

Abstract
Many insights into the mechanisms and signaling pathways underlying aging have resulted from research on the nematode Caenorhabditis elegans. In this paper, we discuss the recent findings that emerged using this model organism concerning the role of reactive oxygen species (ROS) in the aging process. The accrual of oxidative stress and damage has been the predominant mechanistic explanation for the process of aging for many years, but reviewing the recent studies in C. elegans calls this theory into question. Thus, it becomes more and more evident that ROS are not merely toxic byproducts of the oxidative metabolism. Rather it seems more likely that tightly controlled concentrations of ROS and fluctuations in redox potential are important mediators of signaling processes. We therefore discuss some theories that explain how redox signaling may be involved in aging and provide some examples of ROS functions and signaling in C. elegans metabolism. To understand the role of ROS and the redox status in physiology, stress response, development, and aging, there is a rising need for accurate and reversible in vivo detection. Therefore, we comment on some methods of ROS and redox detection with emphasis on the implementation of genetically encoded biosensors in C. elegans.
1. Oxidative Stress Theory of Aging and Correlation of Oxidative Stress with Age
More than fifty years ago, Harman [1] postulated in his free-radical theory of aging that aging results from the accumulation of molecular damage caused by byproducts of the normal oxidative metabolism, called reactive oxygen species (ROS). The discovery of the superoxide dismutase enzyme, which detoxifies the superoxide anion [2], and the detection of the ROS hydrogen peroxide (H2O2) in vivo [3] further gave credibility to the free-radical theory of aging. Harman refined his theory to highlight the role of mitochondria in aging since mitochondria are considered as the main source of ROS [4]. Because there are many ROS that are not free-radicals and that can also cause oxidative damage, the free-radical theory of aging is now referred to as the oxidative stress theory of aging [5]. This new name for the theory also implies that oxidative stress can occur due to an imbalance between ROS production and removal. This imbalance leads to a progressive accumulation of oxidative damage with age, resulting in a decline of the functional efficiency of various cellular processes. Since its formulation, the oxidative stress theory of aging has been the most broadly tested theory of aging. Despite the tremendous effort to verify this theory, experimental studies do not support or remain inconclusive on whether oxidative damage is responsible for aging [6–10]. During the last years, many studies have yielded new insights on the role of ROS in aging. Many of these studies have been performed using the model organism Caenorhabditis elegans and will be discussed in this paper.
As organisms age, a number of behavioral, reproductive, morphological, and biochemical changes occur [11]. Previous studies have shown an age-related increase of oxidative damage in a variety of molecules (DNA, proteins, and lipids) in organisms ranging from invertebrates to humans [5, 12–14]. This finding is in accordance with the oxidative stress hypothesis of aging. In C. elegans, there is an increase in single-strand DNA breaks, 5-methylcytosine [15], and protein carbonyls accumulate with age [16, 17]. However, as in many other invertebrates [18, 19], in subsequent tests on extensively purified DNA of C. elegans, no cytosine methylation was detected during development and senescence [20]. Melov et al. [21] reported an increase in mitochondrial DNA deletions with age. However, these results could not be confirmed in our research group [22].
One way to investigate the role of ROS in aging is to examine oxidative stress resistance in long-lived strains. Two major interventions that greatly influence lifespan are the regulation of insulin/IGF-1 signaling (IIS) and dietary restriction. The IIS pathway is an evolutionary conserved pathway that regulates aging in organisms ranging from nematodes to mammals. In C. elegans, it comprises an insulin-like receptor (daf-2), which negatively regulates a downstream FOXO transcription factor, DAF-16. A reduction of function of daf-2 doubles the lifespan of C. elegans, and this lifespan extension is fully dependent on daf-16. Many of the transcriptional targets of DAF-16 are also activated in the dauer larvae, a special developmental stage of C. elegans that can survive harsh environmental conditions for several months, while the average lifespan of an adult C. elegans is about 2-3 weeks. The IIS pathway and pathways related to IIS and/or DAF-16 are induced by various environmental cues (nutrients, heat, UV, heavy metal, and oxidative stress) [23–26]. These pathways promote oxidative stress resistance and longevity and regulate the expression of ROS detoxification genes. Although oxidative stress resistance is increased in these long-lived strains, this does not mean that increased stress resistance is an essential prerequisite for longevity. To test this, the expression of sod genes was reduced in long-lived daf-2 mutants. Although isolated mitochondria of daf-2 mutants produce more superoxide compared to wild type [27], the loss of sod genes has little or no effect on daf-2 longevity and in some cases, it even further increases lifespan of daf-2 [28–31]. Thus, increased antioxidant defenses in daf-2 do not significantly contribute to the extended lifespan of daf-2 mutants.
A reduction of food intake by 40–60% without malnutrition, called dietary restriction (DR), has remarkable benefits for health and lifespan in many different species, including C. elegans [32]. It was long believed that DR would reduce metabolic rate and thereby decrease ROS production and ROS-induced damage, resulting in extended lifespan [33]. Measurement of the oxygen consumption and heat production rather shows an increase in metabolism in food-restricted worms, however [34, 35]. ROS production in worms imposed to at least one type of DR is not different from wild type [27]. Dietary restriction enhances SOD and catalase activity [36] and increases sod expression (sod-1, sod-2, sod-4, and sod-5), regulated by the FOXA transcription factor PHA-4 [37]. Although diet-restricted worms show increased antioxidant defense and oxidative stress resistance [36, 38], sod deletion does not reduce lifespan in two different types of DR [39, 40]. This suggests that enhanced antioxidant defense may not be essential for DR-induced longevity.
2. Evaluation of the Oxidative Stress Theory in C. elegans
Conform to the oxidative stress theory of aging, oxidatively damaged biomolecules accumulate with age in C. elegans. Moreover, it is often found that manipulations that slow down aging, such as dietary restriction, hormesis (discussed below), or reduced IIS, also increase oxidative stress resistance. Although such observations provide correlative evidence for the oxidative stress theory of aging, they do not allow concluding that this theory is correct. Long-lived strains are resistant to other types of stresses than oxidative stress, such as heat, UV, and pathogenic bacteria. Various studies show that enhanced oxidative stress resistance is not essential for the extended lifespan in long-lived strains [38]. The most conclusive approach to test the causal relation between ROS and aging is to change the antioxidant defense system and to examine the effect on aging. Antioxidant defenses can be altered by genetic (mutation or overexpression of antioxidant enzymes) or pharmacological interventions (SOD/catalase mimetics, vitamins).
2.1. Pharmacological Interventions
Numerous studies have examined the effect of exogenous antioxidant compounds on lifespan in C. elegans [41]. Various noncatalytic antioxidants, such as vitamin E and C, trolox, α-tocopherol, and N-acetylcysteine, affected lifespan differently in distinct studies [42–47]. In some cases, these compounds increased oxidative stress resistance without changing the lifespan [48]. The variability in the outcome of these reports may be explained by differences in dose, experimental conditions, and method of delivery. In C. elegans, the uptake of many drugs is rather poor as they are excluded by the thick cuticle [49]. The use of liposomes ameliorates the uptake of hydrophilic antioxidants, such as ascorbic acid, N-acetyl-cysteine, and reduced glutathione, and prolongs lifespan, while conventional delivery methods do not [50]. It should be noted that this lifespan extension was dose-independent and that liposomes can affect the fat metabolism of the worms, however.
The effect of the SOD/catalase mimetics EUK-8 and EUK-134 on lifespan in C. elegans has been tested by various research groups. Administration of these compounds increases SOD activity in vivo, primarily in the mitochondria, and it also enhances paraquat resistance [51–53]. EUK-8 and EUK-134 treatment extended lifespan in one study [54]. However, other groups detected no increase in lifespan for doses that protect the worm against paraquat [51, 53, 55]. In fact, they even found a dose-dependent decrease in lifespan at higher doses of EUK-8 and EUK-134. This toxic effect might be due to enhanced ROS production when EUK-8 is administered at high doses [56]. Thus, EUK-8 and EUK-134 clearly exhibit antioxidant activity in vivo and can enhance oxidative stress resistance, but without causing lifespan extension.
2.2. Genetic Interventions
C. elegans contains numerous antioxidant genes, and the effect on aging by deleting or overexpressing many of these genes has been examined recently. While most eukaryotes contain 2 or 3 different superoxide dismutase (sod) genes, C. elegans possesses at least six isoforms [64]. sod-1 and sod-5 encode for the cytosolic CuZnSODs [65, 66], while the sod-4 gene expresses two extracellular CuZnSODs that are products of alternative splicing [67] and are either membrane bound or extracellular. sod-2 and sod-3 encode MnSOD enzymes that are localized in the mitochondrial matrix [68, 69]. SOD-1 and SOD-2 contribute to most of the SOD activity during normal development [29, 65], whereas the secondary SODs sod-3 and sod-5 are clearly upregulated in dauers [29, 70], in daf-2 mutants [71] and under oxidative stress [72]. Very recently, a number of groups has examined the effect of sod deletion and overexpression on lifespan and oxidative stress resistance in C. elegans [27, 29–31, 39, 40, 57, 60, 61, 63]. An overview of these studies is represented in Table 1.
Table 1.
Overview of the effect sod knockdown/out or overexpression of C. elegans superoxide dismutases on ROS levels, oxidative damage, and lifespan under stressed and unstressed condition (paraquat or hyperoxia). OE: overexpression; ND: not determined; PQ: paraquat; J: juglone; DR: dietary restriction.
| sod gene | ROS levels | Oxidative damage | Effect on lifespan | Effect on stress survival | Effect on lifespan long-lived worms | |
|---|---|---|---|---|---|---|
| Knockdown/out | ||||||
|
| ||||||
| sod-2 | Increased [57] or no effect [27] | Increased [30, 40] | No effect [29–31, 39] or increased [27, 40, 57] | Decreased (PQ) [29–31, 40] (J) [40] or no effect (hyperoxia) [29] | No effect (daf-2) [29, 30, 40] (DR) [39] or decreased (daf-2) [31] or increased (clk-1) [30, 40] | |
| MnSOD | sod-3 | Increased [57] | ND | No effect [29, 31, 39, 40] or increased [57] | Decreased (pathogenic bacteria) [58] (PQ-J) [40] or no effect (PQ-hyperoxia) [29, 31] or increased [39] | No effect (daf-2) [29] (DR) [39, 40] or increased (daf-2) [31] or decreased (isp-1) [40] |
|
sod-2;
sod-3 |
Increased [59] or decreased [60] | No effect [29, 60] | No effect [29, 31] or increased [40] | Decreased (PQ-hyperoxia) [29, 31, 40] (J) [40] | Increased (daf-2) [31] | |
|
| ||||||
| sod-1 | Increased [61] | Increased [30] or no effect [29] | Decreased [29, 39, 61] or no effect [30, 39, 40] | Decreased (PQ) [29, 30, 39, 40, 61] (J) [40] (hyperoxia) [29] | Decreased (daf-2) [29] or no effect (DR) [39] or increased (daf-2) [30] | |
| sod-4 | ND | ND | No effect [29, 31, 39, 40] | No effect [29] or increased [39] | Increased (daf-2) [29] or no effect (DR) [39] | |
| CuZnSOD | sod-5 | ND | ND | No effect [29, 39, 40] | No effect [29, 39] | No effect (daf-2) [29] (DR) [39] |
| sod-1; | ||||||
| sod-4; | ND | ND | Decreased [29] | ND | ND | |
| sod-5 | ||||||
|
| ||||||
| SOD | sod-12345 | ND | No effect [62] | No effect [62] | Decreased [62] | ND |
|
| ||||||
| OE | ||||||
|
| ||||||
| MnSOD | sod-2 | ND | No effect [63] | Increased [63] | ND | ND |
| CuZnSOD | sod-1 | Increased [63] | Increased [63] | Increased [29, 63] | Decreased (PQ) [29] | ND |
Loss of sod-1 lowers the resistance to oxidative stress [29, 30, 39, 40, 61]. Although SOD-1 contributes to 80% of the total SOD activity in C. elegans [29], its deletion only slightly reduces lifespan [29, 39, 61] or even not at all in other studies [30, 39, 40]. Reduced sod-1 expression increases ROS levels and oxidative damage in some studies [30, 61] but it did not in another one [29]. These contradicting results may reflect that the impact of sod-1 reduction on oxidative stress is relatively subtle and difficult to detect. In contrast to sod-1, loss of sod-4 or sod-5 does not alter the lifespan under stressed or unstressed conditions [29, 31, 39, 40]. Unexpectedly, sod-4 deletion prolongs lifespan in daf-2 mutants [29]. Conform to the findings in mammalian studies [73], it has been proposed that sod-4 may stimulate IIS through the H2O2-dependent inactivation of the phosphatases [6]. This suggests that CuZnSOD is involved in the redox regulation of IIS. Consistent with this statement, mutation of sod-4 or sod-5 enhances dauer larva formation [29]. To resume, loss of CuZnSOD has little negative effect on oxidative stress and lifespan and it may regulate IIS via redox signaling.
While the loss of sod-2, sod-3 or both, reduces oxidative stress resistance in most cases [29–31, 39, 40], it does not shorten lifespan under unstressed conditions [29–31, 39, 40], and in some studies it even increases lifespan [27, 40, 57]. Diminished MnSOD enhances ROS levels and oxidative damage to a minor extent in some reports [30, 40, 57, 59] but not in others [27, 29, 60]. MnSOD knockout/down experiments demonstrated that MnSOD is not required for lifespan extension in long-lived daf-2 mutants and diet-restricted worms [29–31, 39, 40]. In contrast, abolished sod-2 expression largely extends the lifespan of long-lived clk-1 mutants [30, 40]. Consistent with this, sod-2 mutation results in a Clk phenotype, that is, slow growth and defecation rate, and reduced and delayed fecundity [29, 40]. Thus, MnSOD is required for oxidative stress resistance, but it does not limit the lifespan of wild type or long-lived strains. The fact that loss of MnSOD enhances oxidative stress and concurrently prolongs lifespan in certain studies, contradicts a causal role for mitochondrial superoxide in the aging process.
Worms completely devoid of both Mn- and CuZnSOD have been recently obtained and are highly sensitive to multiple stresses (oxidative stress, heat, and osmotic stress), they develop slower and have a reduced brood size [62]. Remarkably, this total loss of SOD activity has no effect on lifespan at all. This lack of effect on lifespan can be explained by a counterbalance between superoxide toxicity on the one hand and an adaptive response to reduced superoxide detoxification on the other hand [62]. Most importantly, it shows that oxidative stress per se has no profound effect on aging in C. elegans.
If the oxidative stress hypothesis of aging is correct, an increase in SOD-1 should result in a decrease in oxidative damage and an extended lifespan. Overexpression of sod-1 indeed prolongs lifespan [29, 63]. But instead of decreasing oxidative stress as expected by the oxidative stress hypothesis, sod-1 overexpression does not reduce lipid oxidation and glycation and actually increases ROS levels and protein oxidation levels [63], and it increases paraquat sensitivity [29]. On the other hand, the lifespan extension of sod-1 overexpressors is DAF-16- and partially HSF-1-dependent and sod-1 overexpression enhances hsp-4 and hsp-6 expression, suggesting an unfolded protein response (UPR) [63]. This idea is further confirmed by the observation that knocking down genes involved in UPR, partly suppresses longevity of sod-1 overexpressors. The exact mechanism by which sod-1 overexpression increases lifespan through DAF-16 remains unclear, however. Like sod-1, sod-2 overexpression also prolongs lifespan in a DAF-16-dependent manner, but it does not change the protein carbonylation levels [63]. These results imply that overexpression of sod-1 or sod-2 extends lifespan, not by reducing oxidative stress, but instead by activating longevity-related transcription factors, such as DAF-16. Therefore, lifespan extension by SOD overexpression does not support the oxidative stress hypothesis. The fact that sod-1 overexpression increases oxidative damage and prolongs lifespan at the same time, even contradicts this hypothesis.
C. elegans contains three catalase enzymes with highly similar sequences in a tandem array: ctl-3, ctl-1, and ctl-2 [74]. The peroxisomal catalase, CTL-2 [75], contributes to ~80% of the total catalase activity and a knockout of ctl-2 can reduce mean lifespan by 16% while decreasing egg-laying capacity [74]. Remarkably, protein carbonyl levels increase more slowly with age in ctl-2 mutants compared to wild type [74]. Moreover, overexpression of all three catalase genes reduces lifespan [29].
C. elegans contains three peroxiredoxins, two 2-Cys peroxiredoxins prdx-2 (CePrx2), and prdx-3 (CePrx1), and one 1-Cys peroxiredoxin prdx-6 (CePrx3). Prdx-3 is probably mitochondrial, and only deletion of prdx-2 results in an altered phenotype, displaying a reduced size, fertility, and oxidative stress resistance [76]. At 25°C, the lifespan of prdx-2 mutants is not reduced, but at 15°C and 20°C prdx-2 knockout worms live shorter than wild-type worms [76–78]. prdx-2 is expressed in pharyngeal neurons and in the reproductive system [76, 78]. Interestingly, overexpression of prdx-2 in the intestine increases oxidative resistance but it does not prolong lifespan [78], indicating that the oxidative stress resistance obtained by tissue-specific prdx-2 expression does not determine lifespan.
Analysis of the C. elegans genome reveals four glutaredoxins (GLRXs), at least eight thioredoxins (TRXs), and two thioredoxin reductases (TRXRs). Deletion of the cytosolic trx-1, expressed in specific pharyngeal neurons and the intestine, decreases lifespan with 19% and increases paraquat sensitivity [79, 80]. Furthermore, trx-1 deletion partially suppresses lifespan extension in daf-2 mutants and completely suppresses longevity induced by two forms of dietary restriction [81]. Overexpression of trx-1 results in a moderate lifespan increase [80]. Deletion of mitochondrial trx-2 and/or trxr-2, expressed in muscles, intestine, and neurons, does not affect lifespan in wild type and daf-2 mutants and has no effect on heat shock and oxidative stress resistance [82]. Deletion of the methionine sulfoxide reductase, encoded by msra-1, reduces paraquat resistance and shortens lifespan of wild type and daf-2 mutant worms [83].
C. elegans contains 57 genes that encode for glutathione-S-transferase (GST). gst-4 is upregulated by oxidative stress, and although its overexpression enhances oxidative stress resistance, it does not extend lifespan [84]. GST-10 catalyzes the detoxification of the lipid peroxidation end-product HNE. gst-10 overexpressors are more resistant to various forms of stress (heat, UV, and oxidative stress) and have an increased lifespan [85]. RNAi of 5 of the 26 tested gst genes causes an increase in HNE-mediated damage, while RNAi of only 2 of these gst genes (gst-5 and gst-10) decreases lifespan [86]. This suggests that an increase in HNE-induced damage is not sufficient to reduce lifespan.
Iron-catalyzed ROS generation can increase protein damage and reduce oxidative stress resistance without affecting lifespan [87]. Similarly, overexpression of the iron storage protein ferritin, ftn-1, reduces protein carbonylation and enhances oxidative stress resistance, but does not increase lifespan. These results suggest that high iron levels can increase oxidative stress, but iron levels under standard culture conditions do not limit lifespan. Moreover, deletion of ftn-1 even increases lifespan and dauer formation of daf-2 mutants, indicating a role for ferritin in IIS.
In general, deletion of some but not all antioxidant genes shortens lifespan to a minor extent. In some cases, loss of antioxidant enzymes can increase oxidative stress sensitivity without affecting lifespan and in some reports, it can even enhance longevity. Overexpression of a few antioxidant genes can moderately increase lifespan, but not necessarily increases oxidative stress resistance. In other instances, overexpression of antioxidant enzymes reduces lifespan. Therefore, genetic interventions in antioxidant defenses do not generally support the oxidative stress theory of aging in C. elegans.
Does this limited effect of antioxidants on aging in C. elegans support studies in higher model organisms, such as Drosophila melanogaster and mice (for review, see [7, 88])? Similar to worms, mice display a slightly reduced lifespan upon loss of CuZnSOD [89], while in the fruit fly, loss of CuZnSOD dramatically reduces lifespan [90, 91]. Heterozygous CuZnSOD+/- Drosophila has an unaltered lifespan, however [90]. Unlike C. elegans, loss of MnSOD is lethal to mice and flies [92–94], but heterozygotic MnSOD mutation has no effect on aging in mice. Interestingly, these heterozygous mice show a 100% increase in tumor incidence [95]. Thus, antioxidant genetic alteration can induce oxidative stress and pathology without affecting lifespan. Overexpression of CuZnSOD, MnSOD, catalase, and glutathione peroxidase does not prolong lifespan in mice, although oxidative stress resistance is increased [88, 96]. It was verified with various parameters, such as mean and maximum lifespan, body weight, and fecundity, that the mice were maintained under optimal health conditions, to avoid stress conditions which may affect lifespan of mice with altered antioxidant defense. In flies, lifespan is not changed or increased in SOD overexpressors, depending on the study [97–102]. This discrepancy between studies can be a result of artifacts, such as transformation method or genetic background [103, 104]. To conclude, like in C. elegans, antioxidant studies in other model organisms do not generally support the oxidative stress theory of aging.
2.3. Redox Signaling Theories on Aging in C. elegans
Since evidence against the oxidative stress theory of aging is accumulating, a few theories have been recently proposed to explain the correlation between ROS and aging. One alternative for the oxidative stress theory of aging is the redox stress hypothesis, which proposes to include a signaling role for ROS in the aging process. It states that functional loss during aging is caused by a progressive pro-oxidizing shift in the redox state of the cell. This leads to overoxidation of redox-sensitive proteins and consequently the disruption of redox-regulated signaling mechanisms [10]. Figure 1 represents an overview of signaling pathways regulated by ROS that we describe below in more detail.
Figure 1.
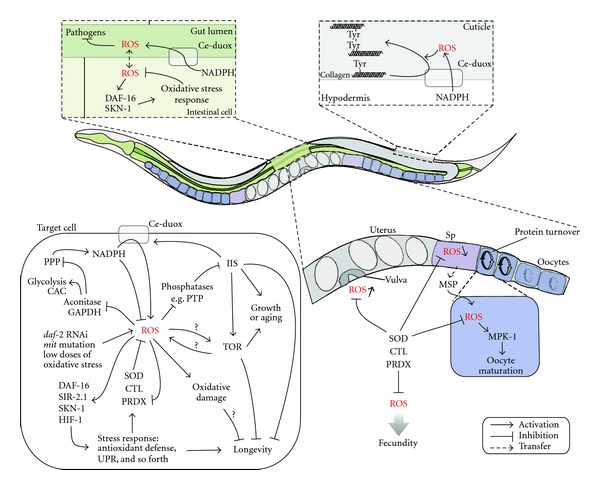
Schematic representation of signaling pathways regulated by ROS in C. elegans. PPP: pentose phosphate pathway; CAC: citric acid cycle; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; Sp: spermatheca.
While high concentrations of the O2 ∙− generators juglone and paraquat are lethal, a life-long exposure to low concentrations of these compounds can extend lifespan [27, 105, 106]. Exposure to low doses of juglone enhances an antioxidant defense (increased GSH levels, SOD, catalase activity, and sod-3 and hsp-16.2 expression). In accordance with these findings, DAF-16, SIR-2.1, and 14-3-3, proteins involved in stress response, are required for the lifespan extension in worms exposed to low concentration of juglone [106]. In short, these results indicate that increased ROS production promotes a stress response, mediated by DAF-16, SIR-2.1 and the redox-dependent transcription factor SKN-1, and thereby extends lifespan. This phenomenon is called hormesis [107] and has also been observed in worms with decreased glucose metabolism, where it is described as mitohormesis more specifically [46]. Reduced glycolysis increases mitochondrial ROS production, which in turn increases oxidative stress resistance and catalase activity, prolonging lifespan [46]. However, pretreatment with antioxidants and vitamins prevents this lifespan extension, demonstrating that the increased ROS formation is an essential signal to activate lifespan extension. Consistent with these findings and the redox stress hypothesis, inhibition of glycolysis enhances stress resistance in C. elegans by activating the pentose phosphate pathway, which is crucial for maintaining the reducing cytosolic NADPH concentration [108]. More recently, it was found that impairment of daf-2 reduces glucose uptake and induces a transient drop in ATP levels, thereby activating the energy sensor AAK-2. In its turn, AAK-2 mediates the generation of a transient ROS signal that ultimately promotes L-proline catabolism and partially extends lifespan. As a result, oxidative phosphorylation is enhanced and ROS levels are increased. This increase in ROS levels induces adaptive response that is partially mediated by the Nrf/SKN-1 transcription factor, resulting in enhanced stress resistance and extended lifespan [109].
Mitohormesis also plays a role in longevity of mutants with an impaired mitochondrial metabolism, or mit mutants. Some mit mutants (clk-1, isp-1, and nuo-6) show increased ROS production [27, 105, 110], an enhanced sod expression [30, 111, 112], and catalase activity [113]. While the increase in antioxidant defenses is dispensable for their long lifespan [30, 74], the increase in ROS levels is a prerequisite for isp-1 and nuo-6 [27] longevity, perhaps by provoking an adaptive response. The hypoxia-inducible transcription factor HIF-1 is activated by mild increases in ROS and HIF-1 is required for the extended lifespans of clk-1 and isp-1 mutants [105]. Thus, HIF-1 appears to link respiratory stress in the mitochondria to a nuclear transcriptional response that promotes longevity. Recently, it was shown that inhibition of mitochondrial respiration triggers the mitochondrial unfolded protein response (UPRmito), increasing hsp-6 expression [112]. This response is needed for the longevity in mit mutants and acts through unidentified signaling molecules, maybe ROS, between different tissues [114]. Like in mit mutants, UPR is essential for the longevity in sod-1 overexpressors [63]. To conclude, according to the (mito)hormesis theory, ROS can be regarded as a molecular signal that triggers a hormetic effect, inducing an overall increase in stress resistance and longevity. The specific antioxidant response may be important to maintain a reduced environment that remains sensitive to subsequent ROS signals.
Mammalian studies have reported that ROS (H2O2) activates the regulator of cell growth and proliferation, TOR, and its target S6 kinase [115, 116]. Moreover, TOR activation increases ROS production [117], whereas TOR inhibition reduces ROS production [118]. According to the TOR-centric model of aging, these multiple links between ROS and TOR indicate that ROS is a messenger molecule rather than a toxic byproduct, that accumulates life-limiting damage [119]. This TOR-centric model postulates that TOR is necessary during development, but that it is not switched off in adults. As a result, cells become hypertrophic and hyperactive. This hyperfunction causes cellular damage and age-related diseases [120]. According to this model, TOR limits lifespan by accelerating age-related diseases before oxidative damage accumulation can cause death.
2.4. Mechanisms of Redox Signaling in C. elegans
While the theories above can explain how ROS influence aging in another way than by causing oxidative damage, they do not provide details on how ROS can transfer a signal in a cellular environment. A few specific mechanisms of ROS or redox signaling has been described in C. elegans and are discussed below.
Protein-tyrosine phosphatases (PTP) contain a reduced cysteine residue that is a well-characterized target of ROS. PTP is oxidized by H2O2 to the sulfenic (–SOH) inactive form, which can be reversed by cellular thiols. The oxidized sulfenic acid rapidly reacts with amide nitrogen of its protein backbone generating inactive sulfenyl-amide, to prevent further irreversible oxidation to sulfonic (–SO2H) and sulfinic species (–SO3H). Inactivation of phosphatases can also occur by glutathionylation. Reactivation of the glutathiolated PTP may be catalyzed by glutaredoxin (GLRX) [73]. Other potential redox-sensitive phosphatases are the PTEN homolog DAF-18, the PP2A homolog PPTR-1, and the MKP-1 homolog VHP-1 [73].
In mammalian studies, insulin is found to stimulate H2O2 production by NADPH oxidase [121, 122]. H2O2 inactivates phosphatases which in turn negatively regulate insulin signaling. This leads to a positive feedback loop: ROS generated in response to insulin facilitate insulin signaling. In C. elegans, loss of sod-4 enhances longevity and loss of either sod-4 or sod-5 enhances dauer formation in daf-2 mutants [29], providing evidence for redox-sensitive regulation of IIS in C. elegans. Moreover, sod-5 is specifically expressed in neurons involved in dauer formation in a daf-16-dependent manner [6, 96]. SOD-4 and SOD-5 generate extracellular and cytosolic H2O2, respectively, which can inactivate phosphatases, such as protein tyrosine phosphatase (PTP), thereby promoting IIS [73]. Therefore, SOD-4 and SOD-5 may facilitate a rapid exit from the dauer larvae stage to ensure further development and offspring during optimal conditions.
Another study that indicates a possible signaling function for ROS in C. elegans, describes redox changes in a limited set of proteins upon H2O2 treatment. A short-term H2O2 treatment causes immediate and reversible behavioral changes, such as reduced mobility, pharyngeal pumping, and reproduction, as well as decreased growth rate, and decreased ATP levels [77]. By using the redox proteomic technique OxICAT, 40 different proteins with redox-sensitive cysteines were identified which are involved in mobility and feeding (oxidative inactivation of MYO-2), protein translation (oxidative inactivation of EFT-1), protein homeostasis (oxidative activation of HSP-1), and ATP regeneration. Proteins involved in glycolysis are oxidatively inactivated, thus, redirecting glucose to the pentose phosphate pathway, which results in enhanced NADPH levels. These observations coincide with the finding that reduced glycolysis enhances oxidative stress resistance [46, 108]. Thus, oxidative stress induces oxidative modifications of specific redox-sensitive proteins to reduce energy consuming processes which are not essential for survival, such as protein synthesis and movement, thereby saving energy to restore redox homeostasis.
2.5. Functional Roles for ROS in C. elegans
Rather than being purely harmful, it has been established that ROS can also play an important role in metabolism. Here we provide a few examples of how ROS contribute to C. elegans metabolism, and therefore may be crucial for maintaining a normal lifespan.
Most organisms, including humans, D. melanogaster, and plants, produce ROS in phagocytic and nonphagocytic cells via NADPH oxidase in response to microbial infection [123–125]. C. elegans also produces extracellular ROS by a dual NADPH oxidase (CeDuox-1) in response to exposure to gut-infecting pathogenic bacteria Enterococcus faecalis or yeast [58, 126–128]. These studies suggest that C. elegans produces ROS in the intestine in response to pathogens while an oxidative stress response mediated by DAF-16 and the redox sensitive transcription factor SKN-1 is induced to protect neighbouring tissues. [58].
Another function for ROS in C. elegans metabolism, mediated by NADPH oxidase, is cuticle biogenesis. C. elegans encodes two dual oxidases, Ce-Duox1 (bli-3) and Ce-Duox2, which contain a NADPH oxidase domain and a peroxidase domain. The NADPH oxidase domain generates extracellular superoxide which dismutates spontaneously or by SOD-4 to H2O2. This peroxide is then used by the peroxidase domain for the cross-linking of tyrosine residues in collagen to create the cuticle of the worm [129].
The redox state of the cell has also been shown to regulate physiological processes during development, such as proliferation and differentiation [130]. Generally, a more reducing environment is associated with proliferation and an oxidizing environment initiates differentiation [131]. Consistent with mammalian studies, where GSH content is high during gametogenesis and fertilization [132], we find that in the spermatheca, where the oocytes are fertilized, GSSG/GSH levels are low [133]. Moreover, in vivo H2O2 levels are low and sod-1 is highly expressed in the spermatheca (unpublished data). It was also reported that protein carbonylation levels are low in spermatheca as they are abruptly reduced by enhanced proteasome activity in the ovaries at the time of oocyte maturation [134]. Before oocytes can be fertilized in the spermatheca, they need to mature, a process that is characterized by their transition into the meiotic metaphase I [135]. The major sperm protein (MSP) secreted by the sperm in the spermatheca promotes ROS production in the most proximal oocytes, which augments MPK-1 activity, essential for this oocyte maturation. SOD-1 is found to inhibit MPK-1 activation [136]. Thus, ROS can act as a secondary messenger for oocyte maturation necessary for fertilization, while these oocytes are cleared from oxidative damage by enhancing proteasomal activity [134].
3. ROS and Redox Detection
A major challenge in establishing the exact function of ROS in metabolism and aging is their accurate detection. Because redox signaling acts through small, local, and transient changes, redox detection should ideally be selective, sensitive, instantaneous, reversible, compartment-specific, noninvasive, and applicable in vivo. Most conventional redox measurements are nonspecific, disruptive (which may create oxidation artifacts), irreversible (which precludes dynamic measurements), and some probes can generate ROS by themselves upon radiation (e.g., H2DCF-DA) [137–139]. These exogenous chemical absorbent, fluorescent or luminescent probes need to be taken up by the biological model. This uptake is rather poor and variable in C. elegans, making comparison between samples complicated. In addition, the uptake of these chemical reporter molecules may not be identical for each subcellular compartment. Recently, small-molecule probes were designed to be targeted to the mitochondria to ensure the probe uptake into the mitochondria [140–142]. An extensive review on these chemical probes can be found elsewhere [137–139].
Many of the limitations of conventional redox probes were overcome by the development of genetically encoded redox sensors based on the green fluorescent protein GFP. Generally, these chimeric proteins contain a regulatory domain that will specifically and reversibly bind the ROS or metabolite of interest, resulting in a conformational change altering the fluorescent properties of the biosensors. As a result, the fluorescent properties of the molecules are a direct measure of the levels of the ROS or metabolite in vivo. A major advantage of these genetically encoded sensors is that they can be targeted to specific tissues, cells or subcellular locations [143]. Hence, when choosing an accurate promoter for expression of the biosensor, real-time and in vivo analysis ranging from a specific single cell to a whole organism can be easily performed. Because of its genetic amenability and its transparency, Caenorhabditis elegans is ideally suited for such approaches, while its size allows analysis by both microplate fluorometry and confocal microscopy. A few examples of genetically encoded sensors that we successfully implemented to measure H2O2 levels and GSH in C. elegans will be further discussed.
H2O2 has emerged as a widespread, physiologically relevant and selective signaling molecule [144–146], and it is relatively, abundant (1–100 nM) [145] and stable compared to other ROS. These features allow H2O2 to serve as an important second messenger. HyPer is a H2O2 (hydrogen peroxide)-specific intracellular biosensor. This sensor consists of a circularly permutated yellow fluorescent protein (cpYFP) inserted into the H2O2-sensitive regulatory domain of the Escherichia coli transcription factor OxyR (OxyR-RD) [147]. HyPer is a H2O2-sensitive, selective, and reversible biosensor, and its fluorescence ratio reflects the balance between H2O2-mediated oxidation and a GLRX/GSH-mediated reduction. Like most cpYFP's, HyPer fluorescence is influenced by pH (6–10): acidification decreases the 500 nm/420 nm excitation ratio [147]. HyPer has already been used in various cell culture [147–151] and zebra-fish [152] studies. Using HyPer, we identified local cells or tissues with distinct in vivo H2O2 levels in C. elegans. For example, we found that, consistent with the role of hypodermal cells in cuticle biogenesis, H2O2 levels are particularly high in these cells [133] (Figure 2).
Figure 2.
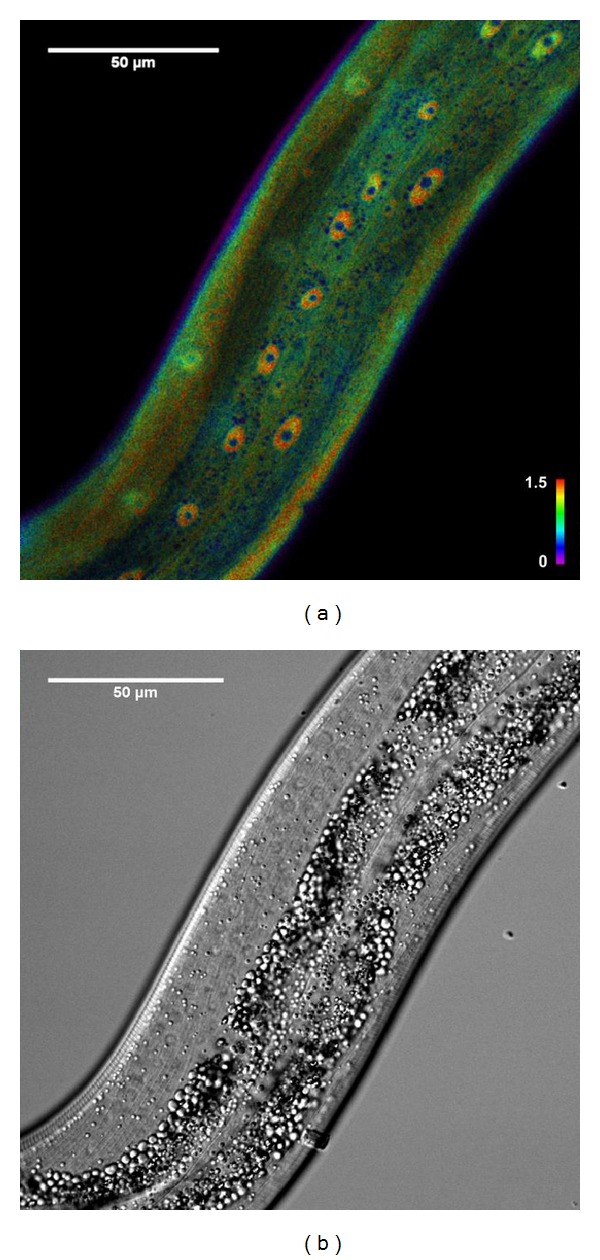
(a) Intensity normalized ratio image of a HyPer transgenic worm. Color represents oxidized/reduced HyPer ratio values, while color saturation represents fluorescence intensities. Hypodermal nuclei clearly show increased levels of H2O2. (b) Corresponding brightfield image. Construction of transgenic strains, confocal microscopy, and image analysis was performed as described in [133].
The redox state of the cell is determined by the redox state of multiple redox pairs in the cell. Because of the high intracellular GSH concentration (1–11 mM) and the high GSH/GSSG ratio (≥100) [131], the redox state of the glutathione couple is a good proxy for the total redox state of the cell. The new generation of redox-sensitive sensors are the redox-sensitive GFP's (roGFP), such as roGFP2. These biosensors are ratiometric by excitation, thus minimizing measurement errors due to variable in vivo probe concentrations and photobleaching [153]. They are able to detect small changes in GSSG within a highly reduced glutathione pool (GSH/GSSG ≥ 100) [131]. The fusion of human glutaredoxin-1 (Grx1) to roGFP2 makes the redox response of the probe faster and more glutathione specific [154, 155]. Ubiquitous overexpression of this sensor in C. elegans shows a reduced state (low levels of GSSG/GSH) of the spermatheca (Figure 3).
Figure 3.
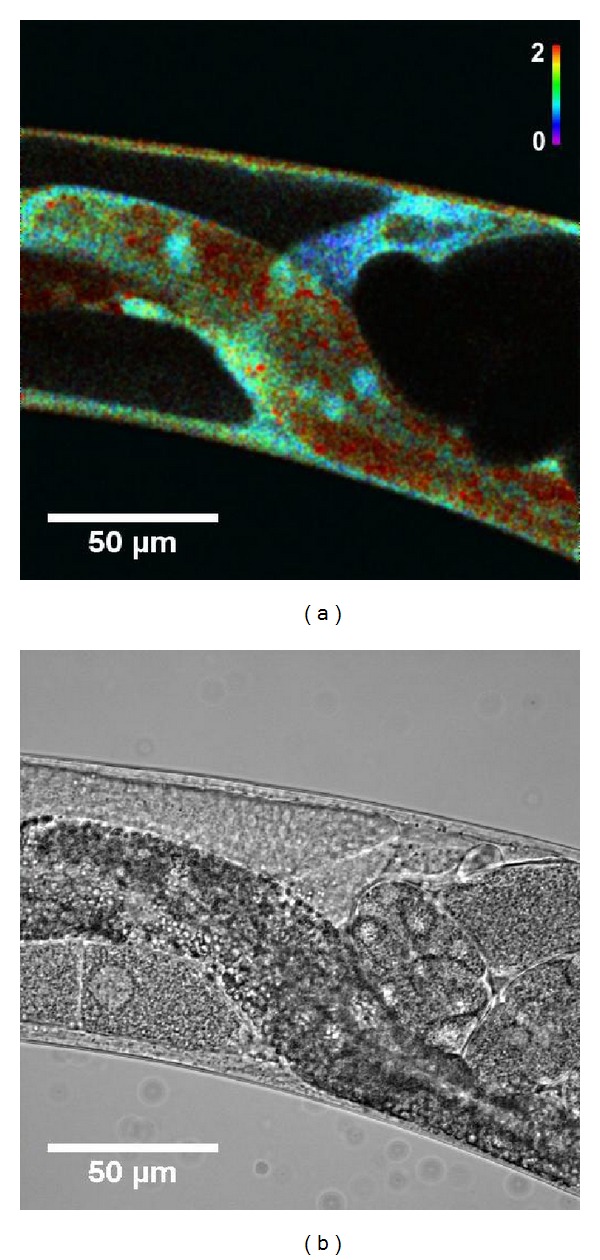
(a) Intensity normalized ratio image of a Grx1-roGFP2 transgenic worm. Color represents oxidized/reduced Grx1-roGFP2 ratio values, while color saturation represents fluorescence intensities. Spermatheca show clearly reduced GSSG/GSH ratios. (b) Corresponding brightfield image. Construction of transgenic strains, confocal microscopy, and image analysis was performed as described in [133].
Consistent with the redox hypothesis of aging, both Hyper and Grx-roGFP2 strains show an increase in H2O2 and GSSG/GSH with age. In dietary restricted populations, this increase is attenuated [133].
The fusion of the yeast peroxidase Orp1 (also known as Gpx3) to roGFP2 converts roGFP2 into a specific probe for H2O2 [156]. H2O2-specific oxidation of Orp1 induces the formation of a disulfide bridge in roGFP2. roGFP2-Orp1 can be selectively and reversibly oxidized by H2O2 [156] and reduced by thioredoxin (TRX) or glutaredoxin (GLRX) [155]. Like all other roGFP-based probes, this probe is independent of pH (5.5–8.0) [157]. roGFP2-Orp1 has been already applied in cell cultures [156] and D. melanogaster [158].
The last few years, many more in vivo ROS, redox, and metabolic sensors have been developed [147, 154, 156, 159–164], and their use and limitations have been reviewed recently [155]. It is very likely that this booming research field will yield more accurate, sensitive, and selective redox sensors in the near future.
4. General Conclusion
In recent years, there is mounting evidence against the oxidative stress theory of aging in Caenorhabditis elegans. Many intervention studies altering its antioxidant system have been performed, and most of them failed to support this theory. Gradually, it has become clear that a general increase in oxidative stress does not limit lifespan in this model organism. However, in the stressful conditions of its natural habitat oxidative stress resistance might be important to ensure a normal lifespan and reproduction. Although it cannot be ruled out that specific targets (proteins, lipids, or nucleic acids) accumulate some type of damage over age, and thereby contribute to the aging process, a global rise in oxidative damage is clearly not a major factor determining lifespan. In contrast, there is a growing body of evidence that ROS and redox signaling may be important in the aging process, and that ROS may exert essential functions in metabolism. However, to fully understand the mechanisms of ROS and redox signaling, it is crucial that the molecular details, the localization, and the regulation of the specific reactive oxygen species involved can be analyzed accurately. This will allow a critical validation of the latest aging theories that try to explain the correlation between ROS and aging. To this end, the recent development and implementation of genetically encoded biosensors is a promising tool that we believe will be highly valuable to further explore ROS and redox biology in aging C. elegans.
References
- 1.Harman D. Aging: a theory based on free radical and radiation chemistry. Journal of Gerontology. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 2.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) Journal of Biological Chemistry. 1969;244(22):6049–6055. [PubMed] [Google Scholar]
- 3.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiological Reviews. 1979;59(3):527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 4.Harman D. The biologic clock: the mitochondria? Journal of the American Geriatrics Society. 1972;20(4):145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 5.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273(5271):59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gems D, Doonan R. Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Cell Cycle. 2009;8(11):1681–1687. doi: 10.4161/cc.8.11.8595. [DOI] [PubMed] [Google Scholar]
- 7.Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radical Biology and Medicine. 2007;43(4):477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 8.Bokov A, Chaudhuri A, Richardson A. The role of oxidative damage and stress in aging. Mechanisms of Ageing and Development. 2004;125(10-11):811–826. doi: 10.1016/j.mad.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Lapointe J, Hekimi S. When a theory of aging ages badly. Cellular and Molecular Life Sciences. 2010;67(1):1–8. doi: 10.1007/s00018-009-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sohal RS, Orr WC. The redox stress hypothesis of aging. Free Radical Biology and Medicine. 2012;52(3):539–555. doi: 10.1016/j.freeradbiomed.2011.10.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins JJ, Huang C, Hughes S, Kornfeld K. WormBook. 2008. The measurement and analysis of age-related changes in Caenorhabditis elegans ; pp. 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bohr VA, Anson RM. DNA damage, mutation and fine structure DNA repair in aging. Mutation Research. 1995;338(1–6):25–34. doi: 10.1016/0921-8734(95)00008-t. [DOI] [PubMed] [Google Scholar]
- 13.Warner HR. Superoxide dismutase, aging, and degenerative disease. Free Radical Biology and Medicine. 1994;17(3):249–258. doi: 10.1016/0891-5849(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 14.Levine RL, Stadtman ER. Oxidative modification of proteins during aging. Experimental Gerontology. 2001;36(9):1495–1502. doi: 10.1016/s0531-5565(01)00135-8. [DOI] [PubMed] [Google Scholar]
- 15.Klass M, Nguyen PN, Dechavigny A. Age-correlated changes in the DNA template in the nematode Caenorhabditis elegans . Mechanisms of Ageing and Development. 1983;22(3-4):253–263. doi: 10.1016/0047-6374(83)90080-5. [DOI] [PubMed] [Google Scholar]
- 16.Adachi H, Fujiwara Y, Ishii N. Effects of oxygen on protein carbonyl and aging in Caenorhabditis elegans mutants with long (age-1) and short (mev-1) life spans. Journals of Gerontology A. 1998;53(4):B240–B244. doi: 10.1093/gerona/53a.4.b240. [DOI] [PubMed] [Google Scholar]
- 17.Yasuda K, Adachi H, Fujiwara Y, Ishii N. Protein carbonyl accumulation in aging dauer formation-defective (daf) mutants of Caenorhabditis elegans . Journals of Gerontology A. 1999;54(2):B47–B51. doi: 10.1093/gerona/54.2.b47. [DOI] [PubMed] [Google Scholar]
- 18.Proffitt JH, Davie JR, Swinton D, Hattman S. 5-methylcytosine is not detectable in Saccharomyces cerevisiae DNA. Molecular and Cellular Biology. 1984;4(5):985–988. doi: 10.1128/mcb.4.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urieli-Shoval S, Gruenbaum Y, Sedat J, Razin A. The absence of detectable methylated bases in Drosophila melanogaster DNA. FEBS Letters. 1982;146(1):148–152. doi: 10.1016/0014-5793(82)80723-0. [DOI] [PubMed] [Google Scholar]
- 20.Simpson VJ, Johnson TE, Hammen RF. Caenorhabditis elegans DNA does not contain 5-methylcytosine at any time during development or aging. Nucleic Acids Research. 1986;14(16):6711–6719. doi: 10.1093/nar/14.16.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melov S, Lithgow GJ, Fischer DR, Tedesco PM, Johnson TE. Increased frequency of deletions in the mitochondrial genome with age of Caenorhabditis elegans . Nucleic Acids Research. 1995;23(8):1419–1425. doi: 10.1093/nar/23.8.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brys K, Castelein N, Matthijssens F, Vanfleteren JR, Braeckman BP. Disruption of insulin signalling preserves bioenergetic competence of mitochondria in ageing Caenorhabditis elegans . BMC Biology. 2010;8, article 91 doi: 10.1186/1741-7007-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henderson ST, Johnson TE. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans . Current Biology. 2001;11(24):1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- 24.Inoue H, Hisamoto N, An JH, et al. The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes & Development. 2005;19(19):2278–2283. doi: 10.1101/gad.1324805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, Tissenbaum HA. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(12):4494–4499. doi: 10.1073/pnas.0500749102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehtinen MK, Yuan Z, Boag PR, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125(5):987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 27.Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans . PLoS Biology. 2010;8(12) doi: 10.1371/journal.pbio.1000556.e1000556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy CT, McCarroll SA, Bargmann CI, et al. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans . Nature. 2003;424(6946):277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 29.Doonan R, McElwee JJ, Matthijssens F, et al. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans . Genes & Development. 2008;22(23):3236–3241. doi: 10.1101/gad.504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang W, Li JJ, Hekimi S. A measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans . Genetics. 2007;177(4):2063–2074. doi: 10.1534/genetics.107.080788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Honda Y, Tanaka M, Honda S. Modulation of longevity and diapause by redox regulation mechanisms under the insulin-like signaling control in Caenorhabditis elegans . Experimental Gerontology. 2008;43(6):520–529. doi: 10.1016/j.exger.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 32.Houthoofd K, Johnson TE, Vanfleteren JR. Dietary restriction in the nematode Caenorhabditis elegans . Journals of Gerontology A. 2005;60(9):1125–1131. doi: 10.1093/gerona/60.9.1125. [DOI] [PubMed] [Google Scholar]
- 33.Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans . Proceedings of the National Academy of Sciences of the United States of America. 1998;95(22):13091–13096. doi: 10.1073/pnas.95.22.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Houthoofd K, Braeckman BP, Lenaerts I, et al. No reduction of metabolic rate in food restricted Caenorhabditis elegans . Experimental Gerontology. 2002;37(12):1359–1369. doi: 10.1016/s0531-5565(02)00172-9. [DOI] [PubMed] [Google Scholar]
- 35.Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans . Nature. 2007;447(7144):545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- 36.Houthoofd K, Braeckman BP, Lenaerts I, et al. Axenic growth up-regulates mass-specific metabolic rate, stress resistance, and extends life span in Caenorhabditis elegans . Experimental Gerontology. 2002;37(12):1371–1378. doi: 10.1016/s0531-5565(02)00173-0. [DOI] [PubMed] [Google Scholar]
- 37.Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans . Nature. 2007;447(7144):550–555. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- 38.Van Raamsdonk JM, Hekimi S. Reactive oxygen species and aging in Caenorhabditis elegans: causal or casual relationship? Antioxidants & Redox Signaling. 2010;13(12):1911–1953. doi: 10.1089/ars.2010.3215. [DOI] [PubMed] [Google Scholar]
- 39.Yen K, Patel HB, Lublin AL, Mobbs CV. SOD isoforms play no role in lifespan in ad lib or dietary restricted conditions, but mutational inactivation of SOD-1 reduces life extension by cold. Mechanisms of Ageing and Development. 2009;130(3):173–178. doi: 10.1016/j.mad.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans . PLoS Genetics. 2009;5(2) doi: 10.1371/journal.pgen.1000361.e1000361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collins JJ, Evason K, Kornfeld K. Pharmacology of delayed aging and extended lifespan of Caenorhabditis elegans . Experimental Gerontology. 2006;41(10):1032–1039. doi: 10.1016/j.exger.2006.06.038. [DOI] [PubMed] [Google Scholar]
- 42.Harrington LA, Harley CB. Effect of vitamin E on lifespan and reproduction in Caenorhabditis elegans . Mechanisms of Ageing and Development. 1988;43(1):71–78. doi: 10.1016/0047-6374(88)90098-x. [DOI] [PubMed] [Google Scholar]
- 43.Ishii N, Senoo-Matsuda N, Miyake K, et al. Coenzyme Q10 can prolong C. elegans lifespan by lowering oxidative stress. Mechanisms of Ageing and Development. 2004;125(1):41–46. doi: 10.1016/j.mad.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 44.Adachi H, Ishii N. Effects of tocotrienols on life span and protein carbonylation in Caenorhabditis elegans . Journals of Gerontology A. 2000;55(6):B280–B285. doi: 10.1093/gerona/55.6.b280. [DOI] [PubMed] [Google Scholar]
- 45.Benedetti MG, Foster AL, Vantipalli MC, et al. Compounds that confer thermal stress resistance and extended lifespan. Experimental Gerontology. 2008;43(10):882–891. doi: 10.1016/j.exger.2008.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metabolism. 2007;6(4):280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Brown MK, Evans JL, Luo Y. Beneficial effects of natural antioxidants EGCG and α-lipoic acid on life span and age-dependent behavioral declines in Caenorhabditis elegans . Pharmacology Biochemistry and Behavior. 2006;85(3):620–628. doi: 10.1016/j.pbb.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 48.Pun PB, Gruber J, Tang SY, et al. Ageing in nematodes: do antioxidants extend lifespan in Caenorhabditis elegans? Biogerontology. 2010;11(1):17–30. doi: 10.1007/s10522-009-9223-5. [DOI] [PubMed] [Google Scholar]
- 49.Burns AR, Wallace IM, Wildenhain J, et al. A predictive model for drug bioaccumulation and bioactivity in Caenorhabditis elegans . Nature Chemical Biology. 2010;6(7):549–557. doi: 10.1038/nchembio.380. [DOI] [PubMed] [Google Scholar]
- 50.Shibamura A, Ikeda T, Nishikawa Y. A method for oral administration of hydrophilic substances to Caenorhabditis elegans: effects of oral supplementation with antioxidants on the nematode lifespan. Mechanisms of Ageing and Development. 2009;130(9):652–655. doi: 10.1016/j.mad.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 51.Keaney M, Matthijssens F, Sharpe M, Vanfleteren J, Gems D. Superoxide dismutase mimetics elevate superoxide dismutase activity in vivo but do not retard aging in the nematode Caenorhabditis elegans . Free Radical Biology and Medicine. 2004;37(2):239–250. doi: 10.1016/j.freeradbiomed.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 52.Sampayo JN, Olsen A, Lithgow GJ. Oxidative stress in Caenorhabditis elegans: protective effects of superoxide dismutase/catalase mimetics. Aging Cell. 2003;2(6):319–326. doi: 10.1046/j.1474-9728.2003.00063.x. [DOI] [PubMed] [Google Scholar]
- 53.Kim J, Takahashi M, Shimizu T, et al. Effects of a potent antioxidant, platinum nanoparticle, on the lifespan of Caenorhabditis elegans . Mechanisms of Ageing and Development. 2008;129(6):322–331. doi: 10.1016/j.mad.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 54.Melov S, Ravenscroft J, Malik S, et al. Extension of life-span with superoxide dismutase/catalase mimetics. Science. 2000;289(5484):1567–1569. doi: 10.1126/science.289.5484.1567. [DOI] [PubMed] [Google Scholar]
- 55.Keaney M, Gems D. No increase in lifespan in Caenorhabditis elegans upon treatment with the superoxide dismutase mimetic EUK-8. Free Radical Biology and Medicine. 2003;34(2):277–282. doi: 10.1016/s0891-5849(02)01290-x. [DOI] [PubMed] [Google Scholar]
- 56.Matthijssens F, Back P, Braeckman BP, Vanfleteren JR. Prooxidant activity of the superoxide dismutase (SOD)-mimetic EUK-8 in proliferating and growth-arrested Escherichia coli cells. Free Radical Biology and Medicine. 2008;45(5):708–715. doi: 10.1016/j.freeradbiomed.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 57.Dingley S, Polyak E, Lightfoot R, et al. Mitochondrial respiratory chain dysfunction variably increases oxidant stress in Caenorhabditis elegans . Mitochondrion. 2010;10(2):125–136. doi: 10.1016/j.mito.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chávez V, Mohri-Shiomi A, Maadani A, Vega LA, Garsin DA. Oxidative stress enzymes are required for DAF-16-mediated immunity due to generation of reactive oxygen species by Caenorhabditis elegans . Genetics. 2007;176(3):1567–1577. doi: 10.1534/genetics.107.072587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Back P, Matthijssens F, Vanfleteren JR, Braeckman BP. A simplified hydroethidine method for fast and accurate detection of superoxide production in isolated mitochondria. Analytical Biochemistry. 2012;423(1):147–151. doi: 10.1016/j.ab.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 60.Gruber J, Ng LF, Fong S, et al. Mitochondrial changes in ageing Caenorhabditis elegans—what do we learn from superoxide dismutase knockouts? PLoS ONE. 2011;6(5) doi: 10.1371/journal.pone.0019444.e19444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yanase S, Onodera A, Tedesco P, Johnson TE, Ishii N. SOD-1 deletions in Caenorhabditis elegans alter the localization of intracellular reactive oxygen species and show molecular compensation. Journals of Gerontology A. 2009;64(5):530–539. doi: 10.1093/gerona/glp020. [DOI] [PubMed] [Google Scholar]
- 62.Van Raamsdonk JM, Hekimi S. Superoxide dismutase is dispensable for normal animal lifespan. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(15):5785–5790. doi: 10.1073/pnas.1116158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cabreiro F, Ackerman D, Doonan R, et al. Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radical Biology and Medicine. 2011;51(8):1575–1582. doi: 10.1016/j.freeradbiomed.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Landis GN, Tower J. Superoxide dismutase evolution and life span regulation. Mechanisms of Ageing and Development. 2005;126(3):365–379. doi: 10.1016/j.mad.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 65.Jensen LT, Culotta VC. Activation of CuZn superoxide dismutases from Caenorhabditis elegans does not require the copper chaperone CCS. Journal of Biological Chemistry. 2005;280(50):41373–41379. doi: 10.1074/jbc.M509142200. [DOI] [PubMed] [Google Scholar]
- 66.Larsen PL. Aging and resistance to oxidative damage in Caenorhabditis elegans . Proceedings of the National Academy of Sciences of the United States of America. 1993;90(19):8905–8909. doi: 10.1073/pnas.90.19.8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fujii M, Ishii N, Joguchi A, Yasuda K, Ayusawa D. A novel superoxide dismutase gene encoding membrane-bound and extracellular isoforms by alternative splicing in Caenorhabditis elegans . DNA Research. 1998;5(1):25–30. doi: 10.1093/dnares/5.1.25. [DOI] [PubMed] [Google Scholar]
- 68.Giglio MP, Hunter T, Bannister JV, Bannister WH, Hunter GJ. The manganese superoxide dismutase gene of Caenorhabditis elegans . Biochemistry and Molecular Biology International. 1994;33(1):37–40. [PubMed] [Google Scholar]
- 69.Hunter T, Bannister WH, Hunter GJ. Cloning, expression, and characterization of two manganese superoxide dismutases from Caenorhabditis elegans . Journal of Biological Chemistry. 1997;272(45):28652–28659. doi: 10.1074/jbc.272.45.28652. [DOI] [PubMed] [Google Scholar]
- 70.Wang J, Kim SK. Global analysis of dauer gene expression in Caenorhabditis elegans . Development. 2003;130(8):1621–1634. doi: 10.1242/dev.00363. [DOI] [PubMed] [Google Scholar]
- 71.Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans . The FASEB Journal. 1999;13(11):1385–1393. [PubMed] [Google Scholar]
- 72.Yanase S, Ishi N. Cloning of the oxidative stress-responsive genes in Caenorhabditis elegans . Journal of Radiation Research. 1999;40(1):39–47. doi: 10.1269/jrr.40.39. [DOI] [PubMed] [Google Scholar]
- 73.Goldstein BJ, Mahadev K, Wu X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005;54(2):311–321. doi: 10.2337/diabetes.54.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petriv OI, Rachubinski RA. Lack of peroxisomal catalase causes a progeric phenotype in Caenorhabditis elegans . Journal of Biological Chemistry. 2004;279(19):19996–20001. doi: 10.1074/jbc.M400207200. [DOI] [PubMed] [Google Scholar]
- 75.Taub J, Lau JF, Ma C, et al. A cytosolic catalase is needed to extend adult lifespan in C. elegans daf-C and clk-1 mutants. Nature. 1999;399(6732):162–166. doi: 10.1038/20208. [DOI] [PubMed] [Google Scholar]
- 76.Isermann K, Liebau E, Roeder T, Bruchhaus I. A peroxiredoxin specifically expressed in two types of pharyngeal neurons is required for normal growth and egg production in Caenorhabditis elegans . Journal of Molecular Biology. 2004;338(4):745–755. doi: 10.1016/j.jmb.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 77.Kumsta C, Thamsen M, Jakob U. Effects of oxidative stress on behavior, physiology, and the redox thiol proteome of Caenorhabditis elegans . Antioxidants & Redox Signaling. 2011;14(6):1023–1037. doi: 10.1089/ars.2010.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oláhová M, Taylor SR, Khazaipoul S, et al. A redox-sensitive peroxiredoxin that is important for longevity has tissue- and stress-specific roles in stress resistance. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(50):19839–19844. doi: 10.1073/pnas.0805507105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jee C, Vanoaica L, Lee J, Park BJ, Ahnn J. Thioredoxin is related to life span regulation and oxidative stress response in Caenorhabditis elegans . Genes to Cells. 2005;10(12):1203–1210. doi: 10.1111/j.1365-2443.2005.00913.x. [DOI] [PubMed] [Google Scholar]
- 80.Miranda-Vizuete A, González JCF, Gahmon G, Burghoorn J, Navas P, Swoboda P. Lifespan decrease in a Caenorhabditis elegans mutant lacking TRX-1, a thioredoxin expressed in ASJ sensory neurons. FEBS Letters. 2006;580(2):484–490. doi: 10.1016/j.febslet.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 81.Fierro-González JC, González-Barrios M, Miranda-Vizuete A, Swoboda P. The thioredoxin TRX-1 regulates adult lifespan extension induced by dietary restriction in Caenorhabditis elegans . Biochemical and Biophysical Research Communications. 2011;406(3):478–482. doi: 10.1016/j.bbrc.2011.02.079. [DOI] [PubMed] [Google Scholar]
- 82.Cacho-Valadez B, Munoz-Lobato F, Pedrajas JR, et al. The characterization of the Caenorhabditis elegans mitochondrial thioredoxin system uncovers an unexpected protective role of thioredoxin reductase 2 in beta-amyloid peptide toxicity. Antioxidants & Redox Signaling. 2012;16(12):1384–1400. doi: 10.1089/ars.2011.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Minniti AN, Cataldo R, Trigo C, et al. Methionine sulfoxide reductase a expression is regulated by the DAF-16/FOXO pathway in Caenorhabditis elegans . Aging Cell. 2009;8(6):690–705. doi: 10.1111/j.1474-9726.2009.00521.x. [DOI] [PubMed] [Google Scholar]
- 84.Leiers B, Kampkötter A, Grevelding CG, Link CD, Johnson TE, Henkle-Dührsen K. A stress-responsive glutathione S-transferase confers resistance to oxidative stress in Caenorhabditis elegans . Free Radical Biology and Medicine. 2003;34(11):1405–1415. doi: 10.1016/s0891-5849(03)00102-3. [DOI] [PubMed] [Google Scholar]
- 85.Ayyadevara S, Engle MR, Singh SP, et al. Lifespan and stress resistance of Caenorhabditis elegans are increased by expression of glutathione transferases capable of metabolizing the lipid peroxidation product 4-hydroxynonenal. Aging Cell. 2005;4(5):257–271. doi: 10.1111/j.1474-9726.2005.00168.x. [DOI] [PubMed] [Google Scholar]
- 86.Ayyadevara S, Dandapat A, Singh SP, et al. Life span and stress resistance of Caenorhabditis elegans are differentially affected by glutathione transferases metabolizing 4-hydroxynon-2-enal. Mechanisms of Ageing and Development. 2007;128(2):196–205. doi: 10.1016/j.mad.2006.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Valentini S, Cabreiro F, Ackerman D, et al. Manipulation of in vivo iron levels can alter resistance to oxidative stress without affecting ageing in the nematode C. elegans . Mechanisms of Ageing and Development. 2012;133(5):282–290. doi: 10.1016/j.mad.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pérez VI, Bokov A, Van Remmen H, et al. Is the oxidative stress theory of aging dead? Biochimica et Biophysica Acta. 2009;1790(10):1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Elchuri S, Oberley TD, Qi W, et al. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24(3):367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 90.Phillips JP, Campbell SD, Michaud D, Charbonneau M, Hilliker AJ. Null mutation of copper/zinc superoxide dismutase in Drosophila confers hypersensitivity to paraquat and reduced longevity. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(8):2761–2765. doi: 10.1073/pnas.86.8.2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reveillaud I, Phillips J, Duyf B, Hilliker A, Kongpachith A, Fleming JE. Phenotypic rescue by a bovine transgene in a Cu/Zn superoxide dismutase-null mutant of Drosophila melanogaster . Molecular and Cellular Biology. 1994;14(2):1302–1307. doi: 10.1128/mcb.14.2.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kirby K, Hu J, Hilliker AJ, Phillips JP. RNA interference-mediated silencing of Sod2 in Drosophila leads to early adult-onset mortality and elevated endogenous oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(25):16162–16167. doi: 10.1073/pnas.252342899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Duttaroy A, Paul A, Kundu M, Belton A. A Sod2 null mutation confers severely reduced adult life span in Drosophila . Genetics. 2003;165(4):2295–2299. doi: 10.1093/genetics/165.4.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li Y, Huang TT, Carlson EJ, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995;11(4):376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 95.Van Remmen H, Ikeno Y, Hamilton M, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiological Genomics. 2003;16(1):29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 96.Pérez VI, Van Remmen H, Bokov A, Epstein CJ, Vijg J, Richardson A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell. 2009;8(1):73–75. doi: 10.1111/j.1474-9726.2008.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seto NO, Hayashi S, Tener GM. Overexpression of Cu—Zn superoxide dismutase in Drosophila does not affect life-span. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(11):4270–4274. doi: 10.1073/pnas.87.11.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster . Science. 1994;263(5150):1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- 99.Parkes TL, Elia AJ, Dickinson D, Hilliker AJ, Phillips JP, Boulianne GL. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nature Genetics. 1998;19(2):171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- 100.Sun J, Tower J. FLP recombinase-mediated induction of Cu/Zn-superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Molecular and Cellular Biology. 1999;19(1):216–228. doi: 10.1128/mcb.19.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Orr WC, Mockett RJ, Benes JJ, Sohal RS. Effects of overexpression of copper-zinc and manganese superoxide dismutases, catalase, and thioredoxin reductase genes on longevity in Drosophila melanogaster . Journal of Biological Chemistry. 2003;278(29):26418–26422. doi: 10.1074/jbc.M303095200. [DOI] [PubMed] [Google Scholar]
- 102.Sun J, Folk D, Bradley TJ, Tower J. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster . Genetics. 2002;161(2):661–672. doi: 10.1093/genetics/161.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kaiser M, Gasser M, Ackermann R, Stearns SC. P-element inserts in transgenic flies: a cautionary tale. Heredity. 1997;78(1):1–11. doi: 10.1038/hdy.1997.1. [DOI] [PubMed] [Google Scholar]
- 104.Spencer CC, Howell CE, Wright AR, Promislow DE. Testing an ’aging gene’ in long-lived Drosophila strains: increased longevity depends on sex and genetic background. Aging Cell. 2003;2(2):123–130. doi: 10.1046/j.1474-9728.2003.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Current Biology. 2010;20(23):2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heidler T, Hartwig K, Daniel H, Wenzel U. Caenorhabditis elegans lifespan extension caused by treatment with an orally active ROS-generator is dependent on DAF-16 and SIR-2.1. Biogerontology. 2010;11(2):183–195. doi: 10.1007/s10522-009-9239-x. [DOI] [PubMed] [Google Scholar]
- 107.Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis) Experimental Gerontology. 2010;45(6):410–418. doi: 10.1016/j.exger.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 108.Ralser M, Wamelink MM, Kowald A, et al. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. Journal of Biology. 2007;6(4, article 10) doi: 10.1186/jbiol61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zarse K, Schmeisser S, Groth M, et al. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metabolism. 2012;15(4):451–465. doi: 10.1016/j.cmet.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yang YY, Gangoiti JA, Sedensky MM, Morgan PG. The effect of different ubiquinones on lifespan in Caenorhabditis elegans . Mechanisms of Ageing and Development. 2009;130(6):370–376. doi: 10.1016/j.mad.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Feng J, Bussière F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans . Developmental Cell. 2001;1(5):633–644. doi: 10.1016/s1534-5807(01)00071-5. [DOI] [PubMed] [Google Scholar]
- 112.Yang W, Hekimi S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans . Aging Cell. 2010;9(3):433–447. doi: 10.1111/j.1474-9726.2010.00571.x. [DOI] [PubMed] [Google Scholar]
- 113.Braeckman BP, Houthoofd K, Brys K, et al. No reduction of energy metabolism in Clk mutants. Mechanisms of Ageing and Development. 2002;123(11):1447–1456. doi: 10.1016/s0047-6374(02)00085-4. [DOI] [PubMed] [Google Scholar]
- 114.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144(1):79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bae GU, Seo DW, Kwon HK, et al. Hydrogen peroxide activates p70(S6k) signaling pathway. Journal of Biological Chemistry. 1999;274(46):32596–32602. doi: 10.1074/jbc.274.46.32596. [DOI] [PubMed] [Google Scholar]
- 116.Huang C, Li J, Ke Q, et al. Ultraviolet-induced phosphorylation of p70S6K at Thr389 and Thr421/Ser424 involves hydrogen peroxide and mammalian target of rapamycin but not Akt and atypical protein kinase C. Cancer Research. 2002;62(20):5689–5697. [PubMed] [Google Scholar]
- 117.Kim JH, Chu SC, Gramlich JL, et al. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood. 2005;105(4):1717–1723. doi: 10.1182/blood-2004-03-0849. [DOI] [PubMed] [Google Scholar]
- 118.Tuñón MJ, Sánchez-Campos S, Gutiérrez B, Culebras JM, González-Gallego J. Effects of FK506 and rapamycin on generation of reactive oxygen species, nitric oxide production and nuclear factor kappa B activation in rat hepatocytes. Biochemical Pharmacology. 2003;66(3):439–445. doi: 10.1016/s0006-2952(03)00288-0. [DOI] [PubMed] [Google Scholar]
- 119.Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008;7(21):3344–3354. doi: 10.4161/cc.7.21.6965. [DOI] [PubMed] [Google Scholar]
- 120.Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006;5(18):2087–2102. doi: 10.4161/cc.5.18.3288. [DOI] [PubMed] [Google Scholar]
- 121.May JM, de Haen C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. Journal of Biological Chemistry. 1979;254(7):2214–2220. [PubMed] [Google Scholar]
- 122.Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxidants & Redox Signaling. 2005;7(7-8):1021–1031. doi: 10.1089/ars.2005.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ha EM, Oh CT, Bae YS, Lee WJ. A direct role for dual oxidase in Drosophila gut immunity. Science. 2005;310(5749):847–850. doi: 10.1126/science.1117311. [DOI] [PubMed] [Google Scholar]
- 124.Cross AR, Segal AW. The NADPH oxidase of professional phagocytes—prototype of the NOX electron transport chain systems. Biochimica et Biophysica Acta. 2004;1657(1):1–22. doi: 10.1016/j.bbabio.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Apel K, Hirt H. Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annual Review of Plant Biology. 2004;55:373–399. doi: 10.1146/annurev.arplant.55.031903.141701. [DOI] [PubMed] [Google Scholar]
- 126.Jain C, Yun M, Politz SM, Rao RP. A pathogenesis assay using Saccharomyces cerevisiae and Caenorhabditis elegans reveals novel roles for yeast AP-1, Yap1, and host dual oxidase BLI-3 in fungal pathogenesis. Eukaryotic Cell. 2009;8(8):1218–1227. doi: 10.1128/EC.00367-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chávez V, Mohri-Shiomi A, Garsin DA. Ce-Duox1/BLI-3 generates reactive oxygen species as a protective innate immune mechanism in Caenorhabditis elegans . Infection and Immunity. 2009;77(11):4983–4989. doi: 10.1128/IAI.00627-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.van der Hoeven R, McCallum KC, Cruz MR, Garsin DA. Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans . PLoS Pathogens. 2011;7(12) doi: 10.1371/journal.ppat.1002453.e1002453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Edens WA, Sharling L, Cheng G, et al. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. Journal of Cell Biology. 2001;154(4):879–891. doi: 10.1083/jcb.200103132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hernández-García D, Wood CD, Castro-Obregón S, Covarrubias L. Reactive oxygen species: a radical role in development? Free Radical Biology and Medicine. 2010;49(2):130–143. doi: 10.1016/j.freeradbiomed.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 131.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biology and Medicine. 2001;30(11):1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 132.Hitchler MJ, Domann FE. An epigenetic perspective on the free radical theory of development. Free Radical Biology and Medicine. 2007;43(7):1023–1036. doi: 10.1016/j.freeradbiomed.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Back P, De Vos WH, Depuydt GG, Matthijssens F, Vanfleteren JR, Braeckman BP. Exploring real-time in vivo redox biology of developing and aging Caenorhabditis elegans . Free Radical Biology and Medicine. 2012;52(5):850–859. doi: 10.1016/j.freeradbiomed.2011.11.037. [DOI] [PubMed] [Google Scholar]
- 134.Goudeau J, Aguilaniu H. Carbonylated proteins are eliminated during reproduction in C. elegans . Aging Cell. 2010;9(6):991–1003. doi: 10.1111/j.1474-9726.2010.00625.x. [DOI] [PubMed] [Google Scholar]
- 135.Greenstein D. WormBook. 2005. Control of oocyte meiotic maturation and fertilization; pp. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang Y, Han SM, Miller MA. MSP hormonal control of the oocyte MAP kinase cascade and reactive oxygen species signaling. Developmental Biology. 2010;342(1):96–107. doi: 10.1016/j.ydbio.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gomes A, Fernandes E, Lima JL. Fluorescence probes used for detection of reactive oxygen species. Journal of Biochemical and Biophysical Methods. 2005;65(2-3):45–80. doi: 10.1016/j.jbbm.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 138.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radical Biology and Medicine. 2007;43(7):995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 139.Bartosz G. Use of spectroscopic probes for detection of reactive oxygen species. Clinica Chimica Acta. 2006;368(1-2):53–76. doi: 10.1016/j.cca.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 140.Dickinson BC, Srikun D, Chang CJ. Mitochondrial-targeted fluorescent probes for reactive oxygen species. Current Opinion in Chemical Biology. 2010;14(1):50–56. doi: 10.1016/j.cbpa.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cochemé HM, Quin C, McQuaker SJ, et al. Measurement of H2O2 within living Drosophila during aging using a ratiometric mass spectrometry probe targeted to the mitochondrial matrix. Cell Metabolism. 2011;13(3):340–350. doi: 10.1016/j.cmet.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Srikun D, Albers AE, Nam CI, Iavarone AT, Chang CJ. Organelle-targetable fluorescent probes for imaging hydrogen peroxide in living cells via SNAP-Tag protein labeling. Journal of the American Chemical Society. 2010;132(12):4455–4465. doi: 10.1021/ja100117u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Souslova EA, Chudakov DM. Genetically encoded intracellular sensors based on fluorescent proteins. Biochemistry. 2007;72(7):683–697. doi: 10.1134/s0006297907070012. [DOI] [PubMed] [Google Scholar]
- 144.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Molecular Cell. 2007;26(1):1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 145.Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antioxidants & Redox Signaling. 2006;8(3-4):243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 146.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nature Reviews Molecular Cell Biology. 2007;8(9):722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- 147.Belousov VV, Fradkov AF, Lukyanov KA, et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nature Methods. 2006;3(4):281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- 148.Markvicheva KN, Bilan DS, Mishina NM, et al. A genetically encoded sensor for H2O2 with expanded dynamic range. Bioorganic and Medicinal Chemistry. 2011;19(3):1079–1084. doi: 10.1016/j.bmc.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 149.Espinosa A, García A, Härtel S, Hidalgo C, Jaimovich E. NADPH oxidase and hydrogen peroxide mediate insulin-induced calcium increase in skeletal muscle cells. Journal of Biological Chemistry. 2009;284(4):2568–2575. doi: 10.1074/jbc.M804249200. [DOI] [PubMed] [Google Scholar]
- 150.Ungvari Z, Labinskyy N, Mukhopadhyay P, et al. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. American Journal of Physiology. 2009;297(5):H1876–H1881. doi: 10.1152/ajpheart.00375.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mishina NM, Tyurin-Kuzmin PA, Markvicheva KN, et al. Does cellular hydrogen peroxide diffuse or act locally? Antioxidants & Redox Signaling. 2011;14(1):1–7. doi: 10.1089/ars.2010.3539. [DOI] [PubMed] [Google Scholar]
- 152.Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459(7249):996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hanson GT, Aggeler R, Oglesbee D, et al. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. Journal of Biological Chemistry. 2004;279(13):13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 154.Gutscher M, Pauleau AL, Marty L, et al. Real-time imaging of the intracellular glutathione redox potential. Nature Methods. 2008;5(6):553–559. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- 155.Meyer AJ, Dick TP. Fluorescent protein-based redox probes. Antioxidants & Redox Signaling. 2010;13(5):621–650. doi: 10.1089/ars.2009.2948. [DOI] [PubMed] [Google Scholar]
- 156.Gutscher M, Sobotta MC, Wabnitz GH, et al. Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. Journal of Biological Chemistry. 2009;284(46):31532–31540. doi: 10.1074/jbc.M109.059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Schwarzländer M, Fricker MD, Müller C, et al. Confocal imaging of glutathione redox potential in living plant cells. Journal of Microscopy. 2008;231(2):299–316. doi: 10.1111/j.1365-2818.2008.02030.x. [DOI] [PubMed] [Google Scholar]
- 158.Albrecht SC, Barata AG, Grosshans J, Teleman AA, Dick TP. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metabolism. 2011;14(6):819–829. doi: 10.1016/j.cmet.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 159.Ostergaard H, Henriksen A, Hansen FG, Winther JR. Shedding light on disulfide bond formation: engineering a redox switch in green fluorescent protein. The EMBO Journal. 2001;20(21):5853–5862. doi: 10.1093/emboj/20.21.5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang W, Fang H, Groom L, et al. Superoxide flashes in single mitochondria. Cell. 2008;134(2):279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kishikawa J, Fujikawa M, Imamura H, et al. MRT letter: expression of ATP sensor protein in Caenorhabditis elegans . Microscopy Research and Technique. 2012;75(1):15–19. doi: 10.1002/jemt.21103. [DOI] [PubMed] [Google Scholar]
- 162.Hung YP, Albeck JG, Tantama M, Yellen G. Imaging cytosolic NADH-NAD+ redox state with a genetically encoded fluorescent biosensor. Cell Metabolism. 2011;14(4):545–554. doi: 10.1016/j.cmet.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Klare JP, de Orue Lucana DO. Conformational changes in the novel redox sensor protein HbpS studied by site-directed spin labeling and its turnover in dependence on the catalase-peroxidase CpeB. Antioxidants & Redox Signaling. 2012;16(7):639–648. doi: 10.1089/ars.2011.4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yano T, Oku M, Akeyama N, et al. A novel fluorescent sensor protein for visualization of redox states in the cytoplasm and in peroxisomes. Molecular and Cellular Biology. 2010;30(15):3758–3766. doi: 10.1128/MCB.00121-10. [DOI] [PMC free article] [PubMed] [Google Scholar]