

Abstract
In asthma, the increase in airway smooth muscle (ASM) can contribute to inflammation, airway wall remodeling and airway hyperresponsiveness (AHR). Targetting peroxisome proliferator-activated receptor γ (PPARγ), a receptor upregulated in ASM in asthmatic airways, may provide a novel approach to regulate these contributions. This review summarises experimental evidence that PPARγ ligands, such as rosiglitazone (RGZ) and pioglitazone (PGZ), inhibit proliferation and inflammatory cytokine production from ASM in vitro. In addition, inhaled administration of these ligands reduces inflammatory cell infiltration and airway remodelling in mouse models of allergen-induced airways disease. PPARγ ligands can also regulate ASM contractility, with acute treatment eliciting relaxation of mouse trachea in vitro through a PPARγ-independent mechanism. Chronic treatment can protect against the loss of bronchodilator sensitivity to β 2-adrenoceptor agonists and inhibit the development of AHR associated with exposure to nicotine in utero or following allergen challenge. Of particular interest, a small clinical trial has shown that oral RGZ treatment improves lung function in smokers with asthma, a group that is generally unresponsive to conventional steroid treatment. These combined findings support further investigation of the potential for PPARγ agonists to target the noncontractile and contractile functions of ASM to improve outcomes for patients with poorly controlled asthma.
1. Introduction
Asthma is a chronic inflammatory lung disease affecting over 300 million people worldwide, with 250,000 deaths per year attributed to the disease [1]. Asthma is characterized by inflammation, airway wall remodeling, and airway hyperresponsiveness (AHR), whereby airways are more sensitive to a variety of stimuli and subsequently contract too easily and too much [2].
A major feature of airway remodeling in asthma is an increase in airway smooth muscle (ASM) mass. This thickened ASM layer can act as both a source and target of inflammatory cytokines and extracellular matrix (ECM) proteins, contributing to persistent inflammation and increased airway narrowing. Proliferative, synthetic, and contractile functions of ASM can therefore play distinct roles in both the pathogenesis of asthma and perpetuation of disease symptoms (Figure 1) [3, 4].
Figure 1.
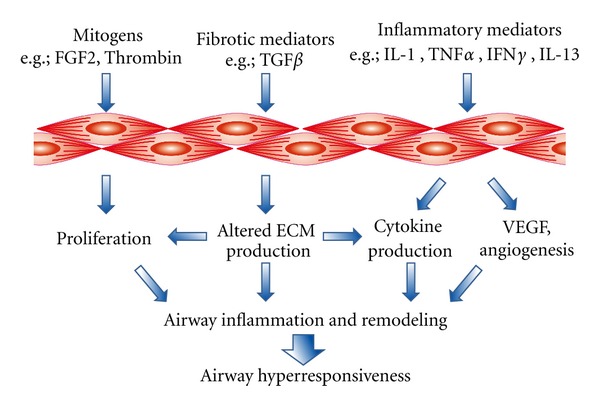
Potential targets for the regulation of noncontractile (proliferative and synthetic) and contractile functions of airway smooth muscle contributing to airway hyperresponsiveness.
In current asthma therapy, inhaled β 2-adrenoceptor agonists are used to reverse ASM contraction while the frequency and severity of asthma attacks can be reduced by combined therapy with β 2-adrenoceptor agonists and glucocorticoids (GCS). However, a significant proportion of patients have poorly controlled symptoms, with variable responses to β 2-adrenoceptor agonists and persistent AHR despite optimal anti-inflammatory treatment. Cigarette smoking in asthma patients also contributes to increased severity of symptoms, with an impaired response to both inhaled and oral corticosteroids [5].
This resistance to therapy is likely to be associated with significant structural changes to the airways, including ASM accumulation, fibrosis, and increased vascularity. These changes have been mechanistically associated with disease severity and accelerated lung function decline [6] but may be difficult to reverse in established asthma. In this context therefore, it is crucial to identify alternative drugs that inhibit the development of AHR, as well as the contribution of ASM to inflammation and remodeling to limit contraction or directly enhance the relaxation of the increased ASM in the airways [7, 8].
A potential novel approach to regulate ASM function in asthma is to target peroxisome proliferator-activated receptor γ (PPARγ). It has been suggested that downregulation of PPARγ signaling may be a contributing factor to the development of AHR in asthma following in utero nicotine exposure [9], while the expression of PPARγ in ASM is upregulated in the airways of asthmatic patients [10].
This paper provides a brief overview of PPARγ pharmacology and describes the contribution of ASM to inflammation, remodeling, and hyperresponsiveness in asthma. Its major focus is to outline the increasing experimental and clinical evidence that PPARγ ligands can regulate ASM function, through both PPARγ-dependent and PPARγ-independent mechanisms.
2. PPARγ Structure and Ligands
PPARγ is a member of the nuclear hormone receptor (NHR) family of ligand-activated transcription factors, which also includes glucocorticoid receptors (GRs) and thyroid hormone receptors. PPARγ is one of three PPAR isoforms designated PPARα (NR1C1), PPARβ (PPARδ, NR1C2), and PPARγ (NR1C3).
Like other NHR, PPARγ possesses a multidomain structure. This includes a DNA binding domain (DBD) containing two zinc finger motifs that recognise specific PPAR response elements (PPREs) sequences. These PPREs consist of hexameric direct repeats of AGGTCA recognition sequences separated by one or two random nucleotide. The DBD is linked via a hinge region to the large ligand binding domain (LBD) that occupies the C-terminal half of the receptor and an activator function (AF)-1 domain is present at the N-terminus [11–13].
PPARγ possesses an unusually large T-shaped ligand-binding pocket that enables interaction with a structurally diverse library of ligands [13]. The most widely studied synthetic agonists are the thiazolidinedione class of drugs, rosiglitazone (RGZ, BRL 49653), pioglitazone (PGZ), troglitazone (TGZ), and ciglitazone (CGZ). RGZ binds the receptor with high affinity (Kd 43 nM), whereas PGZ and CGZ are less potent [14]. Alternative nonglitazone agonists include GW262570 [15] and novel triterpenoid compounds derived from oleanic acid such as 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO) [16]. Despite binding affinities in the nM range, most biological effects of these synthetic PPARγ agonists have been observed at μM concentrations.
Potential natural ligands for PPARγ also show marked structural diversity and include prostaglandin D2 (PGD2) and its metabolites PGJ2 and 15-deoxy-Δ12,14-prostaglandin-J2 (15dPGJ2) [17]. 15dPGJ2 in particular has been widely used for comparisons with glitazones in experimental settings [18]. However, these agonists, and other putative PPARγ ligands such as the oxidised lipids 9- and 13-hydroxyoctadecadienoic acid (HODE) and 12- and 15-hydroxyeicosatetraenoic acid (HETE) [19], have multiple additional sites of action, suggesting that demonstrating their PPARγ-dependent actions is likely to be particularly challenging.
3. Mechanisms of Action of PPARγ and Its Ligands
3.1. PPARγ Activation
Cytosolic PPARγ exists as a monomer, with both the LBD and AF-1 domain regulating interactions with coactivators and corepressors that control activation of the receptor [11–13]. PPARγ does not form homodimers, but can associate with multiple partners to form heterodimers. Its most preferential binding partner is the retinoid X receptor (RXR), with 9-cis retinoic acid acting as its natural ligand [20]. Translocation of the ligand-activated PPARγ-RXR complex to the nucleus and binding to PPRE in the promoter region of target genes can result in either the upregulation or inhibition of gene expression. Multiple PPARγ-responsive genes involved in diverse cellular processes including adipogenesis, insulin sensitivity, and inflammation have been identified [21, 22].
Alternatively, PPARγ can cause the transrepression of transcriptional factors such as NFκB, CAAT/enhancer binding protein (C/EBP), signal transducers and activators of transcription (STAT), or activator protein- (AP-) 1. This transrepression may occur either by direct binding to the transcription factors, by sequestration of shared coactivators of these factors or by Small Ubiquitin-like Modifier (SUMO) ylation of PPARγ and subsequent PPARγ binding to a corepressor complex [23]. These actions also have the potential to inhibit inflammatory responses in the lung.
Given the marked differences between the reported PPARγ binding affinities of the glitazones and the concentrations required to elicit their cellular effects, multiple approaches are required to support claims for PPARγ-dependency. These include confirming PPARγ expression in cells of interest and the use of pharmacological antagonists, with GW9662 being the most commonly used. GW9662 irreversibly inhibits PPARγ by covalently binding to Cys285 in the ligand binding pocket and prevents heterodimerisation with RXR as well as interactions with coactivator and corepressor molecules [24]. GW9662 has been used to confirm the PPARγ-dependence of known PPARγ ligands both in vitro [18] and in vivo [25]. Additional molecular techniques such as the use of dominant-negative constructs or adenoviral PPARγ (AdPPARγ) have been employed to implicate PPARγ in the regulation of cellular responses both in vitro and in vivo [26, 27].
3.2. PPARγ-Independent Mechanisms
The mechanistic complexity underlying responses to PPARγ ligands occurring via PPARγ activation is further complicated by evidence of PPARγ-independent pathways.This may involve PPARγ ligands binding directly to alternative receptors, regulating transcription factor activity, or altering signalling through enzymes or ion channels to mediate their cellular responses.
Glitazones have been shown to activate free fatty acid receptors (FFA1, also known as GPR40) causing phosphorylation of the ERK1/2 mitogen-activated protein (MAP) kinases [28]. RGZ and CGZ can also bind directly to GR independently of PPARγ, evidenced by their stimulation of GR nuclear translocation in a PPARγ-deficient cell line [29], and defining a potential alternative anti-inflammatory mechanism for these ligands.
In addition, 15dPGJ2 has been demonstrated to directly inhibit the activity of the enzyme IκB kinase, thereby reducing the phosphorylation of IκB and its subsequent dissociation from the proinflammatory transcription factor NFκB [30, 31]. Actions of PPARγ ligands may also be mediated by increasing PGE2 levels, subsequent to inhibition of its metabolism via 15-hydroxyprostaglandin dehydrogenase [32].
Both 15dPGJ2 and CDDO have been shown to induce heme-oxidase by PPARγ-independent, glutathione-dependent mechanism, although this antioxidant action was restricted to PPARγ ligands possessing an electrophilic centre [33]. Additional evidence of nongenomic, rapid regulation of enzyme activity by PPARγ ligands includes activation of mitogen-associated protein kinase (MAPK), phosphoinositide-3-kinase (PI3K), and adenosine monophosphate-activated protein kinase (AMPK) pathways [34–36] with implications for regulation of cell proliferation and inflammatory cytokine production.
Some of these nongenomic effects may be mediated through altered calcium signalling. TGZ and PGZ have been shown to mobilize calcium from intracellular stores [37, 38]. Although RGZ rapidly inhibited the activity of sarco/endoplasmic reticulum Ca2+ ATPase (SERCA)-2b [39], chronic treatment with PGZ has been shown to increase SERCA activity via a PPARγ-dependent mechanism [40], suggesting that regulation of calcium homeostasis by PPARγ ligands is likely to depend on temporal and cellular contexts.
These diverse PPARγ-dependent and PPARγ-independent actions define numerous mechanisms whereby PPARγ ligands could regulate the altered proliferative, secretory and contractile functions of ASM contributing to asthma.
4. PPARγ Is Increased in Airway Smooth Muscle in Asthma
Although PPARγ was originally characterised as a regulator of adipocyte differentiation, this receptor is also widely expressed in the lung, in both inflammatory and structural cells implicated in asthma pathophysiology [41].
Regulation of PPARγ expression can occur in response to in vitro exposure to inflammatory cytokines, with acute upregulation occurring in response to interleukin-4 (IL-4) in airway epithelial cells and macrophages [42, 43] during macrophage differentiation and activation [44, 45] or following antigen exposure in sensitised mast cells [46].
PPARγ may provide a target to overcome chronic inflammation and increased airway reactivity in vivo. Higher levels of PPARγ were evident in total lung extracts from mouse models of established allergen-induced inflammation [27, 47] and could be localised to ASM and epithelium, mast cells, and some inflammatory cells [25]. In contrast, perinatal exposure to nicotine appears to decrease PPARγ expression and signaling, with increased alveolar interstitial fibroblast-to-myofibroblast differentiation contributing to the development of an asthma-like phenotype in newborn rats [48, 49].
In human airway biopsies, expression levels of PPARγ in ASM, epithelium, and mucosal eosinophils and macrophages were elevated in asthmatic patients compared with controls [10]. In the same study, asthmatics treated with GCS had lower levels of PPARγ expression compared with untreated asthmatics. Although ASM from asthmatics had lower PPARγ levels compared to healthy controls in vitro, this was reversed in the presence of a mitogenic stimulus [50]. These results suggest that increased PPARγ expression observed in situ may be a product of the inflammatory and mitogenic pathways and may also be sensitive to steroid therapy.
These combined findings suggest that PPARγ expression is increased in response to acute or chronic inflammation in multiple cell types including ASM, and that PPARγ could be targeted to limit inflammation, airway remodeling, and increased ASM contraction in asthma.
5. In Vitro Regulation of ASM Function by PPARγ Ligands
Because of the capacity of ASM to perpetuate airway inflammation, orchestrate airway wall remodelling, and modulate airway tone, it has been suggested that targeting ASM is critical for effective asthma treatment [4, 7, 8]. Accumulating in vitro evidence now supports the efficacy of PPARγ ligands in the regulation of ASM cytokine production, proliferation, and contraction, while their direct effects on the potential contribution of ASM to fibrosis and angiogenesis have yet to be confirmed.
5.1. Regulation of ASM Inflammatory Cytokine Production
In response to inflammatory stimuli, ASM can secrete various cytokines and chemokines contributing to the pathophysiology of asthma [51–53]. These mediators include factors such as granulocyte/macrophage-colony-stimulating factor (GM-CSF) [54], granulocyte-colony stimulating factor (G-CSF) [55], eotaxin [56], regulated on activation, normal T cells expressed and secreted (RANTES) [57], IL-6 [58], monocyte chemotactic protein-1 (MCP-1) [59], and vascular endothelial growth factor (VEGF) [60]. Thus, inhibition of ASM cytokine production has the potential to regulate inflammatory cell growth, survival, and recruitment as well as the autocrine influence of these cytokines to induce ASM proliferation and hyperresponsiveness in the inflamed airways in asthma [4, 61].
The anti-inflammatory efficacy of PPARγ ligands has been established in human-cultured ASM cells, with various glitazones inhibiting the release of multiple cytokines irrespective of stimulus used. IL-1β-induced increases in GM-CSF and G-CSF release were both attenuated by CGZ and to a lesser extent by 15dPGJ2 [55]. Although dexamethasone also completely abolished the increase in GM-CSF release, G-CSF induction was only partially inhibited, suggesting that PPARγ ligands may target steroid-resistant pathways in ASM [55]. Induction of eotaxin and MCP-1 by tumour necrosis factor α (TNFα) was also inhibited by PPARγ agonists, and the expression of these chemokines was further decreased when 15dPGJ2 and TGZ were used in the presence of GCS and/or a long-acting β-adrenoceptor agonists [62], supporting potential benefit when these agents are used in combination.
In a more recent study, TGZ inhibited IL-1β-induced release of IL-6 and VEGF, TNFα-induced release of eotaxin and RANTES, and IL-4-induced release of eotaxin, while RGZ also inhibited TNFα-stimulated release of RANTES. These anti-inflammatory effects were not prevented by the PPARγ antagonist GW 9662 or by PPARγ knockdown using short hairpin RNA [63].
Additional PPARγ-independent mechanisms have been considered. Although PPARγ ligands each caused the activation of AMPK, their effects on cytokine release were not prevented by AMPK inhibitors [63]. Since CGZ increased the IL-1β-induced expression of COX-2 [22], this potentially proinflammatory effect could also contribute to increased PGE2 levels to provide negative feedback to inhibit cytokine release [64]. However, induction of PGE2 synthesis was not a requirement for the anti-inflammatory effect of PPARγ ligands, since CGZ reduced GM-CSF and G-CSF in the presence of indomethacin [55]. Inhibition of NFκB has also been excluded, since CGZ did not regulate NFκB (p65) nuclear translocation in the absence or presence of IL-1β [22].
The qualitative importance of ASM-derived cytokines remains to be clearly established in vivo and in asthmatic subjects. Nevertheless, evidence of the diverse anti-inflammatory actions of PPARγ ligands in ASM, consistent with their reported actions in other cell types [41], supports their therapeutic potential for the treatment of asthma.
5.2. Regulation of ASM Proliferation
A key feature in airway remodeling in asthma is the increased ASM layer associated with increases in both size (hypertrophy) and number (hyperplasia, migration) of myocytes [65] with ASM cell migration also playing a potential role. To assess the potential efficacy of antiremodeling agents, ASM proliferation can be induced in vitro in response to the cocktail of mitogens present in serum, and to specific stimuli such as thrombin or fibroblast growth factor 2 (FGF2), known to be increased in the asthmatic airway [66, 67].
PPARγ ligands have now been shown to inhibit proliferation of human ASM in culture. The increase in [3H]-thymidine incorporation in response to serum was completely abolished by both CGZ and 15dPGJ2 [55], while RGZ and 15dPGJ2 significantly attenuated both FGF2 and thrombin-stimulated increase in ASM cell numbers [18], demonstrating that the antiproliferative effects are mitogen-independent. Unlike GCS, inhibition of proliferation was not associated with reduced cyclin D1 levels [18, 68]. Responses were mediated by both PPARγ-dependent and PPARγ-independent mechanisms, as the PPARγ antagonist GW9662 inhibited the antiproliferative effects of RGZ but not 15dPGJ2 [18], with cell cycle analysis suggesting that neither mediated ASM apoptosis [18]. Although CGZ and 15dPGJ2 had previously been reported to cause nuclear condensation, a characteristic morphological change associated with apoptosis [55], this single finding was not consistent with the known resistance of ASM to apoptosis [69].
Cultured ASM derived from asthmatic patients has been shown to proliferate faster than cells from nonasthmatic patients [70]. Since GCS can only inhibit the in vitro proliferation of ASM from subjects without asthma [68, 71], alternative therapeutic approaches are required to target this steroid-resistant mitogenic response. In a recent study, the effects of CGZ were assessed in cells from nonasthmatic and asthmatic patients cellular proliferation in response to serum by measuring bromodeoxyuridine uptake [50]. Further studies are required to explain why CGZ failed to inhibit serum-induced proliferation in either group [50], since this finding contradicts the previously reported antiproliferative effects of both CGZ [55] and RGZ [18]. CGZ did upregulate PPARγ expression in ASM cells derived from both asthmatic and nonasthmatic subjects, and in ASM from asthmatics in the presence of serum [50], however the functional significance of these changes and their potential impact on ASM in remodeled airways remain to be determined.
5.3. Regulation of Extracellular Matrix Production and Turnover
Airway remodeling in asthma is also characterized by alterations in the amount and composition of ECM proteins, including increases in collagen I and fibronectin deposition [72]. Subepithelial fibrosis is associated with increased transforming growth factor β (TGFβ), with this profibrotic cytokine present at relatively higher levels in BAL fluid from asthmatic subjects compared to healthy subjects [73]. Although fibroblasts are considered the major resident cells contributing to the increased collagen deposition in the asthmatic airway, ASM is also known to produce ECM proteins and to regulate their turnover by secreting matrix modifying enzymes.
In this context, it is important to consider that the ECM exists not only as a structural scaffold in the airways but as a partner in bidirectional interactions with ASM, influencing proliferation and cytokine release as well as contractility [74]. Since in vitro secretion of collagen and fibronectin from ASM derived from asthmatic patients is increased by GCS, and TGFβ-induced ECM protein synthesis is unaffected by GCS, this aspect of remodeling appears to be resistant to steroids [75] and alternative strategies to minimize the impact of the altered ECM on ASM function need to be identified.
Confirmation of the ability of PPARγ ligands to inhibit TGFβ-induced collagen synthesis from ASM would suggest that these agents have the capacity oppose proasthmatic changes associated with increased ASM-ECM interactions. To date, the effects of PPARγ ligands have only been assessed in human lung fibroblasts which express PPARγ, and respond to TGFβ treatment by differentiating into myofibroblasts expressing α smooth muscle actin (αSMA), and increasing their synthesis of fibrillar collagen I [26]. Both differentiation and collagen I secretion were abrogated by treatment with RGZ, CGZ, or 15dPGJ2. These antifibrotic effects of the PPARγ ligands were shown to be at least partially mediated by PPARγ receptor activation as inhibition was attenuated by transfection of TGFβ-treated fibroblasts with a dominant negative PPARγ receptor [26]. Similar antifibrotic properties have also been described for RGZ and CGZ in the regulation of epithelial-mesenchymal transition in alveolar epithelial cells [76].
An alternative way to regulate ASM-ECM interactions would be by regulating the activity of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs). Activation of PPARγ by RGZ or PGZ in human bronchial epithelial cells reduced TNFα-induced MMP-9 gelatinolytic activity via inhibition of NF-κB, but did not alter the expression of its endogenous inhibitor TIMP-1 [77]. These results suggest that limiting the expression of MMP-9 by PPARγ activation might have therapeutic potential in the treatment of chronic inflammatory diseases of the respiratory system. However, the effects of PPARγ ligands on ASM-derived MMPs and TIMPs in the asthma context have yet to be directly assessed.
5.4. Regulation of Angiogenesis
Significant increases in the number and size of blood vessels supplying the remodeled airway wall are seen in asthma [6, 78]. This expanded vascular compartment is likely to contribute to asthma symptoms through tissue swelling and amplification of inflammatory cell trafficking [79]. ASM has the potential to promote angiogenesis as cultured ASM has been shown to constitutively release factors such as VEGF, which can be further increased in response to inflammatory mediators such as IL-1β, TNFα, and TGFβ [80]. Of note, these proangiogenic responses have recently been shown to be further elevated in ASM from asthmatics [81].
Studies examining the effects of PPARγ ligands on this aspect of remodelling are lacking; however, conflicting reports show that the generation of VEGF from vascular smooth muscle cells is increased by CGZ and PGJ2 [82, 83], while TGZ has been shown to inhibit VEGF-induced angiogenic signaling in endothelial cells [84]. Further investigations are required to explore the potential of PPARγ ligands to regulate the contribution of ASM to angiogenesis.
5.5. Regulation of ASM Contraction
The increased contractile response of asthmatic airways which defines AHR is likely to be due to multiple factors (recently reviewed in [4]), including the presence of higher levels of contractile mediators and reduced levels of relaxant mediators. Critically, the increased ASM bulk displays alterations in contractile protein expression that favour contraction [85, 86]. In this context, it is of interest that RGZ and other PPARγ ligands can inhibit the increase in α-smooth muscle actin and calponin associated with both epithelial-mesenchymal transition of alveolar epithelial cells [76] and alveolar interstitial fibroblast-to-myofibroblast differentiation [87].
Increased excitation/contraction coupling may also occur through disruption of calcium homeostasis [88]. Indeed, increased contraction of ASM cells from asthmatic patients has been associated with downregulation in their expression and function of SERCA2 [89]. PPARγ ligands have recently been reported to increase SERCA expression and activity in pancreatic islet cells and platelets [40, 90], with PGZ inhibiting cytokine-induced increases in intracellular calcium by facilitating its reuptake into the SR [40]. In ASM, calcium plays a key role not only in enhancing ASM contractile function, but also in promoting cell proliferation, migration and the secretion of proinflammatory cytokines and chemokines [88]. It will therefore be of particular interest to determine if acute or chronic treatment with PPARγ ligands can also restore SERCA levels and activity in ASM to inhibit the diverse proasthmatic functions that could be driven by elevated intracellular calcium.
There is now evidence that acute treatment with PPARγ ligands may exert direct effects on ASM contractility. In a single study, RGZ has been reported to cause relaxation of mouse tracheal preparations precontracted with carbachol [91]. Since this response was evident within minutes and required μM concentrations, it was likely to be occurring independently of PPARγ activation. Relaxation to RGZ in the static organ bath setting was indomethacin-sensitive and was attributed to accumulation of the dilator prostanoid PGE2 through inhibition of its breakdown rather than an increase in PGE2 synthesis. This interpretation is consistent with the previously reported finding that RGZ can inhibit its metabolism by 15-hydroxyprostaglandin dehydrogenase [32].
Further studies are required to explore acute dilator responses to RGZ and other PPARγ ligands, to compare their efficacy with β 2-adrenoceptor agonists in current clinical use for the relief of asthma symptoms and to test their actions in the disease context when ASM responsiveness is altered.
6. In Vivo Regulation of ASM Function by PPARγ Ligands
6.1. PPARγ Ligands Have Efficacy in Rodent Models of Allergic and Nicotine-Induced Airways Disease
The reported effects of PPARγ ligands on ASM functions in vitro, namely inhibition of proliferation and production of cytokines from human ASM cells as well as regulation of contractile protein expression and direct relaxation intracheal preparations, has provided an impetus for considering their effects in animal models of airways disease, using perinatal exposure to maternal nicotine or chronic ovalbumin (OVA) challenge to trigger asthma-like changes in the airways.
It is well known that cigarette smoking during pregnancy increases the incidence and severity of childhood asthma, and has been associated with increased generation of contractile myofibroblasts in the developing lung (reviewed in [9]). A recent study of newborn rats following in utero nicotine exposure has revealed that increases in airway resistance under basal conditions and reactivity to acetylcholine both in vivo and in vitro are sensitive to RGZ treatment [48].
Chronic allergen challenge models mimic key features of asthma including inflammatory cell infiltration predominantly not only by eosinophils, but also by neutrophils and lymphocytes [92]. Increased inflammatory Th2 cytokines and chemotactic factors can also be detected in bronchoalveolar lavage (BAL) fluid [27, 93, 94]. Airway wall remodeling, with variable changes in goblet cell hyperplasia, thickening of the ASM layer and increased collagen deposition in the lamina propria is also evident [25, 95]. A common feature is the development of AHR to methacholine (MCh), demonstrated both with noninvasive, but now largely discredited [96] measurement of the heuristic variable enhanced pause (Penh) in conscious mice [25, 27, 94, 95] and by measurements of increased airways resistance using invasive plethysmography in anesthetized mice [47, 97, 98].
The efficacy of PPARγ ligands in these OVA challenge models in mice has been confirmed in multiple studies, assessing the effect of chronic treatment with glitazones or other PPARγ selective agonists such as GI 262570, administered either by inhaled, oral or intraperitoneal (i.p.) routes [25, 27, 47, 92–95, 97–100] or the response to transient overexpression of PPARγ via adenoviral delivery (Table 1) [27, 47, 93]. However, since the overall changes in phenotype in these models can vary with the type of OVA used [101], the duration of the challenge protocol and the species of mice in which the model is applied [102], the reported effects of pharmacological intervention with different PPARγ ligands administered by various routes must be considered and interpreted in context. Nevertheless, these models provide compelling evidence that PPARγ ligands can regulate inflammatory cell infiltration, BAL cytokines, airway remodeling particularly ASM thickening and fibrosis, and altered reactivity to MCh.
Table 1.
Effects of PPARγ ligands in mouse OVA models of allergic airways disease.
| Strain | Ligand | Inflammation | Remodeling | AHR | Mechanism | References |
|---|---|---|---|---|---|---|
| Balb/C | CGZ | ↓ IL-4, IL-5, IL-6, IL-13 ↓ eosinophils |
↓ mucus ↓ collagen ↓ wall thickness |
↓ | ↓ GATA-3 | [25, 94] |
| Balb/C | CGZ | ↓ IFNγ, IL-2, IL-4 ↓ eosinophils |
↓ mucus | N.D. | [100] | |
| Balb/C | CGZ | ↓ eosinophils | ↓ ASM thickness | ↓ | [95] | |
| Balb/C | CGZ RGZ | ↓ eosinophils | N.D. | ↑ IL-10 | [99] | |
| Balb/C | RGZ PGZ | ↓ IL-4, IL-5, IL-13, ECP ↓ eosinophils |
↓ | ↑ PTEN ↑ IL-10 |
[27, 47] | |
| Balb/C | GI 262570 | ↓ eosinophils | N.D. | [92] | ||
|
| ||||||
| C57Bl/6 | RGZ PGZ | ↓ IL-4, IL-5, IL-13, ↓ VEGF, eotaxin, RANTES ↓ eosinophils |
↓ | ↑ IL-17 via NFκB | [93, 97] | |
| C57Bl/6 | RGZ | ⇔ eosinophils | ⇔ mucus ⇔ wall thickness |
↓ | [98] | |
ASM: airway smooth muscle; CGZ: ciglitazone; ECP: eosinophil cationic protein; IL: interleukin; N.D.: not determined; PGZ: pioglitazone; PTEN: phosphatase and tensin homologue deleted on chromosome ten; RANTES: regulated upon activation, normal T-cell expressed, and secreted; RGZ: rosiglitazone; VEGF: vascular endothelial growth factor.
6.2. Regulation of Airway Inflammation
Assessment of airway inflammation has consistently shown that RGZ or CGZ treatment attenuated the increase in total and eosinophil cell numbers in BAL fluid in OVA-treated C57Bl/6 or Balb/C mice in a PPARγ-dependent manner (Table 1) [27, 93, 94]. Similar results were observed with GI 262570 administration to Balb/C mice, where eosinophil and lymphocytes, but not neutrophils, were reduced [92]. Although RGZ reduced eosinophilic airway inflammation when administered to Balb/C mice by oral gavage [99], it was ineffective in C57Bl/6 mice administered i.p. [98]. The reason for this discrepancy is therefore more likely to be due to differences in challenge protocols and the administration methods (route, dose, and duration) of different compounds, rather than the mouse strain used.
Regulation of cytokine production in the lung has also been assessed. OVA-induced increases in IL-4, IL-5, IL-13, eosinophil cationic protein (ECP), and eotaxin in lung tissue and BAL fluid were inhibited by administration of RGZ, PGZ, or by PPARγ overexpression [27, 93]. Similar changes were seen with nebulized CGZ, although eotaxin levels were not affected [94], while oral CGZ has been shown to reduce IL-2, IL-4, and interferon γ (IFNγ) [100]. Since cytokine release from ASM is also inhibited by PPARγ ligands in vitro, it is likely that the glitazones can reduce the contribution of ASM-derived cytokines to the levels measured in this in vivo setting.
Several potential mechanisms have been proposed to explain the anti-inflammatory effects of PPARγ ligands in these models. Regulation of NFκB has been considered since PPARγ activation inhibits the function of the proinflammatory transcription factor in vitro [103, 104]. Treatment of OVA-sensitised mice with RGZ, PGZ, or AdPPARγ also reduced the nuclear translocation of NFκB in response to OVA, evidenced by inhibition of increases in NFκB p65 protein in lung extracts [93], suggesting a direct action of PPARγ ligands on NFκB. Inhibition of NFκB activity by PPARγ agonists has also been associated with decreased IL-17 protein and mRNA expression. Since the effects of RGZ or PGZ could be abrogated by coadministration of rIL-17, this implicates a novel mechanism whereby PPARγ agonists regulate NFκB activity by reducing IL-17 to limit inflammation [97].
NFκB-independent mechanisms are also likely to contribute to the anti-inflammatory effects of PPARγ ligands [92]. Alternative mechanisms include PPARγ-mediated inhibition of the increase in GATA-3 expression in response to OVA [94], reducing the local Th2 response elicited by this eosinophil-derived transcription factor. In addition, an increase in IL-10 in response to OVA, thought to occur as part of a negative feedback response to inhibit inflammation, could be further increased by RGZ, PGZ, or ad PPARγ [47]. Increased IL-10 levels could explain the reported reductions in IL-4 and IL-5 as well as the inhibition of eosinophilia, since IL-10 has been shown to downregulate IL-4 and IL-5 expression by Th2 cells and reduce eosinophil survival.
In a separate study, PPARγ expression was increased in response to OVA challenge and further enhanced by the administration of the either PPARγ agonists or AdPPARγ [27]. This was associated with an upregulation of phosphatase and tensin homologue deleted on chromosome ten (PTEN) PTEN expression, correlating with decreased PI3K activity as measured by a reduction in the phosphorylation of Akt. These findings demonstrate a protective role of PPARγ in the pathogenesis of the asthma phenotype through regulation of PTEN expression [27, 93].
6.3. Regulation of Airway Remodeling
In addition to their anti-inflammatory actions in these mouse models, PPARγ ligands have also been shown to inhibit key aspects of airway remodeling, notably fibrosis, mucus production, and thickening of the ASM layer (Table 1).
Inhaled CGZ has been shown to reduce OVA-induced increases in both collagen deposition and basement membrane thickening [25]. This was associated with reduced levels of the profibrotic cytokine TGFβ [25]. Although inhaled CGZ has also been shown to decrease mucus production, based on the intensity and area of epithelial staining [25], there were no detectable effects of i.p. RGZ on goblet cell number or other aspects of airway remodeling [98], suggesting that high local concentrations may be required.
Consistent with the reported antiproliferative effects of PPARγ ligands on human ASM in vitro [18, 50, 55], intranasal administration of CGZ has been shown to reduce not only eosinophilic inflammation, but also to inhibit the thickening of the ASM layer following allergen challenge [95]. This effect appeared to be independent of regulation of TGFβ or VEGF levels, as the increased BAL levels of these potential mitogens were not reduced with CGZ treatment [95]. It would be of interest to measure endogenous factors that could contribute to ASM proliferation in this setting.
6.4. Regulation of Airway Hyperresponsiveness
Studies demonstrating the inhibitory effects of PPARγ ligands on AHR are consistent with the numerous in vitro findings suggesting a role for PPARγ ligands in the regulation of ASM function in asthma (Table 1). The development of AHR to cholinergic agonists subsequent to in utero nicotine exposure or in vivo allergen exposure can be alleviated by chronic treatment with PPARγ ligands, measured either indirectly using Penh [25, 27, 94, 95] or by assessing changes in airway resistance [47, 48, 97, 98].
A recent study has reported that coadministration of RGZ prevented the changes in lung function in rat offspring induced by perinatal nicotine exposure. Inhibition of the development of AHR as measured in vivo and in isolated tracheal preparations was attributed to the ability of RGZ to decrease the lipofibroblast-to-fibroblast transdifferentiation induced by nicotine, minimizing the increased expression and function of mesenchymal markers of contractility [48].
Chronic allergen studies demonstrating PPARγ ligand efficacy suggest a PPARγ-dependent mechanism in opposing ovalbumin-induced AHR, since the inhibitory effects could be mimicked by transient overexpression of PPARγ via adenoviral delivery or prevented by co-treatment with GW9662 [25, 27, 47, 93, 94, 97, 98]. It would be reasonable to attribute this reduction in AHR to the inhibition of inflammation and airway remodeling mediated by the PPARγ ligands used. However, RGZ also reduced AHR measured by invasive plethysmography in OVA-challenged C57Bl/6 mice without detectable effects on markers of inflammation or remodeling [98]. This result suggests that it is possible that PPARγ ligands may also exert a direct effect on ASM contractile function in vivo.
In this context, it is notable that chronic treatment with PPARγ ligands may not only inhibit the development of AHR, but also protect airway dilator responses. In a guinea pig model of in vivo homologous desensitization to salbutamol, chronic treatment with RGZ mitigated AHR to carbachol, preserved salbutamol relaxant activity, and partially restored β 2-adrenoceptor binding sites in tracheal tissues ex vivo [105]. The potential for PPARγ ligands to maintain dilator sensitivity and reverse β 2-adrenoceptor desensitization is of particular interest since GCS can prevent cytokine-induced desensitization [106], but cannot restore sensitivity once tolerance to β 2-adrenoceptor agonists has developed [107].
7. Potential Clinical Benefit of PPARγ Ligands in Asthma
Although several members of the glitazone class of drugs have been used for type 2 diabetes, PGZ is the only PPARγ agonist in current clinical use for this condition, with its potential as a treatment to reduce inflammation in rheumatoid arthritis also being assessed [108]. TGZ was the first glitazone to be marketed for diabetes, but was withdrawn because of serious hepatotoxicity in some patients [109], while RGZ has also recently been withdrawn because of potential cardiovascular risks [110].
There is currently only limited data on the efficacy of glitazones in the treatment of respiratory diseases. Further studies characterizing the effects of PPARγ ligands on lung development as well as nicotine-induced changes in lung function are required to determine whether these agents may provide a new therapeutic approach to minimize, or even reverse, the adverse impacts of maternal smoking that contribute to the development of paediatric asthma.
An isolated report described the effects of PGZ in two case subjects with both diabetes and established asthma [111]. One patient reported reduced wheezing when taking PGZ in addition to his asthma preventer medication, with deterioration of symptoms when PGZ was discontinued. In another, concurrent treatment with the sulfonylurea glibenclamide and PGZ effectively reduced the patient's blood glucose levels and improved pulmonary function test results, increasing both forced vital capacity and force expiratory volume in one second (FEV1).
More recently, a small single-centre trial has been conducted, assessing RGZ in a double-blind, randomised, placebo-controlled, two-period crossover study in the inhaled allergen challenge model of asthma [112]. 32 steroid naïve subjects completed the study, receiving RGZ (4 mg) and placebo twice daily for 28 days in random order. The late asthmatic reaction (LAR) change from postsaline FEV1 from 4–10 hrs after allergen on day 28 was attenuated by 15% compared to the response during placebo-treatment, suggesting an inhibitory effect of RGZ on activated eosinophil recruitment. This reduction was accompanied by trends in several other markers of efficacy and anti-inflammatory activity (e.g., IL-4, IL-6, IL-13). In light of these modest changes, the authors suggested that PPARγ agonist monotherapy is unlikely to represent a clinically useful intervention, at least in the context of relatively mild asthma.
More positive results were obtained in another recently completed exploratory clinical trial, which compared the effects of oral RGZ (8 mg) with inhaled beclometasone in a group of forty-six smokers with asthma, a group that is generally unresponsive to conventional GCS treatment [113]. In measurements taken after two and four weeks, RGZ did not significantly reduce asthma symptoms as determined by the Asthma Control Questionnaire (ACQ) scores and only produced a borderline reduction in sputum IL-8 levels compared to beclometasone-treated patients [113]. However, the patients receiving RGZ experienced significant improvements in FEV1 and forced expiratory flow over beclometasone-treated patients, which may reflect an effect of RGZ to reduce small airway obstruction.
These promising findings support the assessment of the effectiveness of long-term treatment of RGZ in a larger treatment group. The use of substantially higher oral doses may not be associated with a positive benefit/risk profile in asthma since PPARγ agonists are associated with dose-related adverse effects such as weight gain (probably secondary to fluid retention). This suggests that a preferable alternative strategy would be to assess responses to both acute and chronic inhalation of PPARγ agonists. This route of administration would potentially minimize the reported adverse cardiovascular effects that have limited the systemic use of RGZ in diabetes [110]. In addition, it would achieve the higher local airway concentrations that may be required to exert direct effects on ASM contractile function to elicit acute bronchodilation as reported in mouse trachea, and chronic effects to regulate airway inflammation, remodeling, and the development of AHR.
8. Summary
An accumulating body of evidence supports the use of PPARγ ligands for the targeting of PPARγ receptors and other PPARγ-independent mechanisms in ASM for the treatment of inflammatory lung diseases (Figure 2) [9, 41, 114]. In vitro treatment inhibits proliferation of human ASM via PPARγ [18, 55] and also inhibits cytokine release from these cells [55, 62, 63]. Chronic in vivo treatment inhibits the development of nicotine-induced AHR in rat airways [48] as well as OVA-induced increases in ASM mass in mouse airways [95], part of a suite of actions involving inhibition of airway inflammation, remodeling and the development of AHR. PPARγ ligands may also protect dilator responses since they can preserve β 2-adrenoceptor expression and function in a guinea pig model of homologous desensitization to albuterol [105]. The potential for direct bronchodilator actions is supported by the demonstration of acute PPARγ-independent relaxation in mouse trachea [91]. Although clinical trial results are limited, evidence of improved lung function in a difficult-to-treat cohort of smokers with asthma [113] supports further investigation of the potential for PPARγ agonists to target ASM proliferative, inflammatory and contractile functions and their contributions to impaired dilator responses and the consequences of AHR in asthma.
Figure 2.
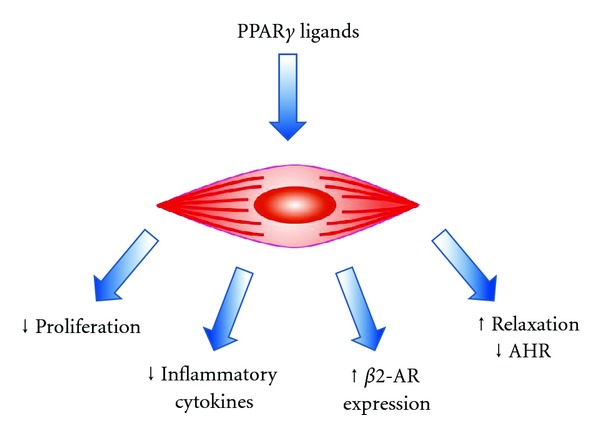
PPARγ ligands regulate noncontractile and contractile functions of airway smooth muscle.
Acknowledgments
This work was supported by the National Health and Medical Research Council (Grant 509239) and ANZ Medical Research and Technology in Victoria Fund.
Abbreviations
- 15dPGJ2:
15-Deoxy-Δ12,14-prostaglandin-J2
- ACQ:
Asthma control questionnaire
- AdPPARγ:
Adenoviral PPARγ
- AF-1:
Activator function-1
- AHR:
Airway hyperresponsiveness
- AMPK:
Adenosine monophosphate-activated protein kinase
- AP-1:
Activator protein-1
- AR:
Adrenoceptor
- ASM:
Airway smoothmuscle
- αSMA:
α smooth muscle actin
- BAL:
Bronchoalveolar lavage
- C/EBP:
CAAT/enhancerbinding protein
- CDDO:
2-Cyano-3,12-dioxoolean-1,9-dien-28-oic acid
- CGZ:
Ciglitazone
- DBD:
DNA binding domain
- ECM:
Extracellular matrix
- ECP:
Eosinophil cationic protein
- FFA:
Free fatty acid
- FGF2:
Fibroblast growth factor
- GCS:
Glucocorticoid
- G-CSF:
Granulocyte-colony stimulating factor
- GM-CSF:
Granulocyte/macrophage-colony-stimulating factor
- GR:
Glucocorticoid receptor
- HETE:
Hydroxyeicosatetraenoic acid
- HODE:
Hydroxyoctadecadienoic acid
- IFNγ:
Interferonγ
- IBκ:
NFκB inhibitory cofactor
- IKK:
IκB kinase
- IL-:
Interleukin
- i.p.:
Intraperitoneal
- LBD:
Ligand binding domain
- MAPK:
Mitogen-associated protein kinase
- MCh:
Methacholine
- MMP:
Matrix metalloproteinases
- NFκB:
Nuclear factor κB
- NHR:
Nuclear hormone receptor
- OVA:
Ovalbumin
- Penh:
Enhanced pause
- PG:
Prostaglandin
- PGZ:
Pioglitazone
- PI3K:
Phosphoinositide-3-kinase
- PPARγ:
Peroxisome proliferator activated receptor
- PPRE:
PPAR response element
- PTEN:
Phosphatase and tensin homologue deleted on chromosome ten
- RANTES:
Regulated on activation, normal T cells expressed and secreted
- RGZ:
Rosiglitazone
- RXR:
Retinoid X receptor
- SERCA:
Sarco/endoplasmic reticulum Ca2+ ATPase
- STAT:
Signal transducers and activators of transcription
- SUMO:
Small Ubiquitin-like Modifier
- TGFβ:
Transforming growth factor β
- TGZ:
Troglitazone
- TNFα:
Tumour necrosis factor α
- VEGF:
Vascular endothelial growth factor.
References
- 1.WHO. Global Surveillance, Prevention and Control of Chronic Respiratory Diseases: A Comprehensive Approach. Geneva, Switzerland: WHO; 2007. [Google Scholar]
- 2.Woolcock AJ, Dusser D, Fajac I. Severity of chronic asthma. Thorax. 1998;53(6):442–444. doi: 10.1136/thx.53.6.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lazaar AL, Panettieri RA., Jr. Airway smooth muscle: a modulator of airway remodeling in asthma. Journal of Allergy and Clinical Immunology. 2005;116(3):488–496. doi: 10.1016/j.jaci.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 4.Ozier A, Allard B, Bara I, et al. The pivotal role of airway smooth muscle in asthma pathophysiology. Journal of Allergy. 2011;2011:20 pages. doi: 10.1155/2011/742710.742710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomson NC, Spears M. The influence of smoking on the treatment response in patients with asthma. Current Opinion in Allergy and Clinical Immunology. 2005;5(1):57–63. doi: 10.1097/00130832-200502000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Wilson JW, Hii S. The importance of the airway microvasculature in asthma. Current Opinion in Allergy and Clinical Immunology. 2006;6(1):51–55. doi: 10.1097/01.all.0000200505.54425.47. [DOI] [PubMed] [Google Scholar]
- 7.Camoretti-Mercado B. Targeting the airway smooth muscle for asthma treatment. Translational Research. 2009;154(4):165–174. doi: 10.1016/j.trsl.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuyderduyn S, Sukkar MB, Fust A, Dhaliwal S, Burgess JK. Treating asthma means treating airway smooth muscle cells. European Respiratory Journal. 2008;32(2):265–274. doi: 10.1183/09031936.00051407. [DOI] [PubMed] [Google Scholar]
- 9.Rehan VK, Asotra K, Torday JS. The effects of smoking on the developing lung: insights from a biologic model for lung development, homeostasis, and repair. Lung. 2009;187(5):281–289. doi: 10.1007/s00408-009-9158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benayoun L, Letuve S, Druilhe A, et al. Regulation of peroxisome proliferator-activated receptor γ expression in human asthmatic airways: relationship with proliferation, apoptosis, and airway remodeling. American Journal of Respiratory and Critical Care Medicine. 2001;164(8):1487–1494. doi: 10.1164/ajrccm.164.8.2101070. [DOI] [PubMed] [Google Scholar]
- 11.Werman A, Hollenberg A, Solanes G, Bjørbæk C, Vidal-Puig AJ, Flier JS. Ligand-independent activation domain in the N terminus of peroxisome proliferator-activated receptor γ (PPARγ). Differential activity of PPARγ1 and -2 isoforms and influence of insulin. The Journal of Biological Chemistry. 1997;272(32):20230–20235. doi: 10.1074/jbc.272.32.20230. [DOI] [PubMed] [Google Scholar]
- 12.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor γ is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. The Journal of Biological Chemistry. 1997;272(8):5128–5132. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- 13.Nolte RT, Wisely GB, Westin S, et al. Ligand binding and co-activator assembly of the peroxisome proliferator- activated receptor-γ . Nature. 1998;395(6698):137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 14.Berger J, Moller DE. The mechanisms of action of PPARs. Annual Review of Medicine. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 15.Heppner TJ, Bonev AD, Eckman DM, Gomez MF, Petkov GV, Nelson MT. Novel PPARγ agonists GI 262570, GW 7845, GW 1929, and pioglitazone decrease calcium channel function and myogenic tone in rat mesenteric arteries. Pharmacology. 2005;73(1):15–22. doi: 10.1159/000081070. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Porter WW, Suh N, et al. A synthetic triterpenoid, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO), is a ligand for the peroxisome proliferator-activated receptor γ . Molecular Endocrinology. 2000;14(10):1550–1556. doi: 10.1210/mend.14.10.0545. [DOI] [PubMed] [Google Scholar]
- 17.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-deoxy-Δ12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ . Cell. 1995;83(5):803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 18.Ward JE, Gould H, Harris T, Bonacci JV, Stewart AG. PPARγ ligands, 15-deoxy-Δ12,14-prostaglandin J2 and rosiglitazone regulate human cultured airway smooth muscle proliferation through different mechanisms. British Journal of Pharmacology. 2004;141(3):517–525. doi: 10.1038/sj.bjp.0705630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. Journal of Medicinal Chemistry. 2000;43(4):527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 20.Moraes LA, Piqueras L, Bishop-Bailey D. Peroxisome proliferator-activated receptors and inflammation. Pharmacology and Therapeutics. 2006;110(3):371–385. doi: 10.1016/j.pharmthera.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 21.Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARs) and their effects on lipid metabolism and adipocyte differentiation. Biochimica et Biophysica Acta. 1996;1302(2):93–109. doi: 10.1016/0005-2760(96)00066-5. [DOI] [PubMed] [Google Scholar]
- 22.Pang L, Nie M, Corbett L, Knox AJ. Cyclooxygenase-2 expression by nonsteroidal anti-inflammatory drugs in human airway smooth muscle cells: role of peroxisome proliferator-activated receptors. Journal of Immunology. 2003;170(2):1043–1051. doi: 10.4049/jimmunol.170.2.1043. [DOI] [PubMed] [Google Scholar]
- 23.Ghisletti S, Huang W, Ogawa S, et al. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARγ . Molecular Cell. 2007;25(1):57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leesnitzer LM, Parks DJ, Bledsoe RK, et al. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 2002;41(21):6640–6650. doi: 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- 25.Honda K, Marquillies P, Capron M, Dombrowicz D. Peroxisome proliferator-activated receptor γ is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. Journal of Allergy and Clinical Immunology. 2004;113(5):882–888. doi: 10.1016/j.jaci.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 26.Burgess HA, Daugherty LE, Thatcher TH, et al. PPARγ agonists inhibit TGF-β induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. American Journal of Physiology. 2005;288(6):L1146–L1153. doi: 10.1152/ajplung.00383.2004. [DOI] [PubMed] [Google Scholar]
- 27.Lee KS, Park SJ, Hwang PH, et al. PPAR-gamma modulates allergic inflammation through up-regulation of PTEN. FASEB Journal. 2005;19(8):1033–1035. doi: 10.1096/fj.04-3309fje. [DOI] [PubMed] [Google Scholar]
- 28.Smith NJ, Stoddart LA, Devine NM, Jenkins L, Milligan G. The action and mode of binding of thiazolidinedione ligands at free fatty acid receptor 1. The Journal of Biological Chemistry. 2009;284(26):17527–17539. doi: 10.1074/jbc.M109.012849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ialenti A, Grassia G, Di Meglio P, Maffia P, Di Rosa M, Ianaro A. Mechanism of the anti-inflammatory effect of thiazolidinediones: relationship with the glucocorticoid pathway. Molecular Pharmacology. 2005;67(5):1620–1628. doi: 10.1124/mol.104.004895. [DOI] [PubMed] [Google Scholar]
- 30.Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflammation Research. 2000;49(10):497–505. doi: 10.1007/s000110050622. [DOI] [PubMed] [Google Scholar]
- 31.Straus DS, Pascual G, Li M, et al. 15-deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho H, Tai HH. Thiazolidinediones as a novel class of NAD+-dependent 15-hydroxyprostaglandin dehydrogenase inhibitors. Archives of Biochemistry and Biophysics. 2002;405(2):247–251. doi: 10.1016/s0003-9861(02)00352-1. [DOI] [PubMed] [Google Scholar]
- 33.Ferguson HE, Thatcher TH, Olsen KC, et al. Peroxisome proliferator-activated receptor-γ ligands induce heme oxygenase-1 in lung fibroblasts by a PPARγ-independent, glutathione-dependent mechanism. American Journal of Physiology. 2009;297(5):L912–L919. doi: 10.1152/ajplung.00148.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris SG, Smith RS, Phipps RP. 15-Deoxy-Δ12,14-PGJ2 induces IL-8 production in human T cells by a mitogen-activated protein kinase pathway. Journal of Immunology. 2002;168(3):1372–1379. doi: 10.4049/jimmunol.168.3.1372. [DOI] [PubMed] [Google Scholar]
- 35.Patel KM, Wright KL, Whittaker P, Chakravarty P, Watson ML, Ward SG. Differential modulation of COX-2 expression in A549 airway epithelial cells by structurally distinct PPARγ agonists: evidence for disparate functional effects which are independent of NF-κB and PPARγ . Cellular Signalling. 2005;17(9):1098–1110. doi: 10.1016/j.cellsig.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 36.LeBrasseur NK, Kelly M, Tsao TS, et al. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. American Journal of Physiology. 2006;291(1):E175–E181. doi: 10.1152/ajpendo.00453.2005. [DOI] [PubMed] [Google Scholar]
- 37.Asano M, Nakajima T, Iwasawa K, et al. Troglitazone and pioglitazone attenuate agonist-dependent Ca2+ mobilization and cell proliferation in vascular smooth muscle cells. British Journal of Pharmacology. 1999;128(3):673–683. doi: 10.1038/sj.bjp.0702818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan YH, Chen H, Natarajan A, et al. Structure-activity requirements for the antiproliferative effect of troglitazone derivatives mediated by depletion of intracellular calcium. Bioorganic and Medicinal Chemistry Letters. 2004;14(10):2547–2550. doi: 10.1016/j.bmcl.2004.02.087. [DOI] [PubMed] [Google Scholar]
- 39.Caddy J, Singh N, Atkin L, et al. Rosiglitazone transiently disturbs calcium homeostasis in monocytic cells. Biochemical and Biophysical Research Communications. 2008;366(1):149–155. doi: 10.1016/j.bbrc.2007.11.095. [DOI] [PubMed] [Google Scholar]
- 40.Kono T, Ahn G, Moss DR, et al. PPAR-γ activation restores pancreatic islet SERCA2 levels and prevents β-cell dysfunction under conditions of hyperglycemic and cytokine stress. Molecular Endocrinology. 2012;26(2):257–271. doi: 10.1210/me.2011-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward JE, Tan X. Peroxisome proliferator activated receptor ligands as regulators of airway inflammation and remodelling in chronic lung disease. PPAR Research. 2007;2007 doi: 10.1155/2007/14983.14983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang JT, Welch JS, Ricote M, et al. Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400(6742):378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- 43.Wang ACC, Dai X, Luu B, Conrad DJ. Peroxisome proliferator-activated receptor-γ regulates airway epithelial cell activation. American Journal of Respiratory Cell and Molecular Biology. 2001;24(6):688–693. doi: 10.1165/ajrcmb.24.6.4376. [DOI] [PubMed] [Google Scholar]
- 44.Chinetti G, Griglio S, Antonucci M, et al. Activation of proliferator-activated receptors α and γ induces apoptosis of human monocyte-derived macrophages. The Journal of Biological Chemistry. 1998;273(40):25573–25580. doi: 10.1074/jbc.273.40.25573. [DOI] [PubMed] [Google Scholar]
- 45.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998;391(6662):79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 46.Sugiyama H, Nonaka T, Kishimoto T, Komoriya K, Tsuji K, Nakahata T. Peroxisome proliferator-activated receptors are expressed in human cultured mast cells: a possible role of these receptors in negative regulation of mast cell activation. European Journal of Immunology. 2000;30(12):3363–3370. doi: 10.1002/1521-4141(2000012)30:12<3363::AID-IMMU3363>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 47.Kim SR, Lee KS, Park HS, et al. Involvement of IL-10 in peroxisome proliferator-activated receptor γ-mediated anti-inflammatory response in asthma. Molecular Pharmacology. 2005;68(6):1568–1575. doi: 10.1124/mol.105.017160. [DOI] [PubMed] [Google Scholar]
- 48.Liu J, Sakurai R, O’Roark EM, Kenyon NJ, Torday JS, Rehan VK. PPARγ agonist rosiglitazone prevents perinatal nicotine exposure-induced asthma in rat offspring. American Journal of Physiology. 2011;300(5):L710–L717. doi: 10.1152/ajplung.00337.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rehan VK, Wang Y, Sugano S, et al. In utero nicotine exposure alters fetal rat lung alveolar type II cell proliferation, differentiation, and metabolism. American Journal of Physiology. 2007;292(1):L323–L333. doi: 10.1152/ajplung.00071.2006. [DOI] [PubMed] [Google Scholar]
- 50.Lau JY, Oliver BG, Moir LM, Black JL, Burgess JK. Differential expression of peroxisome proliferator activated receptor γ and cyclin D1 does not affect proliferation of asthma-and non-asthma-derived airway smooth muscle cells. Respirology. 2010;15(2):303–312. doi: 10.1111/j.1440-1843.2009.01683.x. [DOI] [PubMed] [Google Scholar]
- 51.Shore SA, Moore PE. Effects of cytokines on contractile and dilator responses of airway smooth muscle. Clinical and Experimental Pharmacology and Physiology. 2002;29(10):859–866. doi: 10.1046/j.1440-1681.2002.03756.x. [DOI] [PubMed] [Google Scholar]
- 52.Tliba O, Panettieri RA. Noncontractile functions of airway smooth muscle cells in asthma. Annual Review of Physiology. 2009;71:509–535. doi: 10.1146/annurev.physiol.010908.163227. [DOI] [PubMed] [Google Scholar]
- 53.Panettieri RA, Jr., Kotlikoff MI, Gerthoffer WT, et al. Airway smooth muscle in bronchial tone, inflammation, and remodeling: basic knowledge to clinical relevance. American Journal of Respiratory and Critical Care Medicine. 2008;177(3):248–252. doi: 10.1164/rccm.200708-1217PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saunders MA, Mitchell JA, Seldon PM, et al. Release of granulocyte-macrophage colony stimulating factor by human cultured airway smooth muscle cells: suppression by dexamethasone. British Journal of Pharmacology. 1997;120(4):545–546. doi: 10.1038/sj.bjp.0700998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patel HJ, Belvisi MG, Bishop-Bailey D, Yacoub MH, Mitchell JA. Activation of peroxisome proliferator-activated receptors in human airway smooth muscle cells has a superior anti-inflammatory profile to corticosteroids: relevance for chronic obstructive pulmonary disease therapy. Journal of Immunology. 2003;170(5):2663–2669. doi: 10.4049/jimmunol.170.5.2663. [DOI] [PubMed] [Google Scholar]
- 56.Chung KF, Patel HJ, Fadlon EJ, et al. Induction of eotaxin expression and release from human airway smooth muscle cells by IL-1β and TNFα: effects of IL-10 and corticosteroids. British Journal of Pharmacology. 1999;127(5):1145–1150. doi: 10.1038/sj.bjp.0702660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hallsworth MP, Moir LM, Lai D, Hirst SJ. Inhibitors of mitogen-activated protein kinases differentially regulate eosinophil-activating cytokine release from human airway smooth muscle. American Journal of Respiratory and Critical Care Medicine. 2001;164(4):688–697. doi: 10.1164/ajrccm.164.4.2011004. [DOI] [PubMed] [Google Scholar]
- 58.McKay S, Hirst SJ, Bertrand-De Haas M, et al. Tumor necrosis factor-α enhances mRNA expression and secretion of interleukin-6 in cultured human airway smooth muscle cells. American Journal of Respiratory Cell and Molecular Biology. 2000;23(1):103–111. doi: 10.1165/ajrcmb.23.1.3765. [DOI] [PubMed] [Google Scholar]
- 59.Watson ML, Grix SP, Jordan NJ, et al. Interleukin 8 and monocyte chemoattractant protein 1 production by cultured human airway smooth muscle cells. Cytokine. 1998;10(5):346–352. doi: 10.1006/cyto.1997.0350. [DOI] [PubMed] [Google Scholar]
- 60.Knox AJ, Corbett L, Stocks J, Holland E, Zhu YM, Pang L. Human airway smooth muscle cells secrete vascular endothelial growth factor: up-regulation by bradykinin via a protein kinase C and prostanoid-dependent mechanism. FASEB Journal. 2001;15(13):2480–2488. doi: 10.1096/fj.01-0256com. [DOI] [PubMed] [Google Scholar]
- 61.Amrani Y, Panettieri RA., Jr. Cytokines induce airway smooth muscle cell hyperresponsiveness to contractile agonists. Thorax. 1998;53(8):713–716. doi: 10.1136/thx.53.8.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nie M, Corbett L, Knox AJ, Pang L. Differential regulation of chemokine expression by peroxisome proliferator-activated receptor γ agonists: interactions with glucocorticoids and β 2-agonists. The Journal of Biological Chemistry. 2005;280(4):2550–2561. doi: 10.1074/jbc.M410616200. [DOI] [PubMed] [Google Scholar]
- 63.Zhu M, Flynt L, Ghosh S, et al. Anti-inflammatory effects of thiazolidinediones in human airway smooth muscle cells. American Journal of Respiratory Cell and Molecular Biology. 2011;45(1):111–119. doi: 10.1165/rcmb.2009-0445OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lazzeri N, Belvisi MG, Patel HJ, Yacoub MH, Chung KF, Mitchell JA. Effects of prostaglandin E2 and cAMP elevating drugs on GM-CSF release by cultured human airway smooth muscle cells relevance to asthma therapy. American Journal of Respiratory Cell and Molecular Biology. 2001;24(1):44–48. doi: 10.1165/ajrcmb.24.1.4027. [DOI] [PubMed] [Google Scholar]
- 65.Ebina M, Takahashi T, Chiba T, Motomiya M. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma: a 3-D morphometric study. American Review of Respiratory Disease. 1993;148(3):720–726. doi: 10.1164/ajrccm/148.3.720. [DOI] [PubMed] [Google Scholar]
- 66.Gabazza EC, Taguchi O, Tamaki S, et al. Thrombin in the airways of asthmatic patients. Lung. 1999;177(4):253–262. doi: 10.1007/pl00007645. [DOI] [PubMed] [Google Scholar]
- 67.Redington AE, Roche WR, Madden J, et al. Basic fibroblast growth factor in asthma: measurement in bronchoalveolar lavage fluid basally and following allergen challenge. Journal of Allergy and Clinical Immunology. 2001;107(2):384–387. doi: 10.1067/mai.2001.112268. [DOI] [PubMed] [Google Scholar]
- 68.Fernandes D, Guida E, Koutsoubos V, et al. Glucocorticoids inhibit proliferation, cyclin D1 expression, and retinoblastoma protein phosphorylation, but not activity of the extracellular-regulated kinases in human cultured airway smooth muscle. American Journal of Respiratory Cell and Molecular Biology. 1999;21(1):77–88. doi: 10.1165/ajrcmb.21.1.3396. [DOI] [PubMed] [Google Scholar]
- 69.Freyer AM, Johnson SR, Hall IP. Effects of growth factors and extracellular matrix on survival of human airway smooth muscle cells. American Journal of Respiratory Cell and Molecular Biology. 2001;25(5):569–576. doi: 10.1165/ajrcmb.25.5.4605. [DOI] [PubMed] [Google Scholar]
- 70.Johnson PRA, Roth M, Tamm M, et al. Airway smooth muscle cell proliferation is increased in asthma. American Journal of Respiratory and Critical Care Medicine. 2001;164(3):474–477. doi: 10.1164/ajrccm.164.3.2010109. [DOI] [PubMed] [Google Scholar]
- 71.Roth M, Johnson PRA, Borger P, et al. Dysfunctional interaction of C/EBPα and the glucocorticoid receptor in asthmatic bronchial smooth-muscle cells. The New England Journal of Medicine. 2004;351(6):560–574. doi: 10.1056/NEJMoa021660. [DOI] [PubMed] [Google Scholar]
- 72.Araujo BB, Dolhnikoff M, Silva LFF, et al. Extracellular matrix components and regulators in the airway smooth muscle in asthma. European Respiratory Journal. 2008;32(1):61–69. doi: 10.1183/09031936.00147807. [DOI] [PubMed] [Google Scholar]
- 73.Redington AE, Madden J, Frew AJ, et al. Transforming growth factor-β1 in asthma: measurement in bronchoalveolar lavage fluid. American Journal of Respiratory and Critical Care Medicine. 1997;156(2):642–647. doi: 10.1164/ajrccm.156.2.9605065. [DOI] [PubMed] [Google Scholar]
- 74.Ward JE, Hirst SJ. Airway smooth muscle regulation of matrix proteins and interactions with the extracellular matrix. Airway Smooth Muscle Biology and Pharmacology in Airways Disease. 2007:105–126. [Google Scholar]
- 75.Burgess JK, Oliver BGG, Poniris MH, et al. A phosphodiesterase 4 inhibitor inhibits matrix protein deposition in airways in vitro. Journal of Allergy and Clinical Immunology. 2006;118(3):649–657. doi: 10.1016/j.jaci.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 76.Tan X, Dagher H, Hutton CA, Bourke JE. Effects of PPARγ ligands on TGF-β1-induced epithelial-mesenchymal transition in alveolar epithelial cells. Respiratory Research. 2010;11, article 21 doi: 10.1186/1465-9921-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hetzel M, Walcher D, Grüb M, Bach H, Hombach V, Marx N. Inhibition of MMP-9 expression by PPARγ activators in human bronchial epithelial cells. Thorax. 2003;58(9):778–783. doi: 10.1136/thorax.58.9.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bai TR, Knight DA. Structural changes in the airways in asthma: observations and consequences. Clinical Science. 2005;108(6):463–477. doi: 10.1042/CS20040342. [DOI] [PubMed] [Google Scholar]
- 79.Bischof RJ, Bourke JE, Hirst SJ, Meeusen ENT, Snibson KJ, Van Der Velden J. Measurement and impact of remodeling in the lung: airway neovascularization in asthma. Proceedings of the American Thoracic Society. 2009;6(8):673–677. doi: 10.1513/pats.200907-064DP. [DOI] [PubMed] [Google Scholar]
- 80.Bailey SR, Boustany S, Burgess JK, et al. Airway vascular reactivity and vascularisation in human chronic airway disease. Pulmonary Pharmacology and Therapeutics. 2009;22(5):417–425. doi: 10.1016/j.pupt.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 81.Simcock DE, Kanabar V, Clarke GW, et al. Induction of angiogenesis by airway smooth muscle from patients with asthma. American Journal of Respiratory and Critical Care Medicine. 2008;178(5):460–468. doi: 10.1164/rccm.200707-1046OC. [DOI] [PubMed] [Google Scholar]
- 82.Jozkowicz A, Was H, Taha H, et al. 15d-PGJ2 upregulates synthesis of IL-8 in endothelial cells through induction of oxidative stress. Antioxidants and Redox Signaling. 2008;10(12):2035–2046. doi: 10.1089/ars.2008.2032. [DOI] [PubMed] [Google Scholar]
- 83.Yamakawa K, Hosoi M, Koyama H, et al. Peroxisome proliferator-activated receptor-γ agonists increase vascular endothelial growth factor expression in human vascular smooth muscle cells. Biochemical and Biophysical Research Communications. 2000;271(3):571–574. doi: 10.1006/bbrc.2000.2665. [DOI] [PubMed] [Google Scholar]
- 84.Byung CP, Thapa D, Jong SL, Park SY, Kim JA. Troglitazone inhibits vascular endothelial growth factor-induced angiogenic signaling via suppression of reactive oxygen species production and extracellular signal-regulated kinase phosphorylation in endothelial cells. Journal of Pharmacological Sciences. 2009;111(1):1–12. doi: 10.1254/jphs.08305fp. [DOI] [PubMed] [Google Scholar]
- 85.McVicker CG, Leung SY, Kanabar V, et al. Repeated allergen inhalation induces cytoskeletal remodeling in smooth muscle from rat bronchioles. American Journal of Respiratory Cell and Molecular Biology. 2007;36(6):721–727. doi: 10.1165/rcmb.2006-0409OC. [DOI] [PubMed] [Google Scholar]
- 86.Moir LM, Leung SY, Eynott PR, et al. Repeated allergen inhalation induces phenotypic modulation of smooth muscle in bronchioles of sensitized rats. American Journal of Physiology. 2003;284(1):L148–L159. doi: 10.1152/ajplung.00105.2002. [DOI] [PubMed] [Google Scholar]
- 87.Rehan VK, Sakurai R, Torday JS. Thirdhand smoke: a new dimension to the effects of cigarette smoke on the developing lung. American Journal of Physiology. 2011;301(1):1–8. doi: 10.1152/ajplung.00393.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mahn K, Ojo OO, Chadwick G, Aaronson PI, Ward JPT, Lee TH. Ca2+ homeostasis and structural and functional remodelling of airway smooth muscle in asthma. Thorax. 2010;65(6):547–552. doi: 10.1136/thx.2009.129296. [DOI] [PubMed] [Google Scholar]
- 89.Mahn K, Hirst SJ, Ying S, et al. Diminished sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) expression contributes to airway remodelling in bronchial asthma. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(26):10775–10780. doi: 10.1073/pnas.0902295106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Randriamboavonjy V, Pistrosch F, Bölck B, et al. Platelet sarcoplasmic endoplasmic reticulum Ca2+-ATPase and μ-calpain activity are altered in type 2 diabetes mellitus and restored by rosiglitazone. Circulation. 2008;117(1):52–60. doi: 10.1161/CIRCULATIONAHA.107.719807. [DOI] [PubMed] [Google Scholar]
- 91.Henry PJ, D’Aprile A, Self G, Hong T, Mann TS. Inhibitors of prostaglandin transport and metabolism augment protease-activated receptor-2-mediated increases in prostaglandin E2 levels and smooth muscle relaxation in mouse isolated trachea. Journal of Pharmacology and Experimental Therapeutics. 2005;314(3):995–1001. doi: 10.1124/jpet.105.086124. [DOI] [PubMed] [Google Scholar]
- 92.Trifilieff A, Bench A, Hanley M, Bayley D, Campbell E, Whittaker P. PPAR-α and -γ but not -δ agonists inhibit airway inflammation in a murine model of asthma: in vitro evidence for an NF-κB-independent effect. British Journal of Pharmacology. 2003;139(1):163–171. doi: 10.1038/sj.bjp.0705232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee KS, Kim SR, Park SJ, et al. Peroxisome proliferator activated receptor-γ modulates reactive oxygen species generation and activation of nuclear factor-κB and hypoxia-inducible factor 1α in allergic airway disease of mice. Journal of Allergy and Clinical Immunology. 2006;118(1):120–127. doi: 10.1016/j.jaci.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 94.Woerly G, Honda K, Loyens M, et al. Peroxisome proliferator-activated receptors α and γ down-regulate allergic inflammation and eosinophil activation. Journal of Experimental Medicine. 2003;198(3):411–421. doi: 10.1084/jem.20021384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rhee CK, Lee SY, Kang JY, et al. Effect of peroxisome proliferator-activated receptor-gamma on airway smooth muscle thickening in a murine model of chronic asthma. International Archives of Allergy and Immunology. 2009;148(4):289–296. doi: 10.1159/000170382. [DOI] [PubMed] [Google Scholar]
- 96.Bates J, Irvin C, Brusasco V, et al. The use and misuse of Penh in animal models of lung disease. American Journal of Respiratory Cell and Molecular Biology. 2004;31(3):373–374. doi: 10.1165/ajrcmb.31.3.1. [DOI] [PubMed] [Google Scholar]
- 97.Park SJ, Lee KS, Kim SR, et al. Peroxisome proliferator-activated receptor γ agonist down-regulates IL-17 expression in a murine model of allergic airway inflammation. Journal of Immunology. 2009;183(5):3259–3267. doi: 10.4049/jimmunol.0900231. [DOI] [PubMed] [Google Scholar]
- 98.Ward JE, Fernandes DJ, Taylor CC, Bonacci JV, Quan L, Stewart AG. The PPARγ ligand, rosiglitazone, reduces airways hyperresponsiveness in a murine model of allergen-induced inflammation. Pulmonary Pharmacology and Therapeutics. 2006;19(1):39–46. doi: 10.1016/j.pupt.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 99.Hammad H, De Heer HJ, Soullié T, et al. Activation of peroxisome proliferator-activated receptor-γ in dendritic cells inhibits the development of eosinophilic airway inflammation in a mouse model of asthma. American Journal of Pathology. 2004;164(1):263–271. doi: 10.1016/s0002-9440(10)63116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mueller C, Weaver V, Vanden Heuvel JP, August A, Cantorna MT. Peroxisome proliferator-activated receptor γ ligands attenuate immunological symptoms of experimental allergic asthma. Archives of Biochemistry and Biophysics. 2003;418(2):186–196. doi: 10.1016/j.abb.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 101.Kumar RK, Herbert C, Foster PS. The “ classical” ovalbumin challenge model of asthma in mice. Current Drug Targets. 2008;9(6):485–494. doi: 10.2174/138945008784533561. [DOI] [PubMed] [Google Scholar]
- 102.Kips JC, Anderson GP, Fredberg JJ, et al. Murine models of asthma. European Respiratory Journal. 2003;22:374–382. doi: 10.1183/09031936.03.00026403. [DOI] [PubMed] [Google Scholar]
- 103.Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. Journal of Endocrinology. 2001;169(3):453–459. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- 104.Chung SW, Kang BY, Kim SH, Cho D, Trinchieri G, Kim TS. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-γ and nuclear factor-κB. The Journal of Biological Chemistry. 2000;275(42):32681–32687. doi: 10.1074/jbc.M002577200. [DOI] [PubMed] [Google Scholar]
- 105.Fogli S, Pellegrini S, Adinolfi B, et al. Rosiglitazone reverses salbutamol-induced β 2-adrenoceptor tolerance in airway smooth muscle. British Journal of Pharmacology. 2011;162(2):378–391. doi: 10.1111/j.1476-5381.2010.01021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Barnes PJ. Scientific rationale for inhaled combination therapy with long-acting β 2-agonists and corticosteroids. European Respiratory Journal. 2002;19(1):182–191. doi: 10.1183/09031936.02.00283202. [DOI] [PubMed] [Google Scholar]
- 107.Cooper PR, Panettieri RA. Steroids completely reverse albuterol-induced β 2-adrenergic receptor tolerance in human small airways. Journal of Allergy and Clinical Immunology. 2008;122(4):734–740. doi: 10.1016/j.jaci.2008.07.040. [DOI] [PubMed] [Google Scholar]
- 108.Shahin D, Toraby EE, Abdel-Malek H, Boshra V, Elsamanoudy AZ, Shaheen D. Effect of peroxisome proliferator-activated receptor gamma agonist (pioglitazone) and methotrexate on disease activity in rheumatoid arthritis (experimental and clinical study) Clinical Medicine Insights. 2011;4:1–10. doi: 10.4137/CMAMD.S5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kohlroser J, Mathai J, Reichheld J, Banner BF, Bonkovsky HL. Hepatotoxicity due to troglitazone: report of two cases and review of adverse events reported to the United States food and drug administration. American Journal of Gastroenterology. 2000;95(1):272–276. doi: 10.1111/j.1572-0241.2000.01707.x. [DOI] [PubMed] [Google Scholar]
- 110.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. The New England Journal of Medicine. 2007;356(24):2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 111.Hashimoto Y, Nakahara K. Improvement of asthma after administration of pioglitazone. Diabetes Care. 2002;25(2):p. 401. doi: 10.2337/diacare.25.2.401. [DOI] [PubMed] [Google Scholar]
- 112.Richards DB, Bareille P, Lindo EL, Quinn D, Farrow SN. Treatment with a peroxisomal proliferator activated receptor gamma agonist has a modest effect in the allergen challenge model in asthma: a randomised controlled trial. Respiratory Medicine. 2010;104(5):668–674. doi: 10.1016/j.rmed.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 113.Spears M, Donnelly I, Jolly L, et al. Bronchodilatory effect of the PPAR-γ agonist rosiglitazone in smokers with asthma. Clinical Pharmacology and Therapeutics. 2009;86(1):49–53. doi: 10.1038/clpt.2009.41. [DOI] [PubMed] [Google Scholar]
- 114.Belvisi MG, Mitchell JA. Targeting PPAR receptors in the airway for the treatment of inflammatory lung disease. British Journal of Pharmacology. 2009;158(4):994–1003. doi: 10.1111/j.1476-5381.2009.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]