on an automated ABI3730 DNA analyzer as previously described.13 All patients who appeared homozygous in this assay were further analyzed using a repeat primed PCR method.13 A characteristic stutter amplification pattern on the electropherogram was considered evidence of a pathogenic repeat expansion.
Imaging.
Image acquisition.
Patients underwent structural MRI with previously described sequences on a 3 T scanner at the Neuroscience Imaging Center at UCSF20 and 1.5 T21 and 4 T scanners at the San Francisco Veterans Affairs Medical Center.22 MRI scans were acquired within 1 year of the first UCSF visit.
Image analysis.
Voxel-based morphometry (VBM) analyses of combined gray–white maps preprocessed using standard DARTEL parameters in SPM5 were performed on structural MRIs as described previously.23 Comparisons were analyzed for the following: bvFTD(C9+) vs controls; bvFTD(noncarriers) vs controls; interaction of (C9+) × (bvFTD) vs bvFTD(noncarriers) and controls (controlling for main bvFTD effect); FTD-MND(C9+) vs controls; FTD-MND(noncarriers) vs controls; interaction of (C9+) × (FTD-MND) vs FTD-MND(noncarriers) and controls (controlling for main FTD-MND effect). Controls were selected to best approximate the distribution of scan types represented by all patient cohorts (appendix e-1).
For VBM analysis, a generalized linear model (glm) was fit at each voxel to model the dependence of tissue volume on diagnosis and C9ORF72 genotype for both gray and white matter. Nuisance variables included age at time of scan, total intracranial volume, gender, years of education, and field strength (1.5 T, 3 T, or 4 T), as the distribution of each clinical cohort differed slightly from the normal control group. The T-threshold for familywise error (FWE) correction at pFWE≤0.05 level was established via 1,000 permutations of the error in each analytic model to identify the T-value at p ≤ 0.05 on the error distribution24 (appendix e-1).
Statistical analysis.
Group differences were compared using glm, χ2, or Fisher exact test depending on data type. Kaplan-Meier curves were used with log rank χ2 tests to compare hazard rates of disease survival between groups. To correct for multiple comparisons, statistical significance level was set using the Benjamini and Yekutieli method25,26 to establish a FWE threshold of significance at p < 0.01 for 48 comparisons, and trends reported at p < 0.05.
RESULTS
Demographics.
Of the 648 symptomatic individuals tested, 31 patients were positive for the pathologic repeat expansion within C9ORF72 (C9+). The most common diagnosis among C9+ carriers was bvFTD (48.4%), followed by FTD-MND (35.5%) and ALS without dementia (16.1%, table e-1). Of the bvFTD patients, 23.8% were C9+ while 36.7% of FTD-MND patients and 45.5% of ALS patients were positive for the mutation. No C9+ patients met diagnostic criteria for PPA (any variant), CBS, PSP syndrome, or AD dementia.
A lower proportion (45%) of C9+ FTD-MND patients met FTDC than FTD-MND noncarriers (89%) (p = 0.01) due to later FTD symptom onset in C9+ FTD-MND. Excluding the criterion of initial symptom onset within 3 years increased the number of C9+ meeting FTDC to n = 9 (82%, p = 0.29). Three (27%) C9+ FTD-MND patients met both FTDC criteria and ALS criteria while 6 (55%) met ALS only and 2 (18%) met FTDC only. Eleven (58%) FTD-MND noncarriers met both criteria while 2 (10.5%) met ALS only and 6 (31.5%) met FTDC only.
Although earliest onset age was similar between carriers and noncarriers, no C9+ patient had symptom onset after age 65. Across syndromes, other demographics were similar by genotype with the exception of education (table 1). Review of the family histories of unrelated individuals (table 1 and appendix e-1) revealed that both C9+ (65%) and noncarriers (53%) had family histories of neurodegenerative disease. Because few patients with C9+ pure ALS were identified, subsequent analyses focused on bvFTD and FTD-MND only.
Clinical characteristics at first UCSF evaluation were further compared in the bvFTD and FTD-MND groups separately. Patients with C9+ bvFTD had a trend (p = 0.03) toward fewer years of education, but were otherwise similar to noncarriers. Patients with C9+ FTD-MND displayed longer time to first examination and were younger at symptom onset (table 1).
First symptoms.
First behavioral symptoms were similar by genotype in patients with bvFTD. Delusions were the only symptom more frequently reported at presentation in C9+ bvFTD (p = 0.001, table 2). There was a trend (p = 0.02) toward greater disinhibition in patients with C9+ FTD-MND than in noncarriers. First symptom type (behavioral, cognitive, or motor) did not differ by genotype for the bvFTD or FTD-MND diagnoses.
Table 2.
First symptom, first neuropsychiatric symptom, and NPI scores
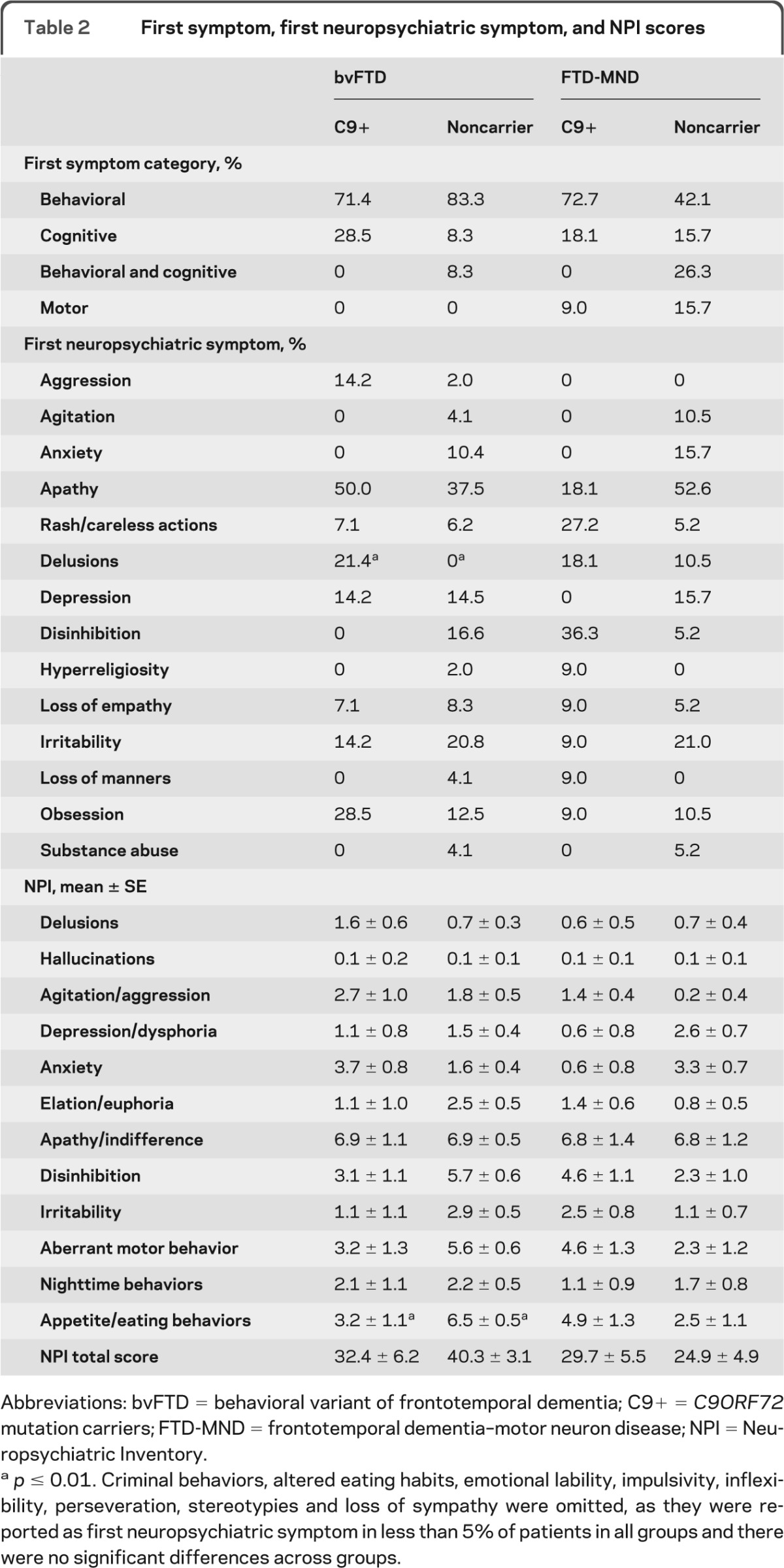
Abbreviations: bvFTD = behavioral variant of frontotemporal dementia; C9+ = C9ORF72 mutation carriers; FTD-MND = frontotemporal dementia–motor neuron disease; NPI = Neuropsychiatric Inventory.
p ≤ 0.01. Criminal behaviors, altered eating habits, emotional lability, impulsivity, inflexibility, perseveration, stereotypies and loss of sympathy were omitted, as they were reported as first neuropsychiatric symptom in less than 5% of patients in all groups and there were no significant differences across groups.
Motor findings.
There was no difference in bulbar ALS frequency in the carrier and noncarrier FTD-MND groups. Likewise, there were no differences in the frequency of parkinsonism, repetitive motor behaviors, tremor, ataxia, or dystonia on examination at first clinic visit when comparing all patients by genotype regardless of clinical syndrome (table 1).
Neuropsychological testing.
There were subtle differences between carriers and noncarriers on neuropsychological evaluations. The C9+ bvFTD group had more impaired working memory (p = 0.012), but was otherwise similar to noncarriers (table e-2). The C9+ FTD-MND group endorsed fewer depressive symptoms than noncarriers on the GDS (p = 0.004) but showed no other differences in neuropsychological performance (table e-2).
Neuropsychiatric findings.
Total NPI scores did not differ by genotype in either group. However, patients with C9+ bvFTD had milder abnormal eating behaviors (p = 0.01) and trends toward higher anxiety (p = 0.03) than bvFTD noncarriers. In contrast, NPI subscores did not differ in patients with FTD-MND by genotype with the exception of trends for lower anxiety (p = 0.02) in carriers (table 2).
Disease course.
To assess whether C9 carrier status affected survival in FTD, we compared time to death in all patients together as well as in the bvFTD (7/15 of C9+ and 12/49 noncarriers deceased at time of analysis) and FTD-MND subgroups (7/11 C9+ and 16/19 noncarriers deceased at time of analysis). Overall, there were no survival differences by genotype, but the C9+ FTD-MND group had a trend toward longer survival (11.7 years) than FTD-MND noncarriers (5.5 years, p = 0.02; figure 2). There were no survival differences in the bvFTD groups based on genotype.
Figure 2. Survival curves for C9+/noncarriers behavioral variant frontotemporal dementia (bvFTD) and frontotemporal dementia–motor neuron disease (FTD-MND).

Survival (means ± standard error) for C9+ bvFTD = 15.1 ± 2.8 years and bvFTD noncarriers = 10.6 ± 0.4 years (p = 0.38). Survival for C9+ FTD-MND = 11.7 ± 2.2 years and FTD-MND noncarriers = 5.5 ± 0.7 years (p = 0.02).
Patterns of C9ORF72-related brain atrophy.
In comparison to healthy controls, both bvFTD groups showed typical atrophy patterns, with decreased gray and white matter volumes in medial, ventral, and dorsal frontal, anterior insular, and anterior temporal lobes (figure 3A). When these atrophy patterns were compared by visual inspection of lesion overlap, C9+ bvFTD showed more parietal and bilateral thalamic (left > right) atrophy than bvFTD noncarriers. By contrast, bvFTD noncarriers demonstrated more medial frontal atrophy (figure 3A). An interaction was found such that patients with C9+ bvFTD showed greater thalamic (left > right) and left posterior insula atrophy than bvFTD noncarriers and controls (both pFWE< 0.05; figure 3C, table e-3).
Figure 3. C9ORF72 differential brain atrophy patterns.

(A, B) Overlay of C9+ and noncarrier atrophy patterns for behavioral variant frontotemporal dementia (bvFTD) and frontotemporal dementia–motor neuron disease (FTD-MND). Atrophy patterns compared for C9ORF72 variant carriers (C9+, in red) and noncarriers (in green) for 2 clinical syndromes, bvFTD (A) and FTD-MND (B). Yellow regions represent overlapping atrophy patterns. Results are shown for T scores thresholded at pFWE ≤ 0.05. Brain sections are indicated on rightmost image. The left side of the image corresponds to left side of the brain for axial sections. (C, D) Interaction effects of the C9ORF72 expansion repeat with bvFTD or FTD-MND diagnoses. bvFTD (C) and FTD-MND (D) C9 carriers (C9+) show differential atrophy as compared to noncarriers of the same diagnostic group and normal controls. Color bars indicate the range of T scores for respective analyses thresholded at pFWE ≤ 0.05. In both sections, the left side of the image corresponds to left side of the brain, and slice Montreal Neurological Institute coordinates are provided below. Results for A–D shown using MRIcron (version 12/2009, http://www.cabiatl.com/mricro/mricron/index.html), overlaid on a high-resolution 1.5 T template image from a single normal subject (MRIcron: ch2.nii.gz).
Both C9+ and noncarrier FTD-MND patients displayed decreased gray and white matter in medial and ventral frontal, anterior insular, and anterior medial temporal lobe regions compared to healthy controls (figure 3B). In comparison with lesion overlap, patients with C9+FTD-MND displayed greater dorsal frontal and posterior (right > left) cortical atrophy, as well as atrophy of the cerebellum (pFWE < 0.05). In contrast, FTD-MND noncarriers showed more ventral and temporal pole involvement than C9+ FTD-MND (pFWE < 0.05, figure 3B). An interaction was found wherein C9+FTD-MND carriers demonstrated more atrophy in the right thalamus than FTD-MND noncarriers and controls (pFWE < 0.05; figure 3D, table e-3). Notably, thalamic atrophy was not evident on structural MRI of individual patients by visual inspection.
DISCUSSION
The major finding of the present work is that patients with bvFTD and FTD-MND show similar clinical and imaging features regardless of genotype. We identified 2 notable exceptions to this general rule. First, we detected a trend toward longer survival in C9+ FTD-MND as compared to FTD-MND noncarriers. This survival difference was not apparent in bvFTD. Second, C9ORF72 mutation carriers displayed more thalamic, posterior insula, and possibly cerebellar atrophy at first assessment than noncarriers. Together, these findings suggest subtle differences in the regional degeneration pattern in C9FTD/ALS that may inform the pathologic mechanisms underlying C9ORF72-related neurodegeneration.
The longer disease course in C9+ FTD-MND reflected a longer time from symptom onset to clinical evaluation and a longer time to develop bvFTD symptoms (patients with C9+ FTD-MND took longer to meet consensus bvFTD criteria). Additionally, patients with C9+ FTD-MND more often presented with an early age at onset (<65 years). All C9+ patients developed symptoms before age 65, whereas 8% of the noncarrier cohort developed symptoms after 65. These findings suggest that C9ORF72 hexanucleotide expansion may bias toward earlier development of bvFTD symptoms.
Delusions as a presenting neuropsychiatric manifestation were more common in patients with C9+ bvFTD. Although previous studies have not associated psychotic features with a specific brain region in FTD,27 psychosis in bvFTD has been associated with FTLD-MND pathology (FTLD-TDP type B).28,29 Because carriers may be more likely to have FTD with underlying FTLD-TDP type B pathology than noncarriers, it is possible that this association may be mediated by FTLD-TDP type B pathology rather than a mutation effect. Supporting this hypothesis is the similar frequency of psychosis in our patients with FTD-MND, regardless of genotype.
The differences in brain atrophy between bvFTD carriers and noncarriers were most pronounced in the thalamus and posterior insula. Thalamic atrophy was unexpected, despite prior reports of thalamic degeneration in FTD.30 Consistent with our results, a recent study found mild to moderate p62 and TDP-43 staining in the thalamus of 4 C9+ carriers with MND.31 Thalamic atrophy has been described in FTLD-TDP type A,5,32 but not in type B, which is typically associated with FTD-MND. Although most FTD-MND cases show FTLD-TDP type B histology, an unexpected feature of C9ORF72 pathology is the frequency of FTLD-TDP type A.33,34 Greater thalamic atrophy demonstrated here could reflect an increased frequency of FTLD-TDP type A in the C9+ group. This finding may prove useful for understanding mechanisms of C9ORF72-related neurodegeneration.
The trend toward greater cerebellar atrophy in C9+ FTD-MND requires further study in a larger sample, although it converges with a previous imaging analysis from our group7 and the pathologic literature, which has described ubiquitin and p62-positive, TDP-43-negative neuronal cytoplasmic inclusions within cerebellar granule cells in C9+ FTD-MND.7,35 Although ataxia has been reported in a patient with chromosome 9p-linked FTD-MND10 and was observed in 1 C9+ patient in this cohort, we found no differences in ataxia by genotype. Cerebellar efferent pathways are connected to prefrontal cortex via the thalamus, and these subcortical structures likely contribute to executive control.35 We hypothesize that cerebellar dysfunction could contribute to the network dysfunction underlying the C9+ neuropsychiatric profile that includes greater disinhibition and working memory impairment.
In this specialized dementia clinic cohort, we found only 3 phenotypes associated with C9ORF72 expansions: bvFTD, FTD-MND, and ALS. It is noteworthy that there were no C9+ patients with svPPA (left or right temporal variants) or nfvPPA, differing from a recent study that described a C9+ patient with nfvPPA-MND phenotype.36 Other clinical presentations that have been associated with C9ORF72 expansions, such as AD dementia phenotype and CBS,7–10,34,37 were not observed in this cohort, suggesting they are rare presentations of the mutation; this is inconsistent with a recent report suggesting that the mutation leads to an amnestic phenotype.37 However, 1 C9+ bvFTD patient in our cohort had a positive amyloid PET scan at age 64, possibly indicative of concurrent AD pathology. Another group has also reported34 a 61-year-old patient with comorbid FTLD-MND and AD among a cohort of 20 C9+ patients, suggesting that comorbid AD pathology may be found in patients with C9ORF72 mutations which could account for amnestic symptoms.
Limitations of this study include small sample size, particularly when C9+ subgroups were divided by syndrome and matched for severity. Due to the rigorous thresholds set for statistical significance, nonsignificant trends found here may reflect true underlying differences between genotype groups and suggest further investigations with larger cohorts. Earlier reports of kindreds of chromosome 9p-linked FTD-MND families suggested that there could be motor, cognitive, or psychiatric distinguishing characteristics of the C9ORF72 mutation.8,10 The lack of these distinctive features seen in our C9+ cohort may partly be due to group analyses of a heterogeneous phenotype or may represent selection bias in our cohort.
The present findings suggest that C9ORF72 mutation carriers are likely to receive a clinical diagnosis of FTD, ALS, or FTD-MND. Future studies focused on neuropathology of the cerebellum and thalamus may elucidate the clinical implications of the present findings and identify novel biologic substrates involved in C9ORF72 dysregulation. Given the substantial contribution of this locus to genetic risk for these syndromes, testing for C9ORF72 expansions may be indicated even without a family history, especially when one of these diagnoses is associated with slow progression or atypical posterior cortical and thalamic atrophy on MRI.
Supplementary Material
GLOSSARY
- AD
Alzheimer disease
- ALS
amyotrophic lateral sclerosis
- bvFTD
behavioral variant frontotemporal dementia
- CBS
corticobasal syndrome
- CDR-SB
Clinical Dementia Rating Scale sum of boxes
- FTD
frontotemporal dementia
- FWE
familywise error
- glm
generalized linear model
- lvPPA
logopenic variant primary progressive aphasia
- MND
motor neuron disease
- nfvPPA
nonfluent variant primary progressive aphasia
- NPI
Neuropsychiatric Inventory
- PSP
progressive supranuclear palsy
- svPPA
semantic variant primary progressive aphasia
- UCSF
University of California, San Francisco
- VBM
voxel-based morphometry
Footnotes
AUTHOR CONTRIBUTIONS
Dr. Sha: design and conceptualization of the study, analysis and interpretation of the data, drafting and revising the manuscript. Dr. Takada: design and conceptualization of the study, analysis and interpretation of the data, drafting and revising the manuscript. Dr. Rankin: design and conceptualization of the study, analysis and interpretation of the data, drafting and revising the manuscript. Dr. Yokoyama: design and conceptualization of the study, analysis and interpretation of the data, drafting and revising the manuscript. N.J. Rutherford: analysis of the data. J.C. Fong: design and conceptualization of the study, analysis and interpretation of the data, revising the manuscript. B. Khan: analysis of the data, revising the manuscript. A. Karydas: design and conceptualization of the study, analysis of the data. M.C. Baker: analysis of the data. M. DeJesus-Hernandez: analysis of the data, revising the manuscript. Dr. Pribadi: analysis of the data, revising the manuscript. Dr. Coppola: analysis and interpretation of the data, revising the manuscript. Dr. Geschwind: analysis of the data and revising the manuscript. Dr. Rademakers: analysis and interpretation of the data, revising the manuscript. Dr. Lee: design of the study, revising the manuscript. Dr. Seeley: interpretation of the data, revising the manuscript. Dr. Miller: design and conceptualization of the study, analysis and interpretation of the data, drafting and revising the manuscript. Dr. Boxer: design and conceptualization of the study, analysis and interpretation of the data, drafting and revising the manuscript.
DISCLOSURE
Dr. Sha and Dr. Takada report no disclosures. Dr. Rankin is funded by NIH/NIA 1R01AG029577–01 and Hillblom 2007/2I. Dr. Yokoyama, N.J. Rutherford, J.C. Fong, B. Khan, A. Karydas, and M.C. Baker report no disclosures. M. DeJesus-Hernandez has a patent pending on Expanded non-coding repeat in C9ORF72 cause frontotemporal dementia and amyotrophic lateral sclerosis. Dr. Pribadi reports no disclosures. Dr. Coppola is funded by NIH/NIA grant: RC1 AG035610. Dr. Geschwind has received institutional support from Alzheimer's Disease Research Center of California (ARCC) grant 03–7527 and is funded by NIH grant R01 AG026938. Dr. Rademakers is funded by NIH grants R01NS065782, R01AG026251, ALS Association, and has a patent pending on Expanded non-coding repeat in C9ORF72 cause frontotemporal dementia and amyotrophic lateral sclerosis. Dr. Lee is funded by K23AG039414–01A1 and the Tau Consortium. Dr. Seeley is funded by NIH grants P50 AG1657303, the John Douglas French Alzheimer's Disease Foundation, Consortium for Frontotemporal Dementia Research, James S. McDonnell Foundation, Larry Hillblom Foundation, has received support for travel by the Alzheimer's Association, and received payment for lectures by the Alzheimer's Association, American Academy of Neurology, and Novartis Korea. Dr. Miller serves as board member on the John Douglas French Alzheimer's Foundation and Larry L. Hillblom Foundation, serves as a consultant for TauRx, Ltd., Allon Therapeutics, the Tau Consortium, and the Consortium for Frontotemporal research, has received institutional support from Novartis, and is funded by NIH grants P50AG023501, P01AG019724, P50 AG1657303, and the state of CA. Dr. Boxer has been a consultant for Bristol Myers Squibb, Genentech, Plexikkon, Phloronol, Registrat-Mapi, Accera, Envivo, TauRx, and Novartis, receives research support from Allon Therapeutics, Bristol Myers Squibb, Janssen, Forest, Pfizer, Medivation, and Genentech, and is funded by NIH grants R01AG038791, R01AG031278, the John Douglas French Foundation, Alzheimer's Drug Discovery Foundation, the Association for Frontotemporal Degeneration, the Silicon Valley Foundation, the Agouron Institute, the Tau Research Consortium, and the Hellman Family Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Knopman DS, Petersen RC, Edland SD, Cha RH, Rocca WA. The incidence of frontotemporal lobar degeneration in Rochester, Minnesota, 1990 through 1994. Neurology 2004;62:506–508 [DOI] [PubMed] [Google Scholar]
- 2. Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology 2002;58:1615–1621 [DOI] [PubMed] [Google Scholar]
- 3. Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 2005;65:586–590 [DOI] [PubMed] [Google Scholar]
- 4. Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology 2003;60:1094–1097 [DOI] [PubMed] [Google Scholar]
- 5. Rohrer JD, Geser F, Zhou J, et al. TDP-43 subtypes are associated with distinct atrophy patterns in frontotemporal dementia. Neurology 2010;75:2204–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet 2011;377:942–955 [DOI] [PubMed] [Google Scholar]
- 7. Boxer AL, Mackenzie IR, Boeve BF, et al. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry 2011;82:196–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Le Ber I, Camuzat A, Berger E, et al. Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology 2009;72:1669–1676 [DOI] [PubMed] [Google Scholar]
- 9. Luty AA, Kwok JB, Thompson EM, et al. Pedigree with frontotemporal lobar degeneration–motor neuron disease and tar DNA binding protein-43 positive neuropathology: Genetic linkage to chromosome 9. BMC Neurol 2008; 8:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pearson JP, Williams NM, Majounie E, et al. Familial frontotemporal dementia with amyotrophic lateral sclerosis and a shared haplotype on chromosome 9p. J Neurol 2011;258:647–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208 [DOI] [PubMed] [Google Scholar]
- 12. Morita M, Al-Chalabi A, Andersen PM, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 2006;66:839–844 [DOI] [PubMed] [Google Scholar]
- 13. Dejesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299 [DOI] [PubMed] [Google Scholar]
- 17. O'Bryant SE, Lacritz LH, Hall J, et al. Validation of the new interpretive guidelines for the Clinical Dementia Rating scale sum of boxes score in the National Alzheimer's Coordinating Center database. Arch Neurol 2010;67:746–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The neuropsychiatric inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994;44:2308–2314 [DOI] [PubMed] [Google Scholar]
- 19. Rankin KP, Kramer JH, Miller BL. Patterns of cognitive and emotional empathy in frontotemporal lobar degeneration. Cogn Behav Neurol 2005;18:28–36 [DOI] [PubMed] [Google Scholar]
- 20. Bettcher BM, Wilheim R, Rigby T, et al. C-reactive protein is related to memory and medial temporal brain volume in older adults. Brain Behav Immun 2012;26:103–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosen HJ, Gorno-Tempini ML, Goldman WP, et al. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 2002;58:198–208 [DOI] [PubMed] [Google Scholar]
- 22. Zhang Y, Schuff N, Ching C, et al. Joint assessment of structural, perfusion, and diffusion MRI in Alzheimer's disease and frontotemporal dementia. Int J Alzheimers Dis 2011;2011:546871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilson SM, Brambati SM, Henry RG, et al. The neural basis of surface dyslexia in semantic dementia. Brain 2009;132:71–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abdulkadir A, Mortamet B, Vemuri P, et al. Effects of hardware heterogeneity on the performance of SVM Alzheimer's disease classifier. Neuroimage 2011;58:785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Statistics 2001;29:1165–1188 [Google Scholar]
- 26. Narum SR. Beyond Bonferroni: less conservative analyses for conservation genetics. Conserv Genet 2006;7:783–787 [Google Scholar]
- 27. Jellinger KA. Cerebral correlates of psychotic syndromes in neurodegenerative diseases. J Cell Mol Med 2012;16:995–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Velakoulis D, Walterfang M, Mocellin R, Pantelis C, McLean C. Frontotemporal dementia presenting as schizophrenia-like psychosis in young people: clinicopathological series and review of cases. Br J Psychiatry 2009;194:298–305 [DOI] [PubMed] [Google Scholar]
- 29. Hodges JR, Davies RR, Xuereb JH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol 2004;56:399–406 [DOI] [PubMed] [Google Scholar]
- 30. Mann DM, South PW. The topographic distribution of brain atrophy in frontal lobe dementia. Acta Neuropathol 1993;85:334–340 [DOI] [PubMed] [Google Scholar]
- 31. Troakes C, Maekawa S, Wijesekera L, et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology Epub 2011. December 19 [DOI] [PubMed] [Google Scholar]
- 32. Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011;122:111–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Snowden JS, Rollinson S, Thompson JC, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 2012;135:693–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murray ME, Dejesus-Hernandez M, Rutherford NJ, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol 2011;122:673–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Al-Sarraj S, King A, Troakes C, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS Acta Neuropathol 2011;122:691–702 [DOI] [PubMed] [Google Scholar]
- 36. Gijselinck I, Van Langenhove T, van der Zee J, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 2011;11:54–65 [DOI] [PubMed] [Google Scholar]
- 37. Majounie E, Abramzon Y, Renton AE, et al. Repeat expansion in C9ORF72 in Alzheimer's disease. N Engl J Med 2012;366:283–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.