

SUMMARY
Glutamate receptors are key mediators of brain communication. Among ionotropic glutamate receptors, kainate receptors (KARs) have been least explored and their relevance to pathophysiology is relatively obscure. This is in part due to the relatively low abundance of KARs, the regulatory function in network activity they play, the lack of specific agonists and antagonists for this receptor subtype, as well as to the absence of striking phenotypes in mice deficient in KAR subunits. Nonetheless, it is now well established that KARs are located presynaptically whereby they regulate glutamate and GABA release, and thus, excitability and participate in short‐term plasticity. In turn, KARs are also located postsynaptically and their activation contributes to synaptic integration. The development of specific novel ligands is helping to further investigate the contribution of KARs to health and disease. In this review, I summarize current knowledge about KAR physiology and pharmacology, and discuss their involvement in cell death and disease. In addition, I recapitulate the available data about the use of KAR antagonists and receptor subunit deficient mice in experimental paradigms of brain diseases, as well as the main findings about KAR roles in human CNS disorders. In sum, subunit specific antagonists have therapeutic potential in neurodegenerative and psychiatric diseases as well as in epilepsy and pain. Knowledge about the genetics of KARs will also help to understand the pathophysiology of those and other illnesses.
Keywords: Antagonists, Epilepsy, Ischemia, Neurodegeneration, Pain, Psychiatric diseases, White matter damage
Introduction
Glutamate activates three subtypes of ligand‐gated ion channel: the N‐methyl‐d‐aspartate (NMDA), (S)‐2‐amino‐3‐(3‐hydroxy‐5‐methyl‐4‐isoxazolyl)propionic acid (AMPA), and kainate receptors (KARs) [1]. KARs are widely expressed in the central nervous system (CNS) and are involved in the regulation of transmitter release, synapse formation, and in the pathophysiology of brain diseases (for earlier reviews see Refs. [2, 3, 4]). Molecular cloning has identified five subtypes which according to the new IUPHAR nomenclature are named as GluK1, GluK2, GluK3, GluK4, and GluK5 which co‐assemble in various combinations to form functional receptors [5]. Accordingly, KAR subunits that were formally known as GluR5‐7, KA1, and KA2 are now named GluK1, GluK2, GluK3, GluK4, and GluK5 respectively, and correspond with the current nomenclature of the gene names GRIK1, GRIK2, GRIK3, GRIK4, and GRIK5.
KARs have different roles in synaptic transmission [2, 3, 4]. At the postsynaptic level, KARs contribute to the synaptic response. In addition, KARs are also present at presynaptic locations, where they regulate transmitter release at both excitatory and inhibitory synapses. Intriguingly, KARs mediate two forms of signaling, a canonical pathway involving ion flux and another, noncanonical signaling pathway which links KAR activation to G protein activation [2].
KARs are expressed in neurons and glial cells throughout the CNS. In glial cells, KARs have the same general characteristics as those present in neurons [6]. However, editing of glutamine/arginine site at KAR subunits GluK1 and GluK2 in glial cells is less extensive than in neurons which suggest that native receptors in these cells have higher calcium permeability.
Finally, KARs have been recently observed in the axonal plasmalemma [7, 8]. Axonal KARs with the GluK1 subunit are coupled to phospholipase C activation. In turn, KARs with the GluK2 subunit induce a small amount of Ca2+ entry that stimulates nitric oxide synthase, as well as a local depolarization which activates L‐type Ca2+ channels and subsequently ryanodine receptors in the axoplasmic reticulum. The functional significance of KARs signaling in axons is unknown but they may serve to amplify axonal Ca2+ signals [8].
Progress in the understanding of the functions of KARs has advanced more slowly than for NMDA and AMPA receptors, due to the lack of highly selective KAR agonists and antagonists and to the fact that the prototypic agonist kainate activates both AMPA receptors and KARs.
In this review, I discuss current information about the potential of KAR antagonists as therapeutic tools to normalize glutamate signaling and as neuroprotective and therapeutic agents in CNS diseases.
KARs Pharmacology
The use of preparations of dorsal root ganglion cells which express KARs but not AMPA receptors has facilitated the development of KAR antagonists (see Ref. [4]; Figure 1 and Table 1). However, this goal has been hindered by the lack of suitable KAR selective agonists acting on native receptors. Because of that, most of the knowledge about KAR agonists and antagonists arises from expression assays carried out with cloned KAR subtypes. These studies have been in turn limited by the fact that GluK4 and GluK5 do not form functional channels. Moreover, glutamate has low agonist potency and rapidly desensitizes GluK3 in a concanavalin A insensitive manner. Consequently, characterization of the affinity of compounds for GluK3, GluK4, and GluK5 relied on the use of radioligand binding assays.
Figure 1.
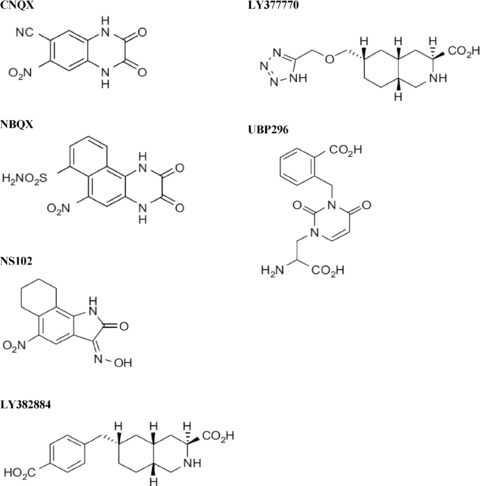
Structures of KARs antagonists.
Table 1.
Kainate receptor antagonists and their activity at recombinant subunits and native receptors
| Antagonist | GluK1 | GluK2 | GluK3 | GluK1/GluK2 | GluK1/GluK5 | GluK2/GluK5 | Native KARs | AMPA receptors# |
|---|---|---|---|---|---|---|---|---|
| CNQX | Potent | Potent | Potent | Potent | Potent | Less potent | Potent | Potent |
| NBQX | Potent | Potent | – | Potent | Potent | Less potent | Potent | Potent |
| NS102 | Potent | Potent | – | – | – | – | Potent | Weak |
| NS3763 | Potent | No effect | – | No effect | No effect | No effect | – | – |
| 2‐arylureidobenzoic III | Potent | Less potent | – | – | – | – | – | Weak |
| 2‐arylureidobenzoic IV | Potent | Potent | – | – | – | – | – | Weak |
| LY382884 | Potent | No effect | No effect | Potent | Potent | No effect | – | No effect |
| LY377770 | Very potent | No effect | No effect | Very potent | Potent | No effect | Very potent | Weak |
| GYKI53655 | – | No effect | Weak | – | – | – | Weak | Potent |
| UBP296 | Very potent | No effect | Weak | Very potent | Potent | No effect | Potent | No effect |
| UBP301 | Potent | – | – | – | – | – | Potent | Weak |
As determined at recombinant subunits; # native or recombinant; –, not determined; very potent, K d < 1 μM; potent, K d 1–30 μM; less potent, K d 30–100 μM; weak or no effect, K d > 100 μM.
Abbreviations: CNQX, 6‐Cyano‐7‐nitroquinoxaline‐2,3‐dione; NBQX, 2,3‐Dioxo‐6‐nitro‐1,2,3,4‐tetrahydrobenzo[f]quinoxaline ‐7‐sulfonamide; NS102, 6,7,8,9‐tetrahydro‐5‐nitro‐1 H‐benz[g]indole‐2,3‐dione‐3‐oxime; NS3763, 4,6‐Bis(benzoylamino)‐1,3‐benzenedicarboxylic acid; LY382884, (3S,4aR,6S,8aR)‐6‐((4‐carboxyphenyl)methyl)‐1,2,3,4,4a,5,6,7,8,8a‐decahydroisoquinoline‐3‐carboxylic acid; LY377770, (3S,4aR,6S,8aR)‐6‐(((1H‐tetrazol‐5‐ylmethyl)oxy)methyl)‐1,2,3,4,4a,5,6,7,8,8a‐decahydroisoquinoline‐3‐carboxylic acid; GYKI53655, 3‐N‐methylcarbamyde‐1‐(4‐aminophenyl)‐4‐methyl‐1.8‐methylen e‐dioxy‐5H‐2.3‐benzodiazepine; UBP296, (RS)‐1‐(2‐amino‐2‐carboxyethyl)‐3‐(2‐carboxybenzyl)pyrimidine‐2,4‐dione; UBP302, (S)‐1‐(2‐amino‐2‐carboxyethyl)‐3‐(2‐carboxybenzyl)pyrimidine‐2,4‐dione; UBP301, (aS)‐a‐Amino‐3‐[(4carboxyphenyl)methyl]‐3,4‐dihydro‐5‐ iodo‐2,4‐dioxo‐1(2H)‐pyrimidinepropanoic acid.
The AMPAR antagonist GYKI53655 has been used extensively to block AMPAR currents to isolate those due to KARs [2]. However, GYKI53655 may marginally block homomeric and heteromeric KARs containing GluK2 and GluK3 at concentrations typically used to block AMPARs [13]. Another important KARs antagonist is LY382884 which is selective for GluK1 and has only weak activity at NMDARs but not at GluA1‐4, GluK2 or GluK3 receptors [13].
A series of KAR allosteric antagonists have also been developed [4]. These include 2‐arylureidobenzoic acids which block selectively and noncompetitively GluK1 (compound III) or both GluK1 and GluK2 (compound IV), while they do not affect AMPARs. Another noncompetitive antagonist is carboxy‐2,4‐di‐benzamido‐benzoic acid (NS3763) which blocks homomeric GluK1 receptors and has no activity on heteromeric GluK1/GluK2 or GluK1/GluK5 receptors, AMPARs or NMDARs [4].
KARs Physiology
Knowledge about the functional significance of KARs has benefited from the development of selective agonists and antagonists in conjunction with the generation of KAR subunit knockouts [2, 3]. A crucial hallmark for understanding the relevance of KARs to brain physiology was the finding of two 2,3‐benzodiazepines (GYKI52466 and GYKI53655) which preferably block AMPA receptors while having marginal effects on KARs [14, 15]. Using kainate as an agonist in the presence of these drugs to prevent AMPA receptor activation it was observed that KARs have a dual role in at hippocampal CA1 synapses depending on the concentration of the agonist [16]. Thus, high affinity KARs facilitate neurotransmission by regulating glutamate release at presynaptic sites whilst low affinity KARs mediate depression of synaptic activity in GABAergic terminals in CA1 (reviewed in Ref. [4]). In turn, KARs are present on presynaptic GABAergic terminals contacting interneurons and their activation increases GABA release which suggest that glutamate selectively control the communication between interneurons by increasing their mutual inhibition by means of KAIRs activation [17].
In addition, KARs are also located at postsynaptic sites whereby they contribute to excitatory postsynaptic potentials as assessed in the presence of AMPA and NMDA receptor antagonists. This contribution is particularly robust at CA3 pyramidal neurons mossy fiber stimulation in agreement with the presence of high concentrations of kainate binding sites (reviewed in Refs. [2, 18]). These pioneer findings were subsequently extended to other CNS regions.
Another turn in the goal to assess KAR function was the discovery of subunit‐selective agonists and antagonists. Thus, 5‐iodowilardine and ATP show preference for activating KARs containing homomeric or heteromeric with GluK1 in hippocampus, cerebral cortex, amigdala, thalamus, spinal cord and dorsal root ganglion neurons, as well as during brain development (reviewed in Refs. [2, 18]).
In addition, the development of LY382884, a selective GluK1 antagonist, has helped to elucidate the contribution of this subunit to native KARs function including its involvement in the induction of long‐term potentiation by mossy fibers in CA3 [19], although this view has later been contested, as well as the participation of this subunit in excitatory and inhibitory autorreceptors [20, 21]. Furthermore, LY382884 was instrumental in assessing the calcium permeability of GluK1‐containing KARs [22].
Learning about KARs in KAR Subunit Knock Out and Genetically Modified Mice
The use of KAR subunit knock out mice has also helped to understand the physiology of KARs. Heinemann and colleagues have generated viable mice that lack each of the KAR subunits and show an apparent normal phenotype ([23, 24, 25]; reviewed in Ref. [10]). GluK2 knock out mice do not have KAR‐mediated currents in hippocampal mossy fiber synapses and in cerebellar Golgi cells, and show reduced long‐term and short‐term plasticity at CA3. In addition, kainate‐mediated upregulation of GABA release on interneuron to interneuron contacts in the hippocampus is absent in these mice. In spite of those alterations, GluK2‐deficient mice display no learning deficits, sensorimotor, behavioral, or neuroanatomical abnormalities. On the other hand, GluK1‐knockout mice show subtle changes in glutamate signaling in that they lack KAR‐dependent currents in dorsal root ganglion neurons, and lack of glutamate release by presynaptic KARs is absent in primary sensory terminals of these mice. More recently, mice deficient for GluK5 revealed that this subunit significantly contributes to functional KARs on both sides of the mossy fiber synapse [24]. GluK3 and GluK4 knock out mice are also normal in appearance and little relevant information is known about them.
KARs signal through both ionotropic and metabotropic pathways. Strikingly, disruption of the Grik4 gene locus results in a significant reduction in synaptic KAR currents and ablation of GluK4 and GluK5 causes complete loss of synaptic ionotropic KAR function, whilst KAR‐mediated inhibition of the slow afterhyperpolarization current, which is dependent on metabotropic pathways, was intact in GluK4/GluK5 knockout mice [26]. These results uncovers the relevance of the high‐affinity subunits for ionotropic KARs function and further demonstrates that KARs participation in metabotropic signaling pathways does not require GluK4 and GluK5 subunits.
Studies on the significance of RNA editing of transcripts that code for KARs have taken advantage of the development of transgenic mice. Thus, mice with mutations at the Q/R‐editing site in the GluK1 subunit have much reduced KAR‐mediated current densities in dorsal root ganglia neurons, but these mice did not show altered responses to thermal and chemical pain stimuli, nor sensorimotor deficits [27]. In turn, mice that lack the GluK2 Q/R editing site have a phenotype showing a more easily induced NMDA‐independent long‐term potentiation and are more vulnerable to kainate‐induced seizures, but did not reveal behavioral differences as compared to wild‐type mice. Thus, it appears that the degree of calcium permeability in GluK2 receptors might modulate both seizure susceptibility and synaptic plasticity.
KARs and Cell Death
Kainate is a potent neuronal excitant and neurotoxin (reviewed in Ref. [28]). Systemic and intracerebral administration of kainate in adult rats induces persistent seizures, seizure‐mediated brain damage syndrome, and degeneration of neurons especially in striatal and hippocampal areas of the brain [29]. In contrast, axons and nerve terminals are more resistant to the destructive effects of kainate. These deleterious actions of kainate are mediated by both AMPA and KARs. In particular, it is important to emphasize that kainate induces nondesensitizing responses at AMPA receptors and that the transmembrane AMPA receptor‐associated protein (TARP) gamma‐2 (or stargazin) and the related TARP gamma‐8 augment responses to kainate and domoate by making these neurotoxins more potent and more efficacious AMPA receptor agonists [30]. Thus, agents interfering with TARPs and TARP‐related species hold neuroprotective value, but are not discuss further since they fall out of the scope of this review which is KARs.
The excitotoxic effects mediated by KARs in neurons have not been fully resolved because of the lack of appropriate selective ligands. Thus, virtually all neurotoxicity studies refer to neuronal toxicity mediated by non‐NMDA receptors or AMPA/KARs [31, 32]. Perhaps, the most relevant studies to KAR‐mediated neurotoxicity are those carried out using domoic acid which was identified as a potent neurotoxin involved in neuronal degeneration and atrophy [33]. KARs responses to domoic acid are characterized by large steady‐state currents and slow deactivation kinetics. Two residues in the GluK2 subunit, Met691 and Val685, are critical for domoic acid binding and for the kinetics and amplitude of the response to agonist, respectively [34]. Domoic acid toxicity results as a consequence of the formation of the retrograde messenger molecule nitric oxide and the production of free radicals, and it is prevented by melatonin as antioxidant in domoic acid‐induced neurotoxicity.
More recently, it was found that kainate in the presence of GYKI53655 (i.e., activating exclusively KARs) is also toxic to oligodendrocytes and myelin, and that the affinity of the KARs involved suggest that these deleterious effects are mediated by two distinct receptors with high and low affinity (Figure 2; [35]). Interestingly, the high‐affinity component shows a dose‐response relation similar to that of KAR‐mediated inhibition of GABA release in hippocampal slices [36]. The study of expression of glutamate receptor subunits in oligodendrocytes indicates that KARs involved in oligodendroglial excitotoxicity are composed of GluK2, GluK3, GluK4, and GluK5 subunits. Given the known affinities of recombinant KARs and the requirement for GluK2 and/or GluK3 to form functional receptors [2], it is likely that the GluK2 subunit is a major constituent of the higher affinity KARs. In addition, activation of low affinity KARs also irreversibly damaged oligodendrocytes. It thus seems that these toxic effects are mediated by receptors composed of GluK3 since the high concentration of kainate required to saturate the toxic effects is compatible with the affinity of GluK3 receptors observed in expression studies [37]. However, the precise contribution of each subunit to native receptors in oligodendrocytes that mediate cell death will require studies in single‐ and double‐subunit knockout mice.
Figure 2.
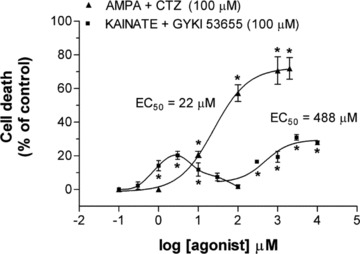
Concentration‐dependent toxicity curves for oligodendrocytes after activation of AMPA and kainate receptors. Three distinct receptor types trigger excitotoxicity: AMPA, and high‐ and low‐affinity kainate receptors. AMPA receptors were activated by AMPA applied in conjunction with cyclothiazide (100 μM). Selective activation of kainate receptors was achieved in the presence of GYKI5365. Adapted from Sánchez‐Gómez and Matute [35]. *P < 0.05.
Axons may also be vulnerable to excitotoxic insults since they express ionotropic glutamate receptors of the AMPA and kainate subtypes [7, 8]. Thus, electrophysiological recordings of the axon resting potential revealed that axons in the dorsal column of the spinal cord are depolarized via activation of AMPA receptors [38]. Consistent with these observations, central axons are damaged by activation of AMPA/KARs [39, 40], and protected by blockers of these receptors in models of white matter injury [41]. However, the lack of the specificity of the antagonists used in those studies prevented from clarifying the contribution of KARs to excitotoxic axonal damage and whether those deleterious effects are secondary to oligodendrocyte loss by excitotoxicity and the ensuing demyelination, rather than by activation per se of KARs in axons.
Relevance of KARs and their Antagonists to Disease and Therapeutics
Neurotoxicity studies suggest that KARs are relevant targets for neuroprotection of both neurons and glia in acute and chronic neurodegenerative diseases. In addition, KARs are also involved in epilepsy, pain and psychiatric disorders. Thus, there are many studies relating KARs to therapeutics in animal models of disease using drugs and genetic manipulations, as well as in genetic analyses of human disorders (2, 3, 4). Moreover, clinical data and detailed analyses of postmortem brain indicate that KARs are involved in CNS diseases (Table 4).
Table 2.
Kainate receptor antagonists in disease models
| Disease, animal model or damage | KAR antagonist | Receptor | Mechanism | Outcome | References |
|---|---|---|---|---|---|
| Ischemia preconditioning | NS102 | GluK2 | Inhibition of GluK2‐PSD assembly | Removes protection | [42] |
| Ischemia hypothermia | NS102 | GluK2 | Inhibition of GluK2‐PSD assembly | Removes protection | [43] |
| Isquemia | NS3763 | GluK1 | Attenuates GABA release | Worsening | [44] |
| Epilepsy | LY382884 | GluK1 | Attenuates GABA and glutamate release | Protection | [45] |
| Pain | LY382884 | GluK1 | Increased inhibiton in substantia gelatinosa | Analgesia | [46, 47] |
| Ischemia | LY377770 | GluK1 | n.d. | Protection | [48] |
| Epilepsy | LY377770 | GluK1 | Attenuates GABA and glutamate release | Protection | [45, 49] |
| Anxiety | LY382884 | GluK1 | n.d | Anxyolysis | [50] |
| EAE | NBQX | Native AMPA and KARs | Protection of oligodendrocytes and axons | Amelioration of symptoms | [51, 52] |
| Oligodendrocyte damage | CNQX | GluK2‐5 | Blockade in the presence of GYKI53655 | Protection of myelin | [35, 53] |
| Axonal damage | NBQX | Native AMPA and KARs | n.d. | Protection of axons | [54, 55, 56] |
| White matter damage | CNQX | Native AMPA and KARs | Reduced destruction of axons and myelin | Protection of tissue | [6, 39] |
n.d., not determined.
Table 3.
Kainate receptor knock out mice in disease models
| Disease, animal model or damage | Receptor | Mechanism | Outcome | Reference |
|---|---|---|---|---|
| Addiction | GluK1 KO | – | Lack of morphine tolerance | [57] |
| Anxiety | GluK1 KO | Reduced GABAergic transmission | Anxiety‐like behavior | [58] |
| Pain | GluK1 KO | – | Analgesia | [59] |
| Epilepsy | GluK2 KO | Reduced KARs at hippocampal mossy synapses | Reduced seizure sensitivity | [25] |
| Mood | GluK2 KO | Sensitive to lithium | mania | [60] |
| Memory | GluK2 KO | – | reduction in fear memory | [59] |
| Ischemia | GluK2 KO | – | Reduced damage | Gottlieb and Matute, unpublished |
| EAE | GluK2 KO | – | Amelioration of symptoms | [61] |
Table 4.
Kainate receptor alterations in human diseases
| Disease | Receptor | Method/Trial | Observation | Reference |
|---|---|---|---|---|
| Migraine pain | Native KARs | Phase II trial with tezampanel | Analgesia | [62] |
| Epilepsy | Native KARs | Clinical case | Domoic acid intoxication and hippocampal atrophy | [63] |
| Native KARs | Binding | Increased in hippocampus | [64] | |
| GluK1 | Genetic analysis | Allele confers risk to juvenile absence epilepsy | [65] | |
| GluK1 | Phase II trial with NS1209 | Alleviates refractory status epilepticus | [62] | |
| GluK1 | Immunohistochemistry | Increase expression in hippocampus | [66] | |
| Alzheimer's | GluK1‐3 | Immunohistochemistry | Lower expression in hippocampal CA1 region | [67] |
| Native KARs | Binding | Higher expression in deep cortical layers | [68] | |
| Native KARs | Binding | Reduced in parahippocampal areas | [69] | |
| Huntington's | Native KARs | Binding | Selective loss of receptors | [70] |
| GluK2 | Genetic analysis | SNP associated to younger onset age | [71, 72] | |
| Multiple sclerosis | GluK1‐3 | Immunohistochemistry | Presence in dystrophic axons | [73] |
| Schizophrenia | GluK1‐3 | Immunohistochemistry | Fewer receptor in prefrontal cortex | [74] |
| GluK4 | Genetic analysis | SNPs confer disease risk | [75] | |
| GluK3 | Genetic analysis | Gene copy variation | [76] | |
| GluK3 | Genetic analysis | GluK3 S310 is a risk allele | [77] | |
| Major depression | GluK3 | Genetic analysis | GluK3 S310 is a risk allele | [78] |
| Bipolar disorder | GluK4 | Genetic analysis | SNPs confers disease protection | [75] |
| GluK3 | Genetic analysis | Gene copy variation | [76] | |
| Mental retardation | GluK2 | Genetic analysis | Deletions and loss of function | [79] |
| Autism | GluK2 | Genetic analysis | Variant in C‐terminal | [80] |
Tezampel and NS1209 are AMPA/kainate receptor antagonists but their therapeutic actions in those particular conditions involve substantially kainate receptors.
Pain
Kainic acid activates nociceptors and consequently KAR antagonists have a potential as analgesics (reviewed in Ref. [18]). KARs are present in nociceptive tracts from dorsal roots, through thalamic neurones to sensory cortices. Blokade of GluK1 with LY382884 increases inhibitory activity in substantia gelatinosa and results in analgesia in the formalin model of chronic pain [46]. Similarly, nociceptive responses of spinothalamic neurones were reduced by the GluK1 antagonist, LY382884 in a primate model of neuropathic pain [47]. These data are in line with findings in GluK1‐deficient mice which show a higher pain threshold [59].
In spite of these well‐characterized properties of KARs, knowledge about these receptors has not been yet translated into therapeutics. However, current Phase II clinical trials using tezampel, an AMPA/receptor antagonist also named NGX424, reduced migraine pain, and effect which is likely attributable to the blockade of KARs based on preclinical evidence with more selective antagonists ([62]; see Table 2).
Epilepsy
Kainic acid is a classical convulsant [81, 82]. The development of selective antagonists of GluK1‐containing receptors has helped to assess the role of this subunit in animal models of epilepsy, neurodegeneration and pain. Thus, GluK1 antagonists LY37770 and LY382884 block epileptiform activity in hippocampal slices and seizures in vivo induced by pilocarpine or electrical stimulation [45, 49], a finding which is consistent with the elevated expression of GluK1 in the hippocampus of patients with temporal lobe epilepsy [66]. Moreover, GluK2‐deficient mice show reduced sensitivity to kainate‐induced seizures or associated cell death, though at high doses animals are indistinguishable from their wildtype counterparts [25]. Promisingly, the AMPA and GluK1 receptor antagonist NS1209 alleviated refractory status epilepticus in small Phase II studies but further research in this molecule was suspended [62]. Nonetheless, these clinical studies provided hints as to the relevance of this and related drugs for further development and clinical testing.
Temporal lobe epilepsy induces induces sprouting of glutamatergic mossy fibers of the hippocampus and aberrant synapses on granule cells from which they originate. KARs are involved in ongoing glutamatergic transmission in granule cells from chronic epileptic and provide a substantial component of glutamatergic activity [83]. Therefore, sprouting of mossy fibers induces a shift in the nature of glutamatergic transmission in granule cells with ectopic expression of KARs that may contribute to the physiopathology of the dentate gyrus in epileptic animals.
Additional evidence of the relevance of KARs to epilepsy was provided by clinical studies on domoic acid intoxication which resulted in seizures and the development of temporal lobe epilepsy 1 year later [63]. Since domoic acid is a more potent and possibly more selective activator of KARs, this clinical case provided a unique human parallel to animal studies of KAR‐induced epilepsy. Further information in humans supporting the involvement of KARs is provided in Table 4. Thus, KARs expression is increase in the hippocampus in patients with medial temporal lobe epilepsy suggesting that these receptors may be an important element in the pathophysiology of epilepsy [64]. Moreover, genetic studies in juvenile absence epilepsy, a common subtype of idiopathic generalized epilepsy, have shown an association between the disease and the presence of a tetranucleotide repeat polymorphism in the noncoding region of GluK1 which suggest that allelic variants in GluK1 confer genetic susceptibility to the pathogenesis of this type of epilepsy [65].
Stroke
GluK1 antagonist LY37770 provides protection in models of global and focal ischaemia [48]. Indeed, the degree of protection with LY37770 is greater than that reported for selective NMDA and AMPAR antagonists [48]. Importantly, this antagonist shows significant neuroprotection even later than 1 h after the onset of ischemia. However, at odds with those earlier findings recent data shows that the GluK1 antagonist NS3763 can worsen the outcome of ischemia by attenuating GABA release [44]. In line with this report, NS102 KAR antagonist removes the protective effects of ischemia preconditioning or hypothermia during ischemic insults [42, 43].
Neurodegenerative Diseases
KARs are also involved in neurodegenerative disorders including Alzheimer's, Huntington's and multiple sclerosis. In contrast, no clear evidence of KARs deregulation is available for Parkinson's disease and cerebellar ataxias despite some recent suggestions [84, 85].
In Alzheimer's disease, kainate binding is significantly reduced in the parahippocampal gyrus [69] and the immunoreactivity of anti‐GluK1‐3 antibodies is diminished in the CA1 hippocampal area [67]. In contrast, KAR binding is increased in deep layers of the frontal cerebral cortex in postmortem brain from Alzheimer's disease patients, and notably, that increase correlates with the plaque loading [68].
The role of KARs in Huntington's disease was suggested by early observations showing that kainic acid lesioning produces a similar pattern of neurodegeneration in the striatum [29], the CNS region most affected in this disease state. Consistent with this observation, selective loss of [3H] kainic acid binding sites has been reported in another study examining postmortem brains with Huntington's disease [70]. More recently, genetic studies have shown that variants of GluK2 contribute to the early‐onset Huntington's disease [71, 72].
Strong evidence supporting a role for KARs is available for cellular and animal models of multiple sclerosis, and for the disease proper. Thus, selective activation of KARs damages oligodendrocytes in vitro and in vivo[35, 39] and sensitizes oligodendrocytes to complement attack [53]. In the latter contribution, complement toxicity was abolished by removing calcium from the medium during glutamate priming, and it was induced by two distinct KAR populations displaying high and low affinities for glutamate. Moreover, toxicity after priming KARs required the formation of the membrane attack complex, which in turn increased membrane conductance and caused calcium overload and mitochondrial depolarization as well as a rise in the level of reactive oxygen species. Treatment with the antioxidant Trolox and inhibition of poly(ADP‐ribose) polymerase‐1, but not of caspases, protected oligodendrocytes against damage induced by complement [53]. These findings indicate that glutamate sensitization to complement attack via activation of KARs in oligodendrocytes may contribute to white matter damage in acute and chronic neurological disorders.
These features suggest that KARs are relevant targets for white matter disorders including multiple sclerosis, as demonstrated for AMPA receptors which mediate myelin damage in experimental autoimmune encephalomyelitis, a model of that disease [51, 52]. Indeed, motor symptoms are less severe in GluK2‐deficient than in wild type mice after induction of experimental autoimmune encephalomyelitis [61]. Finally, AMPA/KAR antagonist may also protect axons from damage by interacting with KARs present along the axonal fibers [39, 54, 55, 56]. Indeed, dystrophic axons expressing KARs have been observed in postmortem brain from multiple sclerosis patients [73].
Together, these data on neurodegenerative diseases hold great promise for GluK antagonists as neuroprotective agents. However, it should be noted that other glutamate receptor antagonists, mostly of the NMDA type, which showed great protective activity in experimental settings failed to deliver significant clinical results. An exception to this bleak situation is memantine which is currently being used to treat advanced Alzheimer's disease with little therapeutic improvement. A major reason for these setbacks relates to the fact that blockade of any kind of glutamate receptors is bound to have side effects due to the variety of functions subserved by these receptors. In the case of KARs, it is important to consider that they have crucial modulatory effects on synaptic transmission and that this feature may compromise the therapeutic value of their antagonists. Highly subunit‐specific GluK antagonists can minimize undesirable interactions.
Psychiatric Diseases
KARs are also relevant to psychiatric diseases as demonstrated in human genetic and post‐mortem brain studies. These include schizophrenia, bipolar disease, major depression, autism, and obsessive‐compulsive disorder (Table 4). Thus, the expression of GluK1‐3 is reduced in the orbitofrontal cortex of patients with schizophrenia [74]. Moreover, multiple genetic studies have associated polymorphic variants of the GluK2 subunit to schizophrenia, autism and obsessive‐compulsive disorder [86]. Also, genetic variants of the GluK3 subunit have been associated to schizophrenia, bipolar disorder and major depression (see Table 4).
Intriguingly, a single nucleotide polymorphism in the GluK4 subunit confers protection against bipolar disorder [75]. In turn, animal experiments GluK2 knock out mice show a mania‐like phenotype which is attenuated with lithium and a reduction in fear memory [59, 60]. These exciting findings indicate that GluK2 antagonists have a great potential to treat bipolar disorder and that overall, KARs are relevant to therapeutic intervention in this disease.
KARs have also therapeutic potential regarding anxiety treatment and addition. Thus, studies with KAR antagonists and KAR subunit‐deficient mice (Tables 2 and 3) have shown that blockade of GluK1 with LY382884 has an anxiolytic effect [50]. However, GluK1‐deficient mice display an anxiety‐like behavior as a consequence of reduced GABAergic transmission [58]. Finally, mice lacking GluK1 display a reduced tolerance to morphine [57] a feature which has relevance to additive behaviors. However, despite these interesting data in animal experiments, there are not as yet clinical correlates of these findings.
Other findings of relevance to on KARs genetics include a complex mutation in GluK2 that cosegregates with moderate‐to‐severe nonsyndromic autosomal recessive mental retardation in a large, consanguineous Iranian family [79]. The predicted gene product lacks the first ligand‐binding domain, the adjacent transmembrane domain, and the putative pore loop, suggesting a complete loss of function of the GluK2 protein. This finding provides relevant support ot the idea that KARs are indispensable for higher brain functions in humans, and that they may be relevant to the pathophysiology of mental retardation.
Finally, transmission disequilibrium test to investigate the linkage and association between GluR6 and autism proved that this receptor is linked to the disease [80]. Importantly, mutation screening in affected individuals, revealed several nucleotide polymorphisms, including one amino acid change (M867I) in a highly conserved domain of the intracytoplasmic C‐terminal region of the protein [80]. This striking finding suggests that GluK2 mutation may contribute to the genetic background of autism.
Conclusions
Overall, the data summarized above reveals that KARs are dysregulated in neurological and psychiatric diseases and that these receptors have emerged in recent years as relevant therapeutic targets. The long awaited development of novel subunit specific antagonists will surely prove critical to translate this potential into clinical applications. In turn, defining more precisely the contribution of KARs to disease will help to develop new therapeutic avenues targeting these receptors. However, it is not obvious currently how the knowledge obtained about the reported genetic alterations in KARs can be directly translated into therapeutics.
Conflict of Interest
The author has no conflict of interest.
Acknowledgments
Work in my laboratory is supported by CIBERNED, Ministerio de Ciencia e Innovación, the European Leukodystrophy Association, Gobierno Vasco and Universidad del País Vasco.
References
- 1. Lodge D. The history of the pharmacology and cloning of ionotropic glutamate receptors and the development of idiosyncratic nomenclature. Neuropharmacology 2009;56:6–21. [DOI] [PubMed] [Google Scholar]
- 2. Lerma J. Kainate receptor physiology. Curr Opin Pharmacol 2006;6:89–97. [DOI] [PubMed] [Google Scholar]
- 3. Vincent P, Mulle C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience 2009;158:309–323. [DOI] [PubMed] [Google Scholar]
- 4. Jane DE, Lodge D, Collingridge GL. Kainate receptors: Pharmacology, function and therapeutic potential. Neuropharmacology 2009;56:90–113. [DOI] [PubMed] [Google Scholar]
- 5. Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand‐gated ion channels. Neuropharmacology 2009;56:2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matute C, Domercq M, Sanchez‐Gomez MV. Glutamate‐mediated glial injury: Mechanisms and clinical importance. Glia 2006;53:212–224. [DOI] [PubMed] [Google Scholar]
- 7. Ouardouz M, Coderre E, Basak A. Glutamate receptors on myelinated spinal cord axons: I. GluR6 kainate receptors. Ann Neurol 2009;65:151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ouardouz M, Coderre E, Zamponi GW, Hameed S, Yin X, Trapp BD, Stys PK. Glutamate receptors on myelinated spinal cord axons: II. AMPA and GluR5 receptors. Ann Neurol 2009;65:160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. More JC, Troop HM, Dolman NP, Jane DE. Structural requirements for novel willardiine derivatives acting as AMPA and kainate receptor antagonists. Br J Pharmacol 2003;138:1093–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lerma J. Roles and rules of kainate receptors in synaptic transmission. Nat Rev Neurosci 2003;4:481–495. [DOI] [PubMed] [Google Scholar]
- 11. Christensen JK, Paternain AV, Selak S, Ahring PK, Lerma J. A mosaic of functional kainate receptors in hippocampal interneurons. J Neurosci 2004;24:8986–8993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Valgeirsson J, Nielsen EO, Peters D, Mathiesen C, Kristensen AS, Madsen U. Bioisosteric modifications of 2‐arylureidobenzoic acids: Selective noncompetitive antagonists for the homomeric kainate receptor subtype GluR5. J Med Chem 2004;47:6948–6957. [DOI] [PubMed] [Google Scholar]
- 13. Perrais D, Pinheiro PS, Jane DE, Mulle C. Antagonism of recombinant and native GluR7‐containing kainate receptors. Neuropharmacology 2009;56:131–140. [DOI] [PubMed] [Google Scholar]
- 14. Paternain AV, Morales M, Lerma J. Selective antagonism of AMPA receptors unmasks kainate receptor‐mediated responses in hippocampal neurons. Neuron 1995;14:185–189. [DOI] [PubMed] [Google Scholar]
- 15. Wilding TJ, Huettner JE. Differential antagonism of alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid‐preferring and kainate‐preferring receptors by 2,3‐benzodiazepines. Mol Pharmacol 1995;47:582–587. [PubMed] [Google Scholar]
- 16. Chittajallu R, Vignes M, Dev KK, Barnes JM, Collingridge GL, Henley JM. Regulation of glutamate release by presynaptic kainate receptors in the hippocampus. Nature 1996;379:78–81. [DOI] [PubMed] [Google Scholar]
- 17. Cossart R, Tyzio R, Dinocourt C, Esclapez M, Hirsch JC, Ben‐Ari Y, Bernard C. Presynaptic kainate receptors that enhance the release of GABA on CA1 hippocampal interneurons. Neuron 2001;29:497–508. [DOI] [PubMed] [Google Scholar]
- 18. Pinheiro P, Mulle C. Kainate receptors. Cell Tissue Res 2006;326:457–482. [DOI] [PubMed] [Google Scholar]
- 19. Bortolotto ZA, Clarke VRJ, Delany CM, et al Kainate receptors are envolved in synaptic plasticity. Nature 1999;402:297–301. [DOI] [PubMed] [Google Scholar]
- 20. Lauri SE, Bortolotto ZA, Bleakman D, Ornstein PL, Lodge D, Isaac JT, Collingridge GL. A critical role of a facilitatory presynaptic kainate receptor in mossy fiber LTP. Neuron 2001;32:697–709. [DOI] [PubMed] [Google Scholar]
- 21. Kidd FL, Coumis U, Collingridge GL, Crabtree JW, Isaac JT. A presynaptic kainate receptor is involved in regulating the dynamic properties of thalamocortical synapses during development. Neuron 2002;34:635–646. [DOI] [PubMed] [Google Scholar]
- 22. Mathew SS, Hablitz JJ. Calcium release via activation of presynaptic IP3 receptors contributes to kainate‐induced IPSC facilitation in rat neocortex. Neuropharmacology 2008;55:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short‐ and long‐term plasticity atmossy fiber synapses in the hippocampus. Neuron 2001;29:209–216. [DOI] [PubMed] [Google Scholar]
- 24. Contractor A, Sailer AW, Darstein M, Maron C, Xu J, Swanson GT, Heinemann SF. Loss of kainate receptor‐mediated heterosynaptic facilitation of mossy‐fiber synapses in KA2‐/‐ mice. J Neurosci 2003;23:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mulle C, Sailer A, Pérez‐Otaño I, et al Altered synaptic physiology and reduced susceptibility to kainate‐induced seizures in GluR6‐deficient mice. Nature 1998;392:601–605. [DOI] [PubMed] [Google Scholar]
- 26. Fernandes HB, Catches JS, Petralia RS, et al High‐affinity kainate receptor subunits are necessary for ionotropic but not metabotropic signaling. Neuron 2009;63:818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sailer A, Swanson GT, Pérez‐Otaño I, et al Generation and analysis of GluR5(Q636R) kainate receptor mutant mice. J Neurosci 1999;19:8757–8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Farooqui AA, Yi Ong W, Lu XR, Halliwell B, Horrocks LA. Neurochemical consequences of kainate‐induced toxicity in brain: Involvement of arachidonic acid release and prevention of toxicity by phospholipase A(2) inhibitors. Brain Res Rev 2001;38:61–78. [DOI] [PubMed] [Google Scholar]
- 29. Coyle JT. Neurotoxic action of kainic acid. J. Neurochem 1983;41:1–11. [DOI] [PubMed] [Google Scholar]
- 30. Tomita S, Byrd RK, Rouach N, et al AMPA receptors and stargazin‐like transmembrane AMPA receptor‐regulatory proteins mediate hippocampal kainate neurotoxicity. Proc Natl Acad Sci U S A 2007;104:18784–18788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choi DW. Calcium: Still center‐stage in hypoxic‐ischemic neuronal death. Trends Neurosci 1995;18:58–60. [PubMed] [Google Scholar]
- 32. Arundine M, Tymianski M. Molecular mechanisms of calcium‐dependent neurodegeneration in excitotoxicity. Cell Calcium 2003;34:325–337. [DOI] [PubMed] [Google Scholar]
- 33. Chandrasekaran A, Ponnambalam G, Kaur C. Domoic acid‐induced neurotoxicity in the hippocampus of adult rats. Neurotox Res 2004;6:105–117. [DOI] [PubMed] [Google Scholar]
- 34. Zhang Y, Nayeem N, Green T. Mutations to the kainate receptor subunit GluR6 binding pocket that selectively affect domoate binding. Mol Pharmacol 2008;74:1163–1169. [DOI] [PubMed] [Google Scholar]
- 35. Sánchez‐Gómez MV, Matute C. AMPA and kainate receptors each mediate excitotoxicity in oligodendroglial cultures. Neurobiol Dis 1999;6:475–485. [DOI] [PubMed] [Google Scholar]
- 36. Rodríguez‐Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron 1998;20:1211–1218. [DOI] [PubMed] [Google Scholar]
- 37. Schiffer HH, Swanson GT, Heinemann SF. Rat GluR7 and a carboxyterminal splice variant, GluR7b, are functional kainate receptor subunits with a low sensitivity to glutamate. Neuron 1997;19:1141–1146. [DOI] [PubMed] [Google Scholar]
- 38. Ouardouz M, Malek S, Coderre E, Stys PK. Complex interplay between glutamate receptors and intracellular Ca2+ stores during ischaemia in rat spinal cord white matter. J Physiol 2006;577:191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Matute C. Characteristics of acute and chronic kainate excitotoxic damage to the optic nerve. Proc Natl Acad Sci USA 1998;95:10229–10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fowler JH, McCracken E, Dewar D, McCulloch J. Intracerebral injection of AMPA causes axonal damage in vivo. Brain Res 2003;991:104–112. [DOI] [PubMed] [Google Scholar]
- 41. Tekkök SB, Goldberg MP. AMPA/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci 2001;21:4237–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Du Y, Li C, Hu WW, Song YJ, Zhang GY. Neuroprotection of preconditioning against ischemic brain injury in rat hippocampus through inhibition of the assembly of GluR6‐PSD95‐mixed lineage kinase 3 signaling module via nuclear and non‐nuclear pathways. Neuroscience 2009;161:370–380. [DOI] [PubMed] [Google Scholar]
- 43. Hu WW, Du Y, Li C, Song YJ, Zhang GY. Neuroprotection of hypothermia against neuronal death in rat hippocampus through inhibiting the increased assembly of GluR6‐PSD95‐MLK3 signaling module induced by cerebral ischemia/reperfusion. Hippocampus 2008;18:386–397. [DOI] [PubMed] [Google Scholar]
- 44. Xu J, Liu Y, Zhang GY. Neuroprotection of GluR5‐containing kainate receptor activation against ischemic brain injury through decreasing tyrosine phosphorylation of N‐methyl‐D‐aspartate receptors mediated by Src kinase. J Biol Chem 2008;283:29355–29366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smolders I, Bortolotto ZA, Clarke VR, et al. Antagonists of GLU(K5)‐containing kainate receptors prevent pilocarpine‐induced limbic seizures. Nat Neurosci 2002;5:796–804. [DOI] [PubMed] [Google Scholar]
- 46. Simmons RM, Li DL, Hoo KH, Deverill M, Ornstein PL, Iyengar S. Kainate GluR5 receptor subtype mediates the nociceptive response to formalin in the rat. Neuropharmacology 1998;37:25–36. [DOI] [PubMed] [Google Scholar]
- 47. Palecek J, Neugebauer V, Carlton SM, Iyengar S, Willis WD. The effect of a kainate GluR5 receptor antagonist on responses of spinothalamic tract neurons in a model of peripheral neuropathy in primates. Pain 2004;111:151–161. [DOI] [PubMed] [Google Scholar]
- 48. O'Neill MJ, Bogaert L, Hicks CA, et al LY377770, a novel iGlu5 kainate receptor antagonist with neuroprotective effects in global and focal cerebral ischaemia. Neuropharmacology 2000;39:1575–1588. [DOI] [PubMed] [Google Scholar]
- 49. Barton ME, Peters SC, Shannon HE. Comparison of the effect of glutamate receptor modulators in the 6 Hz and maximal electroshock seizure models. Epilepsy Res 2003;56:17–26. [DOI] [PubMed] [Google Scholar]
- 50. Alt A, Weiss B, Ornstein PL, Gleason SD, Bleakman D, Stratford RE Jr, Witkin JM. Anxiolytic‐like effects through a GLUK5 kainate receptor mechanism. Neuropharmacology 2007;52:1482–1487. [DOI] [PubMed] [Google Scholar]
- 51. Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 2000;6:67–70. [DOI] [PubMed] [Google Scholar]
- 52. Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med 2000;6:62–66 [DOI] [PubMed] [Google Scholar]
- 53. Alberdi E, Sanchez‐Gomez MV, Torre I, Domercq M, Perez‐Samartin A, Perez‐Cerda F, Matute C. Activation of kainate receptors sensitizes oligodendrocytes to complement attack. J Neurosci 2006;26:3220–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Alix JJ, Fern R. Glutamate receptor‐mediated ischemic injury of premyelinated central axons. Ann Neurol 2009;66:682–693. [DOI] [PubMed] [Google Scholar]
- 55. Baltan S, Besancon EF, Mbow B, Ye Z, Hamner MA, Ransom BR. White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. J Neurosci 2008;28:1479–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci 2005;233:3–13. [DOI] [PubMed] [Google Scholar]
- 57. Bogulavsky JJ, Gregus AM, Kim PT, Costa AC, Rajadhyaksha AM, Inturrisi CE. Deletion of the glutamate receptor 5 subunit of kainate receptors affects the development of morphine tolerance. J Pharmacol Exp Ther 2009;328:579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu LJ, Ko SW, Toyoda H, et al Increased anxiety‐like behavior and enhanced synaptic efficacy in the amygdala of GluR5 knockout mice. PLoS One 2007;2:e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ko S, Zhao MG, Toyoda H, Qiu CS, Zhuo M. Altered behavioral responses to noxious stimuli and fear in glutamate receptor 5 (GluR5)‐ or GluR6‐deficient mice. J Neurosci 2005;25:977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shaltiel G, Maeng S, Malkesman O, et al Evidence for the involvement of the kainate receptor subunit GluR6 (GRIK2) in mediating behavioral displays related to behavioral symptoms of mania. Mol Psychiatry 2008;13:858–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pérez‐Samartín A, Pérez‐Cerdá F, Matute C. 2009 Methodological tools for research in multiple sclerosis. Eur J Anat 2009;13(Suppl 1):27 (Abstract). [Google Scholar]
- 62. Swanson GT. Targeting AMPA and kainate receptors in neurological disease: Therapies on the horizon? Neuropsychopharmacology 2009;34:249–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cendes F, Andermann F, Carpenter S, Zatorre RJ, Cashman NR. Temporal lobe epilepsy caused by domoic acid intoxication: Evidence for glutamate receptor‐mediated excitotoxicity in humans. Ann Neurol 1995;37:123–126. [DOI] [PubMed] [Google Scholar]
- 64. Brines ML, Sundaresan S, Spencer DD, de Lanerolle NC. Quantitative autoradiographic analysis of ionotropic glutamate receptor subtypes in human temporal lobe epilepsy: Up‐regulation in reorganized epileptogenic hippocampus. Eur J Neurosci 1997;9:2035–2044. [DOI] [PubMed] [Google Scholar]
- 65. Sander T, Hildmann T, Kretz R, et al Allelic association of juvenile absence epilepsy with a GluR5 kainate receptor gene (GRIK1) polymorphism. Am J Med Genet 1997;74:416–421. [PubMed] [Google Scholar]
- 66. Li JM, Zeng YJ, Peng F, et al Aberrant glutamate receptor 5 expression in temporal lobe epilepsy lesions. Brain Res 2010;1311:166–174. [DOI] [PubMed] [Google Scholar]
- 67. Aronica E, Dickson DW, Kress Y, Morrison JH, Zukin RS. Non‐plaque dystrophic dendrites in Alzheimer hippocampus: A new pathological structure revealed by glutamate receptor immunocytochemistry. Neuroscience 1998;82:979–991. [DOI] [PubMed] [Google Scholar]
- 68. Chalmers DT, Dewar D, Graham DI, Brooks DN, McCulloch J. Differential alterations of cortical glutamatergic binding sites in senile dementia of the Alzheimer type. Proc Natl Acad Sci USA 1990;87:1352–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dewar D, Chalmers DT, Graham DI, McCulloc J. Glutamate metabotropic and AMPA binding sites are reduced in Alzheimer's disease: An autoradiographic study of the hippocampus. Brain Res 1991;553:58–64. [DOI] [PubMed] [Google Scholar]
- 70. Wagster MV, Hedreen JC, Peyser CE, Folstein SE, Ross CA. Selective loss of [3H]kainic acid and [3H]AMPA binding in layer VI of frontal cortex in Huntington's disease. Exp Neurol 1994;127:70–75. [DOI] [PubMed] [Google Scholar]
- 71. Rubinsztein DC, Leggo J, Chiano M, Dodge A, Norbury G, Rosser E, Craufurd D. Genotypes at the GluR6 kainate receptor locus are associated with variation in the age of onset of Huntington disease. Proc Natl Acad Sci U S A 1997;94:3872–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. MacDonald ME, Vonsattel JP, Shrinidhi J, et al Evidence for the GluR6 gene associated with younger onset age of Huntington's disease. Neurology 1999;53:1330–1332. [DOI] [PubMed] [Google Scholar]
- 73. Newcombe J, Uddin A, Dove R, Patel B, Turski L, Nishizawa Y, Smith T. Glutamate receptor expression in multiple sclerosis lesions. Brain Pathol 2008;18:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Garey LJ, Von Bussmann KA, Hirsch SR. Decreasednumerical density of kainate receptor‐positive neurons in the orbitofrontal cortex of chronic schizophrenics. Exp Brain Res 2006;173:234–242. [DOI] [PubMed] [Google Scholar]
- 75. Pickard BS, Malloy MP, Christoforou A, et al Cytogenetic and genetic evidence supports a role for the kainate‐type glutamate receptor gene, GRIK4, in schizophrenia and bipolar disorder. Mol Psychiatry 2006;11:847–857. [DOI] [PubMed] [Google Scholar]
- 76. Wilson GM, Flibotte S, Chopra V, Melnyk BL, Honer WG, Holt RA. DNA copy number analysis in bipolar disorder and schizophrenia reveals aberrations in genes involved in glutamate signaling. Hum Mol Genet 2006;15:743–749. [DOI] [PubMed] [Google Scholar]
- 77. Begni S, Popoli M, Moraschi S, Bignotti S, Tura GB, Gennarelli M. Association between the ionotropic glutamate receptor kainate 3 (GRIK3) ser310ala polymorphism and schizophrenia. Mol Psychiatry 2002;7:416–418. [DOI] [PubMed] [Google Scholar]
- 78. Schiffer HH, Heinemann SF. Association of the human kainate receptor GluR7 gene (GRIK3) with recurrent major depressive disorder. Am J Med Genet B Neuropsychiatr Genet 2007;144:20–26. [DOI] [PubMed] [Google Scholar]
- 79. Motazacker MM, Rost BR, Hucho T, et al A defect in the ionotropic glutamate receptor 6 gene (GRIK2) is associated with autosomal recessive mental retardation. Am J Hum Genet 2007;81:792–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jamain S, Betancur C, Quach H, et al Paris Autism Research International Sibpair (PARIS) Study. Linkage and association of the glutamate receptor 6 gene with autism. Mol Psychiatry 2002;7:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Olney JW. Excitatory transmitters and epilepsy‐related brain damage. Int Rev Neurobiol 1985;27:337–362. [DOI] [PubMed] [Google Scholar]
- 82. Coyle JT. Kainic acid: Insights into excitatory mechanisms causing selective neuronal degeneration. Ciba Found Symp 1987;126:186–203. [DOI] [PubMed] [Google Scholar]
- 83. Epsztein J, Represa A, Jorquera I, Ben‐Ari Y, Crépel V. Recurrent mossy fibers establish aberrant kainate receptor‐operated synapses on granule cells from epileptic rats. J Neurosci 2005;25:8229–8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Luquin MR, Saldise L, Guillén J, et al Does increased excitatory drive from the subthalamic nucleus contribute to dopaminergic neuronal death in Parkinson's disease? Exp Neurol 2006;201:407–415. [DOI] [PubMed] [Google Scholar]
- 85. Carlson KM, Andresen JM, Orr HT. Emerging pathogenic pathways in the spinocerebellar ataxias. Curr Opin Genet Dev 2009;19:247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bowie D. Ionotropic glutamate receptors & CNS disorders. CNS Neurol Disord Drug Targets 2008;7:129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]