

Abstract
The clinical significance of tumor-infiltrating immune cells has been reported in a variety of human carcinomas including breast cancer. However, molecular signature of tumor-infiltrating immune cells and their prognostic value in breast cancer patients remain elusive. We hypothesized that a distinct network of immune function genes at the tumor site can predict a low risk versus high risk of distant relapse in breast cancer patients regardless of the status of ER, PR, or HER-2/neu in their tumors. We conducted retrospective studies in a diverse cohort of breast cancer patients with a 1–5 year tumor relapse versus those with up to 7 years relapse-free survival. The RNAs were extracted from the frozen tumor specimens at the time of diagnosis and subjected to microarray analysis and real-time RT-PCR. Paraffin-embedded tissues were also subjected to immunohistochemistry staining. We determined that a network of immune function genes involved in B cell development, interferon signaling associated with allograft rejection and autoimmune reaction, antigen presentation pathway, and cross talk between adaptive and innate immune responses were exclusively upregulated in patients with relapse-free survival. Among the 299 genes, five genes which included B cell response genes were found to predict with >85% accuracy relapse-free survival. Real-time RT-PCR confirmed the 5-gene prognostic signature that was distinct from an FDA-cleared 70-gene signature of MammaPrint panel and from the Oncotype DX recurrence score assay panel. These data suggest that neoadjuvant immunotherapy in patients with high risk of relapse may reduce tumor recurrence by inducing the immune function genes.
Keywords: Breast cancer prognosis, Tumor relapse, Tumor microenvironment, Immune response, Neoadjuvant immunotherapy
Introduction
Mechanisms through which breast cancers recur (locally, regionally or distantly) following conventional therapies remain elusive. According to the current knowledge, the patients with triple-negative tumors (ER–/PR–/HER-2/neu–) or HER-2 over-expressing and node-positive tumors are at greater risk for cancer recurrence [1]. Several groups have also identified predictors of outcome with gene expression data derived from tumor specimens comprising both tumor and stroma, although samples judged to possess insufficient tumor cell content were generally excluded [2–5]. In fact, the main focus was on tumor cells rather than on infiltrating host cells. These efforts have resulted in a Food and Drug Administration (FDA)-cleared 70-gene MammaPrint panel and a 21-gene (16 cancer genes and 5 reference genes) Oncotype DX assay panel as prognostic biomarkers for breast cancer recurrence [6, 7]. However, prognostic value of these molecular signatures is limited by the status of the tumor. Studies with focus on stromal gene expression as predictors of clinical outcome in breast cancer patients did not analyze gene signatures expressed by infiltrating immune cells [8]. A few reports with special focus on infiltrating cells emphasized mainly particular immune cells such as lymphocytes or tumor-associated macrophages [9–11], rather than a network of immune function genes. Most recent studies showed that T-cell metagene can predict a favorable prognosis in ER negative and HER2-positive breast cancers [12]. Despite the fact that breast tumors are usually infiltrated and/or surrounded with cells of the immune system, our knowledge as to the network (signature) of immune function genes orchestrating tumor promotion or inhibition is very limited. Without this knowledge, accuracy of predicting breast cancer outcomes following initial therapy would be limited by omitting one of the key components of tumor microenvironment, i.e., infiltrating immune cells.
Recently, we conducted pre-clinical studies using the FVBN202 transgenic mouse model of breast carcinoma and determined that a network of immune function genes in the tumor microenvironment could predict tumor progression, tumor relapse, or relapse-free survival [13]. These findings led to the present retrospective study in breast cancer patients for whom we had outcome data available. Based on our preclinical findings, we hypothesized that distinct networks (signatures) of immune function genes expressed by tumor-infiltrating and/or tumor-associated cells at the time of diagnosis could predict breast cancer recurrence or relapse-free survival following conventional therapies, and could offer immunotherapeutic strategies to overcome tumor relapse.
Materials and methods
Clinical specimens
Tissue specimens were collected from female breast cancer patients before any treatment and maintained in the VCU Massey Cancer Center Tissue & Data Acquisition and Analysis Core (TDAAC) over the past 7 years. According to the follow-up history thus far, we have corresponding annotated patient outcome data available which can stratify the patients into 1–5 years relapse (n = 8) and up to 7 years relapse-free survival (n = 9). Frozen tissues were used for RNA extraction, and paraffin-embedded tissues were used for immunohistochemistry (IHC) staining. Two pathologists identified specimens that contained 10–70% tumor-infiltrating cells for analysis of immune function genes. Variation in the percent infiltrating cells among specimens was due to different methods of tumor collection used. These studies have been reviewed and approved by the Institutional Review Board (HM10920 and 2471-Tissue Acquisition System for Cancer Research) at Virginia Commonwealth University.
RNA amplification, probe preparation, and microarray hybridization
For expression studies based on oligo array techniques, total RNA from tumors was amplified into antisense RNA (aRNA) as previously described [14, 15]. Reference control in human arrays was obtained by pooling peripheral blood mononuclear cells (PBMC) from four normal donors. Both, human reference and test total RNA were amplified into antisense RNA in large amounts using identical conditions [14, 15]. Confidence about array quality was confirmed as previously described [16]. For 36 k human array performances, both reference and test aRNA were directly labeled using ULS aRNA Fluorescent Labeling kit (Kreatech) with Cy3 for reference and Cy5 for test samples. Whole-genome human 36K oligo arrays, representing 25,100 unique genes of the Operon Human Genome Array-Ready OligoSet version 4.0, were printed in house, using oligos purchased from Operon. The design is based on the Ensembl Human Databasebuild NCBI-35c with a full coverage on the NCBI human Refseq dataset (04/04/2005). Hybridization was carried out in a water bath at +42°C for 20 h, and the arrays were then washed and scanned on an Agilent Microarray Scanner. Resulting data files were analyzed using BRB-Array-Tools developed by the Biometric Research Branch, National Cancer Institute, National Institutes of Health and visualized using Cluster and TreeView software. The global gene expression profiling consisted of 17 experimental samples. Subsequent filtering (80% gene presence across all experiments) selected 9797 genes for further analysis. Gene ratios were average corrected across experimental samples and displayed according to uncentered algorithm.
Statistical analysis
Unsupervised analysis was performed for class confirmation using the BRB-Array-Tools and Stanford Cluster Program. Class comparison was performed using parametric unpaired Student’s t test or three-way ANOVA to identify genes differentially expressed among relapse and relapse-free groups using different significance cutoff levels as demanded by statistical power of each comparison. Statistical significance and univariate and multivariate permutation test as previously described [17]. Functional gene network analysis was performed using the Ingenuity Pathway Analysis system (IPA) which transforms large data sets into a group of relevant networks containing direct and indirect relationships between genes based on known interactions in the literature.
Complete leave-one-out cross validation model
Models to predict which breast cancer patients are likely to relapse were developed using BRB-Array-Tools [18]. Complete leave-one-out cross validation-(LOOCV) based prediction accuracy estimates were 100% for the compound covariate predictor (CCP) and the diagonal linear discriminant analysis (DLDA) classifier. CCP is a weighted linear combination of log-ratios for genes that are univariately significant at the specified level. The univariate t-statistics for comparing the classes are used as the weights. DLDA is a version of linear discriminant analysis that ignores correlations among the genes to avoid over-fitting the data. Based on 1000 random permutations, the CCP and the DLDA classifier both had P value of 0.001.
Immunohistochemistry
Immunohistochemistry of paraffin-embedded tumor specimens was performed using Dako automated immunostainer (Dako, Carpinteria, CA). We used anti-human antibodies towards CXCL10 (Santa Cruz Biotechnology, 1:300), signal transducer and activator of transcription 1 (STAT1) (BD Biosciences; 1:100), guanylate binding protein 1 (GBP1) (Abnova, 1:75), granzyme A (GZMA) (SeroTec, 1:50), and CD19 (Abcam, 1:1000) which represent T and B cell responses as well as antigen presentation pathways. The antigen retrieval was achieved using a rice steamer. In order to circumvent the endogenous biotin activity, we used Dako Envision Dual Link System-HRP (Dako, Capinteria CA) in a two-step IHC technique, based on HRP labeled polymer which is conjugated with secondary antibodies. The labeled polymer does not contain avidin or biotin, thereby avoiding the non-specific endogenous avidin–biotin activity in the sections.
Real-time PCR
The RNAs were extracted using Trizol as previously described by our group [13]. The cDNA was prepared from 1 μg of total RNA using the Superscript II Kit (Invitrogen) with a dT18 oligonucleotide primer at 42°C for 2 h. The SensiMix SYBR & Fluorescein Kit (BIOLINE, Taunton, MA) was used according to manufacturer’s instructions, and real-time PCR was performed using the Bio-Rad’s real-time PCR detection system. Primers for GAPDH, CXCL10, interferon regulatory factor 1 (IRF1), GBP1, and IL-7 Rα were described previously (19–22). Other primers included GZMA [5′-GGGACAGCAGCCACAATGAGGAAC-3′ and 5′-AGGTCACCTTCGCGTGTGGC-3′]; GZMB [5′-TGCA GGAAGATCGAAAGTGCG-3′ and 5′-GAGGCATGCCA TTGTTTCGTC-3′]; STAT1 variant beta [5′-ATGGGTGG AGCGGTCCCAGAAC-3′ and 5′-AGCATCTTCAACAGG CCCCAGCC-3′]. Immunoglobulin lambda-like polypeptide 5 (IGLL5) [5′-GGGGGTTTTGGTCTGAGCCTCA-3′ and 5′-TTCATGCGTGACCTGGCAGCTG-3′], immunoglobulin kappa chain locus (IGKC) [5′-TCCAGGCACCCTGTC TTTCTCTCC-3′ and 5′-GCAGGCACACAACAGAGGCA GT-3′], occludin (OCLN) [5′-CCCCCTCCCATCCGAGTT TCAG-3′ and 5′-TCAAGAGGCCTGGATGACATGGCT-3′]. Data were normalized to GAPDH housekeeping gene.
This study was conducted under Institutional Review Board (IRB) protocol# HM10920 at Virginia Commonwealth University. All patients gave informed consent under the IRB protocol #247: Tissue Acquisition System for Cancer Research at Virginia Commonwealth University.
Results
Patients at high risk or low risk of tumor recurrence show distinct clustering of genes in the tumor microenvironment at the time of diagnosis
We have previously shown that microarray analysis of immune function genes in the tumor microenvironment of a mouse model of breast carcinoma had prognostic value for predicting tumor rejection, tumor progression, and recurrence [13]. In this study, we sought to determine whether taking a similar approach with a focus of the specimens with tumor-infiltrating cells may predict disease outcome in breast cancer patients. We performed total RNA extraction from frozen tumor specimens with at least 10% infiltrating cells derived from the patients with no evidence of recurrence up to 7 years follow-up (n = 9) and those with recurrence in the first 5 years of follow-up (n = 8). All patients included in this discovery group had differences in age, ethnicity, tumor stage, and status of ER, PR, or HER-2/ neu in their tumors (Supplementary Table 1). Microarray analyses were performed on the amplified RNA using 36K oligo human arrays. Genes with missing values >80% were excluded from further analysis trimming the final working set to 9797 genes. Unsupervised clustering showed strong differences between the two groups of patients (Fig. 1a). Multiple dimensional scaling based on the complete data set demonstrated that the relapse-free group (red color) segregated completely in Euclidian space from those who had suffered a relapse (green color) (Fig. 1b).
Fig. 1.

Unsupervised gene clustering. a Unsupervised cluster visualization of genes differentially expressed among relapse (n = 8) and relapse-free (n = 9) patients. Tumors were hybridized to 36K oligo human array. Genes with at least 80% presence among all samples (9797) were projected using log2 intensity. Red indicates over-expression; green indicates under-expression; black indicates unchanged expression; gray indicates no detection of expression (intensity of both Cy3 and Cy5 below the cutoff value). Each row represents a single gene; each column represents a single sample. The dendrogram at the left of matrix indicates the degree of similarity among the genes examined by expression patterns. The dendrogram at the top of the matrix indicates the degree of similarity between samples. b Multiple dimensional scaling based on the 36K oligo array human platform comparing Relapse free (Red color) and relapse (Green color)
Differential expression of immune function genes at the tumor microenvironment is associated with breast cancer outcome
In order to determine overall differences between relapse-free and relapsed patients, direct comparison between the two clinical outcomes was performed using Student’s t test with 10000 random permutations test. The differentially expressed genes were selected based on permutation P value < 0.005 and parametric P value < 0.001. The comparison identified 349 genes differentially expressed between the two groups with the zero probabilities of getting at least 349 genes significant by chance (at the 0.001 level) if there are no real differences between the relapse and relapse-free group (Global test P value = 0). Among the 349 genes, 299 were upregulated in relapse-free patients compared to relapsed patients (Fig. 2 and Supplementary Table 2). These genes included a dominant cluster of co-modulated genes involved in T-cell response (CXCL10, CXCL9, GZMA, GZMB, HLA-B, HLA-C, CCR7, AIM2, APOE, GBP1, and IL7R), B cell activation (CD19, C1QA, and CD8A), and antigen presentation (APOE, CD74, CR1, IL23A, and STAT1). In addition, genes such as cytotoxic T lymphocyte antigen 4 (CTLA-4) and IL-23 Rα that are involved in negative regulation of effector immune responses were also upregulated in relapse-free patients (Supplementary Table 2). Conversely, 50 genes that were downregulated in relapse-free patients were not associated with immune function except for a few genes involved in viral defense mechanisms (integrin B5–ITGB5) (Supplementary Table 3).
Fig. 2.
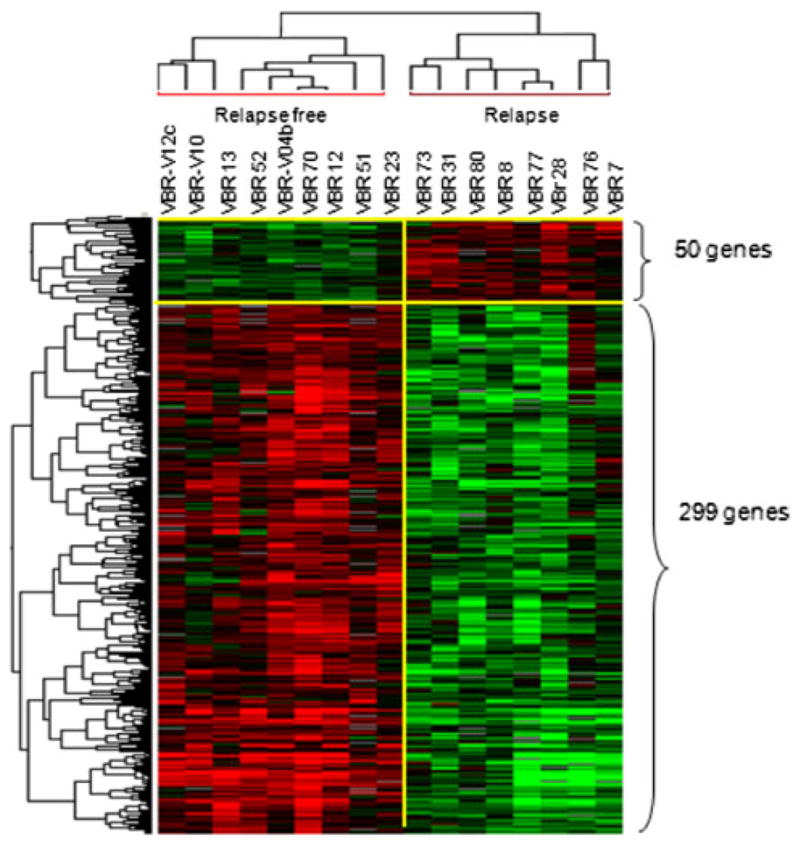
Supervised gene clustering and canonical pathway analysis. Heat map of the 349 genes, identified by Student’s t test (P = 0.001) comparing relapse and relapse-free patients
A heat map of the gene signature, identified by Student’s t test P < 0.001 showed a perfect segregation of the two groups of patients (Fig. 2). Interestingly, none of these genes was found in the 70-gene MammaPrint signature or 16-gene Oncotype DX panel. Moreover, among the selected 9797 genes derived from 80% filtering, we could find 16 (out of 70) and 13 (out of 16) genes belong, respectively, to MammaPrint and Oncotype DX panel. An unsupervised cluster based on the 16 MammaPrint genes (Fig. 3a) and on 13 Oncotype DX genes (Fig. 3b) did not show a clear segregation between relapse and relapse-free groups. On the contrary, Complete LOOCV-based prediction model applied to the 349 genes derived from the Student’s t test p < 0.001 identified the genes IGK@ (IGKC), GBP1, STAT1, IGLL5, and OCLN as best predictor of relapse or relapse-free survival (Fig. 3c). Among these five genes, the first four were selected in all LOOCV models, while OCLN was the only missed one in all LOOCV models, reflecting very stable feature sets. Unsupervised clustering based on these five genes showed a perfect segregation between relapsed and relapse-free patients.
Fig. 3.

Unsupervised gene cluster analysis. a Sixteen genes among Mammaprint panel set differentially expressed among relapse (brown) and relapse-free (red) patients. b Thirteen genes among Oncotype DX assay panel set differentially expressed among relapse (brown) and relapse-free (red) patients. c Five genes selected from the 299 genes by Complete Leave-One-Out Cross Validation (LOOCV) model as best predictors of diagnostic outcome. Red dots under the cluster indicates relapse free and brown dots indicates relapse group
Detection of distinct immune function pathways at the time of diagnosis can predict relapse-free survival in breast cancer patients
The pathway analysis was performed on the 349 genes in the supervised comparisons using the gene set expression comparison kit implemented in BRB-Array-Tools. The human pathway lists determined by “Ingenuity System Database” were selected. Samples with no recurrence showed significant upregulation of genes involved in antigen presentation pathway, allograft rejection, graft-versus-host disease (GVHD), B cell development, dendritic cell maturation, and interferon signaling (Fig. 4). Interestingly, genes involved in T-cell apoptosis, immunodeficiency signaling, CTLA-4 signaling and production of NO, and reactive oxygen species were also upregulated in the tumor specimens of the patients with relapse-free survival. Most relevant pathways are listed in Supplementary Fig. 1a–e. An independent cohort of patients were also included in the validation group, and confirmatory real-time PCR was performed on the selected genes representing IFN-stimulated genes (ISGs) and T-cell response (CXCL10, GZMA, GZMB, IL-7 Rα, and IRF1) as well as the 5-gene signatures identified as best predictor of relapse or relapse-free survival. We used RNAs extracted from tumor lesions of 12 patients who served as validation group (seven with relapse-free survival and five with relapse). Patients with relapse-free survival had significantly higher expression of the immune function genes (>85% with an exception of patient#6) compared to the patients with relapse (Fig. 5).
Fig. 4.

Ingenuity pathway analysis. Forty-six canonical pathways significant at the nominal 0.001 level of the unpaired Student’s t test. The P value for each pathway is indicated by the bar and is expressed as –1 times the log of the P value. The line represents the ratio of the number of genes in a given pathway that meet the cutoff criteria divided by the total number of genes that make up that pathway
Fig. 5.

Real-time PCR analysis of frozen tumor specimens of relapse-free versus relapse patients. Two cohorts of patients were included in the validation group, and their tumors were subjected to confirmatory real-time PCR. Data are presented as average of mean of triplicate wells after normalization to GAPDH
In order to determine cellular sources of genes representing immune function pathways, IHC analysis of paraffin-embedded tumor specimens was performed according to the availability of commercial Abs and also intensity of the genes that would allow detection of their protein products. The IHC further confirmed higher expression of CXCL10, STAT1, GBP1, GZMA, and CD19 in the relapse-free group compared to those from the relapse group: human tonsils are shown as positive controls (Fig. 6a). CXCL10 was expressed both in infiltrating cells and tumor cells of relapse-free patients while it was weakly expressed in tumor cells of patients with relapse. STAT1 showed strong staining in infiltrating cells and tumor cells of relapse-free group while it was expressed to a lesser extent mainly in infiltrating cells of the relapse group. GBP1 was expressed primarily in the infiltrating cells and also in tumor cells of the relapse-free group while it was weakly expressed only in tumor cells of the relapse group. GZMA was barely detectable even in human tonsils, yet it was detected only in tumor-infiltrating cells of the relapse-free group. The CD19-positive infiltrating cells were also present at a higher frequency in the tumor lesions of patients with relapse-free survival compared to only scattered presence in those with tumor relapse. Expression level of the proteins was quantified by showing the proportion of tumor-infiltrating cells that stained positive. As shown in Fig. 6b, there were significant differences between the two groups in the expression of CXCL10 (P = 0.013), STAT1 (P = 0.033), GBP1 (P = 0.005), GZMA (P = 0.010), and CD19 (P = 0.046).
Fig. 6.

IHC analysis of paraffin-embedded tumor specimens of relapse-free versus relapse patients. a Representative data (×400 magnification) from nine patients with relapse-free survival and eight patients with relapse are presented. Human tonsil was stained as positive control. b Cell counts are presented as percent positive cells of tumor-infiltrating cells counted in five fields and averaged using a ×400 magnification
Discussion
We showed that microarray analysis of breast tumor specimens with tumor-infiltrating cells can detect a network of 349 genes which included 299 genes encompassing immune function genes that had a prognostic value in a diverse cohort of breast cancer patients. Importantly, among these genes a 5-gene signature [IGK@ (IGKC), GBP1, STAT1, IGLL5, and OCLN] was identified as best predictor of relapse-free survival with >85% accuracy. Although limitation with the availability of clinical specimens and the patients outcome data did not allow including a larger cohort of specimens in the validation group, such a significant predictive value in a diverse cohort of the patients is promising and has led to an ongoing prospective studies.
The network of immune function genes that were exclusively upregulated in the tumor lesions of breast cancer patients with relapse-free survival included those involved in B cell development, interferon signaling associated with allograft rejection and autoimmune reaction, antigen presentation pathway, and cross talk between adaptive and innate immune responses. On the other hand, these genes were downregulated in tumor specimens of patients with subsequent relapse, compared to those in the standard PBMC. Interestingly, genes involved in primary immunodeficiency signaling, T-cell apoptosis, CTLA4 signaling and production of NO, and reactive oxygen species were also upregulated in the tumor specimens of relapse-free patients. Such paradoxical findings as to simultaneous upregulation of immune effector genes and immune suppressor genes may suggest that tumor-derived factors were responsible for the expression of immune suppressor genes thereby facilitating cancer progression even in the presence of the increased immune effector genes. However, removal of breast tumors by conventional therapy must have eliminated the source of immune suppressive factors and resulted in downregulation of the suppressor genes; subsequently, the immune effector genes may have protected the patients from their residual micrometastases and relapse. This possibility is supported by our preclinical findings [23–25] as well as others [26, 27], showing that primary tumors secrete soluble factors including GM-CSF, VEGF, and MCP-1 that result in increases in myeloid-derived suppressor cells (MDSCs) such that successful immunotherapy was not possible unless MDSCs were depleted or reduced by chemotherapy or shrinking of the primary tumors. Real-time PCR analysis of selected genes in a validation cohort of patients showed significant differences between two groups in the expression of immune function genes. Importantly, variability within a group in the expression of a single immune function gene suggests that a signature rather than a single gene can predict relapse-free survival.
This novel signature associated with favorable outcome included 299 genes encompassing the immune function genes that were distinct from the 70-gene MammaPrint signature and from 16-gene signature of the Oncotype DX panel. Moreover, an unsupervised clustering based on MammaPrint and Oncotype DX genes did not show a clear segregation between relapsed and relapse-free groups. Oncotype DX was originally validated in the patients with ER+ and node-negative tumors, though it is now being expanded to patients with node-positive breast cancer. Therefore, it was not surprising that Oncotype DX could not segregate the patients in this study, because of majority of these patients were ER negative and/or node positive. Interestingly, a perfect segregation was shown by an unsupervised cluster analysis based on 5 genes (IGK@, GBP1, STAT1, IGLL5, and OCLN) among the 299 genes which were identified by the LOOCV prediction model as best predictor of relapse or relapse-free survival. Real-time PCR also confirmed higher expression of these 5 genes in a greater than 85% of patients with relapse-free survival.
The IHC analysis of tumor specimens further confirmed upregulation of the immune function genes representing immune mechanism pathways mainly in tumor-infiltrating cells of the relapse-free group. For instance, CXCL10 and GBP1 are ISGs that showed strong staining in tumor-infiltrating cells of relapse-free patients compared to the relapsed group. CXCL10 binds CXCR3 on DCs, macrophages, and T cells. Increased expression of CXCL10 in tumor lesions of relapse-free patients may suggest CXCL10-induced DC maturation and antigen cross presentation that results in Th1-type immune responses [28]. GBP1 is a key mediator of angiostatic effects of the immune responses, inflammation, in particular, and its expression in the tumors and tumor-infiltrating immune cells is associated with favorable prognosis [29], as was the case in our study. As expected, upregulation of these ISGs in relapse-free patients compared to relapsed groups was associated with higher expression of STAT1 and IRF1 genes as well as an increased expression of nuclear STAT1 in their tumor-infiltrating cells. However, nuclear expression of STAT1 in tumor cells was comparable between the two groups. This may explain progression of primary breast cancer in the two groups. It was shown that increased nuclear STAT1 resulted in the induction of apoptosis in the tumors [30] as well as the metastatic ability of the tumors that escaped from apoptosis [31]. GZMA and GZMB which are involved in T-cell responses and the GVHD pathway as well as CD19 which is involved in B cell response were also uniformly increased in tumor lesions of relapse-free patients. Although there was a significant difference between the two groups in the expression of each molecule, a panel of differentially expressed molecules, i.e., signature of immune function genes provides more valid prognostic marker when compared to one molecule alone.
Our findings as to the prognostic value of immune function genes are consistent with those reported by Rody et al. [12]. However, they found T cells as predictor of a favorable prognosis. Such discrepancies are due to different approaches and experimental design. Rody et al. [12] determined correlation of T cell infiltrates with survival in patients with ER-negative and HER2-positive tumors and validated their findings by IHC only in tissue specimens from the patients with favorable prognosis. We performed comparative analysis between two cohorts of patients and also validated our signature in the two cohorts to determine whether some patients with relapse may also show expression of the immune function genes in their tumors. Our data suggest that the 5-gene signature of immune response (IGK@, GBP1, STAT1, IGLL5, and OCLN) can not only be used as prognostic biomarker for breast cancer patients but it would also offer therapeutic strategies for preventing breast cancer recurrence. Absence of the 5-gene signature in the tumor lesions at the time of diagnosis suggests that the patients may be at a high risk of relapse. Our data suggest that prospective studies need to be conducted to determine whether the patients with high risk of relapse may benefit from neoadjuvant immunotherapy.
Supplementary Material
Acknowledgments
This study was supported by NIH R01 CA104757 Grant (M. H. Manjili), Massey Cancer Center Pilot Project Program 2006FPP-04 (M. H. Manjili), and VCU Technology Transfer Fund. We gratefully acknowledge the support of VCU Massey Cancer Center and the Commonwealth Foundation for Cancer Research. We thank Dr. K. Najarian for his review of the statistical analysis.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s10549-011-1470-x) contains supplementary material, which is available to authorized users.
Contributor Information
Maria Libera Ascierto, Infectious Disease and Immunogenetics Section (IDIS), Department of Transfusion Medicine and Center for Human Immunology, National Institutes of Health, Bethesda, MD 20892, USA.
Maciej Kmieciak, Department of Microbiology & Immunology, Virginia Commonwealth University Massey Cancer Center, Richmond, VA 23298, USA.
Michael O. Idowu, Department of Pathology, Virginia Commonwealth University Massey Cancer Center, Richmond, VA 23298, USA
Rose Manjili, Department of Microbiology & Immunology, Virginia Commonwealth University Massey Cancer Center, Richmond, VA 23298, USA.
Yingdong Zhao, Division of Cancer Treatment and Diagnosis, National Institutes of Health, Bethesda, MD 20892, USA.
Margaret Grimes, Department of Pathology, Virginia Commonwealth University Massey Cancer Center, Richmond, VA 23298, USA.
Catherine Dumur, Department of Pathology, Virginia Commonwealth University Massey Cancer Center, Richmond, VA 23298, USA.
Ena Wang, Infectious Disease and Immunogenetics Section (IDIS), Department of Transfusion Medicine and Center for Human Immunology, National Institutes of Health, Bethesda, MD 20892, USA.
Viswanathan Ramakrishnan, Department of Biostatistics, Virginia Commonwealth University Massey Cancer Center, Richmond, VA 23298, USA.
Xiang-Yang Wang, Department of Human and Molecular Genetics, Virginia Commonwealth University Massey Cancer Center, Richmond, VA 23298, USA.
Harry D. Bear, Department of Surgery, Virginia Commonwealth University Massey Cancer Center, Richmond, VA 23298, USA
Francesco M. Marincola, Infectious Disease and Immunogenetics Section (IDIS), Department of Transfusion Medicine and Center for Human Immunology, National Institutes of Health, Bethesda, MD 20892, USA
Masoud H. Manjili, Email: mmanjili@vcu.edu, Department of Microbiology & Immunology, Virginia Commonwealth University Massey Cancer Center, Richmond, VA 23298, USA
References
- 1.Onitilo AA, Engel JM, Greenlee RT, Mukesh BN. Breast cancer subtypes based on ER/PR and Her2 expression: comparison of clinicopathologic features and survival. Clin Med Res. 2009;7:4–13. doi: 10.3121/cmr.2009.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 3.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, Foekens JA. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 5.Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lønning P, Børresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knauer M, Mook S, Rutgers EJ, Bender RA, Hauptmann M, van de Vijver MJ, Koornstra RH, Bueno-de-Mesquita JM, Linn SC, van ‘t Veer LJ. The predictive value of the 70-gene signature for adjuvant chemotherapy in early breast cancer. Breast Cancer Res Treat. 2010;120:655–661. doi: 10.1007/s10549-010-0814-2. [DOI] [PubMed] [Google Scholar]
- 7.Cronin M, Sangli C, Liu ML, Pho M, Dutta D, Nguyen A, Jeong J, Wu J, Langone KC, Watson D. Analytical validation of the Oncotype DX genomic diagnostic test for recurrence prognosis and therapeutic response prediction in node-negative, estrogen receptor-positive breast cancer. Clin Chem. 2007;53:1084–1091. doi: 10.1373/clinchem.2006.076497. [DOI] [PubMed] [Google Scholar]
- 8.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, Hallett M, Park M. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 9.Pagés F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, Meatchi T, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Galon J. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353:2654–2666. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 10.Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, Ohtani H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58:3491–3494. [PubMed] [Google Scholar]
- 11.Ueno T, Toi M, Saji H, Muta M, Bando H, Kuroi K, Koike M, Inadera H, Matsushima K. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin Cancer Res. 2000;6:3282–3289. [PubMed] [Google Scholar]
- 12.Rody A, Holtrich U, Pusztai L, Liedtke C, Gaetje R, Ruckhaeberle E, Solbach C, Hanker L, Ahr A, Metzler D, Engels K, Karn T, Kaufmann M. T-cell metagene predicts a favorable prognosis in estrogen receptor-negative and HER2-positive breast cancers. Breast Cancer Res. 2009;11(2):R15. doi: 10.1186/bcr2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Worschech A, Kmieciak M, Knutson KL, Bear HD, Szalay AA, Wang E, Marincola FM, Manjili MH. Signatures associated with rejection or recurrence in HER-2/neu-positive mammary tumors. Cancer Res. 2008;68:2436–2446. doi: 10.1158/0008-5472.CAN-07-6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang E, Miller LD, Ohnmacht GA, Liu ET, Marincola FM. High fidelity mRNA amplification. Nat Biotechnol. 2000;18:457–459. doi: 10.1038/74546. [DOI] [PubMed] [Google Scholar]
- 15.Wang E. RNA amplification for successful gene profiling analysis. J Transl Med. 2005;3:28. doi: 10.1186/1479-5876-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin P, Zhao Y, Ngalame Y, Panelli MC, Nagorsen D, Monsurró V, Smith K, Hu N, Su H, Taylor PR, Marincola FM, Wang E. Selection and validation of endogenous endogenous reference genes using a high throughput approach. BMC Genomics. 2004;5:55. doi: 10.1186/1471-2164-5-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang E, Miller LD, Ohnmacht GA, Mocellin S, Perez-Diez A, Petersen D, Zhao Y, Simon R, Powell JI, Asaki E, Alexander HR, Duray PH, Herlyn M, Restifo NP, Liu ET, Rosenberg SA, Marincola FM. Prospective molecular profiling of subcutaneous melanoma metastases suggests classifiers of immune responsiveness. Cancer Res. 2002;62:3581–3586. [PMC free article] [PubMed] [Google Scholar]
- 18.Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-array tools. Cancer Inform. 2007;3:11–17. [PMC free article] [PubMed] [Google Scholar]
- 19.Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor. J Exp Med. 2004;199:731–736. doi: 10.1084/jem.20031482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dominguez F, Martínez S, Quiñonero A, Loro F, Horcajadas JA, Pellicer A, Simón C. CXCL10 and IL-6 induce chemotaxis in human trophoblast cell lines. Mol Hum Reprod. 2008;14:423–430. doi: 10.1093/molehr/gan032. [DOI] [PubMed] [Google Scholar]
- 21.Sun W, Xu W, Snyder M, He W, Ho H, Ivashkiv LB, Zhang JJ. The conserved leu-724 residue is required for both serine phosphorylation and co-activator recruitment for stat1-mediated transcription activation in response to interferon-γ. J Biol Chem. 2005;280:41844–41851. doi: 10.1074/jbc.M505797200. [DOI] [PubMed] [Google Scholar]
- 22.Lundmark F, Duvefelt K, Iacobaeus E, Kockum I, Wallström E, Khademi M, Oturai A, Ryder LP, Saarela J, Harbo HF, Celius EG, Salter H, Olsson T, Hillert J. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat Genet. 2007;39:1108–1113. doi: 10.1038/ng2106. [DOI] [PubMed] [Google Scholar]
- 23.Morales JK, Kmieciak M, Graham L, Feldmesser M, Bear HD, Manjili MH. Adoptive transfer of HER2/neu-specific T cells expanded with alternating gamma chain cytokines mediate tumor regression when combined with the depletion of myeloid-derived suppressor cells. Cancer Immunol Immunother. 2009;58:941–953. doi: 10.1007/s00262-008-0609-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat. 2010;123:39–49. doi: 10.1007/s10549-009-0622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. 2009;9:900–909. doi: 10.1016/j.intimp.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 26.Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rébé C, Ghiringhelli F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–3061. doi: 10.1158/0008-5472.CAN-09-3690. [DOI] [PubMed] [Google Scholar]
- 27.Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. 2010;185:2273–2284. doi: 10.4049/jimmunol.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krathwohl MD, Anderson JL. Chemokine CXCL10 (IP-10) is sufficient to trigger an immune response to injected antigens in a mouse model. Vaccine. 2006;24:2987–2993. doi: 10.1016/j.vaccine.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 29.Naschberger E, Croner RS, Merkel S, Dimmler A, Tripal P, Amann KU, Kremmer E, Brueckl WM, Papadopoulos T, Hohenadl C, Hohenberger W, Stürzl M. Angiostatic immune reaction in colorectal carcinoma: impact on survival and perspectives for antiangiogenic therapy. Int J Cancer. 2008;123:2120–2129. doi: 10.1002/ijc.23764. [DOI] [PubMed] [Google Scholar]
- 30.Thomas M, Finnegan CE, Rogers KM, Purcell JW, Trimble A, Johnston PG, Boland MP. STAT1: a modulator of chemotherapy-induced apoptosis. Cancer Res. 2004;64:8357–8364. doi: 10.1158/0008-5472.CAN-04-1864. [DOI] [PubMed] [Google Scholar]
- 31.Khodarev NN, Roach P, Pitroda SP, Golden DW, Bhayani M, Shao MY, Darga TE, Beveridge MG, Sood RF, Sutton HG, Beckett MA, Mauceri HJ, Posner MC, Weichselbaum RR. STAT1 pathway mediates amplification of metastatic potential and resistance to therapy. PLoS One. 2009;4:e5821. doi: 10.1371/journal.pone.0005821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.