

Abstract
Multiple sclerosis (MS) is a disease which can present in different clinical courses. The most common form of MS is the relapsing-remitting (RR) course, which in many cases evolves into secondary progressive (SP) disease. Autoimmune models such as experimental autoimmune encephalomyelitis (EAE) have been developed to represent the various clinical forms of MS. These models along with clinico-pathological evidence obtained from MS patients have allowed us to propose ‘1-stage’ and ‘2-stage’ disease theories to explain the transition in the clinical course of MS from RR to SP. Relapses in MS are associated with pro-inflammatory T helper (Th) 1/Th17 immune responses, while remissions are associated with anti-inflammatory Th2/regulatory T (Treg) immune responses. Based on the ‘1-stage disease’ theory, the transition from RR to SP disease occurs when the inflammatory immune response overwhelms the anti-inflammatory immune response. The ‘2-stage disease’ theory proposes that the transition from RR to SP-MS occurs when the Th2 response or some other responses overwhelm the inflammatory response resulting in the sustained production of anti-myelin antibodies, which cause continuing demyelination, neurodegeneration, and axonal loss. The Theiler’s virus model is also a 2-stage disease, where axonal degeneration precedes demyelination during the first stage, followed by inflammatory demyelination during the second stage.
1. Introduction
1.1 Homogeneity vs. heterogeneity in MS: Clinical and Neuroimaging studies
Multiple sclerosis (MS) is a chronic demyelinating disease of the human central nervous system (CNS), which mainly affects young adults. The proposed pathogenesis of MS has two main components: inflammatory demyelination and axonal degeneration (Table 1) [1;2]. The demyelination of nerve fibers causes significant neurological impairment including sensory and motor disturbances, vision loss and paralysis. Inflammation is often associated with demyelination. Axonal damage and neuronal loss (neurodegeneration) have also been reported in MS, which results in brain atrophy and permanent neurological damage, particularly cognitive decline [1;3;4] In general, it is hypothesized that inflammatory demyelination causes secondary axonal degeneration [5]. However, there is evidence of axonal degeneration in the absence of demyelination, suggesting that axonal degeneration can precede demyelination [6]. It is also possible that inflammatory demyelination and neurodegeneration run parallel and concurrently.
Table 1.
Two neuropathologies in MS
| Pathology | Cell component | Reference |
|---|---|---|
| Inflammatory demyelination | T cell infiltration Myelin loss, oligodendrocyte apoptosis |
[15–19] |
| Neurodegeneration | Axonal damage Loss of neurons |
[2;20;21] |
MS: multiple sclerosis
The clinical course of MS may present in one of four forms. In the relapsing-remitting (RR) form, relapses and remissions occur with either a full recovery or a partial recovery with residual deficits. In progressive-relapsing (PR) MS, the disease progressively worsens, with intermittent attacks and relapses which may lead to partial or full recovery from the attacks. Secondary progressive (SP) disease occurs when the initial RR course is followed by progressive disease, and the primary progressive (PP) course is characterized by the steady and progressive worsening of neurological condition without any prominent relapses from the onset. [7]. Generally, MS begins as the RR disease which in ~85% of patients eventually evolve into SP-MS within ~10 years of the onset [8]. The onset and clinical course of MS are for the most part unpredictable, although there are some trends [7]. Black Americans represent a group who are less likely to develop MS, but are more likely to exhibit the progressive disease course of MS, and suffer from more rapid neurodegeneration [40]. Caucasian Americans and Europeans are more likely to develop the RR form of disease that will eventually turn progressive [9]. In both Caucasian Americans and Europeans, there is an increased MS incidence which is associated with decreased ultraviolet (UV) exposure [9;10]. Perhaps the most significant factor that contributes to MS incidence and severity is gender: women are 2 to 3 times more likely to develop MS than men [11;12]. Although it is unknown what triggers MS to transform into the progressive disease, neuroimaging studies suggest that MS could be a 2-stage disease, in which inflammatory demyelination is followed by neurodegeneration [13;14].
Inflammation has been linked with RR-MS since active lesions can be seen by gadolinium enhancement during RR-MS [8]. However, gadolinium-enhanced lesions are less prevalent during progressive disease than in RR-MS. Therefore, the progressive form of MS has been speculated to begin when the brain can no longer compensate for neurodegeneration (axonal loss) [3].
Neurodegeneration has been associated with the progressive forms of MS. In the progressive forms of MS, atrophy of gray matter accelerates compared with the RR disease and cannot be blocked by the use of anti-inflammatory drugs [22;23]. Additionally, the regions of the brain where the atrophy occurs differ between RR and the progressive forms of the disease [24]. While ventricular enlargement is predominant in RR-MS, patients with SP-MS develop atrophy in the cortex and deep gray matter. These findings have lead to the hypothesis that inflammatory demyelination in the white matter is a major effector mechanism during the RR stage, while neurodegeneration develops during the progressive stage. Therefore, based on neuroimaging studies, MS could embody two heterogenous pathological events, inflammation and neurodegeneration. However there is currently no etiopathologic mechanism to explain how and whether MS shifts from inflammatory demyelination to neurodegeneration.
1.2 Homogeneity vs. heterogeneity in MS: Pathology studies
Pathology studies on active demyelinating MS lesions suggest heterogeneity in several immunopathological parameters, including T cells, macrophages, immunoglobulin (Ig), and oligodendrocyte apoptosis, among patients (inter-individual heterogeneity) [25]. Lucchinetti et al. classified MS lesions into four patterns, all of which contained T cells and macrophages. Some differences were, however, recognized: pattern I was mediated by T cells and macrophages alone; pattern II was Ig and complement dependent, pattern III exhibited apoptosis of oligodendrocytes in the absence of Ig, complement, and remyelination, and pattern IV showed oligodendrocyte dystrophy with no evidence of remyelination. In these classifications, there appears to be neither an overlap in pattern, nor a change in pattern during the clinical course of individual patients (intra-individual homogeneity). Although the classification by Lucchinetti et al. has been widely used, Breij et al. reported that the pathology is homogenous among all MS patients from the various disease courses (inter-individual homogeneity) [26;27]. All active lesions had antibody, complement, and macrophages associated with them (similar to pattern II in the Lucchinetti scheme).
On the other hand, Barnett et al. observed two lesion types (patterns II and III) in a single patient, consistent with intra-individual heterogeneity, or stage-dependent pathology [26;28;29]. Here the finding may represent the transition from the first stage of disease (oligodendrocyte apoptosis) to the second stage (T cell-mediated inflammation). This conflicts with the reports by Breij and Lucchinetti who only reported single lesion types within individuals (intra-individual homogeneity). These seemingly conflicting reports can be resolved by considering these lesions to be on converging paths where most lesions would ultimately display homogenous characteristics if allowed to progress. Another explanation could be that the different lesion types reflect the heterogeneity of individual’s immune responses and possibly stage-dependent alterations in MS. To this date there is still no general consensus explaining different MS lesion types.
In summary, it remains controversial whether the pathomechanism in MS is homogenous or heterogenous and what triggers the transition from RR to SP disease. Most neuroimaging studies do support a conversion from inflammation to neurodegeneration. In contrast, most neuropathology studies support no stage change in individual MS patients.
We will now consider what experimental models of MS have taught us about the underlying immune etiopathology of MS (Table 2). We will propose 1) that MS can be caused by a single pathomechanism (1-stage disease theory) or 2) that MS pathomechanisms can change during the course of disease (2-stage disease theory).
Table 2.
Effector mechanisms of MS
| Demyelination | Neurodegeneration (axonal damage) |
|---|---|
| Myelin specific Th1 and Th17 cells | Neurotropic virus infection |
| Pro-inflammatory cytokines | Anti-axon immune responses (Th2 mediated; autoantibody) |
| Oligodendrocyte apoptosis | Ca2+ influx leads to axonal damage |
| Secondary demyelination following primary axonal degeneration | Secondary axonal degeneration following primary demyelination |
Th: T helper
2. Immunopathology of MS and EAE
2.1 Role of the immune system
MS is believed to be an immune-mediated disease since alterations of the immune system as well as autoreactive B and T cells have been found in MS patients [26;30–32]. Although the exact cause of aberrant immune activity in MS remains unknown, a microbial infection (viral or bacterial) can trigger 1) cross-reactivity between microorganism’s antigen(s) and CNS antigen(s), so-called ‘molecular mimicry’ or 2) ‘bystander activation’ of autoreactive T cells and 3) ‘epitope (determinant) spreading’ from microbial antigen(s) to CNS antigens leading to destruction of CNS tissues [16]. In addition, there are genetic and environmental factors that affect the immune system, which may contribute to changes in the prevalence and susceptibility to MS [33–36].
Regardless of the initial cause of MS immunopathogenesis, the chronic injury has been proposed to be mediated by direct or indirect immune-mediated attack against myelin antigen(s) and oligodendrocytes, the myelin forming cells. Evidence supporting direct immune attack largely stems from experimental autoimmune (or allergic) encephalomyelitis (EAE), the animal model used to study the autoimmune etiology in MS [37]. EAE is a CD4+ T cell-mediated disease in animals which shares numerous clinical and neuropathological features with MS and is the most common method of testing MS therapeutic approaches [38]. Induction of EAE is conducted by actively sensitizing animals with CNS homogenates, myelin proteins, such as myelin oligodendrocyte glycoprotein (MOG), myelin proteolipid protein (PLP), and myelin basic protein (MBP), or their peptides. In addition, passive transfer of myelin-specific T cells from sensitized animals to naïve animals can cause EAE, known as ‘adoptive’ or ‘passive EAE’ [39]. The pathogenic role of autoimmunity in MS has been supported by the findings that both autoantibodies and autoreactive T cells to myelin protein(s) have been found in MS patients, although anti-myelin T cells can also be found in normal individuals [8;40]. MS patients have also been reported to differ from normal individuals in the numbers and specificities of naïve, memory, and activated T cells in the peripheral circulation, however there are no consistent trends and none of these variations have been successfully applied as markers for disease [15;18;41–43].
2.2 Classical EAE mediated by Th1 and Th17 cells: 1-stage disease theory
Immune responses are differentiated based upon the type of CD4+ T cells that are responsible for directing the response (Table 3). Pro-inflammatory responses are associated primarily with CD4+ T helper (Th) 1 and Th17 cells, as well as the cytokines tumor necrosis factor (TNF)-α, interleukin (IL)-17, and interferon (IFN)-γ. Anti-inflammatory responses are associated with CD4+ Th2, regulatory T (Treg), and natural killer T (NKT) cells as well as the cytokines IL-4, IL-10, and transforming growth factor (TGF)-β [44]. In MS, the disease activity has been associated with the presence of various immune effector cells and cytokines [19;45;46]. During relapses, there is an increase in Th1 and Th17 cells, and inflammatory cytokines; during remission, Th2, Treg, and NKT cells and anti-inflammatory cytokines are more active. Th1 immune responses play a major role in cellular immunity and are characterized by production of proinflammatory mediators such as IL-2, TNF-α, and IFN-γ; whereas Th2 immune responses help humoral immunity and suppress Th1 immune responses and are associated with the cytokines IL-4, IL-5, and IL-13.
Table 3.
Two cellular immune responses
| Immune response | Cell type | Cytokine | Reference |
|---|---|---|---|
| Pro-inflammatory | Th1, Th17 | IFN-γ, IL-17, TNF-α | [17;19;32;47] |
| Anti-inflammatory | Th2, Treg, NKT | IL-4, IL-10, TGF-β | [32;48–52] |
IFN: interferon; IL: interleukin; NKT: natural killer T; TGF: transforming growth factor; TNF: tumor necrosis factor; Treg: regulatory T
Since Th1 immune responses have been associated with disease activity in MS patients, MS has been proposed to be a Th1-mediated disease [53]. For example, CD4+ T-cell lines developed from the cerebrospinal fluid (CSF) of MS patients produce large amounts of IFN-γ and IL-2 [54]. In most EAE models, myelin-specific Th1 cells are associated with immunopathology and can adoptively transfer EAE to recipient animals [55]. Additionally, T-bet (a Th1 transcription factor) deficient C57BL/6 mice sensitized with MOG showed decreased severity of EAE compared with MOG-sensitized wild-type mice [56].
Monophasic EAE can be induced in C57BL/6 mice by sensitization with MOG peptide in complete Freund’s adjuvant (CFA) [57]. During the disease course in monophasic EAE, disability peaks at 3 weeks after sensitization and gradually subsides resulting in complete or incomplete recovery from disease. RR-EAE can be induced in SJL/J mice with PLP139-151, PLP178-191, and MOG92-106 sensitization and results in a RR disease course (Fig. 1a, c). In these monophasic and RR-EAE models, Th1 immune responses appear responsible for relapses, while remissions are induced by Th2 responses (Fig. 1a). IL-4-producing Th2 cells can enhance the expression of GATA-3 (a Th2 transcription factor), leading to the inhibition of Th1 cell differentiation and suppression of Th1 cell function [58]. The adoptive transfer of PLP-specific Th2 cell clones prevented EAE in mice sensitized with PLP in which resistance to EAE was correlated with IL-4 production [59]. Additionally, the enhancement of Th2 cells has been shown to delay the onset and decrease the severity of PLP-induced EAE [60].
Fig. 1.

The 1-stage (a, b) and 2-stage (c, d) disease theories of MS. (a) Classical EAE and RR-EAE are 1-stage diseases where the relapses and remissions of disease courses are due to the balance between inflammatory (Th1 and Th17 cells) versus regulatory (Th2, Treg, and NKT cells) cells. In this scheme, Th1/Th17 cells contribute to relapse and Th2, Treg, and NKT cells support remission. During the SP phase of the 1-stage disease theory, immune responses are skewed toward inflammatory responses. Here, the pathomechanisms of SP-MS remain the same during the disease course (intra-individual homogeneity); excessive anti-myelin inflammatory responses result in secondary axonal degeneration. (b) PP-EAE can be a 1-stage disease, which is mediated by anti-myelin antibody with the participation of Th2 cells. (c) Ataxic SP-EAE could be a 2-stage disease, where the central effector mechanisms change and the RR disease course shifts to the SP course. Here, a new effector mechanism, such as increased autoantibody production, is promoted by Th2 cells, leading to disease progression (intra-individual heterogeneity). (d) TMEV-induced demyelination is also a 2-stage disease. During the first stage, axonal damage, oligodendrocyte apoptosis, and macrophage activation leads to demyelination. During the second stage, epitope spreading results in generation of anti-myelin immune responses, contributing to disease progression.
EAE: experimental autoimmune encephalomyelitis; PP: primary progressive; RR: relapsing-remitting; SP: secondary progressive; TMEV: Theiler’s murine encephalomyelitis virus
Th17 cells which secrete IL-17 are involved in promoting phagocytosis and are considered to be inflammatory [61]. In MS patients, IL-17 has been reported to be expressed at relatively high levels in peripheral leukocytes and CSF mononuclear cells, particularly during relapses [17;62]. Myelin-specific Th17 cells were capable of inducing EAE and can be found in CNS lesions [63]. Treatment of EAE mice with anti-IL-17A antibody reduced the severity and recovery time from peak disease [47]. Th17 cells can be associated with disease, but neutralization of Th17 effectors did not completely abrogate disease; therefore Th17 cells likely contribute to disease activity, but are not the sole cause of disease.
NKT cell stimulation results in a rapid secretion of cytokines, such as IL-4 and IFN-γ that can regulate immune responses [64]. This subset of immune cells expresses natural killer cell markers and an invariant T-cell receptor (TCR, Vα14 in mice and Vα24 in humans) that binds to CD1d molecules on antigen presenting cells that present glycolipid antigens, such as α-galactosylceramide (α-GC) [64]. In MS and EAE, several studies have suggested a beneficial role for NKT cells in pathogenesis [49;65;66]. Mice given α-GC, to activate NKT cells, were protected against EAE in an experiment by Singh et al [51]. In this study, α-GC was unable to protect against EAE in CD1d, IL-4, or IL-10 knockout (KO) mice. This indicates that α-GC presentation on CD1d molecules and secretion of IL-4 and IL-10 were necessary for the suppression of EAE by NKT cells [51]. In this model, the Th1 immune response was suppressed by activating NKT cells with α-GC, which shifted the immune system to a Th2 response. Additionally, IL-4 production could be induced by an α-GC analog, OCH, from NKT cells and modulate EAE [66]. Together these studies suggest that NKT cells play a regulatory role in the disease course and contribute to the remission of MS and EAE.
CD4+CD25+FoxP3+ Treg cells (1–2% and 10% of CD4+ T cells in humans and mice, respectively) produce TGF-β, suppress inflammation, and inhibit the proliferation and cytokine production of effector T cells [67]. In MS, Treg cells have been demonstrated to have impaired function and be present in lower numbers compared with healthy controls [48]. Adoptive transfer of Treg cells isolated from naïve mice has been shown to ameliorate MOG-induced EAE [50]. Co-administration of Treg cells with MOG-specific T cells attenuated the development of EAE. While the exact mechanism of Treg suppression is unknown, IL-10 production by Treg cells may be critical for their immunosuppressive function, since Treg cells from IL-10 deficient mice fail to block the development of EAE [52;68;69].
We propose a “1-stage disease theory” in which a single immune etiopathology during the entire course of disease causes lesion homogeneity. In this theory relapses are precipitated by pro-inflammatory immune responses and remissions are caused by anti-inflammatory immune responses in most cases (Fig. 1a). Here, pro-inflammatory Th1 and Th17 type immune responses contribute to relapses and Th2 type immune responses along with NKT and Treg cells contribute to remissions. Later on, either irreversible neurodegeneration or an overwhelming inflammatory response causes the disease to shift into the progressive phase. The homogeneity of intra-individual and inter-individual pathology in the lesions of MS patient is consistent with the 1-stage theory [25;26].
2.3 2-stage disease theory: Ataxic SP-EAE
Although the 1-stage disease theory is consistent with finding in most MS and EAE models, such as RR-MS and RR-EAE, it cannot explain neuroimaging changes in SP-MS or stage-dependent pathology (intra-individual heterogeneity) in some cases of MS. Here, we propose a ‘2-stage disease’ theory for the transition in MS patients from the RR course to the SP course. Although several scenarios and sets of circumstances could provoke this transition, a change in the effector mechanism from Th1 to autoantibody may play a key role. In this scheme, during the RR course (the initial stage of disease), Th1 responses cause relapses of disease and the Th2 responses cause the remissions. The second stage of the disease or the progressive phase occurs when Th2 immune responses overwhelm Th1 immune responses causing a persistent demyelinating antibody response. Here, the role of Th2 immune responses changes depending on the time course. During the first stage, Th2 cytokines (such as IL-4) play a regulatory role, while in later phases they will drive autoantibody production and contribute to the progressive stage of disease. The contribution of antibody to disease progression is supported by the finding of B cell follicle-like structure, in the meninges in patients with SP-MS [70]. Antibody-mediated disease progression has also been suggested in PP- and SP-MS, in association with Epstein-Barr virus (EBV) infection [31].
The 2-stage disease theory is supported by MOG92-106-induced SP-EAE, experimentally. MOG92-106-immunized A.SW mice develop PP-EAE, however if MOG is supplemented with Bordetella pertussis (BP), the disease begins with a RR course that is followed by SP-EAE (see Table 4 for a summary of disease induction and resulting course) [72]. Clinically, while MOG-sensitized A.SW mice with PP-EAE display ataxia, BP-treated MOG-sensitized A.SW mice do not display ataxia until they reach the secondary progressive stage of disease. Unlike typical EAE lesions, the percentage of CD3+ T cells in CNS lesions of A.SW mice with progressive EAE was surprisingly small, suggesting that T cells do not play an effector role in pathogenesis (Fig. 2d). In MOG-sensitized A.SW mice, Ig deposition was detected on myelin and on the periphery of demyelinating lesions. In A.SW mice with PP-EAE, there was a direct correlation between the anti-MOG IgG2a/IgG1 ratio and survival time where higher ratios increased survival time. Since IgG2a and IgG1 isotypes are linked with Th1 and Th2 type immune responses, respectively, these data suggest that progressive EAE is a Th2-associated disease. The pathogenic role of Th2 cells in progressive EAE has been also studied by Lafaille et al., in which transfer of MBP-specific Th2 cells into RAG-1 KO mice was able to induce EAE [71]. Here, MOG-induced PP-EAE is a 1-stage disease (Fig. 1b), where anti-MOG antibodies play a pathogenic role from disease onset. However, MOG-induced SP-EAE is a 2-stage disease, where T cell-mediated RR disease is followed by antibody-mediated SP disease (Fig. 1c).
Table 4.
One myelin peptide, MOG92-106, induces different disease courses in different mouse strains and within these strains using different supplements
| Mouse strain | Peptide | Supplement | Disease | Ig in lesions | Reference |
|---|---|---|---|---|---|
| A.SW | MOG92-106 | – | PP-EAE | + | [72] |
| A.SW | MOG92-106 | CpG DNA | SP-EAE or no disease | ± | [72] |
| A.SW | MOG92-106 | BP | SP-EAE | + | [72] |
| SJL/J | MOG92-106 | – | RR-EAE | − | [72] |
| SJL/J | MOG92-106 | UV | SP-EAE, RR- | + (SP-EAE) | [9] |
| SJL/J | MOG92-106 | Apoptosis | RR-EAE with 21% SP-EAE | + (SP-EAE) | [73] |
Apoptosis: apoptotic cell injection; BP: Bordetella pertussis; Ig: immunoglobulin; MOG: myelin oligodendrocyte protein; UV: ultraviolet irradiation
Fig. 2.
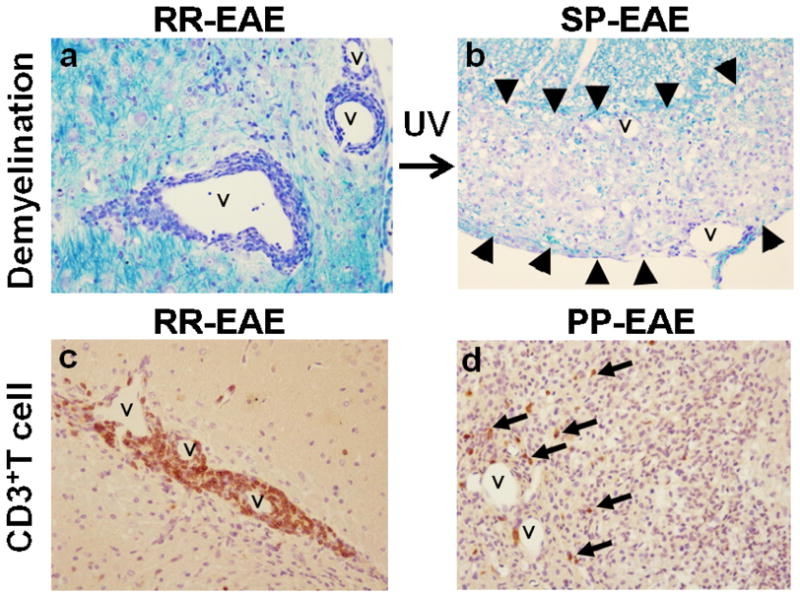
Stage-dependent pathology of EAE induced with MOG92-106. (a, c) In RR-EAE, intense CD3+ T cell infiltration was observed around vessels (v) with mild demyelination. (b, d) In progressive EAE, T cell infiltration was sparse around vessels (v), despite large areas of demyelination (arrowheads). UV irradiation can alter the disease course from RR-EAE to SP-EAE in SJL/J mice sensitized with MOG92-106. PP-EAE was induced in A.SW mice sensitized with MOG92-106. (a, b) Luxol fast blue stain. (c, d) CD3 immunohistochemistry. Magnification × 70.
On the other hand, SJL/J mice normally develop RR-EAE when injected with MOG92-106. However, if the mice were UV irradiated (UV irradiation favors anti-inflammatory Th2 immune responses) after MOG sensitization, some of the mice developed SP-EAE with pathology similar to that of A.SW mice with progressive EAE (Fig. 2a, b) [9;71].
In addition, if apoptotic cells, which alter immune response towards an anti-inflammatory response, were injected into SJL/J mice during RR-EAE, 21% of the mice developed SP-EAE [73]. Histologically, in SJL/J mice that develop RR-EAE after MOG sensitization, Ig deposition was only found in the meninges and endothelial cells. On the other hand, in SJL/J mice that went on to develop SP-EAE, intense Ig deposition could be observed in the white matter. Thus, it appears that Ig is a key contributor to the development of progressive disease (Box 1).
Box 1. Anti-MOG and anti-axon antibodies.
Antibodies have been increasingly recognized as pathological contributors to various forms of CNS diseases. In fact, anti-aquaporin-4 antibody can be used to differentiate neuromyelitis optica from MS [74]. In MS there is both clinical and experimental evidence for antibody-mediated pathology [25;72]. However, the prevalence and impact of autoantibodies in MS patients is extremely heterogeneous. MS lesions may or may not have Ig deposition and this is used in the Lucchinetti scheme to differentiate lesions [75]. For antibodies to mediate disease they must either pass the blood-brain barrier or be generated within the CNS, thus it is likely that antibodies only contribute to disease progression but do not initiate disease.
Antibodies against MOG have been extensively studied since they have been shown to mediate demyelination in EAE [76]. MOG is an oligodendrocyte specific protein found on the outer layer of myelin sheaths. Antibodies in the sera of both MS patients and healthy individuals do not bind to MOG in its native conformation, but do bind to linearized MOG. However, in acute disseminating encephalomyelitis (ADEM), antibodies in the serum can bind to native MOG [21;77]. It appears as if anti-MOG antibodies do not play a major role in MS, but it has been proposed that the myelin surrounding lesions is not in a normal state [78]. In the area surrounding the lesions, MOG may have been partially denatured; this would reconcile the large antibody deposition seen in MS lesions and the lack of sera antibody binding to native MOG. Also, there is evidence of cross-reactivity between MOG-specific T cells to neurofilament antigens, so these antibodies may not be targeting MOG but other CNS antigens [79].
While myelinated axons are largely insulated from the external environment, they remain exposed at the node of Ranvier. Axonal degeneration may be initiated due to autoimmune responses against the exposed areas of the axons. Neurofascin is a glycoprotein expressed by neurons on axons at the nodes of Ranvier. If EAE mice were given anti-neurofascin antibody, disease was exacerbated and severe axonal injury was observed [21]. Other proteins on the nodes of Ranvier such as contactin and various glycoproteins could also serve as targets for anti-axon antibodies and T cells [20;21]. The degeneration of these axons would facilitate axonal degeneration preceding myelin destruction as suggested in the Inside-Out model [5].
Interestingly, if A.SW mice were treated with bacterial DNA rich in CpG that favor Th1 type immune responses, half of the mice showed no clinical signs and only 25% died from SP-EAE [9]. Since both BP and CpG DNA suppress PP-EAE, it is likely that favoring Th1 immune responses inhibits the production of Th2 mediated anti-myelin antibody which limits progression [72]. Here, the roles of BP and CpG in progressive EAE are reversed from those in RR-EAE, where both BP and CpG can exacerbate disease [80]. Similarly, NKT cells that suppress RR-EAE seem to contribute to disease progression in PP-EAE (Martinez and Tsunoda, unpublished data). Additionally, SJL/J mice that generally respond with a Th1 phenotype do not develop progressive disease.
3. Viral pathogenesis of MS and Theiler’s murine encephalomyelitis virus (TMEV) infection
3.1 Viral infection and MS
Viruses and anti-viral immune responses have been detected in the CNS of MS patients, particularly EBV and human herpes virus (HHV)-6, suggesting that viruses may cause or contribute to disease [81–84]. The possibility that MS has an infectious trigger has long been considered since the initial descriptions of the disease [85]. Although there is no consensus as to which microbe causes MS, EBV is the virus most consistently linked with MS pathogenesis. While up to 95% of the population is EBV-infected, ~99% of MS patients are EBV seropositive [86]. Since EBV can infect and immortalize B cells, EBV infection may provoke myelin autoantibody production, although self-reactive B cells should be eliminated in normal individuals [87]. The autoantibodies would lead to the destruction of myelin and the release of antigens to augment the inflammatory process, however convincing evidence of this has not yet been found.
3.2 TMEV infection: 2-stage disease theory
Perhaps the most compelling evidence that MS may have a viral origin is seen in animal models using TMEV and murine hepatitis virus to mimic the disease in mice [37]. TMEV belongs to the family Picornaviridae and has been used for animal models of MS and epilepsy (Box 2). In the TMEV model, mice develop chronic progressive demyelinating disease without remission, one month after infection (Fig. 1d). The disease course of TMEV infection is similar to PP-MS or PP-EAE in MOG-sensitized A.SW mice. However, the pathomechanisms in PP-EAE and TMEV infection seem to be different, which may explain the inter-individual heterogeneity of MS pathology. PP-EAE could be a 1-stage disease, where anti-myelin antibody induces demyelination without oligodendrocyte apoptosis (Fig. 1b, 3b, and d) [72]. In contrast, the chronic demyelinating phase in TMEV infection can be divided into two stages: 1) axonal degeneration and oligodendrocyte apoptosis lead to macrophage activation and demyelination (Fig. 3a, c), and 2) myelin damage that leads to epitope spreading, inducing myelin specific autoimmune responses and demyelination. During the first stage of the chronic phase of TMEV infection, there is evidence that axonal degeneration occurs before demyelination and triggers demyelination [5]. This has lead to an ‘Inside-Out’ model of neurodegeneration, where axonal damage inside the nerve fibers precedes the damage of myelin outside of the nerve fiber [5].
Box 2. Epilepsy and MS.
The occurrence of epilepsy in MS patients is higher than that of the rest of the population, and seizures have been observed and considered to result from the disease since shortly after MS was first described [88]. Since seizures may occur during either relapses or periods of remission in MS, it remains controversial as to whether or not the seizures seen in MS patients are caused by the disease or the two diseases co-occur. However, since the pathology in MS increases over time and other diseases with damage in the CNS are associated with an increased risk of seizures, it is likely that the seizures seen in MS patients are caused by the development of cortical demyelination and not just a coincidence [89].
Interestingly, the same virus used to study MS in SJL/J mice, TMEV, induces seizures in C57BL/6 mice and is being used as a viral model for epilepsy [90]. While C57BL/6 mice are normally resistant to chronic TMEV infection, the mice develop seizures 4 to 8 days post infection. Mice that have the seizures show degeneration in the pyramidal cell layer of the hippocampus, which is consistent with the brain damage observed in epilepsy patients [90].
In both MS and some forms of epilepsy, there is evidence for a potential viral etiology. In children experiencing febrile seizures and in adults with epilepsy there was an increased presence of human herpes virus-6 (HHV-6) [91]. In MS, HHV-6 has been one of the suspected viruses that may contribute to its etiology [92]. Thus, the same neurotropic virus has been associated with MS and epilepsy both epidemiologically and experimentally. Hence, some individuals may develop epilepsy due to the viral infection, others develop MS, some both, and some neither.
Fig. 3.
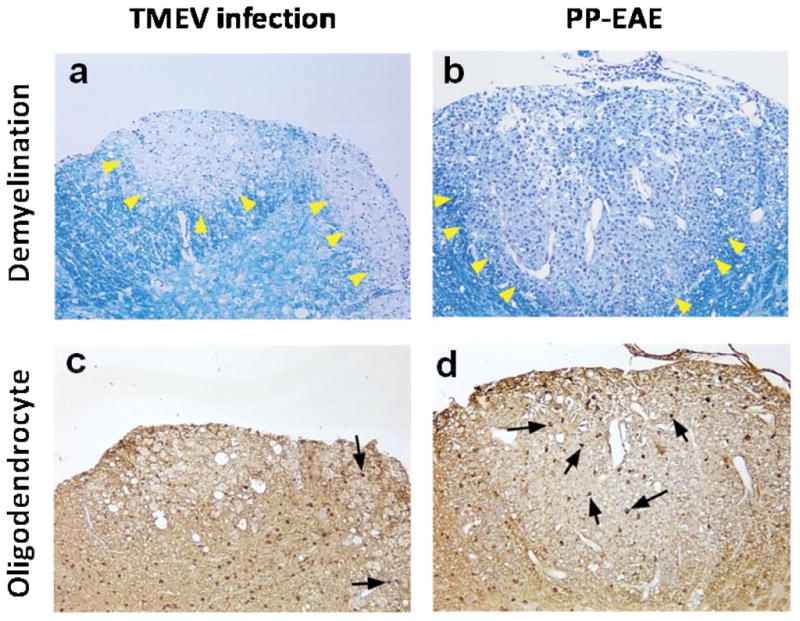
Inter-individual heterogeneity of pathology in animal models for PP-MS. TMEV infection and PP-EAE induced with MOG92-106. (a, b) Both in TMEV infection and PP-EAE, demyelination was observed in the spinal cord (arrowheads, Luxol fast blue stain). (c, d) Although a few oligodendrocytes survived in the lesions in TMEV infection (arrows), many oligodendrocytes were detected in PP-EAE (carbonic anhydrase II immunohistochemistry for oligodendrocytes). Magnification: × 70.
TMEV is a murine neurotropic virus which uses axonal transport to spread in the CNS, thus axonal degeneration by the host could prevent TMEV from spreading and block further disease expansion [93]. This axonal degeneration could provide a physiological barrier against neurotropic virus infection [94]. After infection with the neurovirulent strain of TMEV, the GDVII strain, axonal degeneration can be seen in the white matter of the spinal cord in the absence of inflammation [95]. During infection with a less virulent strain of TMEV, the Daniels (DA) strain, axonal degenerations can be detected ~2–3 weeks post infection, while demyelination and inflammation are not apparent until one month post infection. Thus, axonal degeneration was observed before a demyelinating event. Axonal damage preceding demyelination has also been observed in MS patients, and may occur in a subset of MS patients [5]. Moreover, in EAE and MS, autoantibodies against neurofascin have been found and experimentally shown to bind to the exposed axon at the nodes of Ranvier (Box 2). These autoantibodies can induce axonal damage in the absence of demyelination or inflammation [96].
Axonal degeneration can trigger a cascade reaction, leading to widespread demyelination [94]. The degeneration of axons and oligodendrocyte apoptosis (induced by viral infection or disruption of cross-talk between axons and oligodendrocytes, Fig. 3c) locally activates microglia and macrophages. Chemokine release from activated microglia can lead to increased expression of adhesion molecules in the CNS which drives virus-specific T cell recruitment and inflammation [97]. The activated microglia and macrophages will phagocytize degenerate axons and apoptotic oligodendrocytes, and present neuroantigens to CD4+ T cells.
Here, epitope spreading is initiated where the immune response initially targeted against a viral epitope progressively targets multiple CNS antigens. Indeed, in the late chronic stage of TMEV infection (>100 days post infection), myelin specific T cell responses have been demonstrated [99]. At this point, anti-myelin autoimmune responses can be generated to initiate the conventional ‘Outside-In’ pathomechanism. Thus, the ‘Inside-Out’ model can contribute to or initiate the conventional ‘Outside In’ model, where myelin sheaths outside the nerve fibers are damaged first by anti-myelin T cells, leading to secondary damage of axons inside the nerve fiber (Table 5). Therefore, the TMEV model should be considered a 2-stage disease. During the first stage (1–3 months post infection) axonal damage and oligodendrocyte apoptosis lead to macrophage activation and demyelination mediated by virus-specific T cells. Then, during the second stage (>100 days post infection) myelin-specific T cells contribute to enhancement of demyelination (Fig. 1d).
Table 5.
Lesion development in MS
4. Conclusions: 1-stage versus 2-stage disease theories
The balance of different immune cell types influences the activity of disease (relapse, remission, or progression) in the two ‘stage’ theories (Fig. 1). In the 1-stage disease model, during the RR course of the disease, the inflammatory and anti-inflammatory (Th2, NKT, and Treg) responses balance resulting in relapse and remission, but at some points the inflammatory responses overwhelm the anti-inflammatory immune response, leading to progressive disease (SP-MS). In the 1-stage theory, the cause of pathology remains the same throughout the disease (intra-individual pathological homogeneity). In the 2-stage theory the effector mechanism changes during the time course of disease, during the second stage Th2 responses or some other responses overcome the inflammatory response leading to neurodegeneration, for example, disease mediated by autoantibodies (intra-individual pathological heterogeneity) (Box 1). Since there is both clinical and experimental evidence for both stage theories in MS (Table 6), these patterns may represent different clinical subgroups within MS patients. The presence of subgroupings in MS is strongly supported by the fact that plasmapheresis and B cell depletion appear to be beneficial only in a subset of MS patients [100;101]. What may determine whether the disease transforms into a chronic inflammatory disease or switches to an autoantibody-mediated disease? Experimentally, differences in sensitivity to particular autoantigens and adjuvants are linked with genetic backgrounds, and are known to play important roles in the development of progressive disease.
Table 6.
1-stage vs. 2-stage disease theories
| Theory | Clinical | Pathology | Neuroimaging | Experimental | Reference |
|---|---|---|---|---|---|
| 1-stage | RR-MS PP-MS |
Inter-individual heterogeneity Intra-individual homogeneity |
Gd (+) in RR-MS Gd (−) in PP-MS |
Monophasic EAE RR-EAE |
[25;75] [26] |
| 2-stage | SP-MS PR-MS |
Stage-dependent | Gd (+) or (−) in SP-MS | SP-EAE TMEV |
[28] |
Gd: gadolinium
MOG92-106 can induce RR-, SP-, and PP-EAE depending on the mouse strain and adjuvant, while PLP and MBP induce RR-EAE and monophasic EAE, respectively. Here, the opposing Th cell subsets may have different major histocompatibility complex (MHC)-ligand(s) affinities, where the subset with the stronger affinity ultimately dominates the immune response [102]. Alternatively, one of the subsets of Th cells may become depleted or anergic to the ligand and stops responding [73].
The TMEV infection model can also be classified as a 2-stage disease. However, in the TMEV model, the switch between stages is not based on Th1 or Th2 immune responses, but rather conversion from a viral-mediated to an autoimmune-like pathology. There is no absolute evidence of a direct viral cause of MS. However, newly forming lesions in MS patients, reported by Barnett et al. [28], had characteristics similar to those seen in TMEV infection in mice. For example, the early lesions in TMEV infection (2–3 weeks post infection) resemble MS lesions that display oligodendrocyte apoptosis, early microglial activation, and little or no infiltration by lymphocytes or myelin-laden phagocytes [95]. The lesions observed during the pathology-shift described by Barnett et al. led them to propose that initial oligodendrocyte apoptosis induces microglia activation during the early stages of MS, which eventually leads to recruitment of inflammatory cells into the CNS during the following stage (stage-dependent pathology) [5;28].
A neurotropic virus may infect and spread in one area of the CNS and be eliminated (or at least nearly eliminated) through host-defense mechanisms, including anti-virus immunity and apoptosis of virus-infected neuronal cells. The apoptotic neuronal cells would have to be cleared by phagocytes, which may lead to inappropriate presentation of self-antigens triggering autoimmunity [103]. Then, the autoreactive T cells could be recruited to sites of Wallerian degeneration which are often remote from the original infection site [37]. This could account for the sporadic nature of MS lesions ‘in space’ in the CNS. Alternatively, some of the virus may escape and go on to infect other cell types in the CNS leading to further pathology; for example TMEV has been shown to infect from axons to myelin sheaths [104]. A virus that becomes persistent/latent with a low rate of reactivation could account for the sporadic nature of MS lesions ‘in time’ in the CNS, since reactivation of the virus would trigger new CNS lesions, including recruitment of inflammatory cells, induction of apoptosis, or activation of microglia. These inflammatory events would activate myelin-specific T cells either by epitope spreading or bystander activation perpetuating the vicious cycle [5].
The two disease theories are not mutually exclusive of each other. Evidence from animal experiments shows that sensitization with the same myelin peptide but with different adjuvants/treatments can also induce different clinical courses. Although multiple causative factors have been implicated in MS, these factors may influence the clinical course of MS, while a common cause may be responsible for initiating the seemingly heterogeneous disease. On the other hand, similar clinical courses in MS patients can be caused by different causative factors and adjuvants. Here different causes would be responsible for the development of the homogenous disease in appearance.
5. Future perspective: Treatment strategies
As we discussed above, although immune therapy requires consideration of the disease stage and etiology, neuroprotection and remyelination should be beneficial regardless of the immune effectors or etiology. Additionally, neuroprotection will prevent cascade reactions blocking inflammatory cycles [94]. The two most feasible approaches include 1) promoting remyelination and 2) axonal protection.
5.1 Remyelination
The myelin sheath of nerve fibers allows for rapid saltatory conduction of signals in the CNS and also protects against axonal damage [105]. Demyelinated nerve fibers can be remyelinated. However this process is slow in the CNS, requires the proper conditions, and the remyelinated myelin sheath will be thinner than the original sheath [106]. The two most likely approaches for promoting remyelination are 1) myelin forming cell transplantation and 2) promotion of repair by resident stem- and precursor-cell populations. Methods have been established to transfer several different cell types that result in myelination of the CNS in rodents with myelin deficiencies [107–109]. While this sounds promising, these injections can cause further physical damage to the CNS and with multiple demyelinated plaques in the brains of MS patients this may not be feasible. In addition, it is not clear as to whether or not cell transplants can take hold in the conditions present in active or neurodegenerative plaques.
A safer approach to remyelinating the CNS after attacks in MS would be to use pharmacological agents that promote remyelination. The resident stem- and precursor-cells in the brain are capable of remyelinating axons and this activity can be seen in the plaques of MS patients [110]. Both autoantibodies that react with surface antigens on oligodendrocytes and pregnancy associated with hormones have been shown to actually promote remyelination; however the exact mechanism involved in this is still unclear [111–114]. The recruitment of precursor cells and their differentiation into oligodendrocytes but not astrocytes in lesions is an important consideration in promoting remyelination and not astrogliosis. It has been shown that both chromatin remodeling factors and the redox environment control the fate of neural progenitor cells (NPCs) [115]. Here, pharmacological agents may favorably balance the redox state to block astrogliosis or to activate or deactivate chromatin remodeling factors to promote oligodendrocytes differentiation from NPCs. While a single pharmacological agent that promotes all of these outcomes is unlikely to be found, pharmacologic combination therapy to promote NPC mobility and differentiation into oligodendrocytes would be of great value.
5.2 Axonal protection
When an axon remains demyelinated, it can compensate its function by redistributing its sodium channels to allow for conduction. In addition, the sodium channel that is redistributed along the axon can influence whether or not the axon will degenerate or remain intact. For example, Nav1.2 sodium channels appear to favor axonal survival and Nav1.6 sodium channels are evidence of injury [116]. The theory behind the difference in survival rates between axons with the two sodium channels is that Nav1.6 produces a large sustained sodium conductance that results in calcium and sodium exchange. The increased intracellular calcium concentrations activate proteases resulting in additional damage to the axon. Conversely, Nav1.2, which is present on non-myelinated axons, rapidly activates and inactivates supporting axonal conduction. Thus, a mechanism of either increasing the likelihood of Nav1.2 being redistributed along the axon or inhibiting Nav1.6 conductance may result in neuroprotection and extended life of demyelinated axons. This type of therapy may buy time by preventing the cascade of reactions where axonal degeneration (Inside-Out model) leads to augmentation of demyelination (Outside-In model). However, one must keep in mind that in their natural state axons are myelinated and without remyelination degeneration may be inevitable since longer axons may require myelin for trophic support [117].
Acknowledgments
This work was supported by the National Institutes of Health (R21NS059724, P20-RR018724, and 8 P20GM103433-10). We thank Ashley M. Studt, B.S. for critical comments on this manuscript and Sadie Faith Pearson and Lesya Ekshyyan for excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120(Pt 3):393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 3.Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- 4.Batista S, Zivadinov R, Hoogs M, Bergsland N, Heininen-Brown M, Dwyer MG, Weinstock-Guttman B, Benedict RH. Basal ganglia, thalamus and neocortical atrophy predicting slowed cognitive processing in multiple sclerosis. J Neurol. 2011 doi: 10.1007/s00415-011-6147-1. [DOI] [PubMed] [Google Scholar]
- 5.Tsunoda I, Fujinami RS. Inside-Out versus Outside-In models for virus induced demyelination: axonal damage triggering demyelination. Springer Semin Immunopathol. 2002;24:105–125. doi: 10.1007/s00281-002-0105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geurts JJG, Kooi E-J, Witte ME, van der Valk P. Multiple sclerosis as an “inside-out” disease. Ann Neurol. 2010;68:767–768. doi: 10.1002/ana.22279. [DOI] [PubMed] [Google Scholar]
- 7.Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46:907–911. doi: 10.1212/wnl.46.4.907. [DOI] [PubMed] [Google Scholar]
- 8.Rovaris M, Confavreux C, Furlan R, Kappos L, Comi G, Filippi M. Secondary progressive multiple sclerosis: current knowledge and future challenges. Lancet Neurol. 2006;5:343–354. doi: 10.1016/S1474-4422(06)70410-0. [DOI] [PubMed] [Google Scholar]
- 9.Tsunoda I, Kuang L-Q, Igenge IZ, Fujinami RS. Converting relapsing remitting to secondary progressive experimental allergic encephalomyelitis (EAE) by ultraviolet B irradiation. J Neuroimmunol. 2005;160:122–134. doi: 10.1016/j.jneuroim.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Ebers GC. Environmental factors and multiple sclerosis. Lancet Neurol. 2008;7:268–277. doi: 10.1016/S1474-4422(08)70042-5. [DOI] [PubMed] [Google Scholar]
- 11.Orton SM, Wald L, Confavreux C, Vukusic S, Krohn JP, Ramagopalan SV, Herrera BM, Sadovnick AD, Ebers GC. Association of UV radiation with multiple sclerosis prevalence and sex ratio in France. Neurology. 2011:425–431. doi: 10.1212/WNL.0b013e31820a0a9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jobin C, Larochelle C, Parpal H, Coyle PK, Duquette P. Gender issues in multiple sclerosis: an update. Womens Health (Lond Engl) 2010:797–820. doi: 10.2217/whe.10.69. [DOI] [PubMed] [Google Scholar]
- 13.Yanagawa K, Kawachi I, Toyoshima Y, Yokoseki A, Arakawa M, Hasegawa A, Ito T, Kojima N, Koike R, Tanaka K, Kosaka T, Tan CF, Kakita A, Okamoto K, Tsujita M, Sakimura K, Takahashi H, Nishizawa M. Pathologic and immunologic profiles of a limited form of neuromyelitis optica with myelitis. Neurology. 2009;73:1628–1637. doi: 10.1212/WNL.0b013e3181c1deb9. [DOI] [PubMed] [Google Scholar]
- 14.Kalinski P, Moser M. Consensual immunity: success-driven development of T-helper-1 and T-helper-2 responses. Nat Rev Immunol. 2005;5:251–260. doi: 10.1038/nri1569. [DOI] [PubMed] [Google Scholar]
- 15.Bahbouhi B, Pettre S, Berthelot L, Garcia A, Elong NA, Degauque N, Michel L, Wiertlewski S, Lefrere F, Meyniel C, Delcroix C, Brouard S, Laplaud DA, Soulillou JP. T cell recognition of self-antigen presenting cells by protein transfer assay reveals a high frequency of anti-myelin T cells in multiple sclerosis. Brain. 2010;133:1622–1636. doi: 10.1093/brain/awq074. [DOI] [PubMed] [Google Scholar]
- 16.Deckert M, Sanchez-Ruiz M, Brunn A, Schluter D. Role of CD8 T-cell-mediated autoimmune diseases of the central nervous system. Crit Rev Immunol. 2010;30:311–326. doi: 10.1615/critrevimmunol.v30.i4.10. [DOI] [PubMed] [Google Scholar]
- 17.Durelli L, Conti L, Clerico M, Boselli D, Contessa G, Ripellino P, Ferrero B, Eid P, Novelli F. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol. 2009;65:499–509. doi: 10.1002/ana.21652. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Markovic-Plese S, Lacet B, Raus J, Weiner HL, Hafler DA. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med. 1994;179:973–984. doi: 10.1084/jem.179.3.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172(1):146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Derfuss T, Parikh K, Velhin S, Braun M, Mathey E, Krumbholz M, Kumpfel T, Moldenhauer A, Rader C, Sonderegger P, Pollmann W, Tiefenthaller C, Bauer J, Lassmann H, Wekerle H, Karagogeos D, Hohlfeld R, Linington C, Meinl E. Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc Natl Acad Sci USA. 2009;106:8302–8307. doi: 10.1073/pnas.0901496106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derfuss T, Linington C, Hohlfeld R, Meinl E. Axo-glial antigens as targets in multiple sclerosis: implications for axonal and grey matter injury. J Mol Med(Berl) 2010;88:753–761. doi: 10.1007/s00109-010-0632-3. [DOI] [PubMed] [Google Scholar]
- 22.Filippi M, Rovaris M, Iannucci G, Mennea S, Sormani MP, Comi G. Whole brain volume changes in patients with progressive MS treated with cladribine. Neurology. 2000:1714–1718. doi: 10.1212/wnl.55.11.1714. [DOI] [PubMed] [Google Scholar]
- 23.Fisher E, Lee JC, Nakamura K, Rudick RA. Gray matter atrophy in multiple sclerosis: a longitudinal study. Ann Neurol. 2008:255–265. doi: 10.1002/ana.21436. [DOI] [PubMed] [Google Scholar]
- 24.Pagani E, Rocca MA, Gallo A, Rovaris M, Martinelli V, Comi G, Filippi M. Regional brain atrophy evolves differently in patients with multiple sclerosis according to clinical phenotype. Eur Arch Psychiatry Clin Neurosci. 2005:341–346. [PMC free article] [PubMed] [Google Scholar]
- 25.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 26.Breij EC, Brink BP, Veerhuis R, van den Berg C, Vloet R, Yan R, Dijkstra CD, van der Valk P, Bo L. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol. 2008:16–25. doi: 10.1002/ana.21311. [DOI] [PubMed] [Google Scholar]
- 27.Raine CS. Multiple sclerosis: classification revisited reveals homogeneity and recapitulation. Ann Neurol. 2008:1–3. doi: 10.1002/ana.21314. [DOI] [PubMed] [Google Scholar]
- 28.Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004:458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- 29.Barnett MH, Parratt JD, Pollard JD, Prineas JW. MS: is it one disease? 2009 Aug 13;2009:57–65. [PubMed] [Google Scholar]
- 30.Lalive PH, Menge T, Delarasse C, Della GB, Pham-Dinh D, Villoslada P, von Budingen HC, Genain CP. Antibodies to native myelin oligodendrocyte glycoprotein are serologic markers of early inflammation in multiple sclerosis. Proc Natl Acad Sci USA. 2006;103:2280–2285. doi: 10.1073/pnas.0510672103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pender MP, Greer JM. Immunology of multiple sclerosis. Curr Allergy Asthma Rep. 2007:285–292. doi: 10.1007/s11882-007-0043-x. [DOI] [PubMed] [Google Scholar]
- 32.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 33.Field J, Browning SR, Johnson LJ, Danoy P, Varney MD, Tait BD, Gandhi KS, Charlesworth JC, Heard RN, Stewart GJ, Kilpatrick TJ, Foote SJ, Bahlo M, Butzkueven H, Wiley J, Booth DR, Taylor BV, Brown MA, Rubio JP, Stankovich J. A polymorphism in the HLA-DPB1 gene is associated with susceptibility to multiple sclerosis. PLoS One. 2010;5:e13454. doi: 10.1371/journal.pone.0013454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rudick RA. Multiple sclerosis: is multiple sclerosis caused by venous insufficiency? Nat Rev Neurol. 2010;6:472–474. doi: 10.1038/nrneurol.2010.117. [DOI] [PubMed] [Google Scholar]
- 35.Staples J, Ponsonby AL, Lim L. Low maternal exposure to ultraviolet radiation in pregnancy, month of birth, and risk of multiple sclerosis in offspring: longitudinal analysis. BMJ. 2010;340:c1640. doi: 10.1136/bmj.c1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wood H. Multiple sclerosis: OAS1 genotype linked to multiple sclerosis severity. Nat Rev Neurol. 2010;6:526. doi: 10.1038/nrneurol.2010.134. [DOI] [PubMed] [Google Scholar]
- 37.Sato F, Tanaka H, Hasanovic F, Tsunoda I. Theiler’s virus infection: Pathophysiology of demyelination and neurodegeneration. Pathophysiology. 2011;18:31–41. doi: 10.1016/j.pathophys.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emerson MR, Gallagher RJ, Marquis JG, LeVine SM. Enhancing the ability of experimental autoimmune encephalomyelitis to serve as a more rigorous model of multiple sclerosis through refinement of the experimental design. Comp Med. 2009;59:112–128. [PMC free article] [PubMed] [Google Scholar]
- 39.Tsunoda I, Fujinami RS. Two models for multiple sclerosis: Experimental allergic encephalomyelitis and Theiler’s murine encephalomyelitis virus. J Neuropathol Exp Neurol. 1996;55:673–686. doi: 10.1097/00005072-199606000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Kister I, Chamot E, Bacon JH, Niewczyk PM, De Guzman RA, Apatoff B, Coyle P, Goodman AD, Gottesman M, Granger C, Jubelt B, Krupp L, Lenihan M, Lublin F, Mihai C, Miller A, Munschauer FE, III, Perel AB, Teter BE, Weinstock-Guttman B, Zivadinov R, Herbert J. Rapid disease course in African Americans with multiple sclerosis. Neurology. 2010;75:217–223. doi: 10.1212/WNL.0b013e3181e8e72a. [DOI] [PubMed] [Google Scholar]
- 41.Burns JB, Bartholomew BD, Lobo ST. Isolation of CD45RO+, memory T cells recognizing proteolipid protein from neurologically normal subjects. Cell Immunol. 2001;212:44–50. doi: 10.1006/cimm.2001.1842. [DOI] [PubMed] [Google Scholar]
- 42.Hellings N, Baree M, Verhoeven C, D’hooghe MB, Medaer R, Bernard CC, Raus J, Stinissen P. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J Neurosci Res. 2001;63:290–302. doi: 10.1002/1097-4547(20010201)63:3<290::AID-JNR1023>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 43.Tejada-Simon MV, Hong J, Rivera VM, Zhang JZ. Reactivity pattern and cytokine profile of T cells primed by myelin peptides in multiple sclerosis and healthy individuals. Eur J Immunol. 2001;31:907–917. doi: 10.1002/1521-4141(200103)31:3<907::aid-immu907>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 44.Rouse BT, Sehrawat S. Immunity and immunopathology to viruses: what decides the outcome? Nat Rev Immunol. 2010;10:514–526. doi: 10.1038/nri2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frohman EM, Racke MK, Raine CS. Multiple sclerosis--the plaque and its pathogenesis. N Engl J Med. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 46.Gold R, Luhder F. Interleukin-17--extended features of a key player in multiple sclerosis. Am J Pathol. 2008;172(1):8–10. doi: 10.2353/ajpath.2008.070862. Ref Type: Generic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Costantino CM, Baecher-Allan C, Hafler DA. Multiple sclerosis and regulatory T cells. J Clin Immunol. 2008;28:697–706. doi: 10.1007/s10875-008-9236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng J, Misu T, Fujihara K, Sakoda S, Nakatsuji Y, Fukaura H, Kikuchi S, Tashiro K, Suzumura A, Ishii N, Sugamura K, Nakashima I, Itoyama Y. Ibudilast, a nonselective phosphodiesterase inhibitor, regulates Th1/Th2 balance and NKT cell subset in multiple sclerosis. Mult Scler. 2004;10:494–498. doi: 10.1191/1352458504ms1070oa. [DOI] [PubMed] [Google Scholar]
- 50.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 51.Singh AK, Wilson MT, Hong S, Olivares-Villagomez D, Du C, Stanic AK, Joyce S, Sriram S, Koezuka Y, Van KL. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1801–1811. doi: 10.1084/jem.194.12.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, Weiner HL. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- 53.Hofman FM, von Hanwehr RI, Dinarello CA, Mizel SB, Hinton D, Merrill JE. Immunoregulatory molecules and IL 2 receptors identified in multiple sclerosis brain. J Immunol. 1986;136:3239–3245. [PubMed] [Google Scholar]
- 54.Merrill JE, Kagan JM, Schmid I, Strom SR, Quan SG, Chen IS. T cell lines established from multiple sclerosis cerebrospinal fluid T cells using human retroviruses. J Neuroimmunol. 1989;21:213–226. doi: 10.1016/0165-5728(89)90177-x. [DOI] [PubMed] [Google Scholar]
- 55.Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE. Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989;124:132–143. doi: 10.1016/0008-8749(89)90117-2. [DOI] [PubMed] [Google Scholar]
- 56.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsunoda I, Fujinami RS. Neuropathogenesis of Theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J Neuroimmune Pharmacol. 2010;5(3):355–369. doi: 10.1007/s11481-009-9179-x. Ref Type: Generic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hofer T, Nathansen H, Lohning M, Radbruch A, Heinrich R. GATA-3 transcriptional imprinting in Th2 lymphocytes: a mathematical model. Proc Natl Acad Sci USA. 2002;99:9364–9368. doi: 10.1073/pnas.142284699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, Weiner HL, Nabavi N, Glimcher LH. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- 60.Chakravarti S, Sabatos CA, Xiao S, Illes Z, Cha EK, Sobel RA, Zheng XX, Strom TB, Kuchroo VK. Tim-2 regulates T helper type 2 responses and autoimmunity. J Exp Med. 2005;202:437–444. doi: 10.1084/jem.20050308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Silverpil E, Glader P, Hansson M, Linden A. Impact of interleukin-17 on macrophage phagocytosis of apoptotic neutrophils and particles. Inflammation. 2011;34:1–9. doi: 10.1007/s10753-010-9201-8. [DOI] [PubMed] [Google Scholar]
- 62.Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–104. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- 63.Segal BM. Th17 cells in autoimmune demyelinating disease. Semin Immunopathol. 2010;32:71–77. doi: 10.1007/s00281-009-0186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bendelac A, Savage PB, Teyton L. The Biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 65.O’Keeffe J, Gately CM, Counihan T, Hennessy M, Leahy T, Moran AP, Hogan EL. T-cells expressing natural killer (NK) receptors are altered in multiple sclerosis and responses to alpha-galactosylceramide are impaired. J Neurol Sci. 2008;275:22–28. doi: 10.1016/j.jns.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sakuishi K, Miyake S, Yamamura T. Role of NK cells and invariant NKT cells in multiple sclerosis. Results Probl Cell Differ. 2010;51:127–147. doi: 10.1007/400_2009_11. Ref Type: Generic. [DOI] [PubMed] [Google Scholar]
- 67.Karlsson F, Jackson S, Gray L, Zhang S, Grisham MB. Ex vivo generation of regulatory T cells: Characterizatoin and therapeutic evaluation in a model of chronic colitis. Methods Molecular Biol. 2011;677:47–61. doi: 10.1007/978-1-60761-869-0_4. Ref Type: Generic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Selvaraj RK, Geiger TL. Mitigation of experimental allergic encephalomyelitis by TGF-beta induced Foxp3+ regulatory T lymphocytes through the induction of anergy and infectious tolerance. J Immunol. 2008;180:2830–2838. doi: 10.4049/jimmunol.180.5.2830. [DOI] [PubMed] [Google Scholar]
- 69.Yu P, Gregg RK, Bell JJ, Ellis JS, Divekar R, Lee HH, Jain R, Waldner H, Hardaway JC, Collins M, Kuchroo VK, Zaghouani H. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J Immunol. 2005;174:6772–6780. doi: 10.4049/jimmunol.174.11.6772. [DOI] [PubMed] [Google Scholar]
- 70.Serafini A, Finkielman S. From Fabiola to Fliedner and the first school of nurses. Medicina (B Aires) 2004:265–268. [PubMed] [Google Scholar]
- 71.Lafaille JJ, Keere FV, Hsu AL, Baron JL, Haas W, Raine CS, Tonegawa S. Myelin basic protein-specific T helper 2 (Th2) cells cause experimental autoimmune encephalomyelitis in immunodeficient hosts rather than protect them from the disease. J Exp Med. 1997;186:307–312. doi: 10.1084/jem.186.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsunoda I, Kuang L-Q, Theil DJ, Fujinami RS. Antibody association with a novel model for primary progressive multiple sclerosis: induction of relapsing-remitting and progressive forms of EAE in H2s mouse strains. Brain Pathol. 2000;10:402–418. doi: 10.1111/j.1750-3639.2000.tb00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsunoda I, Libbey JE, Kuang L-Q, Terry EJ, Fujinami RS. Massive apoptosis in lymphoid organs in animal models for primary and secondary progressive multiple sclerosis. Am J Pathol. 2005:1631–1646. doi: 10.1016/S0002-9440(10)61247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, Reuss R, Probst C, Kristoferitsch W, Wandinger KP, Wildemann B. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation. 2010;7:52. doi: 10.1186/1742-2094-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lucchinetti CF, Bruck W, Rodriguez M, Lassmann H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity on pathogenesis. Brain Pathol. 1996;6:259–274. doi: 10.1111/j.1750-3639.1996.tb00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marta CB, Oliver AR, Sweet RA, Pfeiffer SE, Ruddle NH. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc Natl Acad Sci USA. 2005;102:13992–13997. doi: 10.1073/pnas.0504979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.O’Connor KC, McLaughlin KA, De Jager PL, Chitnis T, Bettelli E, Xu C, Robinson WH, Cherry SV, Bar-Or A, Banwell B, Fukaura H, Fukazawa T, Tenembaum S, Wong SJ, Tavakoli NP, Idrissova Z, Viglietta V, Rostasy K, Pohl D, Dale RC, Freedman M, Steinman L, Buckle GJ, Kuchroo VK, Hafler DA, Wucherpfennig KW. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med. 2007:211–217. doi: 10.1038/nm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fu L, Matthews PM, De SN, Worsley KJ, Narayanan S, Francis GS, Antel JP, Wolfson C, Arnold DL. Imaging axonal damage of normal-appearing white matter in multiple sclerosis. Brain. 1998;121(Pt 1):103–113. doi: 10.1093/brain/121.1.103. [DOI] [PubMed] [Google Scholar]
- 79.Krishnamoorthy G, Saxena A, Mars LT, Domingues HS, Mentele R, Ben-Nun A, Lassmann H, Dornmair K, Kurschus FC, Liblau RS, Wekerle H. Myelin-specific T cells also recognize neuronal autoantigen in a transgenic mouse model of multiple sclerosis. Nat Med. 2009;15:626–632. doi: 10.1038/nm.1975. [DOI] [PubMed] [Google Scholar]
- 80.Tsunoda I, Tolley ND, Theil DJ, Whitton JL, Kobayashi H, Fujinami RS. Exacerbation of viral and autoimmune animal models for multiple sclerosis by bacterial DNA. Brain Pathol. 1999;9:481–493. doi: 10.1111/j.1750-3639.1999.tb00537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cepok S, Zhou D, Srivastava R, Nessler S, Stei S, Bussow K, Sommer N, Hemmer B. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J Clin Invest. 2005;115:1352–1360. doi: 10.1172/JCI23661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Levin LI, Munger KL, Rubertone MV, Peck CA, Lennette ET, Spiegelman D, Ascherio A. Temporal relationship between elevation of epstein-barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA. 2005;293:2496–2500. doi: 10.1001/jama.293.20.2496. [DOI] [PubMed] [Google Scholar]
- 83.Pohl D, Krone B, Rostasy K, Kahler E, Brunner E, Lehnert M, Wagner HJ, Gartner J, Hanefeld F. High seroprevalence of Epstein-Barr virus in children with multiple sclerosis. Neurology. 2006;67:2063–2065. doi: 10.1212/01.wnl.0000247665.94088.8d. [DOI] [PubMed] [Google Scholar]
- 84.Virtanen JO, Farkkila M, Multanen J, Uotila L, Jaaskelainen AJ, Vaheri A, Koskiniemi M. Evidence for human herpesvirus 6 variant A antibodies in multiple sclerosis: diagnostic and therapeutic implications. J Neurovirol. 2007;13:347–352. doi: 10.1080/13550280701381332. [DOI] [PubMed] [Google Scholar]
- 85.Murray TJ. The history of multiple sclerosis: the changing frame of the disease over the centuries. J Neurol Sci. 2009;277(Suppl 1):S3–S8. doi: 10.1016/S0022-510X(09)70003-6. [DOI] [PubMed] [Google Scholar]
- 86.Pohl D. Epstein-Barr virus and multiple sclerosis. J Neurol Sci. 2009;286:62–64. doi: 10.1016/j.jns.2009.03.028. [DOI] [PubMed] [Google Scholar]
- 87.Bagert BA. Epstein-Barr virus in multiple sclerosis. Curr Neurol Neurosci Rep. 2009;9:405–410. doi: 10.1007/s11910-009-0059-9. [DOI] [PubMed] [Google Scholar]
- 88.Koch M, Uyttenboogaart M, Polman S, De KJ. Seizures in multiple sclerosis. Epilepsia. 2008;49:948–953. doi: 10.1111/j.1528-1167.2008.01565.x. [DOI] [PubMed] [Google Scholar]
- 89.Kelley BJ, Rodriguez M. Seizures in patients with multiple sclerosis: epidemiology, pathophysiology and management. CNS Drugs. 2009;23:805–815. doi: 10.2165/11310900-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Libbey JE, Kirkman NJ, Smith MC, Tanaka T, Wilcox KS, White HS, Fujinami RS. Seizures following picornavirus infection. Epilepsia. 2008;49:1066–1074. doi: 10.1111/j.1528-1167.2008.01535.x. [DOI] [PubMed] [Google Scholar]
- 91.Laina I, Syriopoulou VP, Daikos GL, Roma ES, Papageorgiou F, Kakourou T, Theodoridou M. Febrile seizures and primary human herpesvirus 6 infection. Pediatr Neurol. 2010;42(1):28–31. doi: 10.1016/j.pediatrneurol.2009.07.016. Ref Type: Generic. [DOI] [PubMed] [Google Scholar]
- 92.Voumvourakis KI, Kitsos DK, Tsiodras S, Petrikkos G, Stamboulis E. Human herpesvirus 6 infection as a trigger of multiple sclerosis. Mayo Clin Proc. 2010;85:1023–1030. doi: 10.4065/mcp.2010.0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tsunoda I, Tanaka T, Terry EJ, Fujinami RS. Contrasting roles for axonal degeneration in an autoimmune versus viral model of multiple sclerosis: When can axonal injury be beneficial? Am J Pathol. 2007;170:214–226. doi: 10.2353/ajpath.2007.060683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsunoda I. Axonal degeneration as a self-destructive defense mechanism against neurotropic virus infection. Future Virol. 2008;3:579–593. doi: 10.2217/17460794.3.6.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tsunoda I, Kuang L-Q, Libbey JE, Fujinami RS. Axonal injury heralds virus-induced demyelination. Am J Pathol. 2003;162:1259–1269. doi: 10.1016/S0002-9440(10)63922-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mathey EK, Derfuss T, Storch MK, Williams KR, Hales K, Woolley DR, Al-Hayani A, Davies SN, Rasband MN, Olsson T, Moldenhauer A, Velhin S, Hohlfeld R, Meinl E, Linington C. Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med. 2007;204:2363–2372. doi: 10.1084/jem.20071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 98.Sato F, Omura NE, Martinez NE, Tsunoda I. Animal models of multiple sclerosis. In: Minager A, editor. Neuroinflammation. Elsevier; Burlington, MA: 2011. pp. 55–79. [Google Scholar]
- 99.Miller SD, Vanderlugt CL, Begolka WS, Pao W, Yauch RL, Neville KL, Katz-Levy Y, Carrizosa A, Kim BS. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat Med. 1997;3:1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- 100.Hawker K, O’Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, Hauser S, Waubant E, Vollmer T, Panitch H, Zhang J, Chin P, Smith CH. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66:460–471. doi: 10.1002/ana.21867. [DOI] [PubMed] [Google Scholar]
- 101.Keegan M, Konig F, McClelland R, Bruck W, Morales Y, Bitsch A, Panitch H, Lassmann H, Weinshenker B, Rodriguez M, Parisi J, Lucchinetti CF. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet. 2005;366:579–582. doi: 10.1016/S0140-6736(05)67102-4. [DOI] [PubMed] [Google Scholar]
- 102.Das MP, Nicholson LB, Greer JM, Kuchroo VK. Autopathogenic T helper cell type 1 (Th1) and protective Th2 clones differ in their recognition of the autoantigenic peptide of myelin proteolipid protein. J Exp Med. 1997;186:867–876. doi: 10.1084/jem.186.6.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rosen A, Casciola-Rosen L, Ahearn J. Novel packages of viral and self-antigens are generated during apoptosis. J Exp Med. 1995;181:1557–1561. doi: 10.1084/jem.181.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brahic M, Roussarie JP. Axon-myelin interactions during a viral infection of the central nervous system. PLoS Pathog. 2009;5:e1000519. doi: 10.1371/journal.ppat.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Taveggia C, Feltri ML, Wrabetz L. Signals to promote myelin formation and repair. Nat Rev Neurol. 2010;6:276–287. doi: 10.1038/nrneurol.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Franklin RJ, Kotter MR. The biology of CNS remyelination: the key to therapeutic advances. J Neurol. 2008;255(Suppl 1):19–25. doi: 10.1007/s00415-008-1004-6. [DOI] [PubMed] [Google Scholar]
- 107.Groves AK, Barnett SC, Franklin RJ, Crang AJ, Mayer M, Blakemore WF, Noble M. Repair of demyelinated lesions by transplantation of purified O-2A progenitor cells. Nature. 1993;362:453–455. doi: 10.1038/362453a0. [DOI] [PubMed] [Google Scholar]
- 108.Imaizumi T, Lankford KL, Waxman SG, Greer CA, Kocsis JD. Transplanted olfactory ensheathing cells remyelinate and enhance axonal conduction in the demyelinated dorsal columns of the rat spinal cord. J Neurosci. 1998;18:6176–6185. doi: 10.1523/JNEUROSCI.18-16-06176.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Windrem MS, Nunes MC, Rashbaum WK, Schwartz TH, Goodman RA, McKhann G, Roy NS, Goldman SA. Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nat Med. 2004;10:93–97. doi: 10.1038/nm974. [DOI] [PubMed] [Google Scholar]
- 110.Zawadzka M, Franklin RJ. Myelin regeneration in demyelinating disorders: new developments in biology and clinical pathology. Curr Opin Neurol. 2007;20:294–298. doi: 10.1097/WCO.0b013e32813aee7f. [DOI] [PubMed] [Google Scholar]
- 111.Bieber AJ, Warrington A, Asakura K, Ciric B, Kaveri SV, Pease LR, Rodriguez M. Human antibodies accelerate the rate of remyelination following lysolecithin-induced demyelination in mice. Glia. 2002;37:241–249. doi: 10.1002/glia.10033. [DOI] [PubMed] [Google Scholar]
- 112.Gregg C, Shikar V, Larsen P, Mak G, Chojnacki A, Yong VW, Weiss S. White matter plasticity and enhanced remyelination in the maternal CNS. J Neurosci. 2007;27:1812–1823. doi: 10.1523/JNEUROSCI.4441-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ibanez C, Shields SA, El-Etr M, Baulieu EE, Schumacher M, Franklin RJM. Systemic progesterone administration results in a partial reversal of the age-associated decline in CNS remyelination following toxin-induced demyelination in male rats. Neuropathol Appl Neurobiol. 2004;30:80–89. doi: 10.1046/j.0305-1846.2003.00515.x. [DOI] [PubMed] [Google Scholar]
- 114.Warrington AE, Asakura K, Bieber AJ, Ciric B, Van K, Kaveri VSV, Kyle RA, Pease LR, Rodriguez M. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc Natl Acad Sci U SA. 2000;97:6820–6825. doi: 10.1073/pnas.97.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Prozorovski T, Schulze-Topphoff U, Glumm R, Baumgart J, Schroter F, Ninnemann O, Siegert E, Bendix I, Brustle O, Nitsch R, Zipp F, Aktas O. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat Cell Biol. 2008;10:385–394. doi: 10.1038/ncb1700. [DOI] [PubMed] [Google Scholar]
- 116.Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci USA. 2004;101:8168–8173. doi: 10.1073/pnas.0402765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci. 2010;11:275–283. doi: 10.1038/nrn2797. [DOI] [PubMed] [Google Scholar]