

Abstract
Exposure of cancer cells to anticancer agents in cultures induces detachment of cells that are usually considered dead. These drug-induced detached cells (D-IDCs) may represent a clinical problem for chemotherapy since they may survive anoikis, enter the circulation, invade other tissues and resume proliferation, creating a metastasis, especially in tissues where the bioavailability of anticancer agents is not enough to eliminate all cancer cells. In this study we evaluated the antiproliferative effect of menadione : sodium orthovanadate (M : SO) combination on A549 lung cancer cells as well as the ability of M : SO to induce cell detachment. In addition, we followed the fate and chemosensitivity of M : SO-induced detached cells. Using transwell chambers, we found that a fraction of the M : SO-induced detached cells were viable and, furthermore, were able to migrate, re-attach, and resume proliferation when re-incubated in drug-free media. The total elimination of A549 detachment-resistant cells and M : SO-induced detached cells were successfully eliminated by equivalent M : SO concentration (17.5 μM : 17.5 μM). Thus, M : SO prevented cell migration. Similar results were obtained on DBTRG.05MG human glioma cells. Our data guarantee further studies to evaluate the in vivo occurrence of D-IDCs, their implications for invasiveness and metastasis and their sensitivity to anticancer drugs.
1. Introduction
In order to preserve tissue integrity, cells adhere tightly to each other and to the surrounding matrix. By cellular interaction with the environment and adaptation to changes through multiple mechanisms, the tissue structure is maintained for proper function and physiology [1]. Important cellular functions including proliferation, motility, migration, and apoptosis are related to cell adhesion [2]. When cells detach from the tissue, they undergo anoikis, a term that defines cell apoptosis induced by inadequate or inappropriate cell-matrix interactions. Cell detachment has been suggested to be part of apoptosis since paclitaxel-induced detached cells showed a full spectrum of apoptotic changes whereas the attached cells only showed activation of caspase-3-like proteases but not poly (ADP-ribose) polymerase (PARP) cleavage [3].
Interestingly, cancer cells show mechanisms that prevent death by anoikis [4–6] making it possible for the detached cells to survive and migrate. This ability of cancer cells to leave home and survive in a foreign tissue is essential for invasion and metastasis. Indeed, suppression of anoikis resulted in rapidly growing tumours in mice that infiltrated lymphatics and blood vessels to colonize distant [7] and also promoted metastasis of esophageal squamous cell carcinoma [8]. Cell detachment has been shown to be the initial step of metastasis [9] and to play an important role for both invasiveness and metastasis [10]. Furthermore, detached cancer cells have been observed to reattach and, under permissive conditions, resume proliferation [9].
Drug-induced cell detachment has been extensively documented and used as a model system to study anoikis [11, 12]. However, to our knowledge, there are few recent studies that address the fate and chemosensitivity of drug-induced detached cells [13, 14]. It has been reported that the acquisition of anoikis in human osteosarcoma cells did not change the sensitivity of the cells to several (etoposide, adriamycin, vinblastine, cisplatin, and paclitaxel) anticancer agents [15]. Nevertheless, the chemosensitivity of drug-induced detached cancer cells (D-IDCs) compared to the chemosensitivity of cancer cells that remain attached is largely unknown. D-IDCs may represent a clinical problem for chemotherapy since such cells may survive anoikis, enter the circulation, invade other tissues, and resume proliferation, creating a metastasis, especially in permissive tissues, that is, where the bioavailability of a given anticancer agent is not enough to eliminate all cancer cells. As an example, lung cancer is the most common type of cancer to spread to the brain, [16–18] where, due to the presence of the blood brain barrier (BBB), the bioavailability of most anticancer drugs can be very low compared to the systemic bioavailability [17]. In addition, most systemic treatments can transiently weaken the BBB which may facilitate for D-IDCs to seed the CNS. This may in part explain, why despite chemotherapy, brain is still a common site for metastasis for lung as well as other cancers.
The fact that detached cancer cells appear to play a fundamental role in invasion and metastasis gives further support to novel models of cancer biology that propose as ultimate goal for successful treatment of the total elimination and at once of all cancer cells. In gliomas, the “complex system model” [19] and “the stemness phenotype model” [20] predicted that in order to completely eradicate glioma all cancer cell types (stem and nonstem cancer cells) should be eliminated at once since the targeting of only one population, for example, cancer stem cells, will unlikely be a successful strategy [21]. If this prediction is true and a similar situation occurs in other types of cancer, a successful treatment will require the use of pankiller drugs, defined by us as a drug or combination of drugs that eliminates 100% of cancer cells in vitro preventing regrowth when the drug is removed [22, 23]. The prefix “pan” comes from the ancient Greek word παν-pan-(all, every). To screen for pankiller drugs we introduced the “Regrowth Concentration zero” parameter (RC0) that defines the minimum drug concentration and time exposure needed to kill 100% of cells [24] in order to prevent cancer relapse.
On the other hand, the effect of anticancer drugs on cell proliferation is usually measured by short-term assays, such as the MTT assay [25, 26]. In that type of assays, attached cells are exposed for different periods of time (usually between 24 and 72 hours) to anticancer drugs. The number of surviving cells is then estimated by the amount of produced formazan which is directly proportional to the number of viable cells. The medium is removed and replaced with DMSO in order to solubilize the water-insoluble formazan dye. Although in these assays some cells may detach, they are usually not accounted for because they are considered dead. If they are alive, the formazan produced by these cells may precipitate, remain in the well, and contribute to the total activity. The use of soluble tetrazolium salts, for example, the WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt), that does not require the solubilization step [27], may minimize the potential underestimation of cell viability due to the loss of detached but viable cells.
In order to efficiently monitor the ability of drugs to eliminate all cancer cells, according to our experience and despite the extended use of short-term assays, the screening of anticancer drugs may require other type, of proliferation assays such as long-term proliferation assays. For this purpose, we recently introduced a proliferation assay where cells are subjected to prolonged (between 7 and 14 days) drug exposure [22–24]. This assay has the potential to detect around 0.01% of resistant cells and is technically easy to perform compared to the clonogenic assay [22] that gives similar information. Using this long-term assay we have reported that combinations of menadione : vitamin C (M : VC) and menadione : sodium orthovanadate (M : SO) have pankiller activity against a panel of glioma cell lines [22, 23]. Menadione alone or in combination with vitamin C has shown anticancer activity likely by its ability to generate oxidative stress and ultimately induce cell death [28–30]. We previously showed in human glioma cells that the M : SO combination exerts cytotoxic effects likely via the generation of oxidative stress and tyrosine kinase stimulation [22]. Sodium orthovanadate is a tyrosine phosphatase inhibitor [31]. In our studies we observed that M : SO induced cell detachment as early as 24 hours of drug exposure. Although the outcome of those cells was not evaluated at that time, we speculate now that it is possible that a fraction of those detached cells remain viable even after 72 hours.
The aim of this study was (i) to investigate the sensitivity of human non-small-cells A549 lung cancer cell line to M: SO in order to expand our previous finding on a panel of glioma cell lines (ii) to study the outcome of D-IDCs as well as their sensitivity to anticancer agents in comparison to detachment resistant cells and (iii) to design and discuss potential strategies for prevention of drug-induced cell detachment (D-ICD).
2. Material and Methods
2.1. Reagents and Drugs
Menadione, sodium orthovanadate, hydroxyurea, aphidicolin, etoposide, and paclitaxel were purchased from Sigma-Aldrich (Stockholm, Sweden). Stock solutions of menadione (100 mM), hydroxyurea (1 M), aphidicolin (2.5 mM), etoposide (25 mM), phenylmethylsulphonyl fluoride (PMSF, 1 mM), marimastat (25 mM), and paclitaxel (10 mM) were prepared in dimethylsulfoxide (DMSO) and stored at −20°C. Stock solution of sodium orthovanadate was prepared in distilled sterile water (1 mM, pH 10) according to previous study guidelines [31, 32] and stored at −80°C. Final drug dilutions were prepared in complete cell culture media just before use. DMSO (<1%) or water (<10%) were used as control.
2.2. Cell Culture
Human lung cancer A549 cell line was provided by Anna Fogdell-Hahn (Department of Clinical Neuroscience, Karolinska Institute, Sweden) while human glioma cell line, DBTRG.05MG, was obtained from the European collection of cell culture (ECACC). Cells were routinely maintained in RPMI-1640 (Sigma, Stockholm, Sweden) medium supplemented with 10% fetal bovine serum (Lonza, Verviers Belgium), 2 mM glutamine (Biochrom AG, Berlin, Germany), 1% HT Media Supplement Hybri-Max (Sigma, Stockholm, Sweden), 1% penicillin-streptomycin solution (Sigma), and 1 mM sodium pyruvate (Sigma), grown at +37°C, 5% CO2 and 95% air and harvested by using 0.25% trypsin-EDTA (Sigma).
2.3. Short-Term Antiproliferative Assay
Cells were plated (~5000 cells per well) into flat bottom 96-well culture plates (Sigma, Stockholm, Sweden) and allowed to adhere for 24 hours. Cells to be tested were incubated with the indicated concentrations of drug while cells used as control were incubated with the highest concentration of vehicle: DMSO (<1%) or dH2O (<10%). After 72 hours cell proliferation was measured by using a cell counting kit (CCK, sigma, Stockholm, Sweden) in a plate reader (Labsystem iEMS reader MF) at a wavelength of 450 nm. Experiments were performed in duplicate and repeated three times in separate occasions.
2.4. Long-Term Antiproliferative Assay
Cells were plated (~5000 cells per well) into flat bottom 96-well culture plates and allowed to reach semiconfluence during 3 days. Afterwards, cells to be tested were treated with the indicated concentration of drug during different time periods (1–4 weeks). After withdrawing the drug, cells were re-incubated into complete cell culture medium for another 2 weeks. Control cells were, instead, incubated with equal concentration of DMSO (<0.1%) or dH2O (<10%) during the same time period as tested cells. In order to retain the potency of the drug and nutrients in the culture medium during the prolonged exposure, complete medium and drugs were changed twice a week. Pankiller activity and RC0 were routinely evaluated by using inverted microscope as previously described [22–24]. Pankiller activity was confirmed by duplicating the experiments three times in different occasions.
2.5. Cell Quantification Assay
Cells were plated (166,500 cells per well, density = 37,000 cells/mL) into flat bottom 6-well culture plates and allowed to reach semiconfluence during 3 days. Cells were, then, incubated with the indicated concentration of drugs or with corresponding vehicle (<0.1% DMSO or <10% dH2O) during 24 hours. Surviving cells were harvested in 0.25% trypsin-EDTA (Sigma, Sweden). Trypsin was neutralized with complete cell culture medium. Cell viability was measured by the Trypan blue exclusion test.
2.6. Cell Staining and Photograph
Cells were cultured (166,500 cell per well, density = 37,000 cell/mL) in 8-well chamber slide (VWR, Stockholm, Sweden) during 3-4 days. After incubation, the cells were washed twice with PBS, fixed with 4% formaldehyde for 5 minutes, and washed twice again with PBS. Cells were then stained with Mayer's hematoxylin and visualized by light microscopy.
2.7. Cell Reattachment Assay
Cells were plated into 6-well culture plate (166,500 cells per well, density = 37,000 cells/mL) and allowed to reach semiconfluence during 3 days. Cells were then incubated overnight with the indicated concentration of drugs. Floating detached cells were collected in a Falcon tube and centrifuged. After centrifugation, the supernatant was discarded and the cells resuspended in 200 μL fresh medium. After gentle mixing, 50 μL of the cell suspensions were used for quantification by trypan blue exclusion method while 100 μL of the cell suspensions were placed into 50 mL cell culture flasks containing fresh culture medium. The flasks were routinely visualized by inverted microscopy to investigate the ability of detached cells to reattach and proliferate.
2.8. Sensitivity to Drugs and Migration Assay
Cells (444,000 cells, density = 37,000 cells/mL) were placed into culture flasks containing 50 mL of culture medium. After overnight incubation cells were treated with M : SO (17.5 μM : 17.5 μM) combination for 24 hours. Floating detached cells were then collected, centrifuged, and resuspended in 2 mL fresh culture medium. Five aliquots of the resuspended cells were prepared and plated into each of the upper compartment of a 24-well transwell plates (Corning, NY, USA) containing an insert. The cells were then treated with 5 different concentrations of M : SO combination during 1 week, followed by 2-week incubation in drug-free media. Viability of detached cells was evaluated by microscopy as the ability of the cells to re-attach onto the filter of the insert in the Transwell plate and resume proliferation. Migration was evaluated by the ability of the cells to cross the insert, reattach and resume proliferation in the lower compartment of the Transwell plate see Figure 6(a) for schematic representation.
Figure 6.
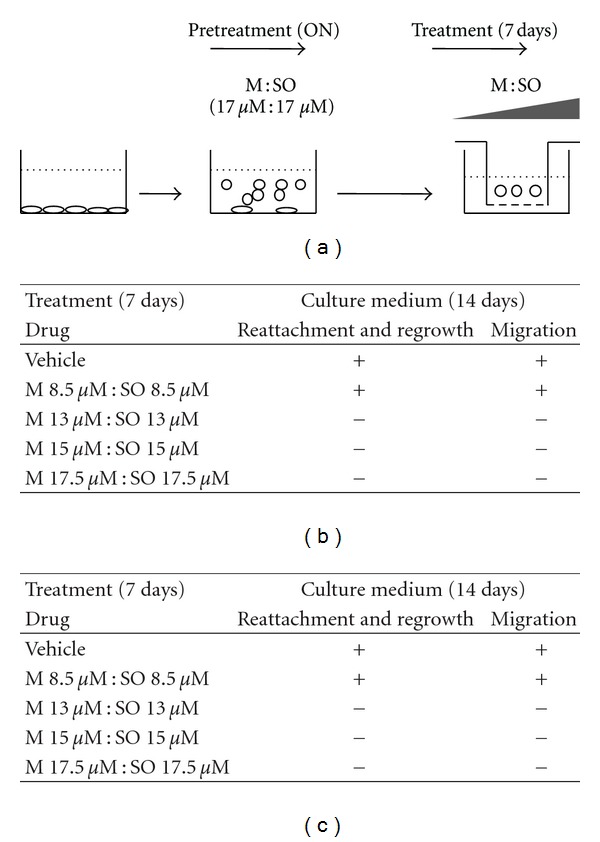
Sensitivity of M : SO-induced detached cells. (a) Schematic representation of the experimental setup. A549 lung cancer cells were pretreated overnight with low concentrations of M : SO (pretreatment, ON). The detached cells were then treated with varying concentrations of M : SO (treatment, 7 days). (b) Effect of M : SO on regrowth and migration of detached A549 lung cancer cells tested at different concentrations. (c) Effect of M : SO on regrowth and migration of detached DBRTG glioma cells tested at different concentrations. (+) and (−) indicate the regrowth and migration of detached cells observed at the indicated M : SO concentration.
2.9. Statistical Analysis
One-way ANOVA with Newman-Keuls multiple comparisons post test was performed using GraphPad Prism version 5.04 for Windows, GraphPad Software, San Diego, California, USA, http://www.graphpad.com/.
3. Results
3.1. Higher Antiproliferative Effect of M : SO Combination on A549 Lung Cancer Cells
In short-term proliferation assay both M and SO, alone or in combination, showed concentration-dependent antiproliferative effect on A549 lung cancer cells. The IC50 for M and SO showed values of 24 μM and 20 μM, respectively, while the IC50 for M : SO combination was 8 μM : 8 μM. The maximum antiproliferative effect for M : SO combination, M alone or SO alone treatment, was reached at the approximate concentrations of 25 : 25 μM, 50 μM, and 100 μM, respectively (Figure 1).
Figure 1.
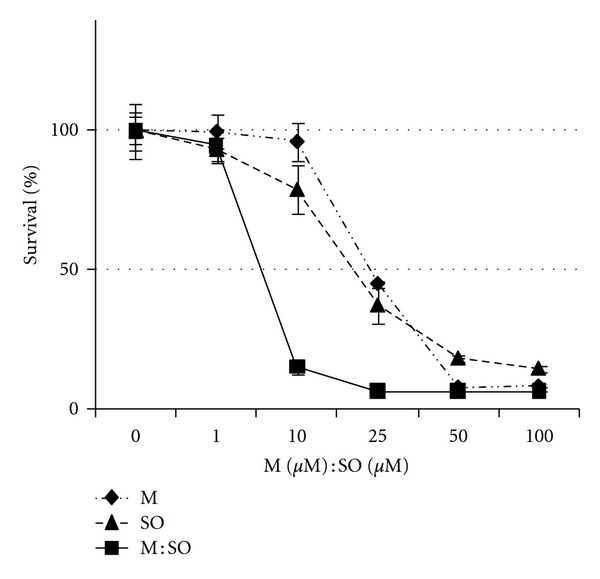
Short-term antiproliferative effect of menadione (M), sodium orthovanadate (SO), and M : SO combination on A549 lung cancer cells. The combined treatment with M : SO shows increased antiproliferative effect compared to single agent treatment.
In long-term proliferation assay the effect of M and SO alone or in combination was evaluated by the ability of the A549 lung cancer cells to resume growth after prolonged exposure (7 days) to different concentrations of the drugs. Exposure to increasing concentrations, in the range 1–50 μM, of M or SO alone for 7 days failed to prevent regrowth (Figure 2, top panel). However, exposure to M : SO combination at concentrations 17.5 μM : 17.5 μM for the same period of time was enough to prevent the regrowth (RC0) of A549 lung cancer cells (Figure 2, bottom panel). The present results confirm our previously observation on the pankiller effect of this combination on a panel of glioma cell lines [22]. Lower concentrations of M or SO in the combination (e.g., M : SO 10 μM : 17.5 μM) failed to prevent regrowth even if incubated for longer time periods (Figure 2, table). In conclusion, the pankiller effect and the ability to prevent regrowth of the M : SO combination were shown to be concentration dependent.
Figure 2.
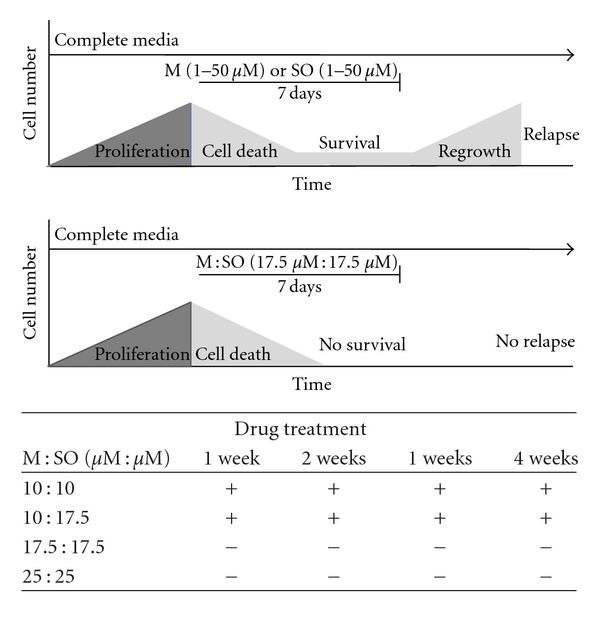
Long-term antiproliferative effect of menadione (M), sodium orthovanadate (SO), and M : SO combination. The combined treatment with M : SO reduced the needed concentration of each agent to kill 100% of A549 lung cancer cells preventing regrowth (RC0) after 7 days. The figure shows the schematic representation of events in a single well. Top panel: cells exposed to increasing concentrations (1–50 μM) of M or SO alone. Bottom panel: combined treatment with M : SO reaches RC0 (17.5 μM : 17.5 μM). Table: effect of M : SO at different concentrations and exposure time. (+) indicates regrowth observed in at least one well. (−) indicates no regrowth observed.
The antiproliferative effect of the selected concentrations of M alone (17.5 μM), SO alone (17.5 μM), or M : SO (17.5 μM : 17.5 μM) was also evaluated by microscopic examination after 4-day exposure. This time point was chosen in order to avoid excessive growth of control (untreated) cells. As observed in Figure 3, these experiments clearly showed that each drug alone had low antiproliferative effect but that the M : SO combination almost completely killed all cells already after 4-day exposure.
Figure 3.
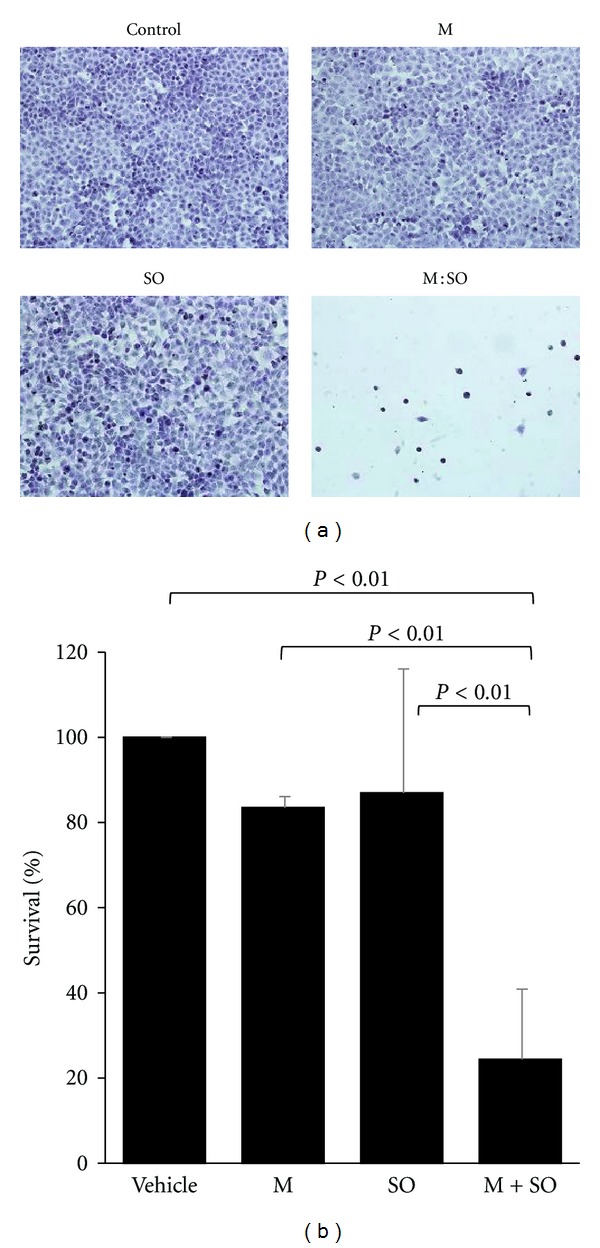
(a) Microscopic examination of A549 lung cancer cells after 4-day treatment with menadione (M), sodium orthovanadate (SO) alone or M : SO combination. (b) Quantitative data from three independent experiments.
3.2. Short Exposure to Anticancer Drugs Induced Cell Detachment
Microscopic observation of A549 lung cancer cells exposed overnight (16 hrs) to M : SO 17.5 μM : 17.5 μM showed intensive cell detachment (Figure 4(a)). The detached cells were able to reattach and resume proliferation when reincubated in drug-free media (Figure 4(b)).
Figure 4.
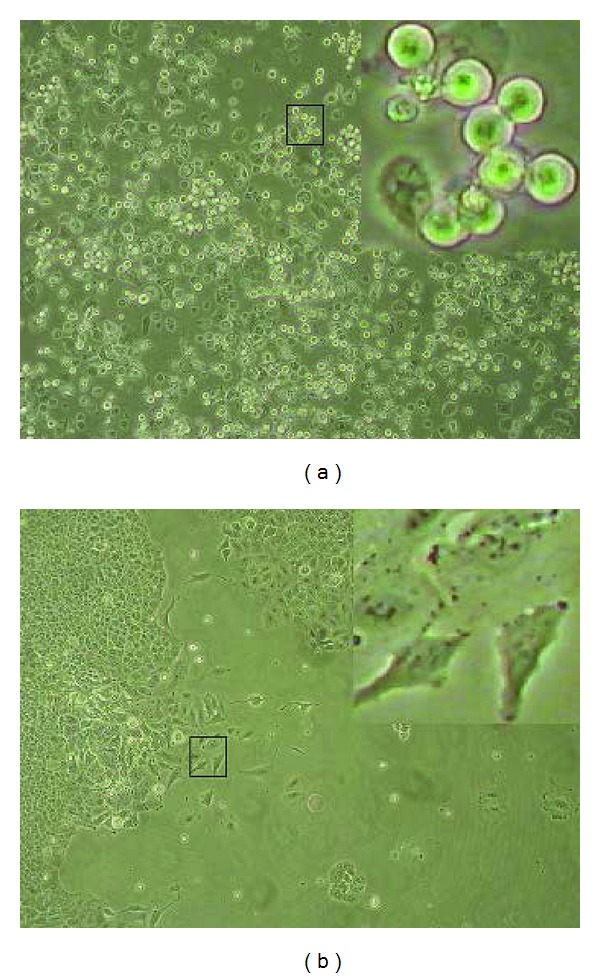
Intensive detachment of A549 lung cancer cell induced by M : SO as well as reattachment of the detached cells. (a) detachment of cells exposed overnight (16 h) to M : SO (17 μM : 17 μM). (b) Regrowing of the detached cells after 1-week incubation in drug-free media. Square insets are zooming out of selected areas showing the morphology of detached cells (a) or reattached cells (b).
For comparison we also tested under the same conditions if other anticancer drugs (2.5 μM aphidicolin, 45 μM etoposide, 250 nM paclitaxel and 10 mM hydroxyurea) also induced cell detachment and if those drugs-induced detached cells were able to reattach and resume proliferation. All the previously mentioned substances induced extensive cell detachment. However, while cells detached by aphidicolin, paclitaxel, and hydroxyurea were able to reattach and resume proliferation, the cells detached by etoposide were unable to resume proliferation (see Supplementary Material avalible online at doi:10.5402/2012/307102, Figure S1) although few of them were viable as observed by the trypan blue exclusion test. The concentrations were chosen since they are close to their IC50 for the A549 cell line, and, therefore, the specific effect on cell detachment is unlikely due to difference in the concentration used for each compound.
3.3. Protease Inhibitors Partially Prevented Drug-Induced Cell Detachment
Since cell detachment requires degradation of extracellular matrix proteins [33], we tested the potential preventive effect of PMSF (serine proteinase inhibitor) and marimastat (matrix metalloproteinase inhibitor) on M : SO-induced A549 lung cancer cell detachment. Based on data obtained by the short (72 hours) proliferation assay we selected 2.5 mM PMSF and 100 and 250 μM marimastat for further experiments (Figure 5(a)). We shall point out that the antiproliferative effect of these protease inhibitors by themselves, at the concentrations used in this experiment, was not significantly different from that observed with DMSO in control cultures (Figure 5(b)). As described previously, overnight exposure to M : SO combination (17.5 μM : 17.5 μM) produced intensive cell detachment. Interestingly, the addition of PMSF or marimastat significantly reduced the number of detached cells (Figure 5(c)). The strongest effect of marimastat at lower concentration (100 μM) seen in Figure 5(c) is likely because marimastat at higher concentrations (250 μM) has higher antiproliferative effect when used alone (Figure 5(a)) or in combination with with M : SO. It is then expected that the number of detached cells (although viable) will be higher in marimastat 250 μM + M : SO treated cultures.
Figure 5.
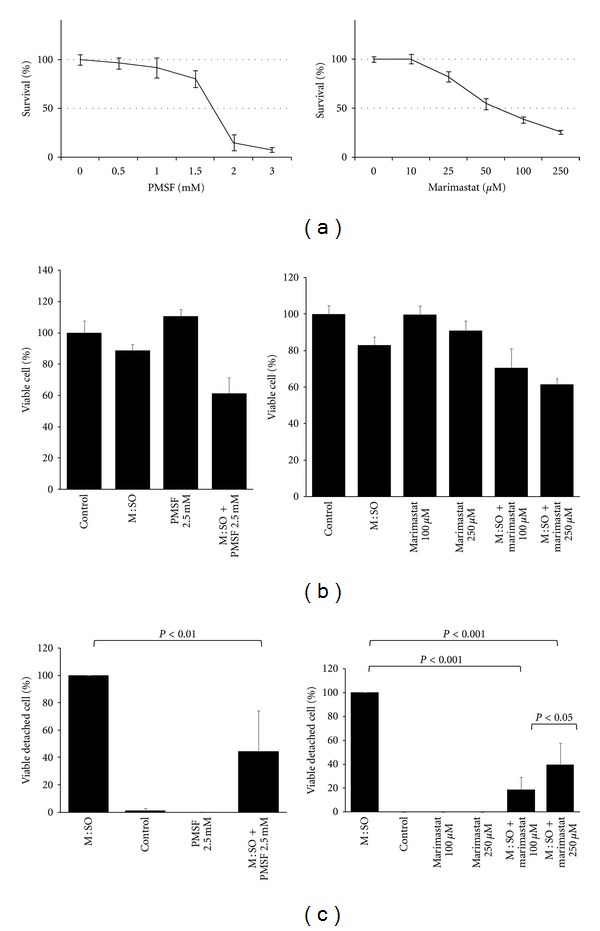
Protease inhibitors partially prevented drug-induced cell detachment. (a) Antiproliferative effect of short-term (72 hours) exposure to PMSF and marimastat on A549 lung cancer cells. (b) Antiproliferative effect of PMSF and marimastat alone or in combination with M : SO on A549 lung cancer cells. (c) PMSF and marimastat prevented M : SO-induced cell detachment.
3.4. The Drugs Sensitivity of M : SO-Induced Detached Cells and Detachment Resistant Cells Were Similar
The elimination of A549 lung cancer cells needed an M : SO concentration of 17.5 μM : 17.5 μM (see long-term antiproliferative assay results). Similarly, M : SO-induced detached cells were also eliminated at this concentration and at even lower (≥13 μM : 13 μM) M : SO concentrations (as shown in Figure 6(b)). When reincubated with vehicle alone or with M : SO 8 μM : 8 μM, the detached cells were able to reattach, resume proliferation, and migrate (Figure 6(b)).
Similar results were obtained when M : SO-induced detached glioma cells were tested (Figure 6(c)).
4. Discussion
In this work, we confirmed our previous observation that M : SO combination has stronger antiproliferative effect than any of those two drugs used alone [22]. We found that the M : SO needed to kill all cancer cells (pankiller) induced cell detachment and that these M : SO-induced detached cells were able to migrate, reattach and proliferate. The sensitivity of M : SO detached cells compared to detachment-resistant cells (DRCs) was similar since equivalent concentration of M : SO were able to eliminate them. This indicates that detached cells, although resistant to anoikis, have actually similar sensitivity to those cells that remain attached. In other words, resistance to anoikis does not necessarily translate into increased chemotherapy resistance.
In short-term assay (72 hours) the IC50 of the M : SO combination on A549 cells (human lung adenocarcinoma epithelial cell line) was around 8 μM (Figure 1). The maximum antiproliferative effect for M : SO combination, M alone or SO alone treatment was reached at the approximate concentrations of 25 : 25 μM, 50 μM, and 100 μM, respectively (Figure 1). In long-term assay, the M : SO combination showed pankiller activity against the A549 human lung cancer cell line after 7-day exposure (M : SO, 17.5 μM : 17.5 μM) (Figure 2). The sensitivity of A549 cell line to M : SO combination was similar to that previously reported for glioma cells which were eliminated after exposure to M : SO (10 μM : 17.5 μM) during 10 days [22].
In the current study we have revealed that M : SO as well as four other anticancer drugs induce extensive cell detachment (Figures 4(a) and S1)). Since cell detachment has been reported to require degradation of extracellular matrix proteins [33] we evaluated the effect of protease inhibitors on M : SO-induced cell detachment. Both, PMSF and marimastat reduced the number of detached cells (Figure 5(c)) suggesting a role of MMPs in D-IDC, in agreement with results obtained in other systems [34, 35].
The fate of the D-IDCs was evaluated by following their ability to migrate and resume proliferation. As seen in Figure 4(b), M : SO detached cells were able to migrate, reattach, and resume proliferation when reincubated in drug-free media. More important, we found that detached cells showed similar sensitivity to the drugs cells showed similar sensitivity to the drugs than detachment resistant cells. The elimination of detachment resistant cells required exposure to pankiller concentrations (17.5 μM : 17.5 μM) of M : SO for 7 days (Figure 2). D-IDCs were not able to resume proliferation (and migrate) when exposed to 13 μM : 13 μM of M : SO during the same time (Figure 6). This small difference that may suggest that D-IDCs are more sensitive than DRCs may be due to the overnight exposure to 17.5 μM : 17.5 μM of M : SO and is likely clinically irrelevant.
As we observed in this study in vitro, anticancer agents may well in vivo induce cell detachment within a tumor (Figure S1). This may occur if the anticancer agents do not reach pankiller concentrations (RC0 required to kill 100% cells) due to, for example, insufficient tissue bioavailability or insufficient administration time. We shall point out that we also observed a small fraction of cells that detached spontaneously (Figure 5(c), controls bar) similarly to what it is observed in vivo where circulating tumor cells (CTCs) are commonly present in many types of cancer [36–38]. CTCs have been shown to have decreased adhesiveness and resistance to anoikis [39]. Similarly, drug-induced detached cancer cells may survive, migrate, reattach, and resume proliferation in permissive tissues [40], that is, where the concentration of the anticancer agent is lower, creating a metastasis. Thus, D-ICD may represent a problem during cancer treatment even in the event that the primary tumor is (apparently) successfully treated.
One proposed strategy to eliminate detached cells has been to find drugs that promote anoikis, [6]. We propose here that in order to avoid metastasis caused by detached cells, at least four additional strategies should be considered.
To prevent D-ICD, for example, by concomitant administration of marimastat, to existing chemotherapy regimens. This approach was shown to be partially successful since marimastat reduced the tumor spread and metastasis in experimental animal models [41, 42]. However, since not all cancer types express the MMP, important for cell detachment [33], it is anticipated that use of MMP inhibitor will be useful only in a fraction of patients. In addition, other mechanisms and signaling pathways, such as the PI3K/Akt pathway, [13] are involved in cell detachment.
To deliver anticancer agents (after the established chemotherapy) at concentrations enough to eliminate viable D-IDCs in all tissues. Our finding that pankiller concentrations of M : SO not only prevent migration of but also eliminate D-IDC is important regarding metastasis prevention. Pankiller concentration, enough to deplete detached cells, during or immediately after local treatment with anticancer drugs may be necessary and enough to prevent metastasis. If D-IDCs were more resistant than DRCs, the elimination of these cells will require “supra-pankiller” concentrations that will be more difficult to achieve due to toxicity issues. On the other hand, plasmatic pankiller concentrations are more likely to be achieved during systemic treatment with anticancer agents but it will be a difficult task in certain tissues such as brain due to presence of the blood brain barrier. Targeted drug delivered by the use of nanoparticles [43, 44] will aid the selective load anticancer drugs at “pankiller” concentrations to cancer cells and likely overcome the blood brain barrier [45] with tolerable toxicity to normal cells.
To use drugs that, despite the fact that they induce cell detachment, the detached cells are non-viable (as reported for etoposide (This article, Figure S1) or for other drugs such as safingol [46]).
To identify anticancer substances that do not induce cell detachment at all.
In conclusion, since most patients die from the progressive growth of metastases [9], D-ICD may represent an important problem and limitation of chemotherapy.
In the present study we have shown the occurrence of D-ICD and, in addition, that cells that detached were able to migrate, reattach, and resume proliferation in vitro. These results have important implications for metastasis development. The heterogeneous response of cancer cells to anticancer drugs shown in Figure S1 regarding the ability of D-IDCs to reattach and regrow, although in vitro, clearly guarantees further evaluation of the in vivo occurrence of these phenomena as well as the development of strategies to prevent, minimize, and/or eliminate this risk.
Supplementary Material
Fate of detached A549 lung cancer induced by aphidicolin, etoposide, paclitaxel or hydroxyurea. Figure S1 shows that these drugs are all able to induce cell detachment and, except for etoposide, they are able to re-attach and proliferate when re-incubated in drug-free media.
Conflict of Interests
The authors declared that they have no conflict of interests.
Acknowledgments
This study was supported by grants from the Swedish Research Council and the Karolinska Institute. The authors thank Dr. Anna Fogdell-Hahn for providing the A549 cell line and Rasmus Gustafsson for technical help.
References
- 1.Nelson CM, Tien J. Microstructured extracellular matrices in tissue engineering and development. Current Opinion in Biotechnology. 2006;17(5):518–523. doi: 10.1016/j.copbio.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 2.Kim SH, Turnbull J, Guimond S. Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. Journal of Endocrinology. 2011;209(2):139–151. doi: 10.1530/JOE-10-0377. [DOI] [PubMed] [Google Scholar]
- 3.Au JL, Kumar RR, Li D, Wientjes MG. Kinetics of hallmark biochemical changes in paclitaxel-induced apoptosis. The AAPS Journal. 1999;1(3, article 8):7–14. doi: 10.1208/ps010308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. Journal of Cell Biology. 1994;124(4):619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horbinski C, Mojesky C, Kyprianou N. Live free or die: tales of homeless (cells) in cancer. American Journal of Pathology. 2010;177(3):1044–1052. doi: 10.2353/ajpath.2010.091270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakamoto S, Kyprianou N. Targeting anoikis resistance in prostate cancer metastasis. Molecular Aspects of Medicine. 2010;31(2):205–214. doi: 10.1016/j.mam.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430(7003):1034–1039. doi: 10.1038/nature02765. [DOI] [PubMed] [Google Scholar]
- 8.Wang XC, Wu YP, Ye B, et al. Suppression of anoikis by SKP2 amplification and overexpression promotes metastasis of esophageal squamous cell carcinoma. Molecular Cancer Research. 2009;7(1):12–22. doi: 10.1158/1541-7786.MCR-08-0092. [DOI] [PubMed] [Google Scholar]
- 9.Langley RR, Fidler IJ. The seed and soil hypothesis revisited—the role of tumor-stroma interactions in metastasis to different organs. International Journal of Cancer. 2011;128(11):2527–2535. doi: 10.1002/ijc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marcus AI, Zhou W. LKB1 regulated pathways in lung cancer invasion and metastasis. Journal of Thoracic Oncology. 2010;5(12):1883–1886. doi: 10.1097/JTO.0b013e3181fbc28a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keledjian K, Kyprianou N. Anoikis induction by quinazoline based α1-adrenoceptor antagonists in prostate cancer cells: antagonistic effect of bcl-2. The Journal of Urology. 2003;169(3):1150–1156. doi: 10.1097/01.ju.0000042453.12079.77. [DOI] [PubMed] [Google Scholar]
- 12.Wahl ML, Owen CS, Grant DS. Angiostatin induces intracellular acidosis and anoikis in endothelial cells at a tumor-like low pH. Endothelium. 2002;9(3):205–216. doi: 10.1080/10623320213633. [DOI] [PubMed] [Google Scholar]
- 13.Kočí L, Hýžd’alová M, Vaculová A, Hofmanová J, Kozubík A. Detachment-mediated resistance to TRAIL-induced apoptosis is associated with stimulation of the PI3K/Akt pathway in fetal and adenocarcinoma epithelial colon cells. Cytokine. 2011;55(1):34–39. doi: 10.1016/j.cyto.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 14.Zeeli T, Langberg M, Rotem C, David M, Koren R, Ravid A. Vitamin D inhibits captopril-induced cell detachment and apoptosis in keratinocytes. British Journal of Dermatology. 2011;164(1):62–67. doi: 10.1111/j.1365-2133.2010.10044.x. [DOI] [PubMed] [Google Scholar]
- 15.Díaz-Montero CM, McIntyre BW. Acquisition of anoikis resistance in human osteosarcoma cells does not alter sensitivity to chemotherapeutic agents. BMC Cancer. 2005;5, article 39 doi: 10.1186/1471-2407-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blanchard P, Le Péchoux C. Prophylactic cranial irradiation in lung cancer. Current Opinion in Oncology. 2010;22(2):94–101. doi: 10.1097/CCO.0b013e32833500ef. [DOI] [PubMed] [Google Scholar]
- 17.Schuette W. Treatment of brain metastases from lung cancer: chemotherapy. Lung Cancer. 2004;45(supplement 2):S253–S257. doi: 10.1016/j.lungcan.2004.07.967. [DOI] [PubMed] [Google Scholar]
- 18.Yamanaka R. Medical management of brain metastases from lung cancer (review) Oncology Reports. 2009;22(6):1269–1276. doi: 10.3892/or_00000564. [DOI] [PubMed] [Google Scholar]
- 19.Laks DR, Visnyei K, Kornblum HI. Brain tumor stem cells as therapeutic targets in models of glioma. Yonsei Medical Journal. 2010;51(5):633–640. doi: 10.3349/ymj.2010.51.5.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cruz M, Siden Å, Tasat DR, Yakisich JS. Are all glioma cells cancer stem cells? Journal of Cancer Science and Therapy. 2010;2(4):100–106. [Google Scholar]
- 21.Hatiboglu MA, Wei J, Wu ASG, Heimberger AB. Immune therapeutic targeting of glioma cancer stem cells. Targeted Oncology. 2010;5(3):217–227. doi: 10.1007/s11523-010-0151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delwar ZM, Avramidis D, Follin E, et al. Cytotoxic effect of menadione and sodium orthovanadate in combination on human glioma cells. Investigational New Drugs. 2012;30(4):1302–1310. doi: 10.1007/s10637-011-9680-y. [DOI] [PubMed] [Google Scholar]
- 23.Vita MF, Nagachar N, Avramidis D, et al. Pankiller effect of prolonged exposure to menadione on glioma cells: potentiation by vitamin C. Investigational New Drugs. 2011;29(6):1314–1320. doi: 10.1007/s10637-010-9489-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avramidis D, Cruz M, Sidén Å, Tasat DR, Yakisich JS. Regrowth concentration zero (RC0) as complementary endpoint parameter to evaluate compound candidates during preclinical drug development for cancer treatment. Journal of Cancer Science & Therapy. 2009;1:19–24. [Google Scholar]
- 25.Carmichael J, Mitchell JB, DeGraff WG, et al. Chemosensitivity testing of human lung cancer cell lines using the MTT assay. British Journal of Cancer. 1988;57(6):540–547. doi: 10.1038/bjc.1988.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods. 1983;65(1-2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 27.Miyamoto T, Min W, Lillehoj HS. Lymphocyte proliferation response during Eimeria tenella infection assessed by a new, reliable, nonradioactive colorimetric assay. Avian Diseases. 2002;46(1):10–16. doi: 10.1637/0005-2086(2002)046[0010:LPRDET]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 28.Akiyoshi T, Matzno S, Sakai M, Okamura N, Matsuyama K. The potential of vitamin K3 as an anticancer agent against breast cancer that acts via the mitochondria-related apoptotic pathway. Cancer Chemotherapy and Pharmacology. 2009;65(1):143–150. doi: 10.1007/s00280-009-1016-7. [DOI] [PubMed] [Google Scholar]
- 29.Jamison JM, Gilloteaux J, Venugopal M, et al. Flow cytometric and ultrastructural aspects of the synergistic antitumor activity of vitamin C-vitamin K3 combinations against human prostatic carcinoma cells. Tissue and Cell. 1996;28(6):687–701. doi: 10.1016/s0040-8166(96)80072-3. [DOI] [PubMed] [Google Scholar]
- 30.Venugopal M, Jamison JM, Gilloteaux J, et al. Synergistic antitumor activity of vitamins C and K3 on human urologic tumor cell lines. Life Sciences. 1996;59(17):1389–1400. doi: 10.1016/0024-3205(96)00466-3. [DOI] [PubMed] [Google Scholar]
- 31.Gordon JA. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods in Enzymology. 1991;201:477–482. doi: 10.1016/0076-6879(91)01043-2. [DOI] [PubMed] [Google Scholar]
- 32.Capella MAM, Capella LS, Valente RC, Gefé M, Lopes AG. Vanadate-induced cell death is dissociated from H2O2 generation. Cell Biology and Toxicology. 2007;23(6):413–420. doi: 10.1007/s10565-007-9003-4. [DOI] [PubMed] [Google Scholar]
- 33.Bonomi P. Matrix metalloproteinases and matrix metalloproteinase inhibitors in lung cancer. Seminars in Oncology. 2002;29(1, supplement 4):78–86. doi: 10.1053/sonc.2002.31528. [DOI] [PubMed] [Google Scholar]
- 34.Charvat S, Le Griel C, Chignol MC, Schmitt D, Serres M. Ras-transfection up-regulated HaCaT cell migration: inhibition by Marimastat. Clinical and Experimental Metastasis. 1999;17(8):677–685. doi: 10.1023/a:1006709403193. [DOI] [PubMed] [Google Scholar]
- 35.Tonn JC, Kerkau S, Hanke A, et al. Effect of synthetic matrix-metalloproteinase inhibitors on invasive capacity and proliferation of human malignant gliomas in vitro. International Journal of Cancer. 1999;80(5):764–772. doi: 10.1002/(sici)1097-0215(19990301)80:5<764::aid-ijc22>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 36.Bonmassar L, Fossile E, Scoppola A, et al. Detection of circulating tumor cells is improved by drug-induced antigen up-regulation: preclinical and clinical studies. Anticancer Research. 2010;30(11):4721–4730. [PubMed] [Google Scholar]
- 37.Koga T, Tokunaga E, Sumiyoshi Y, et al. Detection of circulating gastric cancer cells in peripheral blood using real time quantitative RT-PCR. Hepato-Gastroenterology. 2008;55(84):1131–1135. [PubMed] [Google Scholar]
- 38.Wang JY, Wu CH, Lu CY, et al. Molecular detection of circulating tumor cells in the peripheral blood of patients with colorectal cancer using RT-PCR: significance of the prediction of postoperative metastasis. World Journal of Surgery. 2006;30(6):1007–1013. doi: 10.1007/s00268-005-0485-z. [DOI] [PubMed] [Google Scholar]
- 39.Howard EW, Leung SCL, Yuen HF, et al. Decreased adhesiveness, resistance to anoikis and suppression of GRP94 are integral to the survival of circulating tumor cells in prostate cancer. Clinical and Experimental Metastasis. 2008;25(5):497–508. doi: 10.1007/s10585-008-9157-3. [DOI] [PubMed] [Google Scholar]
- 40.Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD. The seed and soil hypothesis: vascularisation and brain metastases. The Lancet Oncology. 2002;3(1):53–57. doi: 10.1016/s1470-2045(01)00622-2. [DOI] [PubMed] [Google Scholar]
- 41.Kimata M, Otani Y, Kubota T, et al. Matrix metalloproteinase inhibitor, marimastat, decreases peritoneal spread of gastric carcinoma in nude mice. Japanese Journal of Cancer Research. 2002;93(7):834–841. doi: 10.1111/j.1349-7006.2002.tb01326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maekawa KI, Sato H, Furukawa M, Yoshizaki T. Inhibition of cervical lymph node metastasis by marimastat (BB-2516) in an orthotopic oral squamous cell carcinoma implantation model. Clinical and Experimental Metastasis. 2002;19(6):513–518. doi: 10.1023/a:1020329411957. [DOI] [PubMed] [Google Scholar]
- 43.Kumar Khanna V. Targeted delivery of nanomedicines. ISRN Pharmacology. 2012;2012:9 pages. doi: 10.5402/2012/571394.571394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rahman M, Ahmad MZ, Kazmi I, et al. Emergence of Nanomedicine as cancer targeted magic bullets: recent development and need to address the toxicity apprehension. doi: 10.2174/157016312803305898. Current Drug Discovery Technologies. In press. [DOI] [PubMed] [Google Scholar]
- 45.Staquicini FI, Ozawa MG, Moya CA, et al. Systemic combinatorial peptide selection yields a non-canonical iron-mimicry mechanism for targeting tumors in a mouse model of human glioblastoma. The Journal of Clinical Investigation. 2011;121(1):161–173. doi: 10.1172/JCI44798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noda T, Iwai S, Hamada M, Fujita Y, Yura Y. Induction of apoptosis of detached oral squamous cell carcinoma cells by safingol. Possible role of Bim, focal adhesion kinase and endonuclease G. Apoptosis. 2009;14(3):287–297. doi: 10.1007/s10495-009-0319-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fate of detached A549 lung cancer induced by aphidicolin, etoposide, paclitaxel or hydroxyurea. Figure S1 shows that these drugs are all able to induce cell detachment and, except for etoposide, they are able to re-attach and proliferate when re-incubated in drug-free media.