

Abstract
The hepatotoxicity of bromobenzene (BB) is directly related to the covalent binding of both initially formed epoxide and secondary quinone metabolites to at least 45 different liver proteins. 4-Bromophenol (4BP) is a significant BB metabolite and a precursor to reactive quinone metabolites, yet when administered exogenously it has negligible hepatotoxicity compared to BB. The protein adducts of 4BP were thus labeled as non-toxic (Monks, T. J.; Hinson, J. A.; Gillette, J. R. (1982) Life Sci. 30, 841–848). To help identify which BB-derived adducts might be related to its cytotoxicity, we sought to identify the supposedly non-toxic adducts of 4BP and eliminate them from the BB target protein list. Administration of [14C]-4BP to phenobarbital-induced rats resulted in covalent binding of 0.25, 0.33 and 0.42 nmol-eq 4BP/mg protein in the mitochondrial, microsomal and cytosolic fractions, respectively. These values may be compared to published values of 3–6 nmol/mg protein from a comparable dose of [14C]-BB. After subcellular fractionation and 2D electrophoresis, 47 radioactive spots on 2D gels of the mitochondrial, microsomal and cytosolic fractions were excised, digested and analyzed by LC-MS/MS. Twenty nine of these spots contained apparently single proteins, of which 14 were non-redundant. Nine of the 14 are known BB targets. Incubating freshly-isolated rat hepatocytes with 4BP (0.1–0.5 mM) produced time- and concentration-dependent increases in lactate dehydrogenase release and changes in cellular morphology. LC-MS/MS analysis of the cell culture medium revealed rapid and extensive sulfation and glucuronidation of 4BP as well as formation of a quinone-derived glutathione conjugate. Studies with 7-hydroxycoumarin (7HC), (−)-borneol or D-(+)-galactosamine (DGN) showed that inhibiting the glucuronidation/sulfation of 4BP increased the formation of a GSH-bromoquinone adduct, increased covalent binding of 4BP to hepatocyte proteins and potentiated its cytotoxicity. Taken together, our data demonstrate that protein adduction by 4BP metabolites can be toxicologically consequential, and provide a mechanistic explanation for the failure of exogenously administered 4BP to cause hepatotoxicity. Thus the probable reason for the low toxicity of 4BP in vivo is that rapid conjugation limits its oxidation and covalent binding and thus its toxicity.
Introduction
The hepatotoxicity of bromobenzene (BB), first reported in 1935,1 was shown by Brodie et al.2 to stem from the covalent binding of chemically reactive metabolites to hepatocellular proteins. Subsequent work in several laboratories demonstrated that pretreatment of rats with phenobarbital accelerates BB metabolism and its glutathione-depleting activity, increases the rate and extent of its protein covalent binding and potentiates its hepatotoxicity both in vivo and in isolated rat hepatocytes.3–6 Based on the structures of known stable metabolites of BB (Scheme 1) it was proposed that BB-3,4-oxide (2) was the critical reactive intermediate that covalently bound to proteins leading to cytotoxicity. Although BB-3,4-oxide has never been isolated or synthesized, its intermediacy in BB metabolism and covalent binding is strongly supported by the isolation of 3–7 as BB metabolites.7–9 Analogous studies with chlorobenzene10 and naphthalene11, 12 have provided additional support for arene oxides as critical cytotoxic metabolites.
Scheme 1.

Metabolism and covalent binding of bromobenzene and 4-bromophenol.
4-Bromophenol (4BP) is a major metabolite of bromobenzene.7 It is derived primarily by the rapid non-enzymatic rearrangement of BB-3,4-oxide13 Microsomal metabolism studies of 4BP,14 naphthol15 and phenol16 have shown that phenols are efficiently activated to protein covalent binding species, probably quinones. These observations naturally raised a question about the relative ability of quinone binding vs. epoxide binding to cause cytotoxicity. To address this question, Monks et al.17 compared the protein covalent binding and hepatotoxicity of BB vs. 4BP in non-induced rats. They found that at equimolar doses, 4BP bound 62% as much as BB, yet only BB caused hepatotoxicity. From this they concluded that the hepatotoxic effects of BB derive mainly from its 3,4-oxide metabolite and that 4BP metabolites were non-toxic despite their ability to covalently modify proteins.
From the 1970s through the 1990s, as the amount of information linking covalent binding to cytotoxicity grew, attention began to shift away from the identity and chemistry of small molecule electrophiles toward the identity and significance of the individual protein targets to which they bind.18 In an attempt to gain insight into cellular mechanisms of reactive metabolite toxicity, our laboratory has identified numerous hepatocellular proteins that become covalently labeled by metabolites of [14C]-BB.19–21 It was thus of interest to identify proteins targeted by reactive metabolites of 4BP since these adduction events are not associated with hepatotoxic consequences.17 It was thought that their identification as a specific subset of proteins targeted by BB metabolites could help focus attention on those targets that are unique to BB and thus more likely to be more directly associated with cytotoxicity.
To this end we treated phenobarbital-induced rats with [14C]-4BP, isolated liver subcellular fractions, separated radioactive proteins by 2D gel electrophoresis, and identified the proteins in radioactive spots by tryptic digestion and mass spectrometry. Although the total amount of [14C]-4BP binding was low compared to that usually observed with BB, we identified 29 proteins (14 non-redundant) in single-protein spots, nine of which were previously known BB targets. The unexpectedly low binding of 4BP metabolites in vivo prompted us to re-examine the earlier literature on this subject and to re-evaluate the relative toxicity of BB and 4BP experimentally using isolated hepatocytes. Our observations suggest that the low toxicity of 4BP may actually stem from its much lower level of metabolic activation and covalent binding relative to BB, and that this in turn may be due to the rapid removal of 4BP via glucuronidation and sulfation. In support of this hypothesis we demonstrate herein that when the latter processes are inhibited, the covalent binding and cytototoxicity of 4BP are both significantly potentiated.
Materials and Methods
Materials
4-Bromophenol, (−)-borneol, D-(+)-galactosamine (DGN), umbelliferone (7-hydroxycoumarin, 7HC), 4-vinylpyridine, phenylmethylsulfonyl fluoride (PMSF), collagenase type IV and Percoll were from Sigma-Aldrich (www.sigmaaldrich.com/). HPLC grade solvents and analytical grade inorganic salts were obtained from Fisher (www.fishersci.com). Dulbecco’s Modified Eagle Medium (DMEM) and phosphate buffered saline (PBS, pH 7.4) were purchased from Cellgro (www.cellgro.com), Williams’ Medium E (WME) was purchased from Gibco-Invitrogen (www.invitrogen.com), and lactate dehydrogenase (LDH) assay kits were purchased from Takara-Bio (www.clontech.com/takara). Sequencing grade trypsin was obtained from Roche (www.rocheusa.com). Sequenal grade urea and CHAPS were obtained from Pierce Biotechnology (www.piercenet.com). Tris, SDS, glycine, Sequi-blot PVDF membranes (0.2 μm), Bradford reagent, broad range isoelectric focusing (IEF) Standards and Precision Protein Standards were obtained from Bio-Rad (www.bio-rad.com). All other electrophoresis supplies were obtained from GE-HealthCare (www.gehealthcare.com). Deionized water (resistivity 18.2 MΩ/cm) was used for preparation of all solutions and buffers. [U-14C]-4BP (42.6 Ci/mol, >98% radiochemical purity) was kindly provided by Dr. Larry Hall (Bayer Crop Science) and mixed with unlabeled 4BP to a final specific activity of 8.05 Ci/mol. Chromatography paper 3MM was purchased from Whatman (www.whatman.com).
Animals
All animal husbandry protocols were in accordance with the NIH Guide for the Care and Use of Laboratory Animals.22 Experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Kansas. Male Sprague-Dawley rats from Charles River Laboratories (www.criver.com) were housed in a temperature and humidity controlled room with a 12 h light/dark cycle and ad libitum access to food and water.
Preparation of Subcellular Fractions of Liver Tissue
After acclimating for at least 3 days, animals were given 3 daily ip injections of sodium phenobarbital (80 mg/kg) in 0.9% saline (1.0 mL/kg) in order to induce cytochrome P450 2B1/2 and increase 4BP metabolism and the yield of covalent adducts. After the third dose the animals weighed 224–268 g. Food was withheld overnight and the next morning four animals were injected with [14C]-4BP (8.05 mCi/mmol, 1 mmol/kg, ip) in corn oil (2.4 mL/kg). Five hr after injection the animals were anesthetized by CO2 narcosis and killed by decapitation. Their livers (41 g wet weight total) were removed and subcellular fractions isolated essentially as described by Koen et al.19, 20 The livers were rinsed with ice-cold buffer A (50 mM potassium phosphate buffer, pH 7.4, containing 0.15 M KCl, 5 mM EDTA and 1 mM phenylmethanesulfonyl fluoride), weighed, and then minced with scissors and homogenized in buffer A (4 mL/g tissue). The homogenates were pooled and centrifuged at 800 × g for 10 min. The resulting pellet was discarded, and the supernatant was further centrifuged at 9,000 × g for 20 min, to give a pellet (P9-fraction or crude mitochondria) and supernatant S9. The P9-fraction was resuspended in buffer A and centrifuged at 6,500 × g for 20 min. The resulting pellet was resuspended in buffer B (100 mM potassium phosphate, pH 7.4, containing 0.5 mM EDTA, 0.5 mM DTT and 20% glycerol), to obtain a final mitochondrial sample. The S9-fraction (above) was further centrifuged at 12,000 × g for 20 min; the resulting pellet was discarded, and supernatant (S12) was centrifuged at 100,000 × g for 60 min, to obtain a pellet P100 (microsomes) and supernatant S100 (cytosol). The microsomal fraction was further purified by resuspending in 0.1 M sodium pyrophosphate buffer, pH 8.2 (1.3 mL/g original tissue weight), followed by centrifugation at 100,000 × g; the final microsomal pellet was resuspended in buffer B. The cytosol fraction was purified by re-centrifugation at 100,000 × g followed by dialysis against 20 mM potassium phosphate (pH 7.4), 0.2 mM DTT, to remove soluble radiolabel. Protein CVB in rat liver fractions in vivo was determined as described earlier.19, 20, 23 All final samples were stored in small aliquots at −80°C.
Two-Dimensional Electrophoresis, Phosphorimaging and In-gel Digestion
These procedures were carried out essentially as described earlier.19–21
Mass-Spectrometry of Tryptic Digests and Protein Identification
Of the 52 digest samples, half were analyzed by matrix-assisted laser desorption-ionization time-of-flight (MALDI-TOF/TOF) MS on a Proteomics Analyzer 4700 (Applied Biosystems, www.appliedbiosystems.com/) as described.19 The other half of the samples were submitted to a capillary LC-MS/MS using an LTQ-FT hybrid linear quadrupole ion trap Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (ThermoFinnigan, Bremen, Germany) as described.23 Raw data files were processed using BioWorks 3.2 software followed by peptide/protein identification using Sequest (ThermoFinnigan), Mascot (Matrix Science, version 2.2) and X! Tandem (www.thegpm.org) database-searching programs with the IPI rat 20080908 database (40041 entries) with a fragment ion mass tolerance of 0.20 Da and a parent ion tolerance of 10.0 PPM. S-Pyridylethyl (+105), bromophenyl (+154), and bromohydroquinone (+186) derivatives of cysteine residues were specified as variable modifications. Scaffold software (Proteome Software Inc., version 2.06) was used to combine and validate MS/MS based peptide and protein identifications. Peptide identifications with greater than 50% probability as specified by the Peptide Prophet algorithm24 were accepted for reporting protein coverage.
Preparation and Incubation of Isolated Hepatocytes
Hepatocytes from non-induced male Sprague-Dawley rats (175–275 g) were isolated using a previously described protocol.25 Initial cell viability (85–95%) was assessed using trypan blue exclusion. After isolation cells were transferred to medium A (DMEM supplemented with 5% FBS, 1% (v/v) Pen-Strep, insulin (4 mg/L), 100 nM dexamethasone, and 2 mM L-glutamine). For metabolism studies 4BP (0.2 mM) was incubated with 4.0 × 106 freshly isolated rat hepatocytes suspended in medium A (2.0 mL) with or without inhibitors of glucuronidation/sulfation in 50 mL round bottom screw-cap culture tubes. Incubations were shaken at 100 Hz at 37 °C under an atmosphere of oxygen:carbon dioxide (95:5) for 2 hr. Aliquots (150 μL) were removed at 0, 10, 20, 30, 40, 60, 90 and 120 min and added to 150 μL of cold acetonitrile. After centrifuging for 5 min at 2,000 × g the supernatant was removed and stored at −20 °C until LC-MS/MS analysis. Incubations were repeated a minimum of three times using different batches of hepatocytes.
For covalent binding studies, hepatocytes (2.4–4.0 × 106 live cells/mL) were incubated with [14C]-BP (0.05–0.2 mM) in medium A lacking the FBS in 16 × 100 mm screw-capped glass culture tubes in a final volume of 0.5 mL, at 37 °C for 90 min. In some cases 7HC (0.5 mM), borneol (0.45 mM) or DGN (2 mM) were added as inhibitors of glucuronidation/sulfation. At 0, 10, 20, 30 and 90 min incubations were quenched by adding 55 μL of an aqueous solution containing 10% Triton X-100 and 5 mM 4BP, and placing the tubes on ice. For analysis of CVB in incubated hepatocytes we applied the principle of protein precipitation on filter paper.26, 27 Aliquots (100 μL) of quenched incubations were applied on 12 × 80 mm strips of absorbent paper 3MM. After drying briefly in air the strips were washed by immersion for 10–15 min in each of a series of four baths (ca. 250 mL each) comprised of 10% TCA, 95% ethanol, 95% ethanol again, and finally ethyl acetate. The strips were then dried in air and submitted to liquid scintillation counting. CVB is reported as pmol-equiv./mg protein.
Cytotoxicity Studies
Freshly isolated hepatocytes were plated on duplicate collagen-coated 6-well plates at a density of 7×105 live cells/well in 2.0 mL of medium A as noted above. After holding the plates for 3 h at 37 °C in a CO2 incubator to allow attachment, the cells were washed twice with pre-warmed phosphate-buffered saline (PBS, pH 7.4). After attachment, cell viability was >98% according to Hoechst/PI staining. Incubations were started by adding solutions of 4BP and/or inhibitors in pre-warmed medium B (WME supplemented with 1% ITS, 1% Pen-Strep, 2 mM L-glutamine, and 100 nM dexamethasone; 3 mL/well) and returning the plates to the incubator. At appropriate times aliquots of culture medium (100 μL) were removed for analysis of LDH according to the assay kit manufacturer’s directions, but with all volumes reduced by half. For measurement of total cellular LDH content, 200 μL of 10% Triton X-100 was added directly to wells containing untreated cells immediately after the attachment period.
For time-lapse photomicrography freshly isolated cells were allowed to attach for 3 hr in collagen-coated wells (300,000 live cells/well in a 12-well plate) in medium A (1 mL/well). Cells were then washed twice with pre-warmed PBS and incubations were started by adding pre-warmed medium B (1 mL/well) with or without 4BP (0.2 mM). Plates were observed and photographed using an Olympus IX-81 epi-fluorescence microscope with a computer-controlled mechanical stage, automated shutter and filters (Sutter Instrument), temperature, humidity, and CO2 control. Images were collected every 10 minutes for 18 hr using a 20X objective with a numerical aperture of 0.45 (Olympus), an EMCCD camera (Hamamatsu Photonics) and the Slidebook image acquisition and processing package (Intelligent Imaging Innovations). After the last photo the cells were stained with Hoechst 33342 and propidium iodide to assess the live/dead ratio. The collected images (ca. 110 total) were then compiled into movies at a playback speed of 0.3 sec/frame.
LC-MS/MS Analysis of 4BP and Its Metabolites
The LC-MS/MS system consisted of a Waters (Milford, MA) Acquity UPLC with a Micromass (Manchester, UK) Quattro Ultima triple quadrupole mass spectrometer equipped with an electrospray ionization interface. Chromatographic separation was performed on an Intersil ODS-3 column (2.1 × 50 mm, 5 μm, GL Sciences, Inc.). The mobile phase consisted of solvent A (15 mM ammonium acetate in 0.1% formic acid in water) and solvent B (15 mM ammonium acetate and 0.1% formic acid in CH3CN/water (10:1 v/v). The gradient started at 1% solvent B for 0.5 min, increased linearly to 95% solvent B for 4.5 min, was held for 95% solvent B for 1.0 min, and then returned to the starting composition over 4 min. For all MS analyses, the electrospray block and the desolvation temperatures were set at 100° and 250°, respectively. The cone voltage was 25V, Q1 and Q3 resolution were set to 0.8u FWHH and for MS/MS collision energy was 20 eV for 4BP conjugates and 5 eV for phenols. For analysis of 4BP and its phase II metabolites, the mass spectrometer was operated in negative mode. 4BP, 4BP-glucuronide, 4BP-sulfate and 4-chlorophenol (internal standard) were quantitatively analyzed in multiple reaction monitoring mode (MRM) using the following transitions: m/z 171.0 → 171.0, 346.9 → 171.0, 250.9 → 171.0 and 127.0 → 127.0, respectively. The bromine isotope pattern was utilized to qualitatively confirm the identity of 4BP, 4BP-glucuronide and 4BP-sulfate using transitions: m/z 173.0 → 173.0, 348.9 → 173.0 and 252.9 → 173.0, respectively. The unusual use of MRM to analyze 4BP and 4-chlorophenol was due to the fact that these analytes did not fragment efficiently and thus gave only weak signals in traditional MRM while in Q1-only mode high backgrounds resulted in poor S/N ratios. However, transmitting their intact precursor ions in MRM mode afforded very good S/N ratios. Calibration curves for 4BP were prepared by plotting the appropriate peak area ratios of analyte/internal standard against the known concentrations (2.5–100 μM) of 4BP in blank incubation medium using a linear regression with 1/x weighting using the Waters Quanlynx data processing tool. For detection and characterization of GSH-trapped reactive metabolites of 4BP, we first used a precursors of m/z 272 negative ion scan with further structural identification accomplished by product ion scan in positive mode.28
Results and Discussion
Isolation and Identification of Rat Liver Proteins Adducted by Metabolites of [14C]-4-Bromophenol
Five hr after ip injection of [14C]-4BP (1 mmol/kg body wt) a homogenate of liver tissue contained 0.83 μCi 14C/g liver wet weight. After stepwise centrifugation to isolate mitochondria, microsomes and cytosol, these fractions were found to contain covalently bound 14C corresponding to 0.25, 0.33 and 0.42 nmol-eq. 4BP/mg protein. For comparison, in a similar experiment with [14C]-BB we previously observed the level of covalent binding in these fractions to be 6.1, 3.5 and 3.9 nmol-eq./mg protein, respectively (Table 2).20 We were surprised that the CVB we observed with 4BP was not much higher given 1) the large dose of 4BP that we used, and 2) the prior report of Monks et al.17 indicating that the CVB of 4BP in vivo was 62% of that resulting from an equimolar dose of BB.
Table 2.
Covalent Binding of Bromobenzene vs. 4-Bromophenol in Liver vs. Isolated Hepatocytes.
| # | Systema | Agent | Dose (mmol/kg) | Time (hr) | CVB (nmol-eq./mg protein) | Ref. |
|---|---|---|---|---|---|---|
| 1 | UT rat | 4BP | 3 | 10 | 0.23 ± 0.04 | 17 |
| 2 | PB rat | 4BP | 1 | 5 | 0.25–0.42 | this work |
| 3 | PB rat hepatocytes | 4BP | 0.25 mM | 2 | 0.08, 0.11 | 39 |
| 4 | UT rat | BB | 3 | 10 | 0.37 ± 0.07 | 17 |
| 5 | UT rat | BB | 7.16 | 24 | 1.33 ± 0.11 | 3 |
| 6 | 3MC rat | BB | 7.16 | 24 | 0.75 ± 0.03 | 3 |
| 7 | UT mouse | BB | 4.8 | 24 | 1.89 | 38 |
| 8 | UT mouse | BB | 6.53 | 16 | 2.5 | 38 |
| 9 | UT mouse | BB | 4.06 | 5 | 1.09 | 38 |
| 10 | UT mouse | BB | 4.06 | 24 | 2.52 | 38 |
| 11 | UT mouse | BB | 1.5 | 5 | 0.79 ± 0.14 | 38 |
| 12 | PB mouse | BB | 1.5 | 5 | 0.99 ± 0.23 | 38 |
| 13 | UT rat | BB | 10 | 5 | 2.3 | 4 |
| 14 | PB rat | BB | 2.5 | 4 | 0.68 ± 0.59 | 31 |
| 15 | PB rat | BB | 2.5 | 4 | 2.9 | 32 |
| 16 | PB rat | BB | 2 | 4 | 3.5–6.1 | 20 |
| 17 | PB rat hepatocytes | BB | 3 mM | 2 | 2.9 | 39 |
| 18 | PB rat hepatocytes | BB | 5 mM | 2 | 3.6 | 34 |
UT means untreated (uninduced); PB means pretreated (induced) with phenobarbital.
Despite the low total yield of covalent binding, aliquots of the three subcellular fractions were subjected to 2D gel electrophoresis (4–6 gels/fraction). After Coomassie staining, the gels of the mitochondrial, microsomal and cytosolic fractions revealed ca. 500, 200 and 800 protein spots, respectively (see Supporting Information). Radioactive protein spots on the 2D gels were located by phosphorimaging a polyvinylidene difluoride transblot of one gel from each subcellular fraction. A total of 17, 9 and 21 weakly radioactive spots were observed in mitochondrial, microsomal and cytosolic fractions, respectively. The corresponding spots on the replicate Coomassie-stained gels were excised, pooled and processed for tryptic digestion as described previously.19, 20, 23 We also excised and analyzed a number of other spots corresponding to familiar and relatively abundant “landmark” proteins (e.g. [Cu, Zn]-superoxide dismutase, cytochrome b5, aldehyde dehydrogenase 2 and others) to ensure good registry of spot locations from one gel to the next. Twenty nine of the 47 radioactive spots contained only one detectable protein. However, these 29 spots comprised only 14 non-redundant target proteins because several of these spots contained the same protein appearing in multiple spots of the same apparent molecular weight but different apparent pI. This phenomenon is commonly observed in the 2D gel analysis of hepatocellular proteins.19, 20, 29, 30 As indicated in Table 1, all the proteins listed were observed as a single protein in at least one spot, even though many of them were also observed in other spots mixed with other proteins.
Table 1.
Liver protein targets of 4-bromophenol metabolites.
| 4BP Target protein | # of spotsa | Matchesb | Coverage (%)c | Fractiond | Also targeted bye |
|---|---|---|---|---|---|
| Protein maturation or stabilization | |||||
| 56 kDa selenium-binding protein, Selenium-binding protein 1, SP56 | 2 | 6–9 | 13–21 | cytosol | AMAP, APAP, BB, BHT, MYCO, NAPH |
| Cellular thyroid hormone binding protein, PDI, PDI A1, Prolyl 4-hydroxylase beta subunit, Protein disulfide-isomerase [Precursor] | 4 | 8–18 | 14–34 | microsomes, mitochondria | 1,4-BQ, 1,4-NQ, AMAP, APAP, BB, BENZ, HALO, NAPH, MCTP, TB |
| 58 kDa microsomal protein, Disulfide isomerase ER-60, ERp57, ERp60, p58, PDI A3, Protein disulfide isomerase A3 [Precursor] | 6 | 3–15 | 5–32 | microsomes, mitochondria | 1,4-BQ, 1,4-NQ, APAP, ATRZ, BB, DACT, MCTP, MYCO, NAPH, TB |
| 78 kDa glucose-regulated protein [precursor], BiP, GRP 78, Heat shock 70 kDa protein 5, Immunoglobulin heavy chain binding protein, Steroidogenesis-activator polypeptide | 1 | 6 | 12 | microsomes | BB, HALO, NAPH, TB, TEUC |
| 60 kDa chaperonin, 60 kDa heat shock protein (mitochondrial [precursor]), CPN60, Heat shock protein 60, HSP-60, HSP-65, Hsp60, Mitochondrial matrix protein P1 | 1 | 6 | 18 | mitochondria | BQ, COC, DACT, NAPH, 1,4-NQ, TASO, TEUC, TFEC |
| 75 kDa glucose regulated protein, GRP 75, Heat shock 70 kDa protein 9, Mortalin, MTHSP70, PBP74, Peptide-binding protein 74, Stress-70 protein (mitochondrial [precursor]) | 1 | 4 | 9 | mitochondria | 1,4-BQ, 1,4-NQ, DACT, NAPH, TFEC |
| Enzymes of intermediary metabolism | |||||
| Beta-diketonase, FAA, Fumarylacetoacetase, Fumarylacetoacetate hydrolase | 1 | 5 | 13 | cytosol | BB, TB |
| Cytoplasmic malate dehydrogenase, Malate dehydrogenase (cytoplasmic) | 1 | 3 | 9 | cytosol | TB |
| hypoxanthine-guanine phosphoribosyltransferase, HGPRT | 1 | 5 | 25 | cytosol | (none) |
| TIM, Triose-phosphate isomerase, Triosephosphate isomerase, triosephosphate isomerase 1 | 2 | 2–6 | 12–29 | cytosol | BB, BHT, DACT, MYCO |
| Electrophile or peroxide defense | |||||
| Contraception-associated protein 1, Fertility protein SP22, Protein CAP1, Protein DJ-1 | 1 | 6 | 27 | cytosol | BB |
| Glutathione S-transferase Mu 2, Glutathione S-transferase Yb-2, GST 4-4, GST Yb2, GSTM2-2 | 4 | 3–9 | 13–42 | cytosol | BB, TB |
| Catalase | 2 | 5–8 | 10–18 | mitochondria | TB, TEUC |
| Binding proteins | |||||
| Albumin | 2 | 6 | 12 | mitochondria | ATRZ, BB, DCE, FCXC, NAPH, TB |
Many proteins appear in more than one spot on a 2D gel (see text). Each of the proteins listed in this table were observed in at least one spot which contained no other detectable proteins, even though some of them were also observed in other spots containing one or more other proteins.
Number of peptides sequenced and matched to the identified protein.
Percent of entire protein sequence covered by observed peptides.
Subcellular fraction in which the target protein was observed.
Other chemicals whose reactive metabolites are also known to target the same protein. Abbreviations: 1,4-BQ, 1,4-benzoquinone; 1,4-NQ, 1,4-naphthoquinone; AMAP, m-hydroxyacetanilide; APAP, p-hydroxyacetanilide; ATRZ, atrazine; BB, bromobenzene; BENZ, benzene; BHT, butylated hydroxytoluene; COC, cocaine; DACT, diaminochlorotriazine; DCE, 1,1-dichloroehtylene; FCXC, flucloxacillin; HAL, halothane; MCTP, monocrotaline pyrrole; MYCO, mycophenolic acid; NAPH, naphthalene; TB, thiobenzamide; TEUC, teucrin; TFEC, S-(1,1,2,2-tetrafluoroethyl) cysteine. Data from the Reactive Metabolite Target Protein Database (http://tpdb.medchem.ku.edu:8080/protein_database/).
Since 4BP is not hepatotoxic in rats, it was suggested that the covalent labeling of proteins by its oxidative metabolites is not toxicologically significant.17 It was therefore of interest to identify these proteins and compare them to the proteins targeted by BB (which is hepatotoxic). Of the 14 proteins identified in Table 1, nine were previously identified as targets of BB metabolites in rat liver. This is perhaps not surprising since 4BP is a significant metabolite of BB comprising 21–36% of total metabolites depending on the dose.7 The important distinction, however, is that while 4BP can be oxidized further to electrophilic quinone metabolites, 4BP can not revert back to BB-3,4-oxide, the putative toxic metabolite of BB. In earlier studies we found direct evidence for the alkylation of cysteine and histidine side chains by bromobenzene-2,3- and -3,4-epoxides,31, 32 but it was clear that the vast majority of the identifiable adducts present were in fact quinone derived.33–35 In the present work with 4BP we observed many tryptic peptides derived from target proteins but none of them carried detectable 4BP-derived adducts. Nevertheless, there can be little doubt that the adducts formed from 4BP are quinone-derived. In view of the low net covalent binding of 4BP, the similarity of its target protein profile to that of BB, and the major role for quinone metabolites in protein adduction from both compounds, we began to speculate that the non-toxicity of 4BP might be more a function of its low extent of adduct formation rather than an intrinsic lack of adduct toxicity. We will return to this point later in the manuscript.
Of the 4BP target proteins that we identified, three were from the mitochondrial fraction, three were from the microsomal fraction and eight were found in the cytosolic fraction. Six of these proteins are known to function in connection with protein maturation or stabilization within the cell, four are enzymes of intermediary metabolism, and three function in defense against electrophiles and peroxides. The final protein, albumin, is a plasma protein well known for its ability to bind non-covalently many lipophilic molecules, both xenobiotic and endogenous. It is synthesized in the liver in large quantities and exported through the endoplasmic reticulum. This may explain why it is observed as a target protein in liver, even though the vast majority of albumin in the body is found in the bloodstream.
As shown in Table 1, most of the 4BP target proteins listed are also known to be targets for electrophilic metabolites of several other xenobiotics.36 For example, the endoplasmic reticulum lumenal proteins PDI A1 and PDI A3 are known as targets for reactive metabolites for 13 other compounds. Overall, a total of 18 different agents, nearly all of which are metabolically activated pro-hepatotoxins, target one or more of the 14 targets listed in Table 1. It is interesting to ask whether some of these frequently targeted proteins could serve as harbingers or “biomarkers” for potential cytotoxicity due to reactive metabolites. This is a difficult question to answer for many reasons, not the least of which is the general absence of target protein data for “negative control” compounds, i.e., compounds that covalently bind relatively extensively but are truly non-toxic. We are aware of only one well-documented example of such a compound, namely, the meta isomer of acetaminophen (3-hydroxyacetanilide).37 It was thought that 4BP might be such a compound, but as discussed below, this view requires re-consideration.
Since we observed only low levels of covalent binding from 4BP in vivo, we reexamined the original comparison of its covalent binding to that of bromobenzene.17 As shown in Table 2 (lines 1 vs. 4), the net covalent binding of 4BP was ca. 62% that of BB. However, by comparison to numerous other measurements of bromobenzene covalent binding,3, 4, 20, 31, 32, 38, 39 the value in line 4 of Table 2 appears to be too low by as much as a factor of 10. Because the toxic effects of reactive metabolites generally correlate with their degree of covalent binding, it appeared that the lack of toxicity observed for exogenously administered 4BP might be due to the fact that it produces very little covalent binding, rather than to its adducts being intrinsically non-toxic. We further hypothesized that the low binding of 4BP could be due to efficient glucuronidation and/or sulfation in preference to oxidation to reactive quinone metabolites (see Scheme 1). In other words, the introduction of the hydroxyl group to BB may ablate its toxicity by directing its metabolism toward conjugation (detoxication) rather than oxidation (bioactivation). As an example of this, phenylthiourea is metabolized by S-oxidation to a very potent pneumotoxin, but its p-hydroxyphenyl analog is at least five times less toxic and its major metabolites are the glucuronide and sulfate conjugates rather than oxidative desulfurization products.40 Similarly, while the hepatotoxicity of thiobenzamide is greatly increased by electron-donating methyl or methoxy substituents in the meta or para positions,41 p-hydroxythiobenzamide is essentially non-toxic despite the strong electron-donating effect of the p-OH group.42 Conversely, pretreatment of hamsters with a regimen of 3-methylcholanthrene (to induce P450), borneol (to inhibit glucuronidation and sulfation) and diethyl maleate (to deplete glutathione), greatly potentiates the covalent binding and hepatotoxicity of acetaminophen.43 Thus, it is reasonable to propose that rapid glucuronidation and/or sulfation of 4BP could seriously limit its metabolic activation and covalent binding and thereby thwart its potential toxicity.
Metabolism, Covalent Binding and Toxicity of 4-Bromophenol in Isolated Hepatocytes
In view of the uncertainties noted above in regard to the relative covalent binding of 4BP vs. BB in vivo, we sought to examine the toxicity of 4BP using isolated hepatocyte systems in which the metabolism, covalent binding and toxicity of BB has been thoroughtly studied.5, 44 As shown by the LDH release assay data in Figure 1, 4BP is definitely toxic to isolated hepatocytes in a time- and concentration-dependent manner. As the concentration of 4BP in the medium increases from 0.1 mM to 0.5 mM, the onset of toxicity becomes earlier and the overall severity greater. Time-lapse photomicrography (see Supporting Information) shows that hepatocytes exposed to 0.2 mM 4BP undergo dramatic changes in their morphology and their inter- and intracellular motility. Over a period of hours the attached cells reverse their tendency to spread and establish cell-cell contacts and instead begin to contract, round up, extend numerous and sometimes quite large cytoplasmic protrusions (blebs) and finally become shriveled and motionless. By this time Hoechst/PI staining shows that essentially all of the cells are stained by PI even though LDH release is not yet maximal.
Figure 1.

Toxicity of 4BP to isolated rat hepatocytes. Plated cells were washed as described in Materials and Methods and then incubated with the indicated concentrations of 4BP. Untreated (UT) control cells were incubated in medium lacking 4BP. At the indicated times aliquots of medium were withdrawn for analysis of LDH leakage.
We next examined whether inhibitors of glucuronidation and/or sulfation would potentiate the toxicity of 4BP by increasing its availability within the cell for further oxidation to reactive quinone intermediates as implied by Scheme 1. (−)-Borneol and 7HC are both alternate substrate inhibitors of glucuronidation that are well tolerated in vivo45–47 while DGN blocks the synthesis of the UDP-glucuronic acid cosubstrate required for glucuronide formation.48 The latter can be toxic at higher concentration over longer times, but we found, as did Moldeus et al.,45 that 2 mM DGN was well tolerated by hepatocytes (Figure 2A). Figures 2B-2D show the effect of these three inhibitors on the toxicity of 4BP to isolated rat hepatocytes. At a concentration of 0.1 mM, 4BP shows little if any toxicity as measured by LDH release, but in the presence of the inhibitors its toxicity becomes apparent (Figure 2B). As the concentration of 4BP increases to 0.2 or 0.3 mM (Figures 2C and 2D), its toxicity becomes increasingly apparent and the potentiation of its toxicity by the conjugation inhibitors becomes more pronounced.
Figure 2.

Effect of conjugation inhibitors on the toxicity of 4BP to isolated rat hepatocytes. Plated cells were washed as described in Materials and Methods and then incubated with the indicated concentrations of 4BP ± inhibitors. Untreated (UT) control cells were incubated in medium lacking 4BP and inhibitors. At the indicated times aliquots of medium were withdrawn for analysis of LDH leakage. A) Effect of inhibitors alone. B–D) Effect of inhibitors on increasing concentrations of 4BP.
To verify that the glucuronidation inhibitors actually decreased the conjugation of 4BP hepatocytes were incubated in suspension and the supernatant sampled and analyzed by LC-MS/MS. Putative metabolite peaks were identified by manual surveys of LC/MS data and confirmed by MS/MS analysis of fragmentation patterns in positive and/or negative ion mode. Briefly, glucuronide conjugates showed strong neutral loss ions at [M-176]− while sulfates were monitored by [M-80]− ions, as shown by the composite “MRM chromatogram” in Figure S4. To monitor quantitatively the disappearance of 4BP during incubations an internal standard (4-chlorophenol) was added to aliquots of incubation medium withdrawn at various times.
As shown in Figure 3A, 4BP (initial concentration 0.2 mM) was completely consumed over the course of a 2 hour incubation, and its disappearance followed first order kinetics throughout (0.026 min−1). The addition of either 7HC or borneol slowed the rate considerably (to 0.0061 and 0.0086 min−1, respectively). Borneol depressed the rate of glucuronidation of 4BP to almost the same degree as 7HC (Figure 3B), but was noticeably less effective than 7HC at inhibiting the sulfation of 4BP (Figure 3C). Increasing the sulfate concentration in the medium from the basal level of 0.9 mM to 10 mM by adding sodium sulfate had no effect, indicating that the availability of sulfate was not rate limiting (data not shown).
Figure 3.

Effects of (−)-borneol and 7-hydroxycoumarin on glucuronidation and sulfation of 4BP by isolated rat hepatocytes. 4BP (0.2 mM) was incubated with 4 × 106 hepatocytes in 2 mL medium, either alone or with 0.5 mM borneol or 0.5 mM 7HC. A) Disappearance of 4-bromophenol. B) Formation of 4-bromophenyl glucuronide. C) Formation of 4-bromophenyl sulfate.
Because we had hypothesized that inhibiting the conjugation of 4BP would make it more available for oxidation to reactive metabolites (viz. Scheme 1), we also searched the incubation media by LC/MS for glutathione conjugates derived from 4BP. In addition to manual examination of the data we scanned in negative ion mode for precursors of m/z = 272 (corresponding to the tripeptide γ-glutamyl-dehydroalanyl-glycine), as this fragmentation is diagnostic for GSH conjugates.28, 49 In the LC/MS analysis of the medium from incubation of 4BP alone, a negative ion scan for parents of m/z 272 showed a small peak of ion current at ca. 2.9 min retention time. The negative ion mass spectrum of the eluate under this peak showed peaks of m/z = 506 and 508 suggestive of a bromine-containing molecule corresponding to the composition [4BP + O + GSH + CH2 − 2H], derived by the 4-electron oxidation of 4BP to 4-bromo-1,2-benzoquinone followed by Michael addition of GSH and methylation of the resulting catechol moiety (Scheme 2). Bromophenols are known to deplete glutathione in hepatocytes50 and the unmethylated form of this bromoquinone-GSH adduct was previously observed in microsomal incubations with 4BP.51 Positive ion scans for products of m/z 508 and 510 (MH+ for 79Br and 81Br, respectively) showed identical fragmentation patterns that confirmed this structural assignment (Figure S5) but did not allow us to determine which oxygen was methylated. A related example of the COMT-mediated methylation of a catechol moiety in a large complex polar metabolite was reported by Xie et al.52 To quantitate the relative amount of this metabolite in the incubations we generated MRM chromatograms for the transitions m/z 508 → 362 and m/z510 → 364 (see Figure S5) and integrated the peaks. These integrals showed that adding borneol, 7HC or both to the incubation medium increased the amount of this GSH-trapped reactive metabolite by factors of 2.3-, 5.2- and 5.7-fold, respectively. In parallel with the increased glutathione trapping of a reactive quinone metabolite when glucuronidation and sulfation are inhibited, 7HC and DGN increase the net covalent binding of 4BP to cellular proteins as shown in Figure 4. This increase parallels the increases in cytotoxicity observed when 4BP conjugation is inhibited (Figure 2). Although borneol potentiated the toxicity of 4BP it did not increase the CVB under the conditions tested. In contrast DGN increased the CVB of BP but only weakly potentiated toxicity, whereas 7HC clearly potentiated both. These differences probably reflect the intrinsic complexity of the metabolic pathways leading to bioactivation vs. detoxication (Scheme 1), and the relative concentration, Km and Ki values for the various compounds being investigated, but more significantly, they emphasize the importance of using multiple probes when attempting to manipulate specific enzymes in complex systems like hepatocytes.
Scheme 2.

Structure and mass spectral fragmentation of glutathione-trapped reactive metabolite of 4BP.
Figure 4.
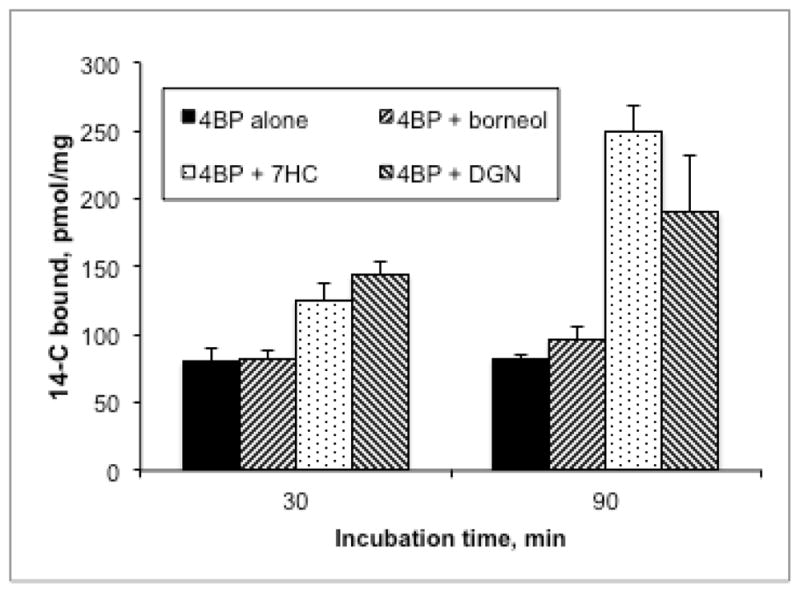
Effect of glucuronidation inhibitors on covalent binding of 4BP (50 μM; specific activity 8.05 Ci/mol) in isolated hepatocytes (1.25 × 106 cells suspended in 0.5 mL medium). Data shown are the mean ± SD (n=3). Compared to 4BP alone, the increases caused by 7-HC and DGN are significant (p < 0.01).
Summary
4BP, which is not hepatotoxic in the rat, was reported to undergo metabolic activation and covalent binding only 38% less than an equimolar (but decidedly hepatotoxic) dose of its metabolic precursor BB.17 This observation was interpreted to suggest that the covalent binding derived from 4BP metabolites was not toxicologically significant. BB metabolites covalently modify at least 45 liver proteins in vivo.36 By identifying proteins also targeted by reactive metabolites of 4BP, we sought to reduce the size and complexity of the list of BB targets and thereby focus attention on those proteins whose modification was more likely to be associated causally with the toxicity of BB. After administering a large dose of [14C]-4BP to rats, we observed only very low levels of covalent binding. Nevertheless, we were able to identify single proteins in 29 radioactive spots on 2D gels of the mitochondrial, microsomal and cytosolic fractions of rat liver. These 29 spots comprised only 14 non-redundant proteins, nine of which were already known as targets of reactive BB metabolites. Re-examination of earlier literature (Table 2) indicated that the value for BB covalent binding reported by Monks et al.17 was much lower than other published values. Compared to this low value (but not to other published values) the covalent binding of 4BP appeared to be comparatively high, which in turn lead to the erroneous conclusion that adducts derived from 4BP were not toxic like those of BB.
In isolated hepatocytes and presumably in vivo, 4BP undergoes rapid conjugation by glucuronidation and sulfation. These processes compete with 4BP oxidation to reactive quinone metabolites, thus limiting its covalent binding and thereby also limiting its toxicity. However, when these conjugating pathways are inhibited, the clearance of 4BP is markedly decreased, the formation of glutathione conjugates of reactive quinone metabolites of 4BP rises almost 6-fold, and both protein covalent binding and cytotoxicity increase substantially. We therefore suggest that the main reason that 4BP is not significantly hepatotoxic is not that protein adduction by its reactive metabolites is not harmful, but rather that 4BP simply doesn’t give rise to very much covalent binding in vivo because of its rapid conjugation. As demonstrated above, 4BP is definitely cytotoxic at moderately low concentrations, and treatments that block its conjugation both increase its covalent binding and potentiate its cytotoxicity. These results support the hypothesis that most covalent binding probably does have toxicological consequences, if the extent to which it occurs reaches a high enough level. Evidently that level is not reached by metabolites of 4BP following its exogenous administration to rats.
Supplementary Material
Acknowledgments
Funding
This research was supported in part by NIH grant GM-21784 (to R.P.H.). The Micromass Ultima was purchased with support from the KU Research Development Fund and the Acquity UPLC with support from K-INBRE (NIH P20 RR016475).
We thank Dr. Larry Hall (Bayer Crop Science) for providing a sample of [14C]-4BP, and Dr. David Moore and Ms. Heather Shinogle for expert technical training and assistance with microscopy and photomicrography.
Abbreviations used
- 4BP
4-bromophenol
- 7HC
7-hydroxycoumarin
- BB
bromobenzene
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- CVB
covalent binding
- DGN
D-(+)-galactosamine
- DMEM
Dulbecco’s Modified Eagle Medium
- FBS
fetal bovine serum
- LDH
lactate dehydrogenase
- MRM
multiple reaction monitoring
- WME
Williams’ Medium E
Footnotes
Three figures of blots and gels; MRM chromatogram; MS/MS spectra of a GHS-bromoquinone conjugate; two time-lapse movies of hepatocytes (control and 4BP-treated).
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.White A, Jackson RW. The effect of bromobenzene on the utilization of cystine and methionine by the growing rat. J Biol Chem. 1935;111:507–513. [Google Scholar]
- 2.Brodie BB, Reid WD, Cho AK, Sipes G, Krishna G, Gillette JR. Possible mechanism of liver necrosis caused by aromatic organic compounds. Proc Nat Acad Sci (USA) 1971;68:160–164. doi: 10.1073/pnas.68.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reid WD, Christie B, Krishna G, Mitchell JR, Moskowitz J, Brodie BB. Bromobenzene metabolism and hepatic necrosis. Pharmacology. 1971;6:41–55. doi: 10.1159/000136226. [DOI] [PubMed] [Google Scholar]
- 4.Jollow DJ, Mitchell JR, Zampaglione N, Gillette JR. Bromobenzene-induced liver necrosis. Protective role of glutathione and evidence for 3,4-bromobenzene oxide as the hepatotoxic metabolite. Pharmacology. 1974;11:151–169. doi: 10.1159/000136485. [DOI] [PubMed] [Google Scholar]
- 5.Thor H, Moldeus P, Kristoferson A, Hogberg J, Reed DJ, Orrenius S. Metabolic activation and hepatotoxicity - Metabolism of bromobenzene in isolated hepatocytes. Arch Biochem Biophys. 1978;188:114–121. doi: 10.1016/0003-9861(78)90363-6. [DOI] [PubMed] [Google Scholar]
- 6.Hayes MA, Roberts E, Roomi MW, Safe SH, Farber E, Cameron RG. Comparative influences of different PB-type and 3-MC-type polychlorinated biphenyl-induced phenotypes on cytocidal hepatotoxicity of bromobenzene. Toxicol Appl Pharmacol. 1984;76:118–127. doi: 10.1016/0041-008x(84)90035-8. [DOI] [PubMed] [Google Scholar]
- 7.Zampaglione N, Jollow DJ, Mitchell JR, Stripp B, Hamrick M, Gillette JR. Role of detoxifying enzymes in bromobenzene-induced liver necrosis. J Pharmacol Exp Therap. 1973;187:218–227. [PubMed] [Google Scholar]
- 8.Zheng J, Hanzlik RP. Premercapturic acid metabolites of bromobenzene derived via its 2,3- and 3,4-oxide metabolites. Xenobiotica. 1991;21:535–546. doi: 10.3109/00498259109039493. [DOI] [PubMed] [Google Scholar]
- 9.Monks TJ, Pohl LR, Gillette JR, Hong M, Highet RJ, Ferretti JA, Hinson JA. Stereoselective formation of bromobenzene glutathione conjugates. Chem-Biol Interactions. 1982;41:203–216. doi: 10.1016/0009-2797(82)90090-4. [DOI] [PubMed] [Google Scholar]
- 10.Reid WD. Mechanism of renal necrosis induced by bromobenzene or chlorobenzene. Experimental and Molecular Pathology. 1973;19:197–214. doi: 10.1016/0014-4800(73)90079-8. [DOI] [PubMed] [Google Scholar]
- 11.Richieri PR, Buckpitt AR. Efflux of naphthalene oxide and reactive naphthalene metabolites from isolated hepatocytes. J Pharmacol Exp Therap. 1987;242:485–492. [PubMed] [Google Scholar]
- 12.Buonrati M, Morin D, Plopper C, Buckpitt AR. Glutathione depletion and cytotoxicity by naphthalene 1,2-oxide in isolated hepatocytes. Chem-Biol Interactions. 1989;71:147–165. doi: 10.1016/0009-2797(89)90031-8. [DOI] [PubMed] [Google Scholar]
- 13.Hanzlik RP, Hogberg K, Judson CM. Microsomal hydroxylations of specifically deuterated monosubstituted benzenes. Evidence for direct aromatic hydroxylation. Biochem. 1984;23:3048–3055. doi: 10.1021/bi00308a031. [DOI] [PubMed] [Google Scholar]
- 14.Hesse S, Wolff T, Mezger M. Involvement of phenolic metabolites in the irreversible protein-binding of 14C-bromobenzene catalysed by rat liver microsomes. Arch Toxicol Suppl. 1980;4:358–362. doi: 10.1007/978-3-642-67729-8_77. [DOI] [PubMed] [Google Scholar]
- 15.Hesse S, Mezger M. Involvement of phenolic metablites in the irreversible protein binding of aromatic hydrocarbons: Reactive metabolites of [14C]-naphthalene and [14C]-1-naphthol formed by rat liver microsomes. Mol Pharmacol. 1979;16:667–675. [PubMed] [Google Scholar]
- 16.Tunek A, Oesch F. Multi-step metabolic activation of benzene in rat liver microsomes. Adv Exper Med Biol. 1981;136(Part A):319–329. doi: 10.1007/978-1-4757-0674-1_19. [DOI] [PubMed] [Google Scholar]
- 17.Monks TJ, Hinson JA, Gillette JR. Bromobenzene and p-bromophenol toxicity and covalent binding in vivo. Life Sci. 1982;30:841–848. doi: 10.1016/0024-3205(82)90598-7. [DOI] [PubMed] [Google Scholar]
- 18.Hanzlik RP, Fang J, Koen YM. Filling and mining the reactive metabolite target protein database. Chem-Biol Interactions. 2009;179:38–44. doi: 10.1016/j.cbi.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koen YM, Gogichaeva NV, Alterman MA, Hanzlik RP. A proteomic analysis of bromobenzene reactive metabolite targets in rat liver cytosol in vivo. . Chem Res Toxicol. 2007;20:511–519. doi: 10.1021/tx6003166. [DOI] [PubMed] [Google Scholar]
- 20.Koen YM, Hanzlik RP. Identification of seven proteins in the endoplasmic reticulum as targets for reactive metabolites of bromobenzene. Chem Res Toxicol. 2002;15:699–706. doi: 10.1021/tx0101898. [DOI] [PubMed] [Google Scholar]
- 21.Koen YM, Williams TD, Hanzlik RP. Identification of three protein targets for reactive metabolites of bromobenzene in rat liver cytosol. Chem Res Toxicol. 2000;13:1326–1335. doi: 10.1021/tx000165l. [DOI] [PubMed] [Google Scholar]
- 22.(http://grants1.nih.gov/grants/guide/notice-files/not96-208.html))
- 23.Ikehata K, Duzhak T, Galeva NA, Ji T, Koen YM, Hanzlik RP. Protein targets of reactive metabolites of thiobenzamide in rat liver in vivo. Chem Res Toxicol. 2008;21:1432–1442. doi: 10.1021/tx800093k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller A, Nesvizhskii AI, Kolker E, Aebersold RH. Empirical statistical model to estimate the accuracy of peptide identifications made by ms/ms and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 25.Mudra DR, Parkinson A. Preparation of hepatocytes. Current Protocols in Toxicology. 2001;(Supplement 8):14.12.11–14.12.13. doi: 10.1002/0471140856.tx1402s08. [DOI] [PubMed] [Google Scholar]
- 26.Fisher R, Brendel K, Hanzlik RP. Correlation of metabolism, covalent binding and toxicity for a series of bromobenzene derivatives using rat liver slices in vitro. Chem-Biol Interactions. 1993;88:191–208. doi: 10.1016/0009-2797(93)90091-c. [DOI] [PubMed] [Google Scholar]
- 27.Wallin H, Schelin C, Tunek A, Jergil B. A rapid and sensitive method for determination of covalent binding of benzo[a]pyrene to proteins. Chem-Biol Interactions. 1981;38:109–118. doi: 10.1016/0009-2797(81)90157-5. [DOI] [PubMed] [Google Scholar]
- 28.Dieckhaus CM, Fernandez-Metzler CL, King R, Krolikowski PH, Baillie TA. Negative ion tandem mass spectrometry for the detection of glutathione conjugates. Chem Res Toxicol. 2005;18:630–638. doi: 10.1021/tx049741u. [DOI] [PubMed] [Google Scholar]
- 29.Qiu Y, Burlingame AL, Benet LZ. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem. 1998;273:17940–17953. doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- 30.Meier BW, Gomez JD, Kirichenko OV, Thompson JA. Mechanistic basis for inflammation and tumor promotion in lungs of 2,6-di-tert-butyl-4-methylphenol-treated mice: Electrophilic metabolites alkylate and inactivate antioxidant enzymes. Chem Res Toxicol. 2007;20:199–207. doi: 10.1021/tx060214f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bambal RB, Hanzlik RP. Bromobenzene-3,4-oxide alkylates histidine and lysine side chains of rat liver proteins in vivo. Chem Res Toxicol. 1995;8:729–735. doi: 10.1021/tx00047a013. [DOI] [PubMed] [Google Scholar]
- 32.Weller PE, Hanzlik RP. Isolation of S-(bromophenyl)cysteine isomers from liver proteins of bromobenzene-treated rats. Chem Res Toxicol. 1991;4:17–20. doi: 10.1021/tx00019a002. [DOI] [PubMed] [Google Scholar]
- 33.Narasimhan N, Weller PE, Buben JA, Wiley RA, Hanzlik RP. Microsomal metabolism and covalent binding of [3H/14C]-bromobenzene. Evidence for quinones as reactive metabolites. Xenobiotica. 1988;18:491–499. doi: 10.3109/00498258809041686. [DOI] [PubMed] [Google Scholar]
- 34.Slaughter DE, Hanzlik RP. Identification of epoxide- and quinone-derived bromobenzene adducts to protein sulfur nucleophiles. Chem Res Toxicol. 1991;4:349–359. doi: 10.1021/tx00021a015. [DOI] [PubMed] [Google Scholar]
- 35.Slaughter DE, Zheng J, Harriman S, Hanzlik RP. Identification of covalent adducts to protein sulfur nucleophiles by alkaline permethylation. Anal Biochem. 1993;208:288–295. doi: 10.1006/abio.1993.1048. [DOI] [PubMed] [Google Scholar]
- 36.(http://tpdb.medchem.ku.edu:8080/protein_database/)
- 37.Myers TG, Dietz EC, Anderson NL, Khairallah EA, Cohen SD, Nelson SD. A comparative study of mouse liver proteins arylated by reactive metabolites of acetaminophen and its nonhepatotoxic regioisomer, 3′-hydroxyacetanilide. Chem Res Toxicol. 1995;8:403–413. doi: 10.1021/tx00045a012. [DOI] [PubMed] [Google Scholar]
- 38.Dankovic DA, Billings RE. The role of 4-bromophenol and 4-bromocatechol in bromobenzene covalent binding and toxicity in isolated rat hepatocytes. Toxicol Appl Pharmacol. 1985;79:323–331. doi: 10.1016/0041-008x(85)90354-0. [DOI] [PubMed] [Google Scholar]
- 39.Reid WD, Krishna G. Centrolobular hepatic necrosis related to covalent binding of metabolites of halogenated aromatic hydrocarbons. Exper Mol Pathology. 1973;18:80–99. doi: 10.1016/0014-4800(73)90009-9. [DOI] [PubMed] [Google Scholar]
- 40.Scheline RR, Smith RL, Williams RT. The metabolism of arylthioureas – ii. The metabolism of 14C- and 35S-labelled 1-phenyl-2-thiourea and its derivatives. J Med Pharm Chem. 1964;4:109–135. doi: 10.1021/jm50017a009. [DOI] [PubMed] [Google Scholar]
- 41.Cashman JR, Parikh KK, Traiger GJ, Hanzlik RP. Relative hepatotoxicity of ortho and meta monosubstituted thiobenzamides in the rat. Chem-Biol Interactions. 1983;45:341–347. doi: 10.1016/0009-2797(83)90080-7. [DOI] [PubMed] [Google Scholar]
- 42.Hanzlik RP, Cashman JR, Traiger GJ. Relative hepatotoxicity of substituted thiobenzamides and thiobenzamide S-oxides in the rat. Toxicol Appl Pharmacol. 1980;55:260–272. doi: 10.1016/0041-008x(80)90088-5. [DOI] [PubMed] [Google Scholar]
- 43.Roberts SA, Price VF, Jollow DJ. Acetaminophen structure-toxicity studies: In vivo covalent binding of a nonhepatotoxic analog, 3-hydroxyacetanilide. Toxicol Appl Pharmacol. 1990;105:195–208. doi: 10.1016/0041-008x(90)90181-s. [DOI] [PubMed] [Google Scholar]
- 44.Thor H, Moldeus P, Hermanson R, Hogberg J, Reed DJ, Orrenius S. Metabolic activation and hepatotoxicity - toxicity of bromobenzene in hepatocytes isolated from phenobarbital- and diethylmaleate-treated rats. Arch Biochem Biophys. 1978;188:122–129. doi: 10.1016/0003-9861(78)90364-8. [DOI] [PubMed] [Google Scholar]
- 45.Moldeus P, Andersson B, Gergely V. Regulation of glucuronidation and sulfate conjugation in isolated hepatocytes. Drug Metab Dispos. 1979;7:416–419. [PubMed] [Google Scholar]
- 46.Sadeghi-Aliabadi H, Chan K, Lemler HJ, Robertson LW, O’Brien PJ. Molecular cytotoxic mechanisms of catecholic polychlorinated biphenyl metabolites in isolated rat hepatocytes. Chem-Biol Interactions. 2007;167:184–192. doi: 10.1016/j.cbi.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 47.Antoine B, Magdalou J, Siest G. Kinetic properties of UDP-glucuronlytransferase(s) in different membrates of rat liver cells. Xenobiotica. 1984;14:575–579. doi: 10.3109/00498258409151451. [DOI] [PubMed] [Google Scholar]
- 48.Sherratt AJ, Damani LA. The metabolism of N,N-dimethylaniline by isolated rat hepatocytes: Identification of a novel N-conjugate. Xenobiotica. 1989;19:379–388. doi: 10.3109/00498258909042280. [DOI] [PubMed] [Google Scholar]
- 49.Jian W, Yao M, Zhang D, Zhu M. Rapid detection and characterization of in vitro and urinary n-acetyl-l-cysteine conjugates using quadrupole-linear ion trap mass spectrometry and polarity switching. Chem Res Toxicol. 2009;22:1246–1255. doi: 10.1021/tx900035j. [DOI] [PubMed] [Google Scholar]
- 50.Thor H, Svensson S-A, Hartzell P, Orrenius S. Biotransformation of bromobenzene to reactive metabolites by isolated hepatocytes. Adv Exp Med Biol. 1982;136:287–299. doi: 10.1007/978-1-4757-0674-1_17. [DOI] [PubMed] [Google Scholar]
- 51.Monks TJ, Lau SS, Highet RJ. Formation of nontoxic reactive metabolites of p-bromophenol: Identification of a new glutathione conjugate. Drug Metab Dispos. 1984;12:432–437. [PubMed] [Google Scholar]
- 52.Xie C, Zhong D, Chen X. Identification of the ortho-benzoquionone intermediate of 5-O-caffeoylquinic acid in vitro and in vivo: Comparison of bioactivation under normal and pathological situations. Drug Metab Dispos. 2012;40:1628–1640. doi: 10.1124/dmd.112.045641. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.