

Abstract
Galactose-1-phosphate uridylyltransferase (GALT) catalyses the conversion of galactose-1-phosphate to UDP-galactose, a key step in the galactose metabolism. Deficiency of GALT activity in humans caused by deleterious variations in the GALT gene can cause a potentially lethal disease called Classic Galactosemia. In this study, we selected 14 novel nucleotide sequence changes in the GALT genes found in galactosemic patients for expression analysis and molecular modeling. Several variants showed decreased levels of expression and decreased abundance in the soluble fraction of the Escherichia coli cell extracts, suggesting altered stability and solubility. Only six variant GALT enzymes had detectable enzymatic activities. Kinetic studies showed that their Vmax decreased significantly. To further characterize the variants at molecular level, we performed static and dynamic molecular modeling studies. Effects of variations on local and/or global structural features of the enzyme were anticipated for the majority of variants. In-depth studies with molecular dynamic simulations on selected variants predicted the alteration of the protein structure even when static models apparently did not highlight any perturbation. Overall, these studies offered new insights on the molecular properties of GALT enzyme, with the aim of correlating them with the clinical outcome.
Keywords: Galactosemia, GALT, Heterologous expression, Molecular modeling
Introduction
In all living cells, productive metabolism of galactose begins with the phosphorylation of galactose to galactose-1-phosphate (gal-1P) by galactokinase (GALK) (Cardini and Leloir, 1953), the first metabolic enzyme of the Leloir pathway of galactose metabolism (Leloir, 1951). In the presence of galactose-1-phosphate uridylyltransferase (GALT), gal-1P will react with uridine diphosphate-glucose (UDP-Glu) to form uridine diphosphate-galactose (UDP-Gal) and glucose-1-phosphate (glu-1P) (Kalckar et al., 1953). Although the reaction mechanism was revealed through intensive study of the Escherichia coli (E. coli) GALT enzyme, the highly conserved nature of the GALT enzymes of the ubiquitous Leloir pathway suggested that similar reaction scheme applies to GALT enzymes of other species, including humans. In E. coli, the GALT enzyme catalyzes the above-mentioned reaction through a double displacement mechanism (Arabshahi et al., 1986) by which the enzyme will bind to UDP-Glu, followed by the attack of the β–phosphate of UDP-Glu through His166 of the enzyme to form an uridylyl-enzyme intermediate and to release glu-1P. The double-displacement reaction completes when the uridylyl-enzyme intermediate binds gal-1P and mediates the transfer of the uridine monophosphate (UMP) group to gal-1P to form UDP-Gal (Wong et al., 1977; Arabshahi et al., 1986; Field et al., 1989; Geeganage and Frey, 1998). Moreover, structural analysis of E. coli GALT reveals that the enzyme is a homodimer, and each monomer composes six α–helices and a β–sheet formed by nine anti-parallel strands (Wedekind et al., 1995; Thoden et al., 1997). Two active sites exist in the dimer, each one formed by amino acid residues contributed by both monomers (Wedekind et al., 1995; Thoden et al., 1997). Therefore, correct inter-subunit interactions are needed for the correct activity of the enzyme (Thoden et al., 1997).
In humans, deficiency of GALT activity caused by deleterious variations of the GALT genes can result in a potentially lethal disorder called Classic Galactosemia (MIM# 230400) (Isselbacher et al., 1956; Segal and Berry, 1995). If the affected neonates are not treated in time, they will suffer from severe hepatic and renal failure, bleeding diatheses and E. coli sepsis, which can lead to death within days of birth (Goppert, 1917; Mason et al., 1935; Isselbacher et al., 1956; Segal and Berry, 1995). The exact pathophysiology of these acute symptoms remains uncertain, partially because of the lack of experimental animal model for this disease (Leslie et al., 1996), but the accumulation of gal-1P is regarded as one of the most important pathogenic factors (Gitzelmann et al., 1967; Gitzelmann, 1995). The best treatment for Classic Galactosemia to-date is the removal of galactose from the patients’ diet to prevent the manifestation of the acute toxicity syndrome (Mason et al., 1935; Salt et al., 1955; Jones and Leak, 1959; Bosch, 2006). However, long-term complications such as IQ deficits, ataxia, speech dyspraxia, and premature ovarian insufficiency persist in many patients with a galactose-restricted diet (Waggoner et al., 1990; Waisbren et al., 2011).
For the past few decades, the inclusion of Classic Galactosemia in the newborn screening programs in many countries greatly facilitates the identification, diagnosis and timely treatment of the affected patients. Furthermore, useful information about distribution and types of GALT gene variations among different populations were revealed. The human GALT gene maps to chromosome 9p13, and to-date, over 180 variations have been discovered; most of them are nucleotide substitutions (http://www.arup.utah.edu/database/GALT/GALT_display.php) (Calderon et al, 2007). In the past, majority of studies about GALT variations focused on the identification of new variations as well as their effects on patients’ red cell GALT activity (Singh et al., 2011), the distributions of different variant alleles among populations (Lai et al., 1996) and the predictive use of these variations in the prognosis of the disease (Guerrero et al., 2000). Few groups reported in-depth studies about how variations in GALT gene affect GALT enzyme activity at the structural level (Lai et al., 1999; Facchiano and Marabotti, 2010). Here we report 14 novel GALT gene variations and examine how they cause abnormal GALT activity in patients by in vitro enzymatic studies, as well as molecular modeling and molecular dynamic simulation analysis.
Materials and Methods
Site-directed mutagenesis of human GALT cDNA
The cDNA of human GALT gene was cloned into a bacterial expression vector pET-30 (EMD Chemicals Darmstadt, Germany). Each individual variation was introduced into the construct through site-directed mutagenesis using QuickChange II Site-directed Mutagenesis Kit (Stratagene Inc., La Jolla, CA). The introduced sequence changes were confirmed by DNA sequencing. DNA variation numbering system used here is based on the reference cDNA sequence from GenBank (BC015045.2 GI:34783207). Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence.
Over-expression, purification, and quantification of recombinant GALT enzymes
The wild-type GALT and variant proteins were expressed and purified using methods similar to those previously described (Lai et al., 1999). Generally, the plasmids mentioned above were transformed into Escherichia coli HMS174 (DE3) (Novagen). The bacterial culture was grown in 37°C to reach OD600=0.6, then the culture was cooled down to room temperature (19°C), and 1mM of IPTG was added to induce protein expression. Induction took place overnight at room temperature. Protein purification was conducted at 4°C throughout. Briefly, cell pellets were re-suspended in lysis buffer (50mM HEPES, 300mM NaCl, 10mM imidazole, pH 8.0). Cells were then lysed using a microfluidizer and clarified by centrifugation, and the lysate was loaded onto a chromatography column containing Nickel affinity resin. The resin was washed with buffer mentioned above but in 20mM imidazole and bound protein was eluted using the same buffer containing 200mM imidazole.
The purified proteins and the whole cell lysates were separated by SDS-PAGE, and then transferred from gel to nitrocellulose membrane. After blocking the membrane with Li-Cor blocking buffer (Li-Cor, Inc., Lincoln, NE) for one hour at room temperature, primary mouse anti-His6 antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA) was added at a dilution of 1:1000 to the membrane, and the mixture was incubated for one hour at room temperature. After washing with 1X PBS + 0.15% Tween-20 for three times (five minutes each), secondary antibody, Alexa Fluor® 680—allophycocyanin goat anti-mouse IgG (H+L) (Invitrogen Inc., Carlsbad, CA), was then added at a dilution of 1:5000 for another hour. After another wash, the membrane was scanned with Odyssey infra-red scanner (Li-Cor Inc., Lincoln, NE).
GALT enzyme activity measurement
The specific activity of the purified wild-type and variant GALT proteins were assayed in the glycine buffer (100mM, pH 8.7) containing 0.6mM UDP-Glu, 5mM MgCl2, 5mM dithiothreitol, 0.8mM NADP, 1.2mM gal-1P, 5μM glucose-α-1,6-diphosphate, phosphoglucomutase (0.5IU/ml), glucose-6-phosphate dehydrogenase (0.5IU/ml). The formation of NADPH was quantified by monitoring change in absorbance of the reaction mixture at 340nm for half an hour using BioTek plate reader (BioTek Inc., Winooski, VT). The relationship between increase in NADPH production and glu-1P released was quantified using the Beer-Lambert equation.
To assay for the Vmax and the KM for individual substrate, the substrate concentration was varied and the other substrate was held at saturated level (8mM for gal-1P and 4mM for UDP-Glu). The initial velocity of the reaction was measured by monitoring the change of absorbance at 340nm, and was plotted against the substrate concentration. Curve-fitting was accomplished using the equation V= Vmax S/ (KM+S) by Sigma Plot 10.0 (Systat Software Inc., San Jose, CA), and the Vmax and KM were calculated from the above-stated equation. All biochemicals and enzymes used in the in vitro biochemical assays were purchased from Sigma-Aldrich Inc., St. Louis, MO.
Static structure modeling
The 3D model of human GALT enzyme (Marabotti and Facchiano, 2005; PDB code 1R3A) was used as the starting point to simulate the effect of variations. The strategy used to model and analyze variant GALT enzymes, as well as the criteria to define the impact of a variation on the static protein structure, are the same as reported previously (Facchiano and Marabotti, 2010). Each variant structure was submitted to two different Web servers to predict the variation-induced change of protein stability with respect to the wild-type enzyme: PoPMuSiC v.2.1 (Dehouck et al., 2011) and CUPSAT (Parthiban et al., 2006), and we decided to consider reliable only results for which both predictors reached a consensus, otherwise, the effect of variation on protein stability is classified as “not determined”. We also checked the variations for their conservation score, which was previously calculated (Facchiano and Marabotti, 2010) and stored in the GALT protein database (http://bioinformatica.isa.cnr.it/GALT) (d’Acierno et al., 2009).
Molecular dynamics simulation
Molecular dynamics (MD) simulations were performed on selected GALT variants using Gromacs v.4.5.3 (Hess et al., 2008). Simulations were hosted on a Linux Cluster of 32 nodes, each SuperMicro equipped of two processors: the 2.50GHz INTEL(R) Quad-core and the 16GB Xeon(R), for a 256 total processors interconnected with the Infiniband 4X network and 512 GB of RAM. The GROMOS96 43a1 force field (van Gunsteren et al., 1996) was used throughout simulations. When the ligand UDP-Gal was included in calculation, its topology was created with the aid of the server PRODRG 2.5Beta (Schuttelkopf and van Aalten, 2004), but charges were assigned on the basis of charges of ribose, uridylyl and galactose moieties available in the database of the force field.
Each dimeric protein was simulated in a rhombic dodecahedric box, setting a distance of 1.5nm between the solute (the protein) and the box walls, so that the dimensions of the box are set to the diameter of the protein plus twice the specified distance. The protein was centered in the box. Approximately 25,600 water molecules (spc model) (Berendsen et al., 1984) were added to each system, and either six or ten Na+ ions were used respectively to neutralize the net negative charge, in the absence or presence of the ligand into the protein. Periodic boundary conditions were used to exclude surface effects.
For each selected variant, a preliminary energy minimization step with a gradient limit of 500 kJ/mol/nm was run with the Steepest Descent method, constraining all bonds with the P-LINCS method (Hess, 2008). Then, a 20ps-long MD simulation with position restraints using the NVT ensemble, followed by 100ps-long simulation with position restraints using NPT ensemble was applied to each system to soak the solvent into the macromolecule. A time step of 2fs was used in all cases. Each system was coupled to a temperature bath at 37°C using a velocity rescaling thermostat with a stochastic term (Bussi et al., 2007) and, in the case of the NPT ensemble, to a barostat at a pressure of 1atm using Berendsen’s method (Berendsen et al., 1984). Long-range electrostatics were handled using the PME method (Essmann et al., 1995). Cut-offs were set at 1.0nm for Coulombic interactions and at 1.4nm for van der Waals interactions. Finally, 30ns-full MD simulations were carried out with the same settings adopted for the short NPT simulation, but without any position restraints. To check for the quality of simulations, the energy components were analyzed to verify the stabilization of the systems, and the root mean square deviation (RMSD) of the structures in the trajectory with respect to the starting structure and to the average structure of the simulation was evaluated to check the convergence of the simulations towards an equilibrium state. The convergence of all the holo enzymes (i.e., with bound UDP-Gal) towards an equilibrium state was achieved after 5ns of simulation. 10ns of simulations for apo (i.e., without UDP-Gal in the active site) enzymes but variant Q188R were necessary to reach an equilibrium state. The equilibrium state of the apo form of Q188R was reached after 20ns of simulation. Therefore, this last simulation was prolonged to 40ns, in order to obtain 20ns of stable simulation for the analysis, in analogy to all other variants. In addition, for each simulation, minimum distance between periodic images was evaluated and found to be always not less than 2.0nm (a distance higher than the cut-off used to evaluate electrostatic and van der Waals interactions). This ensured that no non-physical self-interactions took place between periodic images of the protein during simulations.
Analyses of MD simulations were conducted using programs built within the GROMACS package. Results were visualized and elaborated with the aid of the freeware program Grace (http://plasma-gate.weizmann.ac.il/Grace). The cluster analysis was made using the clustering method of Daura (Daura et al., 1999), with the default cut-off of 0.25nm. RMSD calculation and least square fitting were performed on the protein without hydrogens. Starting from this cluster analysis, a representative structure was calculated for each simulation.
Results
Clinical laboratory findings of patient GALT gene variations and enzyme activity
In this study, we chose 14 variations published at the ARUP GALT gene variation database for further characterization (Supp. Table S1), as none of the 14 variations have been previously characterized in any depth at protein level. All variations are single nucleotide substitutions and only I170T (509T>C) is close to a splicing site (one nucleotide away from the splicing site between Intron 5 and Exon VI). Only one variation (V168L) was found to be homozygous in a patient and it resulted in zero enzyme activity. Four variations (I170T, R223S, E291V, L327P) were found to be in compound heterozygosity with the common Q188R variation, but only two of them (I170T and L327P) resulted in zero enzyme activity, while the other two (R223S and E291V) had some residual activity. One variation, R259Q, was found to be in compound heterozygosity with the S135L variation in a patient, and the patient had no detectable enzyme activity in the red blood cells. Three variations (H132Q, L227P and Y34N) were found to be in compound heterozygosity with the Duarte 2 haplotype, and the patients’ enzyme activities were consistent with Duarte/Classical Galactosemia compound heterozygote (D/G). Another three variations (E220K, I278N and L289F) were found in heterozygous state with enzyme activity around 10μmol/h/gHb. One patient was found to be compound heterozygous for the P185H and R201C variations, which resulted in minute enzyme activity.
Based on the information above, we can safely conclude that variations V168L, I170T, R259Q and L327P result in no enzyme activity, and R223S, E291V variations result in reduced enzyme activity. As for variations Y34N, H132Q and L227P, since they were found to be in compound heterozygosity with the Duarte 2 haplotype and the patients’ enzyme activities were consistent with the D/G biochemical phenotype, it is highly likely these variations render no enzyme activity in the red blood cells.
Heterologous expression of variant GALT proteins
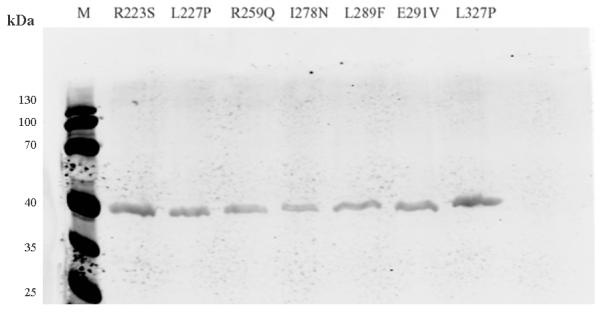
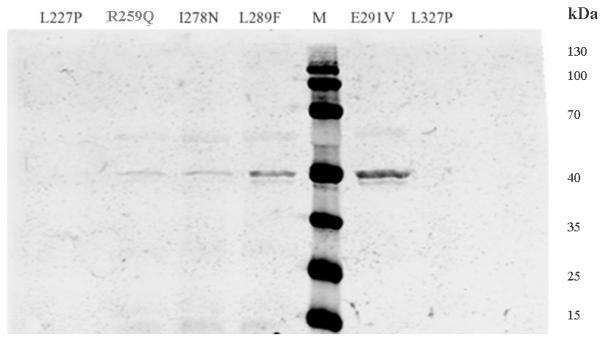
In order to understand how they affect the patient red cell GALT enzyme activities at the mechanistic level, we expressed all of the 14 variant GALT proteins in a bacterial expression system. As shown in Fig. 1a, except for the Y34N- and H132Q-GALT variants, the protein level of all the variants and wild-type in the whole cell lysates were comparable. However, when we separated the insoluble cell debris from the whole cell lysates and purified the variant GALT proteins from the soluble (cytoplasmic) fractions, we detected, except for the E291V variant, significantly less amount of all variant GALT proteins (Fig. 1b). Two variants, L227P and L327P, resulted in no detectable protein in the soluble fraction. Since all the variant and wild-type GALT enzymes were expressed, purified and analyzed under identical experimental conditions, one can propose that except for the Y34N-, H132Q- and E291V-GALT variants, all the variations studied resulted in decreased solubility of the GALT enzymes. For the Y34N and H132Q variants, it appears that these two variations might have adverse effects on protein translation and/or protein stability, especially in the case of Y34N.
Fig. 1. Expression and purification of GALT wild-type and variant proteins.
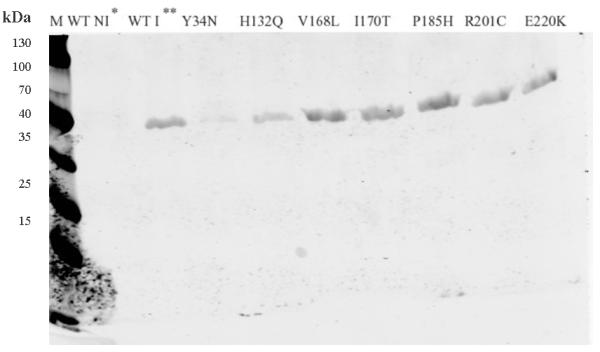
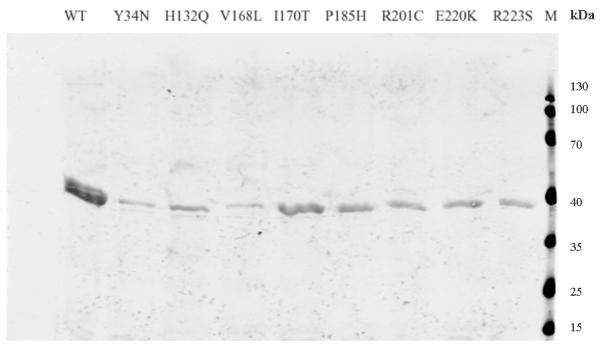
(a) Whole cell lysates containing corresponding GALT proteins (wild-type and variants) from same amount of cell culture were separated by SDS-PAGE and detected by anti-His6 tag antibody. * Wild-type, non-induced. ** Wild-type, induced. M, molecular weight marker.
(b) GALT proteins (wild-type and variants) purified from same amount of cell culture were separated and detected as in (a).
Kinetic analysis of purified variant GALT enzymes
Assay for activities of the 14 variant GALT enzymes revealed that only six of them (R201C, E220K, R223S, I278N, L289F, E291V) have GALT activity close to 5% or above of the control value (Fig. 2). Among the six “active” variants, except for the I278N-GALT, the GALT activity detected in all others was substantial (>30% of control) (Fig. 2).
Fig. 2. Enzymatic activity measurement of purified wild-type and variant GALT proteins.
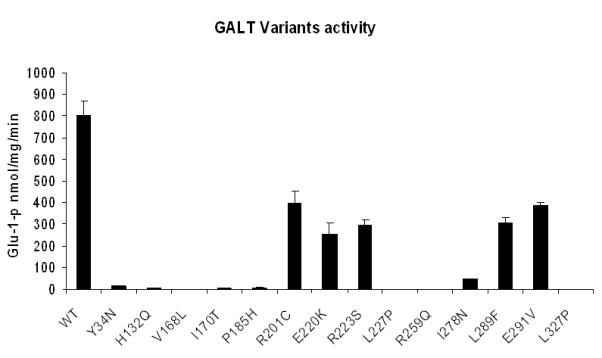
0.1μg of purified protein was tested in a total volume of 100μl glycine buffer. The activity values were generated from averaging three experimental results, error bars indicated standard deviations calculated from the triplicates.
A detailed analysis of kinetic parameters of these variants showed that the major cause of their reduced activities was a compromised Vmax (Table 1). All six active variants had their Vmax decreased more than two-fold. The KM for UDP-Glu of these variants didn’t change significantly, only those of I278N and E291V increased more than two-fold. As for the KM for gal-1P, three variants, E220K, L289F and E291V, with their KM increased more than 1.7 fold as compared with that of the control. Among all six active variants, I278N was the most affected; its Vmax decreased about 20-fold and KM for both substrates increased significantly.
Table 1.
Enzymatic parameters of six active variants
| KM gal-1-p (mM) | KM UDP-Glu (mM) | Vmax (nmol/mg/sec) | |
|---|---|---|---|
| WT | 1.25±0.36 | 0.43±0.09 | 804±65 |
| R201C | 1.89±0.62 | 0.58±0.13 | 396±59 |
| E220K | 2.34±0.42 | 0.69±0.16 | 253±53 |
| R223S | 1.12±0.31 | 0.76±0.09 | 297±25 |
| I278N | 1.98±0.35 | 1.23±0.28 | 45±3 |
| L289F | 2.14±0.21 | 0.48±0.13 | 306±23 |
| E291V | 2.68±0.16 | 0.95±0.43 | 385±18 |
Mean values and standard deviations presented here were calculated from three separate experiments.
Analysis on static variant GALT structures
In order to explain the reduced activity of the variant GALT enzymes at the structural level, we constructed and analyzed the models of the variant GALT enzymes for potential perturbations on protein structure and enzymatic function, as we did previously for other GALT variations (Facchiano and Marabotti, 2010). In Fig. 3, we show the modeled structure of wild-type GALT enzyme highlighting the amino acid residues that were changed in the variants.
Fig. 3. Schematic view of the modeled 3D structure of wild-type GALT enzyme.

The backbone of the protein is shown as a ribbon. UDP-Gal is shown in CPK mode and residues that undergo the variations are represented in ball & stick mode and labeled. Hydrogen atoms are not represented for sake of clarity. (In the color version online, the two subunits forming the dimer are colored in yellow and cyan for monomer A and B, respectively. Color codes for atom types are: carbon: green, oxygen: red, nitrogen: blue, phosphorus: magenta).
The results of our investigations on the static protein structure are summarized in Table 2. From the conservation score attributed to each residue, only L327P variation affected a highly conserved residue, not only in the eukaryotic organisms, but also in the prokaryotic species. The other variant residues in general were conserved in eukaryotic organisms only.
Table 2.
Summary of the structural effects of single-point variations deduced from analysis of static GALT structure
| Variant | Conservation score§ |
Impact of variation on active site of the enzyme |
Impact of variation on intersubunit interactions |
Impact of variation on secondary structure of the variant residue |
Impact of variation on solvent accessibility of the variant residue |
Impact of variation on H-bond network of the variant residue |
Impact of variation on salt bridge pattern of the variant residue |
Impact of variation on overall stability of the enzyme |
|---|---|---|---|---|---|---|---|---|
| Y34N | 0.415 | NO | YES | NO | YES | YES | NO | YES |
| H132Q | 0.498 | YES* | NO | NO | NO | NO | NO | N.D. |
| V168L | 0.513 | NO | NO | NO | NO | NO | NO | N.D. |
| I170T | 0.587 | NO | NO | NO | NO | YES | NO | YES |
| P185H | 0.510 | YES* | NO | YES | NO | YES | NO | YES |
| R201C | 0.446 | NO | YES | NO | NO | YES | YES | YES |
| E220K | 0.457 | NO | NO | NO | NO | YES | YES | NO |
| R223S | 0.374 | NO | NO | NO | NO | NO | YES | YES |
| L227P | 0.458 | NO | NO | YES | NO | YES | NO | YES |
| R259Q | 0.340 | NO | NO | NO | NO | NO | YES | YES |
| I278N | 0.338 | NO | NO | NO | NO | NO | NO | YES |
| L289F | 0.426 | NO | NO | NO | NO | NO | NO | N.D. |
| E291V | 0.479 | NO | NO | NO | NO | YES | NO | YES |
| L327P | 0.809 | NO | NO | NO | YES | YES | NO | N.D. |
Analyses were made on the static modelled structures of each homodimeric variant. “YES” and “NO” indicate if the variation is able to perturb each structural feature represented in columns, or not, following the criteria described previously (Facchiano and Marabotti, 2010). N.D indicates that the effect of the variation on protein stability is not determined because the predictors did not reach a consensus.
The higher the value, the most conserved the residue (for further information see http://bioinformatica.isa.cnr.it/GALT).
These residues are not included into the active site of the enzyme, but may affect the active site indirectly since they interact with active site residues.
In general agreement with experimental results, the two Web servers used to predict changes of GALT stability concur to indicate most variations as able to destabilize the enzyme. No variation reside into the GALT active site (as defined in (Facchiano and Marabotti, 2010)), although two variations (H132Q and P185H) are predicted to perturb the active site because both His132 and Pro185 interact with residues of the wild-type enzyme active site. His132 forms an H-bond with His184, which is spatially close to the catalytic residue His186 in human GALT. His184 is also part of the signature HhHhHhh motif (where H represents histidine and h represents hydrophobic amino acid) (Brenner, 2002) that characterizes the histidine triad superfamily members. Variation H132Q causes the loss of the H-bond between His132 and His184, which in turn could alter the active site. The P185H variation affects another residue of the signature motif, which is also covalently linked to the catalytic residue His186. Moreover, this variation was predicted to perturb the secondary structure of the segment in which the residue resides.
Variation I170T affects a residue in the β-strand adjacent to the one that includes His186 and Gln188. Residue 170 is not interacting with them, but the variation causes the formation of new H-bonds with Gln169. Due to its spatial proximity, this alteration of the H-bond network may also affect, indirectly, the catalytic active site.
Two variations, namely Y34N and R201C, affected residues that are involved in intersubunit interactions. Tyr34 forms H-bonds with Gln118 of the other subunit, and Arg201 is involved in an intersubunit network of salt bridges and H-bonds with Glu40, as discussed previously (Facchiano and Marabotti, 2010). Interestingly, based on the experimental data shown in Fig. 2, disrupting the interaction between Tyr34 and Gln118 might be more severe as Y34N resulted in no enzyme activity at all.
Variations E220K, R223S, L227P, R259Q, E291V and L327P were predicted to affect the H-bond and/or the salt bridge networks in which the wild-type residues are involved. Glu220 and Arg223 are part of a network of H-bonds and salt bridges that allow the interaction between the two biggest helices of the enzyme. In addition, the replacement of a negative charge with a positive one in E220K might have additional negative impact on protein structure. In fact, in addition to Arg223, the nearest residue to Glu220 is His213 and therefore, variation E220K could cause an accumulation of positive charged residues, with electrostatic repulsive effect. Leu227 is at the end of a helix and its backbone nitrogen interacts with the carbonyl oxygen of Arg223 with the typical helix-stabilizing H-bond. The replacement of Leu227 with Pro, the typical “helix-breaking” residue, could potentially produce negative effects on the local secondary structure, as well as on the overall stability of GALT. The salt bridge between Arg259 and Glu271 allows the interaction between one strand belonging to the central β-sheet and helix 8, and this interaction is lost in variant R259Q. Glu291 interacts with Asn288 through an H-bond involving the atoms of the main chain; and the replacement with Val could have altered this interaction, particularly since the new residue could potentially interact with Asp287. Finally, L327P showed limited effects on H-bond network and on solvent accessibility.
For three variations (V168L, I278N and L289F), no structural effects were predicted from the static models. The L289F variant, however, retains substantial residual activity (Fig. 2) and therefore it can be inferred that variation does not dramatically affect its structural features. On the contrary, variants V168L and I278N showed minimal to no enzymatic activity (Fig. 2). Val168 resided on the same β-strand as Ile170, spatially close to His186 and Gln188, but not interacting with them. Ile278 is far from the active site. In these last two cases, static models alone appear to be insufficient to predict the perturbations caused to the structural features of the protein.
Molecular dynamics (MD) simulations
In order to obtain more information on those variations that apparently did not cause any structural effect on GALT enzyme in static molecular modeling, we conducted MD simulations on the two variants V168L and I278N. Moreover, we performed MD simulation on variants H132Q and P185H that affect indirectly the active site of the enzyme, in order to gain more information about the effects of these alterations. All simulations were done both in absence and presence of the ligand UDP-Gal, and results were compared with those obtained on the wild-type protein and on the most known variant of GALT enzyme, i.e. Q188R, in the same conditions. The simulations were conducted at 37°C in order to mimic the human body temperature and for a timescale sufficient to infer the perturbation of short-term interactions.
A preliminary observation on the MD simulations can be made by evaluating the rate of convergence of the systems towards an equilibrium state (see Materials and Methods). The longer time of equilibration needed for the apo form of variant Q188R is in agreement with the known strong deleterious effects of this variation on protein structure (Marabotti and Facchiano, 2005).
The results of the analyses made on MD simulations are summarized in Table 3. Parameters related to the global fold, shape and conformation of the protein, and parameters related to structural effects, such as perturbations of secondary structures and H-bonds, were taken into account. Finally, analyses were made on the interactions of the ligand into the active site. A brief explanation of each analysis is reported below.
Table 3.
Summary of the effects of selected single point variations deduced from analysis of MD simulations, compared to the wild type protein (Variation Q188R was used as reference)
| Variant | Impact on conformation variability |
Impact on Rg | Impact on secondary structures |
Impact on H-bond pattern |
Impact on ligand binding by Q188 |
||||
|---|---|---|---|---|---|---|---|---|---|
| apo form | holo form | apo form | holo form | apo form | holo form | apo form | holo form | holo form | |
| H132Q | YES | NO | YES | NO | YES | YES | A: YES I: NO |
A: YES I: NO L: YES |
YES |
| V168L | YES | NO | YES | YES | YES | YES | A: YES I: NO |
A: YES I: YES L: YES |
NO |
| P185H | YES | YES | YES | NO | NO | YES | A: YES I: YES |
A: YES I: YES L: YES |
YES |
| I278N | NO | YES | YES | YES | NO | YES | A: NO I: YES |
A: NO I: NO L: YES |
YES |
| Q188R | YES | NO | YES | YES | YES | NO |
A: YES
I: NO |
A: YES
I: YES L: YES |
(This variant
lacks Q188) |
“YES” and “NO” indicate if the variation is able to perturb each feature represented in columns, or not. A: H-bond pattern of the whole protein; I: H-bond pattern between chain A and chain B; L: H-bond pattern between protein and ligand
The cluster analysis gives information about the range of conformations accessible to the protein. The analysis revealed the presence of a single cluster of conformations for each simulation made. However, the analysis of RMS distributions of the clusters suggests the presence of sub-populations of conformations with a different distribution between wild-type and variant enzymes, in both apo and holo forms (Supp. Figure S1). In particular, looking at the apo forms (Supp. Figure S1, panel A), the shape of RMS distribution for wild-type protein and for I278N is quasi-Gaussian, with only minor shoulders detectable, whereas for P185H, H132Q and Q188R it is possible to see the presence of distinct peaks. Apo V168L shows a distribution with a very large plateau formed probably by multiple equivalent underlying conformations. Also in holo systems (Supp. Figure S1, panel B), P185H and I278N, and to a minor extent H132Q, V168L and Q188R, show the presence of sub-populations of conformations in their RMS distribution.
The radius of gyration (Rg) is a parameter related to the global shape and volume of the protein. The analysis of Rg along the different trajectories revealed that the wild-type enzyme in apo form (Supp. Figure S2, panel A, black line) has Rg value higher than in the holo form (Supp. Figure S2, panel B, black line), and therefore is slightly more “relaxed”. All the apo form of variants show Rg values lower than that of wild-type enzyme (Supp. Figure S2, panel A). In the holo forms, the Rg values of V168L and Q188R are higher than that of the wild-type enzyme, whereas the other variants have Rg values more similar to the wild-type enzyme (Supp. Figure S2, panel B).
In order to detect if variations are able to affect secondary structures of GALT enzyme, we analyzed the representative structures, obtained by cluster analysis, with the program DSSP (Kabsch and Sander, 1983). Results are shown in Supp. Table S2. Differences are relatively slight, but it is possible to note that in general the variations analyzed consistently decreased the percentage of residues included in more organized secondary structures such as helices or strands, and increased the percentage of residues included in less organized secondary structures or in random coils. The most affected secondary structures are the strands forming the central β-sheet of the protein. We would like to point out that the timescale of simulations we made is not sufficient to show the complete unfolding of secondary structures; however these variations are good indicators of perturbations exerted by variations on the secondary structure of the enzyme (see for example Marabotti et al., 2008; Sciré et al., 2009, 2010).
We also performed an analysis on the variation of H-bonds calculated along the trajectory (Supp. Table S3), taking into account H-bonds internal to the protein (for both apo and holo systems), between the two subunits (for both apo and holo systems) and between protein and ligand (for holo systems only). The average number of H-bonds into the protein is generally reduced in all variants but I278N. V168L in both apo and holo forms is the variant with the largest decrease of this parameter. An increase in the average number of intersubunit H-bonds per timeframe is present in the apo forms of the variants P185H, H132Q and I278N, and a decrease is evident mainly for variants V168L, P185H and Q188R in holo forms. Finally, the average number of H-bonds per timeframe between the protein and the ligand increases in all variants but Q188R (the only one with a variation into the active site of the enzyme), with respect to the wild-type enzyme.
Finally, we monitored the distances between Gln188 and UDP-Gal atoms O2A and O1B in the variants (with the exception of Q188R, which lacks Gln188), and compared the results with those of wild-type enzyme. Results are shown in Supp. Figure S3. In the wild-type enzyme, these interactions between Gln188 and the substrate (which are involved in the stabilization of the reaction intermediate) are conserved during the simulations, whereas in the variant enzymes, especially P185H and H132Q, these interactions are more unstable or even completely lost during simulations.
Discussion
Although there have been numerous mutational analyses performed on the human GALT gene, few studies combined the techniques of in vitro expression analysis and in-depth molecular modeling, not to mention any attempt to correlate among these analyses and the clinical data. Therefore, our efforts to study the 14 uncharacterized variations presented here are both unique and novel.
In the heterologous expression analysis, not only did we confirm the deleterious nature of the amino acid changes in all 14 variations, but also offered novel insights into the effects of the variations on the stability and solubility of the GALT protein. For instance, in the cases of Y34N and H132Q, we showed that these variant proteins were present in significantly lower abundance in the whole cell lysates (Fig. 1a). When we tested the purified variant proteins for specific activity, it appeared that these two variants have less than 1% of control activity well (Fig. 2). Except for R201C, E220K, R223S, I278N, L289F and E291V, the rest of the variations did not exhibit any significant residual activity in their pure forms (Fig. 2). Further, many of them, including the six above-mentioned variations demonstrated reduced solubility in the bacterial lysate (Fig. 1b). Overall, the results from the in vitro expression analysis correlate well with the clinical laboratory measurements of the patients’ red blood cell GALT activity (Supp. Table S1). In fact, the in vitro separate assays of P185H and R201C variant proteins allowed us to conclude that residual GALT activity of the patient, who is compound heterozygous for both of these variations (Supp. Table S1), is likely contributed by the residual activity of R201C-GALT enzyme. Yet, based on the decreased solubility of this variant enzyme as shown in the bacterial expression system (Fig. 1b), it was highly possible that this variant also had solubility issue in the patient and therefore, significantly compromised the total enzymatic activity. The same issue of reduced solubility could also explain why the patient, who is compound heterozygous for the R223S and Q188R variations, displays only minute GALT activity, despite the fact that the R223S-GALT manifested close to 37% of control activity (Fig. 2, Table 1). Lastly, it should be noted that not all variations are as deleterious as Y34N. The E291V variation, which appears to have the least impact on solubility among the 14 variations, also retains nearly 50% of control activity (Fig. 2, Table 1). Consequently, this explains very well why the patient’s biochemical phenotype was ascertained as D/G (Supp. Table S1).
To have a better understanding how these variations affected the protein from a structural point of view, we applied the technique of static molecular modeling for further analysis. Nine of the 14 variations were predicted to have stability problem (Table 2). The data correlated well with the fact that decreasing solubility was common among these variations (Fig. 1b). Also, the studies gave more hints how some of the variations inactivated the enzyme in addition to the solubility issue mentioned above. For instance, variation H132Q and P185H were shown indirectly disrupting the active site of the enzyme (Table 2), resulting in inactive enzyme (Fig. 2). Y34N and R201C interfered with the intersubunit interactions (Table 2), which is crucial for the formation of the active center of the enzyme. Yet, the R201C change was more tolerable than Y34N (Fig. 2), suggesting Tyr34 plays a more important role than Arg201. Also, L227P introduced a “helix-breaking” residue to the end of the helix, disrupting a helix-stabilizing H-bond which resulted in inactive enzyme.
For the variations which retain substantial residual activities, such as E220K, R223S and E291V, our studies showed that they affected networks of H-bond and salt bridges (Table 2). Since this resulted in local structural alterations, it appears in agreement with the experimental evidence that they still had some residual activity (Fig. 2).
Static structure modeling alone, however, was not sufficient to explain the deleterious effects of two variations: V168L and I278N. Therefore, in these cases, we decided to perform MD simulations to observe phenomena that can be appreciated only following the evolution of structural features during the time. In addition, we performed MD simulations also to H132Q and P185H, in order to gain more information about the effects of these variations. From the analysis of MD simulations, it appears that even those variations with no apparent effects on the static model could have caused perturbations in the protein’s structure and dynamics.
The cluster analyses highlighted a different distribution of conformations between wild-type enzyme and these variants, with the presence of distinct sub-populations of conformations (Supp. Figure S1). In the past, the presence of different clusters of conformations was correlated to experimentally detected partially unfolded/molten globule states (Scirè et al., 2009). In this study, the presence of these unfolded states cannot be inferred directly, since only one cluster is identified for each trajectory, but this analysis can indicate higher conformational variability of the variants with respect to the wild-type enzyme. This phenomenon, in association with the decrease of highly organized secondary structure elements and of the internal H-bonds, with respect to the wild-type protein (Supp. Table S2 and S3), can be, ultimately, at the basis of the decreased stability and solubility of variants detected in the in vitro expression analysis. In fact, aggregation and precipitation tend to occur more frequently when the proteins show kinetic instability and/or the lack of ordered structures.
In wild-type enzyme, the apo form has a higher Rg and a lower number of interchain H-bonds with respect to the holo form (Supp. Figure S2 and Supp. Table S3). This suggests that the enzyme in the apo form is an “open” structure which favors the accessibility of the active sites, located at the interface between the two subunits. The binding of the ligand promotes the “closure” of the enzyme to reach a more compact conformation, stabilized by more intersubunit interactions. This condition is probably necessary for the enzymatic reaction to occur. In variant V168L, similar to variant Q188R, the comparison of Rg and intersubunit H-bond pattern between the apo and holo forms shows that the binding of ligand does not promote either the increase of compactness in the protein or the increase in the number of intersubunit H-bonds (Supp. Figure S2 and Supp. Table S2). Therefore, one might deduce that these two variants cannot promote the further phases of the enzymatic reaction. This is in agreement with the full inactivation of the enzyme carrying this mutation (Fig. 3). For the other variants simulated, the apo forms are more compact than that of the wild-type, because they show lower Rg values and higher number of intersubunit H-bonds (Supp. Figure S2 and Supp. Table S3). This suggests that their apo forms are not fully open, hindering the access of ligand to the active site.
It is interesting to note that the number of average H-bonds per timeframe between protein and UDP-Gal is higher in all the variants than in wild-type (Supp. Table S3). The increased strength of enzyme-substrate interactions could prevent the release of the product, thus inactivating the enzyme. Moreover, the simulations show in most cases, especially in variants P185H and H132Q, a perturbation of the interactions between UDP-Gal and Gln188 that stabilize the intermediate of reaction. Therefore, confirming deductions from static models, these variations can deeply alter the active site of the enzyme, impairing its activity.
In conclusion, combined use of the techniques of in vitro expression analysis and molecular modeling offers novel and important insights into mutational analysis of the human GALT gene. The computational costs of MD simulations and analyses are still high and do not allow a routinely application of these procedures, but these procedures can be used as a further source of information in those cases in which the static model is not sufficient to reveal the impact of mutations on protein structure and functions. Results of these studies appear to correlate well with clinical data and tender useful explanations for the observed activities measured in patient samples.
Supplementary Material
Acknowledgments
This work was supported by a Parents of Galactosemic Children Research Grant (to A.M.), and by project FIRB MIUR ITALBIONET (RBPR05ZK2Z and RBIN064YAT_003) (to A.M.). This work has been made in the frame of the Flagship Project InterOmics and of the project CNR-Bioinformatics. A.M. wishes also to acknowledge the support of people of Gromacs user list, in particular J. Lemkul, T. Wassenaar and M. Abraham, for useful suggestions. Research grant support to K.L. includes U.S. NIH grants 5R01 HD054744-04 and 3R01 HD054744-04S1.
Grant Support to A.M.: (1) Parents of Galactosemic Children Research Grant (2) Project FIRB MIUR ITALBIONET (RBPR05ZK2Z and RBIN064YAT_003) Grant support to K.L.: U.S. NIH grants 5R01 HD054744-04 and 3R01 HD054744-04S1.
Footnotes
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
References
- Arabshahi A, Brody RS, Smallwood A, Tsai TC, Frey PA. Galactose-1-phosphate uridylyltransferase. Purification of the enzyme and stereochemical course of each step of the double-displacement mechanism. Biochemistry. 1986;25:5583–5589. doi: 10.1021/bi00367a036. [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, Di Nola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- Bosch AM. Classical galactosaemia revisited. J Inherit Metab Dis. 2006;29:516–525. doi: 10.1007/s10545-006-0382-0. [DOI] [PubMed] [Google Scholar]
- Brenner C. Hint, Fhit, and GalT: function, structure, evolution, and mechanism of three branches of the histidine triad superfamily of nucleotide hydrolases and transferases. Biochemistry. 2002;41:9003–9014. doi: 10.1021/bi025942q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Calderon FR, Phansalkar AR, Crockett DK, Miller M, Mao R. Mutation database for the galactose-1-phosphate uridyltransferase (GALT) gene. Hum Mutat. 2007;28(10):939–943. doi: 10.1002/humu.20544. [DOI] [PubMed] [Google Scholar]
- Cardini CE, Leloir LF. Enzymic phosphorylation of galactosamine and galactose. Arch Biochem Biophys. 1953;45:55–64. doi: 10.1016/0003-9861(53)90404-4. [DOI] [PubMed] [Google Scholar]
- d’Acierno A, Facchiano A, Marabotti A. GALT protein database, a bioinformatics resource for the management and analysis of structural features of a galactosemia-related protein and its mutants. Genomics Proteomics Bioinformatics. 2009;7:71–76. doi: 10.1016/S1672-0229(08)60035-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, Mark AE. Peptide folding: When simulation meets experiment. Angew Chem Int Ed Engl. 1999;38:236–240. [Google Scholar]
- Dehouck Y, Kwasigroch JM, Gilis D, Rooman M. PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics. 2011;12:151. doi: 10.1186/1471-2105-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth Particle Mesh Ewald potential. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- Facchiano A, Marabotti A. Analysis of galactosemia-linked mutations of GALT enzyme using a computational biology approach. Protein Eng Des Sel. 2010;23:103–113. doi: 10.1093/protein/gzp076. [DOI] [PubMed] [Google Scholar]
- Field TL, Reznikoff WS, Frey PA. Galactose-1-phosphate uridylyltransferase: identification of histidine-164 and histidine-166 as critical residues by site-directed mutagenesis. Biochemistry. 1989;28:2094–2099. doi: 10.1021/bi00431a019. [DOI] [PubMed] [Google Scholar]
- Geeganage S, Frey PA. Transient kinetics of formation and reaction of the uridylyl-enzyme form of galactose-1-P uridylyltransferase and its Q168R-variant: insight into the molecular basis of galactosemia. Biochemistry. 1998;37:14500–14507. doi: 10.1021/bi9815546. [DOI] [PubMed] [Google Scholar]
- Gitzelmann R. Galactose-1-phosphate in the pathophysiology of galactosemia. Eur J Pediatr. 1995;154:S45–49. doi: 10.1007/BF02143803. [DOI] [PubMed] [Google Scholar]
- Gitzelmann R, Curtius HC, Schneller I. Galactitol and galactose-1-phosphate in the lens of a galactosemic infant. Exp Eye Res. 1967;6:1–3. doi: 10.1016/s0014-4835(67)80047-2. [DOI] [PubMed] [Google Scholar]
- Goppert F. Galaktosurie nach Milchzuckergabe bei angeborenem, familiaerem chronischem Leberleiden. Klin Wschr. 1917;54:473–477. [Google Scholar]
- Guerrero NV, Singh RH, Manatunga A, Berry GT, Steiner RD, Elsas LJ., 2nd Risk factors for premature ovarian failure in females with galactosemia. J Pediatr. 2000;137:833–841. doi: 10.1067/mpd.2000.109148. [DOI] [PubMed] [Google Scholar]
- Hess B, Kutzner C, Van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulations. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Hess B. P-LINCS: A parallel linear constraint solver for molecular simulations. J Chem Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- Isselbacher KJ, Anderson EP, Kurahashi K, Kalckar HM. Congenital galactosemia, a single enzymatic block in galactose metabolism. Science. 1956;123:635–636. doi: 10.1126/science.123.3198.635. [DOI] [PubMed] [Google Scholar]
- Jones NL, Leak D. The treatment of congenital galactosaemia. Arch Dis Child. 1959;34:307–311. doi: 10.1136/adc.34.176.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kalckar HM, Braganca B, Munch-Petersen HM. Uridyl transferases and the formation of uridine diphosphogalactose. Nature. 1953;172:1038. [PubMed] [Google Scholar]
- Lai K, Langley SD, Singh RH, Dembure PP, Hjelm LN, Elsas LJ., 2nd A prevalent mutation for galactosemia among black Americans. J Pediatr. 1996;128:89–95. doi: 10.1016/s0022-3476(96)70432-8. [DOI] [PubMed] [Google Scholar]
- Lai K, Willis AC, Elsas LJ., 2nd The biochemical role of glutamine 188 in human galactose-1-phosphate uridyltransferase. J Biol Chem. 1999;274:6559–6566. doi: 10.1074/jbc.274.10.6559. [DOI] [PubMed] [Google Scholar]
- Leloir LF. The enzymatic transformation of uridine diphosphate glucose into a galactose derivative. Arch Biochem. 1951;33:186–190. doi: 10.1016/0003-9861(51)90096-3. [DOI] [PubMed] [Google Scholar]
- Leslie ND, Yager KL, McNamara PD, Segal S. A mouse model of galactose-1-phosphate uridyl transferase deficiency. Biochem Mol Med. 1996;59:7–12. doi: 10.1006/bmme.1996.0057. [DOI] [PubMed] [Google Scholar]
- Marabotti A, Facchiano AM. Homology modeling studies on human galactose-1-phosphate uridylyltransferase and on its galactosemia-related mutant Q188R provide an explanation of molecular effects of the mutation on homo- and heterodimers. J Med Chem. 2005;48:773–779. doi: 10.1021/jm049731q. [DOI] [PubMed] [Google Scholar]
- Marabotti A, Lefèvre T, Staiano M, Crescenzo R, Varriale A, Rossi M, Pézolet M, D’Auria S. Mutant bovine odorant-binding protein: Temperature affects the protein stability and dynamics as revealed by infrared spectroscopy and molecular dynamics simulations. Proteins. 2008;72:769–778. doi: 10.1002/prot.21966. [DOI] [PubMed] [Google Scholar]
- Mason HH, Turner ME. Chronic galactosemia: report of case with studies on carbohydrates. Am. J. Dis. Child. 1935;50:359–374. [Google Scholar]
- Parthiban V, Gromiha MM, Schomburg D. CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res. 2006;34(Web Server issue):W239–242. doi: 10.1093/nar/gkl190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt HB, Ross CA, Gerrard JW. Low-lactose milk for congenital galactosaemia. Lancet. 1955;268:1177–1178. doi: 10.1016/s0140-6736(55)90673-5. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Sciré A, Marabotti A, Staiano M, Briand L, Varriale A, Bertoli E, Tanfani F, D’Auria S. Structure and stability of a rat odorant-binding protein. Another brick in the wall. J Proteome Res. 2009;8:4005–4013. doi: 10.1021/pr900346z. [DOI] [PubMed] [Google Scholar]
- Scirè A, Marabotti A, Staiano M, Iozzino L, Luchansky MS, Der BS, Dattelbaum JD, Tanfani F, D’Auria S. Amino acid transport in thermophiles: characterization of an argininebinding protein in Thermotoga maritima. 2. Molecular organization and structural stability. Mol Biosystems. 2010;6:687–698. doi: 10.1039/b922092e. [DOI] [PubMed] [Google Scholar]
- Segal S, Berry GT. Disorders of galactose metabolism. In: Beaudet AL, Scriver CR, Sly WS, Valle D, editors. The Metabolic Basis of Inherited Diseases. McGraw-Hill; New York: 1995. pp. 967–1000. [Google Scholar]
- Singh R, Kaur G, Thapa BR, Prasad R. A case of classical galactosemia: identification and characterization of 3 distinct mutations in galactose-1-phosphate uridyl transferase (GALT) gene in a single family. Indian J Pediatr. 2011;78:874–876. doi: 10.1007/s12098-010-0348-y. [DOI] [PubMed] [Google Scholar]
- Thoden JB, Ruzicka FJ, Frey PA, Rayment I, Holden HM. Structural analysis of the H166G site-directed mutant of galactose-1-phosphate uridylyltransferase complexed with either UDP-glucose or UDP-galactose: detailed description of the nucleotide sugar binding site. Biochemistry. 1997;36:1212–1222. doi: 10.1021/bi9626517. [DOI] [PubMed] [Google Scholar]
- van Gunsteren WF, Billeter SR, Eising AA, Hunenberger PH, Kruger P, Mark AE, Scott WRP, Tironi IG. The GROMOS96 manual and user guide. Vdf Hochschulverlag AG an der ETH Zurich. 1996:1042. [Google Scholar]
- Waggoner DD, Buist NR, Donnell GN. Long-term prognosis in Galactosemia: results of a survey of 350 cases. J Inherit Metab Dis. 1990;13:802–818. doi: 10.1007/BF01800204. [DOI] [PubMed] [Google Scholar]
- Waisbren SE, Potter NL, Gordon CM, Green RC, Greenstein P, Gubbels CS, Rubio-Gozalbo E, Schomer D, Welt C, Anastasoaie V, D’Anna K, Gentile J, Guo CY, Hecht L, Jackson R, Jansma BM, Li Y, Lip V, Miller DT, Murray M, Power L, Quinn N, Rohr F, Shen Y, Skinder-Meredith A, Timmers I, Tunick R, Wessel A, Wu BL, Levy H, Elsas L, Berry GT. The adult galactosemic phenotype. J Inherit Metab Dis. 2012;35:279–286. doi: 10.1007/s10545-011-9372-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedekind JE, Frey PA, Rayment I. Three-dimensional structure of galactose-1-phosphate uridylyltransferase from Escherichia coli at 1.8 Å resolution. Biochemistry. 1995;34:11049–11061. doi: 10.1021/bi00035a010. [DOI] [PubMed] [Google Scholar]
- Wong LJ, Sheu KFR, Lee SL, Frey PA. Galactose-1-phosphate uridylyltransferase: isolation and properties of a uridylyl-enzyme intermediate. Biochemistry. 1977;16:1010–1016. doi: 10.1021/bi00624a032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.