

Abstract
Background
Desulfovibrio vulgaris Hildenborough is a sulfate-reducing bacterium (SRB) that is intensively studied in the context of metal corrosion and heavy-metal bioremediation, and SRB populations are commonly observed in pipe and subsurface environments as surface-associated populations. In order to elucidate physiological changes associated with biofilm growth at both the transcript and protein level, transcriptomic and proteomic analyses were done on mature biofilm cells and compared to both batch and reactor planktonic populations. The biofilms were cultivated with lactate and sulfate in a continuously fed biofilm reactor, and compared to both batch and reactor planktonic populations.
Results
The functional genomic analysis demonstrated that biofilm cells were different compared to planktonic cells, and the majority of altered abundances for genes and proteins were annotated as hypothetical (unknown function), energy conservation, amino acid metabolism, and signal transduction. Genes and proteins that showed similar trends in detected levels were particularly involved in energy conservation such as increases in an annotated ech hydrogenase, formate dehydrogenase, pyruvate:ferredoxin oxidoreductase, and rnf oxidoreductase, and the biofilm cells had elevated formate dehydrogenase activity. Several other hydrogenases and formate dehydrogenases also showed an increased protein level, while decreased transcript and protein levels were observed for putative coo hydrogenase as well as a lactate permease and hyp hydrogenases for biofilm cells. Genes annotated for amino acid synthesis and nitrogen utilization were also predominant changers within the biofilm state. Ribosomal transcripts and proteins were notably decreased within the biofilm cells compared to exponential-phase cells but were not as low as levels observed in planktonic, stationary-phase cells. Several putative, extracellular proteins (DVU1012, 1545) were also detected in the extracellular fraction from biofilm cells.
Conclusions
Even though both the planktonic and biofilm cells were oxidizing lactate and reducing sulfate, the biofilm cells were physiologically distinct compared to planktonic growth states due to altered abundances of genes/proteins involved in carbon/energy flow and extracellular structures. In addition, average expression values for multiple rRNA transcripts and respiratory activity measurements indicated that biofilm cells were metabolically more similar to exponential-phase cells although biofilm cells are structured differently. The characterization of physiological advantages and constraints of the biofilm growth state for sulfate-reducing bacteria will provide insight into bioremediation applications as well as microbially-induced metal corrosion.
Background
It is becoming increasingly clear that a mode of attached growth more closely resembles in situ conditions for many microorganisms in different environments and might likely be a universal feature that presents an important physiology to explore in addition to the typically conducted studies on planktonic cells [1,2]. The subsurface is a physically dynamic habitat where fluxes in water, nutrients, temperature, pH, and osmolarity can create challenges for microorganisms to survive and thrive, and dehydration events can inhibit motility and limit nutrient availability that can result in decreased microbial activity [3]. Biofilms can provide a protective habitat for bacterial cells to exist, and recent work has demonstrated in situ biofilms in a variety of habitats [4-7]. Biofilm matrices can retain water, sorb nutrients, and protect against rapid changes in salinity, osmolarity, pH, nutrient availability, and redox [3], and we have observed the formation of biofilms (i.e., surface-attached cells) that contain Desulfovibrio species in sediments placed down-well at bioremediation field sites (Bowen-DeLeon and Fields, unpublished results).
Biofilms produced by sulfate-reducing bacteria (SRB) have been studied under the context of metal exposure and corrosion. For example, work done with Desulfovibrio desulfuricans G20 not only demonstrated the precipitation of hexavalent uranium and lead in the periplasm of the cell but also within the biofilm matrix [8-11]. SRB populations have been observed to adhere to enteric bacteria and other aerobes that have formed biofilms on underground pipelines, allowing further corrosion to occur [12]. Different corrosion studies indicate that the biofilms produced by SRB populations can be composed mainly of proteins, with minimal exopolysaccharide present [13], and this coincides with our study of D. vulgaris biofilms on glass in that the EPS (exopolymeric substance) was not enriched for carbohydrate compared to other bacteria [14]. Although these studies provide insights into corrosion and metal-reducing mechanisms of SRB biofilms, little is known about the general response in SRBs that underlie physiological adaptation between the biofilm and planktonic growth modes.
Previous studies done on aerobic or facultative pathogens have shown differential gene expression and protein profiles for biofilm cells compared to planktonic cells in different microorganisms [15-23], and the results suggest that biofilm formation is a complex and highly regulated process [24]. In order to understand molecular mechanisms of D. vulgaris survival within the subsurface and the development of SRB biofilms in different environments, we compared D. vulgaris Hildenborough biofilm and planktonic cell transcript and protein levels. Our results indicated that D. vulgaris biofilm cells altered transcript and protein abundances involved in carbon and energy metabolism, amino acid metabolism, stress response, proteases, and ribosomal proteins. These results suggested that biofilm cells were not merely analogous to planktonic cells in stationary-phase. Characterization of physiological advantages and constraints of the biofilm growth state may lead to a better understanding of how to stimulate (subsurface) or inhibit (corrosion) SRB biofilms.
Results and discussion
Growth and sampling
Three replicates of D. vulgaris cultures were grown in a continuous reactor system (CDC biofilm reactors, Biosurface Technologies) with lactate (60 mM) as the growth-limiting energy and carbon source and sulfate (50 mM) as the electron acceptor. The CDC reactors were maintained at a dilution rate of 0.084 h-1. Planktonic and biofilm biomass was collected 70 h post-coupon insertion, and both protein and carbohydrate levels had approached steady-state under the tested growth conditions at this time point (Figure 1). By 70 h the biofilm had 12 ± 0.03 μg/cm2 protein and the biofilm maintained a carbohydrate:protein ratio (C:P) of approximately 0.13 (μg/μg). Planktonic cultures had a protein level of 250 ± 2.90 μg/ml with an approximate C:P ratio of 0.10 (μg/μg). These results are consistent with the fact that D. vulgaris does not produce a carbohydrate-rich biofilm matrix under the described growth conditions [14].
Figure 1.

Protein (●,○) and carbohydrate (■,□) levels for biofilm and planktonic cells, respectively from continuous culture with lactate and sulfate.
Biofilms were compared via both transcriptomics and proteomics to the planktonic counterparts grown continuously within the reactor system and with planktonic cells grown under batch conditions. Based upon the majority of studies with aerobic and facultative bacteria, the notion of heterogeneity and complexity within biofilms has become increasingly appreciated [25] and can play a role in the interpretation of overall expression analyses. Heterogeneity issues remain to be resolved for biofilm studies; however, models predicted that the D. vulgaris biofilms in the described study were not limited for lactate or sulfate at the given biofilm depths (10-30 μm, Additional file 1) based upon substrate diffusivity with a biofilm accumulation model [26]. In addition, complete correlation between the transcriptomic and proteomic data sets cannot be expected [23,27,28] ( Additional file 2), although some genes showed similar transcript and protein trends. Therefore each data set (transcript and protein) revealed key aspects of the physiological state of the cells. While a recent study provided an estimate of translation-related sequence features to protein levels, it is still not known to what degree different mRNA and protein molecules differ with respect to in vivo half-lives, although Nie et al. [27] predicted that protein stability only accounted for 5% of the variation.
Comparison of biofilm to planktonic cells
The mRNA levels between biofilm and planktonic cells were compared using hierarchical clustering analysis (HCL) and principle components analysis (PCA). HCL compared similarities between all significantly differentially expressed genes (i.e. log2 ratio ≥ 2.0 or ≤ -2.0) between biofilm cells and the different planktonic cells. The growth-phase differences of batch, planktonic growth have been previously described [29]. The gene expression profile for biofilm cells did not cluster with exponential- and stationary-phase planktonic cells (Figure 2a). PCA showed a similar result when genes with significantly altered expression were considered (Figure 2b).
Figure 2.
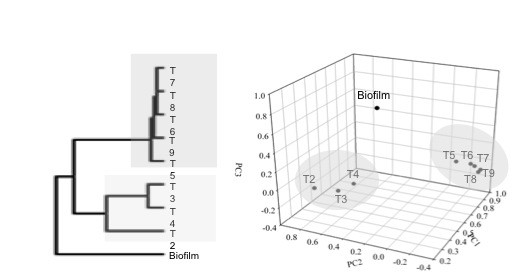
Hierarchal clustering (a) and principal components analysis (b) of the nine samples from planktonic and biofilm (T2-T4 represent exponential-phase and T5-T9 represent stationary-phase) grown on lactate and sulfate for Desulfovibrio vulgaris. The samples are grouped based upon similarities in the expression patterns of all genes with significant changes.
The biofilm mRNA data were then compared to planktonic cells grown with different carbon/energy sources via a gene expression correlation matrix with a centered Pearson correlation based upon expression of all detected genes (Figure 3). Biofilm cells were distinct when compared to multiple physiological states and were not highly correlated with cells grown planktonically with pyruvate/sulfate (r = 0.002 to 0.03), hydrogen/sulfate (r = -0.08 to -0.2), formate/sulfate (r = 0.2), or fermentatively (r = 0.1). In addition, the comparison of biofilm cells to either reactor- or batch-planktonic cells were more similar to each other (r = 0.70) than the comparison of biofilm cells to the other growth states (i.e., growth-phase and substrates). These results suggested that the biofilm is a physiological state distinct from lactate-grown, planktonic cells (exponential or stationary-phase), and that many genes in biofilm cells had similar expression levels regardless of being compared to reactor- or batch-planktonic cells. In addition, the lactate/sulfate-grown biofilm cells had a disticnt expression profile when compared to planktonic cells grown with different electron/carbon sources (e.g., lactate, hydrogen, formate, pyruvate).
Figure 3.

Correlation matrix for D. vulgaris gene expression that compares biofilm cells to planktonic cells grown with different substrates using a centered Pearson correlation. Cells were grown in a defined S4D medium with sulfate (except for ‘pyruvate only’), and provided with a different carbon and energy source (lactate, pyruvate, hydrogen, or formate). The correlation matrix was generated with MicrobesOnline functional genomics analysis (microbesonline.org).
Transcripts and proteins with increased abundances
A total of 231 and 108 transcripts had increased expression in biofilm cells when compared to batch- or reactor-grown planktonic cells, respectively (Table 1 and 2; whole genome expression data in Additional file 3 and Additional file 4). When compared to either reactor- or batch-planktonic cells, most of the same hypothetical and conserved hypothetical genes had altered expression. More motility genes were up-expressed in biofilm cells when compared to batch cells; however, the five genes up-expressed in biofilm cells when compared to reactor-planktonic cells were also up-expressed when compared to batch-planktonic cells. Current work includes the elucidation of flagellar genes needed for biofilm development and maintenance. Regardless of biofilm cells being compared to reactor- or batch-planktonic cells, significantly up-expressed genes in biofilm cells were involved in amino acid metabolism, energy conservation, signal transduction, and translation (Table 1, Table 2, Figure 4).
Table 1.
COG functional categories with altered expression (z score > 2.0) in biofilm cells compared to batch, planktonic cells (exponential-phase)
| COG Function Categories | Up | Down |
|---|---|---|
| Amino acid transport and metabolism |
19 |
20 |
| Carbohydrate transport and metabolism |
11 |
3 |
| Cell cycle control, cell division, chromosome partitioning |
- |
- |
| Cell motility |
15 |
2 |
| Cell wall/membrane/envelope biogenesis |
8 |
5 |
| Chromatin structure and dynamics |
- |
- |
| Coenzyme transport and metabolism |
11 |
8 |
| Defense mechanisms |
- |
3 |
| Energy production and conversion |
44 |
17 |
| Function unknown |
13 |
9 |
| General function prediction only |
18 |
12 |
| Inorganic ion transport and metabolism |
12 |
8 |
| Intracellular trafficking, secretion, and vesicular transport |
5 |
6 |
| Lipid transport and metabolism |
- |
10 |
| Nucleotide transport and metabolism |
3 |
4 |
| Posttranslational modification, protein turnover, chaperones |
11 |
3 |
| Replication, recombination and repair |
5 |
5 |
| Secondary metabolites biosynthesis, transport and catabolism |
3 |
3 |
| Signal transduction mechanisms |
40 |
9 |
| Transcription |
11 |
9 |
| Translation, ribosomal structure and biogenesis | 2 | 44 |
Table 2.
COG functional categories with altered expression (z score > 2.0) in biofilm cells compared to reactor, planktonic cells (exponential-phase)
| COG Function Categories | Up | Down |
|---|---|---|
| Amino acid transport and metabolism |
6 |
28 |
| Carbohydrate transport and metabolism |
4 |
1 |
| Cell cycle control, cell division, chromosome partitioning |
- |
2 |
| Cell motility |
5 |
1 |
| Cell wall/membrane/envelope biogenesis |
5 |
5 |
| Chromatin structure and dynamics |
- |
- |
| Coenzyme transport and metabolism |
4 |
11 |
| Defense mechanisms |
- |
2 |
| Energy production and conversion |
22 |
24 |
| Function unknown |
7 |
5 |
| General function prediction only |
10 |
15 |
| Inorganic ion transport and metabolism |
4 |
5 |
| Intracellular trafficking, secretion, and vesicular transport |
2 |
4 |
| Lipid transport and metabolism |
1 |
5 |
| Nucleotide transport and metabolism |
- |
1 |
| Posttranslational modification, protein turnover, chaperones |
8 |
4 |
| Replication, recombination and repair |
1 |
9 |
| Secondary metabolites biosynthesis, transport and catabolism |
1 |
3 |
| Signal transduction mechanisms |
22 |
3 |
| Transcription |
3 |
6 |
| Translation, ribosomal structure and biogenesis | 1 | 21 |
Figure 4.

Cell-wide depiction of significantly up-expressed transcripts for mature D. vulgaris biofilms compared to both batch and chemostat planktonic cells. The following genes were also detected as significantly up-expressed proteins: SodB, KatA, Hsp20 (DVU2441), Hsp20 (DVU2442), UspA (DVU0423).
In the iTRAQ proteome analysis, a total of 560 proteins passed the data filtering criteria ( Additional file 5), and there were 11 and 17 proteins that had significantly increased levels in the biofilm cells when compared to the batch or reactor planktonic samples, respectively. Energy conversion, stress response, and unknown function proteins had increased abundances and amino acid metabolism and signal transduction had decreased abundances in biofilm cells when compared to either batch- or reactor-planktonic cells (Table 3 and 4).
Table 3.
Comparison of biofilm to batch culture
| DVU Gene # | Gene Name | Description | Log2ratio (z-score) |
|---|---|---|---|
| 0429 |
echF |
Ech hydrogenase, putative subunit |
3.41 (3.91) |
| 0799 |
- |
conserved hypothetical protein |
3.02 (3.46) |
| 2342 |
- |
amino acid ABC transporter |
2.34 (2.68) |
| 1817 |
cyf |
cytochrome c553 |
2.31 (2.64) |
| 2781 |
- |
hypothetical protein |
2.30 (2.63) |
| 0797 |
- |
conserved hypothetical protein |
2.15 (2.46) |
| 0752 |
- |
amino acid ABC transporter |
1.95 (2.22) |
| 0588 |
hybA |
formate dehydrogenase, beta subunit |
1.94 (2.22) |
| A0108 |
- |
- |
1.87 (2.14) |
| 2982 |
leuC |
3-isopropoylmalate dehydratase |
1.78 (2.03) |
| 2333 |
ndk |
nucleoside dephosphate kinase |
1.77 (2.02) |
| 0465 |
trpE |
anthrnilate synthase, component I |
-1.72 (-2.00) |
| 1207 |
fabH |
3-oxyacyl-acyl-carrier protein synthase |
-1.72 (-2.00) |
| 0107 |
glnH |
glutamine periplasmic binding protein |
-1.75 (-2.04) |
| 1873 |
ppiB-2 |
peptidyl-prolyl-isomerase |
-1.78 (-2.07) |
| 0997 |
metF |
5,10-methylenetetrahydrofolate red. |
-1.79 (-2.09) |
| 3029 |
pta |
phosphate acetyltransferase |
-1.87 (-2.17) |
| 2449 |
metK |
S-adenosylmethionen synthetase |
-1.88 (-2.18) |
| 0470 |
trpB-2 |
tryptophan synthase |
-1.94 (-2.25) |
| 1095 |
argG |
argininosuccinate synthase |
-1.96 (-2.28) |
| 0462 |
- |
chorismate mutase |
-1.99 (-2.31) |
| 0607 |
ahcY |
adenosylhomocysteinase |
-2.06 (-2.40) |
| 2590 |
- |
sensory box protein |
-2.38 (-2.76) |
| 1241 |
- |
conserved hypothetical protein |
-2.55 (-2.96) |
| 3371 |
metE |
S-methyltransferase |
-2.83 (-3.28) |
| 0494 | - | aminotransferase V | -3.30 (-3.82) |
This table lists all proteins that were significantly increased or decreased (|z| ≤ 2) when biofilm cells were compared to batch, planktonic cells.
Table 4.
Comparison of biofilm to reactor, planktonic culture
| DVU Gene # | Gene Name | Description | Log2ratio (z-score) |
|---|---|---|---|
| 0799 |
- |
- |
3.65 (4.78) |
| 0797 |
- |
- |
3.08 (4.02) |
| 0587 |
fdnG-1 |
formate dehydrogenase, alpha subunit |
2.59 (3.38) |
| 1817 |
cyf |
cytochrome c553 |
2.44 (3.18) |
| 2135 |
- |
hypothetical protein |
2.39 (3.11) |
| 2442 |
- |
Hsp family |
2.30 (2.99) |
| 0251 |
- |
putative membrane protein |
2.20 (2.86) |
| 0588 |
hybA |
formate dehydrogenase, beta subunit |
2.11 (2.74) |
| 2410 |
sodB |
superoxide dismutase, Fe |
2.02 (2.63) |
| 2100 |
- |
universal stress protien |
1.85 (2.40) |
| A0115 |
- |
- |
1.81 (2.34) |
| 2543 |
b0873 |
hybrid cluster protein |
1.80 (2.34) |
| 0429 |
echF |
EchF |
1.75 (2.27) |
| 0430 |
echE |
EchE |
1.66 (2.15) |
| 2441 |
hspC |
Hsp20 family |
1.63 (2.10) |
| 1067 |
- |
Bmp family membrane protein |
1.60 (2.07) |
| 1876 |
dnaJ |
DnaJ |
1.56 (2.02) |
| 0467 |
trpD |
anthrnilate phosphoribosyltransferase |
-1.52 (-2.06) |
| 0415 |
pepA |
cytosol aminopeptidase |
-1.55 (-2.10) |
| 3027 |
glcD |
glycolate oxidase |
-1.80 (-2.43) |
| 2590 |
- |
sensory box protein |
-1.81 (-2.44) |
| 0494 |
- |
aminotransferase V |
-1.82 (-2.47) |
| 1257 |
- |
RNA-binding protein |
-2.00 (-2.69) |
| 0318 | - | TPR domain protein | -2.23 (-3.00) |
This table lists all proteins that were significantly increased or decreased (|z| ≤ 2) when biofilm cells were compared to reactor, planktonic cells.
When comparing the transcriptomic and proteomic data, some corresponding mRNAs and proteins both displayed increased abundances. For example, a presumptive c553 cytochrome (DVU1817), echF/EchF (DVU0429), hybrid cluster (DVU2543), and hybA/HybA (DVU0588) (Tables 3, 4, Additional file 3 and Additional file 4), and these respective results corroborated the other (discussed below). However, as expected, there were instances that elevated mRNA levels did not correspond to increased protein abundances (e.g., DVU0586, DVU3133, DVU2100, DVU0251). In addition, there were detected proteins with elevated levels and not corresponding mRNA levels when compared to planktonic, reactor cells (e.g., DVU0797, DVU0799, DVU2781, DVU0430-echE, DVU0587-fdnG1, DVUA0108, DVUA0115). These results are most likely a result of differences in mRNA and protein half-lives and highlight the utility of both transcriptomic and proteomic analyses.
Transcripts and proteins with decreased abundances
A total of 358 transcripts were down-expressed in biofilm cells compared to the planktonic counterparts grown in either the batch or reactor systems. Similar to the up-expression patterns, numerous down-expressed genes (Table 1 and 2) were annotated as hypothetical or conserved hypothetical proteins. Interestingly, 10% of the down-expressed genes were predicted to be involved with translation and ribosomal structure/biogenesis. Genes involved with translation were down-expressed for biofilm cells compared to either planktonic state (batch or reactor).
In a previous study, ribosomal gene expression decreased by 3-fold during the exponential/stationary-phase transition for planktonic cells and remained 2-fold down throughout the stationary-phase [29]. Correspondingly, several ribosomal genes were identified in the transcriptomic and proteomic data in the current study and displayed a similar trend in decreased abundances ( Additional file 6). However, when the log-ratios of ribosomal protein transcripts with significant expression changes (n = 15; Additional file 6) were compared between biofilm and planktonic cells in different growth phases, there was less down-expression for biofilm cells compared to stationary-phase cells (Figure 5; -0.8 ± 0.7 vs. -2.1 ± 0.8).
Figure 5.

Transcript levels for 15 ribosomal proteins compared among four (expontial, transition, stationary, and biofilm) different growth stages. The genes are sorted highest to lowest with respect to the levels observed for exponential-phase cells.
Because the production of ribosomal transcripts/proteins typically correlates with metabolic activity, we sought to confirm this result by measuring the activity levels of planktonic and biofilm cells with the respiratory indicator, 5-cyano-2,3,-ditolyl tetrazolium chloride (CTC) as previously described [30]. Biofilm cells were only 2-fold less active than exponential-phase cells. However, stationary-phase cells were approximately 500-fold less active than exponential-phase cells and 300-fold less active than biofilm cells. The metabolic activity of biofilm cells might not result in ‘exponential’ cell growth (and/or biomass accumulation) per se, but may provide the energy required for nutrient uptake and biofilm maintenance. Indeed, a recent study that developed a model to explain variation in growth rate optimization and cellular stoichiometry hypothesized that resource allocation and nutrient-use efficiency was a result of distinct growth and uptake activities [31]. Our results suggested that the biofilm physiological state should be further evaluated in the context of ‘growth’ and resource allocation in comparison to planktonic cells.
The notion that biofilm cells were different from stationary-phase cells contradicts previous work that estimates the strong gene expression similarity between these growth states for different bacteria, albeit mostly aerobes [16,32]. These results corroborated the increased expression of ribosomal transcripts and protein levels that indicated biofilm cells were active and that slower cell division (i.e., steady-state biofilm) does not equate to lower metabolism. In order to determine possible functions that contributed to the distinct state of D. vulgaris biofilms, we further analyzed various functional categories of genes and proteins in biofilm cells with altered abundances.
(i) Energy conversion
The expression of genes involved with energy conversion and carbon flow within the biofilm is a major contributor to the uniqueness of the biofilm growth state regardless of being compared to batch- or reactor-planktonic cells. Biofilm cells had different mRNA and protein levels related to the pyruvate to acetate and formate metabolic nodes. Two annotated pyruvate:ferredoxin oxidoreductases, which convert pyruvate to acetyl-CoA, had differential expression in biofilm cells with up-expression of oor (DVU1944, DVU1945, DVU1946, and DVU1947) and down-expression of por (DVU1569 and DVU1570) compared to planktonic cells (Figure 6 and 7). Expression of the mRNA was verified via qPCR, and oor and por genes were up- and down-expressed, respectively.
Figure 6.

Conceptual model (a) of the transcriptional responses in energy metabolism and pyruvate-acetate metabolic node for a mature D. vulgaris biofilm (red is up-expressed and blue is down-expressed) compared to planktonic cells.
Figure 7.

Expression levels of representative genes involved in carbon and energy flow that displayed altered expression distinct to biofilm cells when compared to reactor planktonic cells, batch exponential-phase, or batch stationary-phase.
Although expression for the annotated pyruvate-formate lyase was not altered, the abundance of formate dehydrogenase (DVU0588-hybA transcript and protein) was increased (Figure 6, Table 3 and 4). However, only protein abundance was increased for fdnG1 (DVU0587) (Table 4). A cytochrome c553 (DVU1817), which is the electron transfer partner for the formate dehydrogenase in D. vulgaris[33], had increased abundances for both mRNA and protein levels (Figure 6, Table 3 and 4). Because formate dehydrogenase was increased in biofilms, enzymatic assays were done to compare activity within biofilm and planktonic cells. Crude extracts of biofilm cells had an approximate 26-fold higher rate of formate-dependent methyl viologen reduction than reactor planktonic cells, and these results indicated that the increased gene expression corresponded to increased enzymatic activity.
Formate, like hydrogen, has been proposed to be a mechanism of electron flow in Desulfovibrio[34,35]. Formate cycling within lactate-respiring biofilm cells could explain the increase in expression and activity of the formate dehydrogenases and might suggest that biofilm cells are better able to cycle formate compared to planktonic cells in a stirred, homogenous environment. Additional hydrogenases were also differentially expressed within the biofilm cells compared to planktonic cells (Figure 6; DVU3145). The possible route of proton and electron cycling in SRB has long been debated and biofilm cells could be an additional physiological state to provide significant insights.
(ii) Carbohydrate metabolism
D. vulgaris biofilm cells had several genes involved with glycerol uptake and utilization shift in expression (Figure 4, Additional file 5). These results suggested that biofilm cells may use phospholipids released from lysed neighboring cells and that these may possibly serve as a carbon backbone for amino acid biosynthesis because the overall glycolytic pathway was not significantly up-expressed. In addition, an annotated fructose-1,6-bisphosphatase (DVU1539) was transcriptionally down-expressed in biofilm cells compared to reactor planktonic cells and suggested a decrease in gluconeogenesis. Decreased gluconeogenic activity may indicate a decreased requirement for new amino sugars for cell wall synthesis, and this observation corresponded with the decreased expression of genes involved with cell wall synthesis and cell division. It should be noted that biofilm cells did not appear to be smaller based upon electron microgrpahs (data not shown).
(iii) Nitrogen metabolism
The increased expression of nrfA (DVU0625) may indicate the available nitrogen source (i.e., ammonium) might be limited within some biofilm cells. nrfA, annotated as cytochrome c nitrite reductase (c552), had increased mRNA expression within biofilm cells compared to planktonic cells. This result is interesting because ammonia was added as the nitrogen input into the system and nitrite reduction has not been linked to growth [35,36]. D. vulgaris also up-expressed nrfA when cells were exposed to acetone, ethanol, nitrite, and alkaline pH [37]; He and Zhou, unpublished results], and these results suggested that nrfA expression may be part of a more general response. D. vulgaris can utilize nitrogen via the nitrogenase that is encoded on the 200-kb plasmid (pDV1), but nifU gene expression did not change as detected via qPCR (data not shown).
(iv) Stress response
Various chaperone proteins, including uspA (DVU0423) and three hsp20 genes (DVU2442, 2241, and 1471), had increased transcripts within the biofilm compared to both reactor and batch planktonic cells (Figure 4). D. vulgaris biofilms had increased transcripts for the stress response proteins superoxide dismutase (sodB DVU2410), alkyl hydroperoxide reductase (ahpC DVU2247), msrB (DVU0576) and ahpC/TSA (thiol specific antioxidant) and an increase in protein levels for SodB (Table 4). However, preliminary results indicated that D. vulgaris biofilm cells do not have increased tolerance to dissolved oxygen in terms of growth initiation (Sundararajan and Fields, unpublished results).
Although mRNA levels for stress response genes were elevated in the biofilms of other organisms (e.g.E. coli, P. aeruginosa, Legionella pneumophila and Staphylococcus aureus), the anaerobic, hyperthermophile Thermotoga maritima showed decreased expression in similar genes [16-18]. In addition, D. vulgaris grown on steel slides showed a similar trend in down-expression of related genes [38]. For aerobic bacteria, oxidative stress within biofilms is thought to be a result of slow growth in conjunction with a shift away from oxygen respiration at different biofilm depths [25]. For D. vuglaris biofilms, the relatively slower growth may be associated with lower intracellular levels of reducing equivalents that would leave the cell more susceptible to potential oxidizing agents, and a defense mechanism could be elevated levels for genes and proteins involved in potential detoxification.
These responses are also thought to increase during iron limitation due to the increase in iron sequestration and intracellular accumulation [39]. This is not surprising given the ability of some metals to form hydroxyl radical, metallo-oxo, and metallo-peroxo species [40]. The elevated abundances for genes and proteins involved in detoxification of oxidative agents also coincided with the up-expression of iron acquisition mechanisms in biofilm cells (see below) and the possible connection between iron storage and oxidative damage response may at least partially explain the observed expression profiles.
Interestingly, more genes annotated as proteases were down-expressed, including lon protease (DVU1337), clpX (DVU1336), clpB (DVU1874), htrA peptidase (DVU3278), htpG (DVU2643), and hflC (DVU0683) than were up-expressed. A decrease in proteases for D. vulgaris biofilm cells contrasted with observations for stationary-phase planktonic cells [29]. Biofilm cells might simply have a slower turnover of proteins due to the biofilm growth mode, but further research is needed to determine this characteristic.
Biofilm cells had altered transcript expression profiles for methyl-accepting chemotaxis proteins, histidine kinases, response regulators, and sensory box proteins (Figure 4, Table 1, 2, 3, and 4, Additional file 5). D. vulgaris contains a number of two-component signal transduction systems and methyl-accepting chemotaxis proteins; however, little is known about the required signals and regulation that these proteins may impose on the cells nor how that regulation contributes to biofilm formation and/or disassembly.
(v) Iron acquisition
D. vulgaris biofilms may be iron limited as indicated by the increase in transcripts for the subunit feoA (DVU2572), part of the iron transporter Feo, and fepC (DVU0103), a presumptive ABC transporter for iron uptake. However, similar genes were up-expressed in D. vulgaris stationary-phase, planktonic cells [29], and it is possible that an iron acquisition response is the result of increased sulfides associated with D. vulgaris growth in general. The iron storage protein bacterioferritin (DVU1397) had increased transcripts within biofilm cells along with the ferritin storage protein (DVU1568). Increased abundances of iron sequestration proteins has been reported in other biofilm studies [17,38,39] but iron transport systems like feoAB were transcriptionally down-expressed in D. vulgaris biofilm cells grown on steel [38]. Several studies have indicated that iron plays different roles within biofilm formation of various organisms. P. aeruginosa uses iron as a signal to produce thick biofilms with mushroom morphology while excess iron is detrimental to S. aureus and L. pneumophila biofilms [39,41,42].
(vi) Transporters
Biofilm cells up-expressed four presumptive ABC transporter genes (DVU2387, DVU2384, DVU0484, and DVU2385) and increased abundances for two transporter proteins (DVU0752 and DVU2342) (Figure 4, Table 3). Differential expression of various ABC transporters has commonly been reported in different biofilms (e.g., P. aeruginosa, E. coliB. subtilisThermotoga maritimeStreptococcus mutans and S. aureus) [16,17,19,20]. The putative substrates have yet to be determined, but biofilm cells may transport different small molecules and solutes compared to planktonic cells.
For D. vulgaris biofilm cells, various subunits of the Sec transport system were transcriptionally down-expressed including secE (DVU2922), secY (DVU1323), and secG (DVU1676), and SecD at the protein level. Interestingly, a preprotein translocase, YajC (DVU1820), was increased at the protein level. Different types of secretion may be increased as indicated by increased transcripts of a type I system (DVU1013) and increased protein levels of an annotated type III protein secretion system (DVUA0115). The ORF, DVUA0115, is located on the D. vulgaris plasmid (pDV1), and a strain cured of the plasmid is biofilm deficient [14].
Biofilm cells differentially expressed two putative sulfate permeases at the mRNA level; DVU0053 was up-expressed and DVU0279 was down-expressed. A putative lactate permease mRNA (DVU2285) was down-expressed in biofilm cells, and a similar trend of expression was observed with exponential-phase, planktonic cells [29].
(vii) Extracellular proteins
In-gel digestion analysis of the extracellular samples revealed 188 proteins that passed the proteomic analysis cutoff criteria. The list was screened against signal and transmembrane domain predictions generated by Phobius [43] and revealed a general enrichment for proteins with signal sequences and few proteins that contained transmembrane domains were identified ( Additional file 7). The most highly abundant protein in the sample was DVU1012, annotated to have a vonWillebrand factor domain (vWF). Interestingly, vWF proteins are large, multimeric glycoproteins that can have a variety of roles with different ligands that include cell adhesion, pattern formation, and signal transduction [44]. However, the presumptive protein is also annotated to contain a hemolysin-type calcium-binding domain and only has 21% homology with the closest homolog. Interestingly, a paralog of DVU1012, DVU1545, was also enriched in the extracellular protein fraction from biofilms. In addition, two presumptive proteins that had increased protein but not mRNA levels, DVU0797 and DVU0799, were also detected at significant levels in the extracellular, biofilm fraction. Hence, though the function of these proteins in D. vulgaris remains obscure, participation in extracellular matrix formation in biofilm structures could be a potential role. Further work is underway to determine both structural and enzymatic proteins in the extracellular biofilm.
(viii) Fatty acid and lipid synthesis
Numerous genes and proteins involved in fatty acid synthesis had lower abudances in biofilm cells (Table 1, 2, 3 and 4, Additional file 3, Additional file 4, Additional file 5). The gene lpxC (lipid A production) that encodes UDP-3-0-acyl N-acetylglucosamine deacetylase (DVU2917) was decreased in biofilm cells. Transcripts for a TolB protein, which is part of the peptidoglycan-associated lipoprotein (PAL) multiprotein complex and associates with OmpA and Lpp to contribute to membrane integrity and peptidoglycan turnover, were decreased. There were also decreased protein levels for undecaprenyl diphosphate synthase (DVU0869). These results corroborated the suggestion that biofilm cells were in a different growth state relative to exponential-phase cells in terms of cellular division (i.e., slower cell wall and membrane synthesis).
(ix) Amino acid and nucleotide production
The decrease in amino acid and nucleotide biosynthesis transcripts and/or proteins within biofilm cells corresponded well with the reduction of protein synthesis and cell division (Tables 1, 2, 3 and 4). Only two genes annotated as enzymes involved with amino acid synthesis were increased in biofilm cells, gltA (DVU2476) and cysK/CysK (DVU0663) that encode putative glutamate and cysteine synthases, respectively. Cysteine synthase converts O-acetyl-serine and hydrogen sulfide to cysteine. Transcriptional analysis of T. maritima biofilms revealed an increase in cysteine biosynthesis enzymes as well as an increase in iron and sulfur uptake systems and proteins used for the incorporation of iron-sulfur clusters [17]. Since sulfates are usually incorporated into cells by the cysteine biosynthesis pathway, the authors suggested that an increase in transcripts for cysteine biosynthesis was a result of the demand for iron-sulfur containing proteins [17]. Several iron-sulfur containing proteins had elevated abundances at both the transcript and protein levels within D. vulgaris biofilms. In addition, increasing amounts of cysteine are toxic to D. vulgaris planktonic cells, and growth effects are observed above 1 mM (data not shown). Whether the increase in enzymes involved with cysteine biosynthesis is due to a demand for sulfur acquisition and/or a means to help regulate local sulfide concentrations is yet to be determined.
Genes annotated for enzymes involved in tryptophan biosynthesis were down-expressed within biofilm cells (8 of the 12 genes in the operon were significantly down-expressed) (Figure 8). Two enzymes that contribute to aromatic amino acid and quinone synthesis, 3-octaprenyl-4-hydroxybenzoate carboxy-lyase (DVU3307) and type II 3-dehydroquinate dehydratase (aroQ DVU1665) were also down-expressed. Coinciding with these data, seven of thirteen enzymes involved with converting phosphoenolpyruvate and erythrose 4-phophate into tryptophan had decreased protein abundances ( Additional file 3, Additional file 4, Additional file 5). In previous studies, enzymes involved in tryptophan biosynthesis were elevated in young E. coli biofilms but declined during biofilm maturation [45,46]. In this study, the decreased levels of tryptophan genes/proteins corresponded with the decreased abundances in enzymes involved with converting tryptophan to indole, which is thought to inhibit biofilm formation in E. coli[46,47]. Although D. vulgaris has an annotated tryptophanase, expression did not change significantly within the biofilms. The effect and/or role of indole on D. vulgaris biofilm formation and maturation are not known; however, hydroxyindole increased the formation of D. vulgaris batch biofilms 2-fold (Ramsay and Fields, unpublished results).
Figure 8.

Graphical representation of the biofilm to planktonic comparison via log2transcript ratios. As an example, Ech hydrogenase genes and cytochrome c-553 components had increased expression in biofilm cells and tryptophan biosynthesis genes were down expressed.
Conclusions
Our study compared multiple D. vulgaris transcriptomes and proteomes from different growth phases in the same defined sulfate medium with the same carbon and energy source (lactate). The data indicated that the D. vulgaris biofilm growth mode was distinct when compared to both exponential- and stationary-phases in the planktonic state. The biofilm transcriptome did have a cluster of genes (n = 70) with similar expression patterns to stages of stationary-phase that included putative proteins involved in DNA modification, protein turnover, signal transduction, and phage genes, but over half of the genes were annotated as hypothetical or conserved hypothetical proteins. The role of the hypothetical proteins (proteins of unknown function) are not known but likely play a role in the distinct physiological state of the biofilm [48].
In comparison, most of the genes that had similar expression values between biofilm and exponential-phase cells displayed a trend of down-expression (e.g., phage genes) or no significant change. In addition, the transcript levels for 15 rRNAs were more similar to exponential-cells as were metabolic indicator assays. These results indicated that the biofilm growth-phase was a distinct mode of growth that was not analogous to stationary-phase and this result contradicts previous studies with aerobic microorganisms.
Gene expression responses in the biofilm cells included the down-expression of an operon that encoded por (pyruvate:ferredoxin oxidoreductase), acetate kinase, a possible lactate dehydrogenase, a phosphotransacetylase, and ech/Ech. Interestingly, preliminary results with a ΔechA mutant indicated a deficiency in biofilm formation (Sabalowsky, Wall and Fields, unpublished results). Also related to carbon flow, mRNA for three of the four subunits (oorABD) of a separate PFO were up-expressed in the biofilm cells (Figure 6 and 7). The putative por gene encodes a large (1215 aa), multi-domain protein whereas the oor genes encode putative subunits of a multimeric protein [49]. Both por and oor are predicted to catalyze decarboxylation of pyruvate to acetyl-CoA, but the altered levels may result from different regulatory features of the different PFOs (i.e., multimeric vs. mutlidomain) or different kinetic characteristics.
The respective increase and decrease of presumptive electron transfer systems suggested that electron flow is altered in biofilms compared to both exponential- and stationary-phase cells. The different patterns of gene expression for modules in electron transfer systems may reflect differences in rates of nutrient acquisition and incorporation. These results indicate that D. vulgaris biofilm has an altered metabolism not a lower metabolism. With respect to hydrogen, a recent study showed that biofilm cells grown on an electrode up-expressed hyn-1 and hyd, [NiFe] and [Fe] hydrogenases, respectively, as well as hmc genes [50]. In comparison, D. vulgaris biofilms on glass slides did not up-express the same hydrogenases nor the hmc genes when grown as biofilms with sulfate and lactate.
Future work is needed to discern protein interactions that control metabolic flux and cell behavior through particular nodes in order to elucidate the relationships between genotype and phenotype in the context of distinct cellular states, including young and mature biofilms. While the expression of other systems is certainly involved in biofilm physiology, the presented results indicated that sulfate-reducing biofilm cells use a unique combination of genes and proteins to establish distinct energy flow and cell associations different from sulfate-reducing planktonic cells. Similar developmental states have been described in P. aeruginosa and Candida albicans biofilms [18,51]. This study provides insights into the formation, growth, and development of anaerobic, sulfate-reducing biofilms, and the improved understanding of the genetic control for SRB biofilms will have implications for biotechnology, the environment, and human health.
Methods
Bacterial strains and growth conditions
Cultures of Desulfovibrio vulgaris ATCC 29579 were acquired from Dr. T.C. Hazen (Lawrence Berkeley National Laboratory). Three CDC reactors that contained LS4D defined medium [29] were used to propagate biofilms, and the CDC reactors (continuous culture conditions) were maintained at 30°C, continuously stirred at 125 rpm, and nitrogen was sparged into the head-space at a rate of 0.85 kPa/min to ensure anaerobic conditions. Growth was initiated with an exponential-phase culture and cells were incubated for 24-h in a batch-mode. After the 24-h incubation, eight modified coupon holders were inserted into each CDC reactor that held glass slides and fresh medium was pumped into the reactor from an anaerobic reservoir (D = 0.084 h-1). Biofilms were allowed to form for 70-h and the reactor monitored for cell, lactate, acetate, and sulfate levels. At the end of the 70-h biofilm growth period, coupons were removed from each reactor and scrapped in order to collect the biofilm biomass. Biofilm growth in reactors showed that under these conditions, steady-state biofilm growth was reached within 70-h with consistant protein and carbohydrate levels (Figure 1).
Sample collection for transcripts and proteins
Three coupons from each reactor were pooled together and scraped into ice-cold phosphate buffered saline (1 ml PBS; pH 7.2) to represent three biological replicates for transcriptomic analysis. Samples were kept on ice and were centrifuged immediately after scraping at 5,000 x g for 6 min. The PBS supernatant was removed after centrifugation and pellets were immediately frozen in liquid nitrogen. Concurrently, planktonic cells (60 ml) were collected from each reactor and immediately chilled as described previously [29] for transcriptomic analysis. The chilled samples were pelleted via centrifugation (4,000 x g; 6 min; 4°C). The supernatant from these samples was removed and pellets were immediately frozen in liquid nitrogen. All pelleted samples were stored at -70°C.
The remaining five coupons from each reactor were rinsed briefly with ice-cold 50 mM HEPES and pooled in ice-cold 500 mM TEAB 4 M urea solution (1 ml). The sample was sonicated for 10 min on ice and pelleted via centrifugation (30 min at 10,000 x g at 4°C). Concurrently, planktonic-phase culture (33 ml from each reactor, VT = 100 ml) was collected and pelleted via centrifugation (4,000 x g; 10 min; 4°C). The pellets were rinsed in 50 mM HEPES (1 ml) by vortexing and pelleted again via centrifugation (5,000 x g; 8 min; 4°C). The pellets were resuspended in 500 mM TEAB 4 M urea (1 ml) and sonicated on ice for 10 min. After sonication, samples were pelleted via centrifugation (10,000 x g, 30 min; 4°C) and supernatants were saved. A separate planktonic sample grown under batch conditions in LS4D was also collected and sonicated as described above. All samples were stored at -70°C.
Chemical analysis
Protein levels were measured via the Lowry Method [52] with bovine serum albumin (Pierce Biochemicals) as the standard. Lactate, acetate, and sulfate concentrations were determined by ion chromatography (Metrohm-Peak) with a Metrosep organic acid column and a Metrosep Anion Supp 5 column as described previously [29]. All measurements were done in duplicate.
Oligonucleotide probe design and microarray construction
DNA microarrays that included 3,482 of the 3,531 annotated protein-encoding sequences of the D. vulgaris genome were constructed with 70-mer oligonucleotide duplicate probes as previously described [28,29,53]. Following examination of the entire probe set using the oligonucleotide probe design criteria [52], 3,471 (97.1%) specific oligonucleotide probes were obtained, and 103 probes (2.9%) were nonspecific. In addition, 10 oligonucleotides for 10 human genes and 10 oligonucleotides for 10 Arabidopsis genes were used with the D. vulgaris genome for positive (with mRNA spiked) or negative (without mRNA spiked) controls. Each slide contained duplicates of each oligonucleotide probe. Six concentrations of genomic DNA were spotted (eight duplicates on a single slide) as positive controls.
Total RNA extraction, purification, and labeling
RNA was isolated, transcribed and labeled as described previously [29,53,54]. Briefly, total cellular RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA) and was purified with an RNeasy mini kit (QIAGEN Valencia, CA) with an on-column DNase treatment. cDNA probes were generated by reverse transcription (RTase) of 10 μg purified RNA with random hexamers (Invitrogen) labeled with Cy3-dUTP or Cy5-dUTP (Amersham Biosciences, Piscataway, NJ). RNA was removed by NaOH treatment after labeling and cDNA was immediately purified with a QIAGEN PCR mini kit. Two samples of each total RNA preparation were labeled, one with Cy3-dUTP and the other with Cy5-dUTP, for microarray hybridization.
Microarray hybridization, washing, and scanning
For determination of the overall hybridization signals, the sensitivity, and the number of genes detected, a genomic DNA or RNA sample labeled with a single dye was used as previously described [28,29,53,54]. Hybridization was performed using a slide for each biological replicate.
Microarray image quantification and data analysis
To determine the signal fluorescence intensity for each spot, 16-bit TIFF scanned images were analyzed with ImaGene v6.0 (Biodiscovery, Marina Del Rey, CA) to quantify the spot signal, spot quality, and background fluorescent intensities. Empty spots, poor spots, and negative spots (e.g., spots without deposited probe) were flagged according to the instruction of the software and removed in the subsequent analysis as previously described [28,29,53,54]. The data files were subjected to Lowess intensity-based normalization and were analyzed further with the MicrobesOnline (microbesonline.org) workbench as previously described with a statistical model that incorporated both per-gene variance (z values) and operon structure (OpWise) to compute the posterior probability that the expression level of each gene changed in the direction indicated by the mean value [55,56]. Data for members of operons without a consistent signal for replicates were not used and genes with log2 ratios of | z | > 2.0 were considered significant. The microarray expression data will be publicly available within the microbesonline workbench (microbesonline.org) as well as GEO (GSE35883).
Protein preparation, labeling, and analysis
For the planktonic cell sample, 35-mL aliquots of planktonic cells were harvested from three separate bioreactors in separate falcon tubes. The resulting cell pellets were pooled into 1 mL of lysis buffer (500 mM triethylammonium bicarbonate with 4 M urea). To harvest biofilm, the glass slides were rinsed gently with 50 mM phosphate buffered saline, pH 7.2, before being scraped off the glass slides directly into 1 mL of lysis buffer. To provide a non-biofilm associated control, a separate 100 mL D. vulgaris aliquot was grown in a serum bottle at 30°C for 30 hours, reaching a maximum OD600 of 0.86. The sample was pelleted serially into a single 50 mL falcon tube. The pellet was washed gently in 1 mL of 1 M HEPES buffer, pH 7.2 and the sample was pelleted again. The final pellet was resuspended in 1 mL of lysis buffer. The three samples (biofilm, planktonic, and batch) were lysed by pulsed sonication on ice for 10 minutes and the samples were clarified by centrifugation at 10,000 × g for 30 min. Samples were shipped on dry ice overnight. iTRAQ labeling was carried out as previously described [57]. Briefly, samples were iTRAQ labeled and pooled as per manufacturers instructions (Applied Biosystems, Framingham, MA, USA). The labeled samples were acidified (pH 3) and fractionated via strong cation exchange (Ultimate HPLC, Famos Autosampler, Dionex-LC Packings, Sunnyvale, CA, USA) using a PolyLC Polysulfoethyl A column (4.6 mm × 100 mm) and a three step 0-100% gradient in buffer B (800 mM KCl, 25% ACN, and 0.1% FA). Fractions were collected at a flow rate of 700 μl/min on the basis of the UV trace at 214 nm. Several fractions were pooled post-collection to yield a total of 20 fractions. Each fraction was desalted (C18 MacroSpin Columns (Nest Group, Southborough, MA), dried using a vacuum centrifuge, reconstituted in 86 μL 0.1% FA and subjected to reverse phase separation, (Ultimate HPLC, Famos Autosampler, Dionex-LC Packings, Sunnyvale, CA) using a PepMap100 column (75 μm × 15 cm). A flow rate of 200 nL/min with buffers 2% ACN, 0.1% FA (A) and 80% ACN, 0.1% FA (B) was used at a gradient of 0-30% B in 120 min, 30-100% B in 5 minutes, 100% B for 10 minutes, 100-0% B in 5 minutes, and 0% B for 20 minutes. Elutions from the reverse phase column were labeled as follows: tag114, biofilm; tag115, batch culture; tag116, biofilm replicate; tag117, planktonic cells from bioreactor. Because tag114 and tag116 were technical replicates, the reported ratios are the average of log2 (114/115) and log2 (116/115). The internal error was defined as the value at which 95% of all proteins had no deviation from each other, where the deviation was the absolute value (0.28) of the difference between log2 (114/115) and log2 (116/115).
Extracellular protein analysis
To examine the protein content of the extracellular matrix of the biofilm via scraping the glass slides from a single bioreactor into 1.5 mL of chilled 50 mM HEPES buffer (pH 7.2). The resuspended samples were pipetted up and down through a 12-gauge needle for ten repetitions to break apart the extracellular matrix, and then the sample was filtered through a 0.22-μm polyethersulfone (PES) filter to remove cells and other extracellular matrix material. The remaining soluble fraction was concentrated to approximately 300 μL using a Millipore centricon concentrator. Due to the dilute nature of the extracellular fraction, the proteins were separated on an SDS-PAGE gel, as previous in-solution digestions did not yield protein identifications. Sample extracted from 30 biofilm slides was loaded into 3 lanes on a 12-well, 4-12% NuPage Bis-Tris gels (Invitrogen, Carlsbad, CA) and run in MOPS buffer for 1 hour. The gel was stained with Coomassie Brilliant Blue, and destained with 45% methanol and 10% acetic acid solution overnight. Bands were excised from the gel across all three lanes and were subjected to an in-gel digestion protocol. Prepared samples were split into three equal portions, one of which loaded onto a reverse-phase column as previously described [57]. Reverse-phase separation was completed as described except that the gradient was run as follows: 0-30% B in 30 min, 30-100% B in 5 minutes, 100% B for 10 minutes, 100-0% B in 5 minutes, and 0% B for 20 minutes. Data analysis proceeded as previously described [57].
Transcriptome comparisons
Microarray data sets for different times were analyzed using average linkage hierarchical cluster analysis with Euclidean distance matrices and were visualized with TreeView as previously described [28,29,37]. If there were expression data for a gene for all sample times and the differential expression of log2 (ratio) was ≥2.0 or ≤ -2.0 at one or more times, the gene was used for clustering analysis (ratio of the biofilm gene expression versus different planktonic time points from a batch culture). Gene expression with a log2 (ratio) of ≥2.0 was considered up-expression, and gene expression with a log2 (ratio) of ≤ -2.0 was considered down-expression. The same data set was used for principal components analysis (PCA). The correlation matrix of multiple transcriptomes was generated with a centered Pearson correlation using MicrobesOnline functional genomics analysis (microbesonline.org). The data sets for growth with the different substrates will be described in separate manuscripts, but all growth was done in the same defined medium as used for biofilm growth in this study.
Quantitative PCR
Biofilm and reactor planktonic cells were grown and harvested as described above for quantitative PCR (qPCR). Four biofilm coupons and 20 mL of reactor planktonic cells were collected at 35 and 70 h post coupon insertion and nutrient flow. RNA was extracted and purified as described above except that in addition to the on-column DNase treatment a second DNase treatment using Turbo DNase (Ambion) was done following the manufactures instruction. cDNA was prepared by reverse transcription with appropriate controls as previously described [29]. Briefly, 10 μg total RNA was mixed with 6 μg of random primers (Invitrogen) and was incubated at 70°C for 10 min followed by a cooling on ice for approximately 1 min. After cooling, 1 μl of reverse transcriptase (Invitrogen), 4 μl of 5X reverse transcription buffer, 2 μl of 0.1 M dithiothreitol, 1 μl of RNase-Out inhibitor (Invitrogen), and 1 μl of a solution containing each deoxynucleoside triphosphate at a concentration of 10 mM were added to each tube. The tubes were incubated at 42°C for 2 h, 70°C for 15 min, and then cooled on ice. A 2.5-μl sample of cDNA (4 ng/μl) was used for qPCR.
The primers used for qPCR were diluted to a concentration of 12.5 μM (Table 1). Each qPCR mixture (final volume, 25 μl) contained 12.5 μl of SYBR Green Master Mix (Applied Biosystems), 0.7 μl of 12.5 μM solution of the forward primer, 0.7 μl of a 12.5 μM solution of the reverse primer, 2.5 μl of cDNA template, and 8.6 μl of nuclease-free water. The real-time PCR was carried out with a Smart Cycler II (Cepheid, Sunnyvale, CA) using the following conditions: one cycle of 95°C for 10 min and 45 cycles of 95°C for 15 s, 55°C for 30 s, and 60°C for 30 s. A serial dilution of genomic DNA was used to generate a standard curve. A negative control that did not contain RTase was included for all samples. Real-time PCR analysis was used to determine the levels of expression of seven genes. The results from the microarray and real-time PCR analysis revealed a high degree of correlation (R = 0.82). Similar results were observed in previous studies that used similar microarray procedures and techniques [28,29,37,53].
CTC Staining
Metabolic activity was measured with the redox dye 5-cyano-2,3-ditolyl tetrazolium chloride (CTC) as previously described [58]. Briefly, biomass was collected aseptically under anaerobic conditions for exponential planktonic, stationary planktonic, and biofilm cells. Samples were pelleted via centrifugation (2,000 x g at room temperature) and washed 2 times with anoxic PBS buffer (10 mM NaPO4, 138 mM NaCl, 2.7 mM KCl, pH 7.4). After the final wash and centrifugation, cell pellets were suspended in 2 ml of anoxic PBS. A total volume of 2.5 ml was used for the assay. For each phase of growth, three separate dilutions were tested: 1.2, 6, and 12-fold. Each diluted sample received 1.9 ml of a 6.5 mM stock solution of CTC in PBS (final concentration approximately 5 mM CTC). Each dilution was done in triplicate.
CTC-treated samples were incubated at 37°C for 4 h with gentle agitation and kept in the dark. Samples were collected on a 25 mm diameter white polycarbonate filter with a 0.22 μm pore size. The filter was then placed in a vial containing 2.5 ml 95% EtOH and agitated for 12 h at room temperature in the dark. After 12 h, the EtOH was filtered through a 0.22 μm pore size filter and the solution absorbance was measured (450 nm). The amount of CTC-formazan within the solution was calculated using a molar extinction coefficient of 1.63 X 104 M-1 cm-1 and the amount calculated was normalized to protein.
Formate Dehydrogenase activity
Formate dehydrogenase activity was measured for biofilm and reactor planktonic cells using a methyl viologen reduction assay as described previously [59]. Briefly, biofilm biomass was collected by scraping 4 slides from the CDC reactor into Tris HCl buffer (50 mM Tris-HCl pH 8.0) within an anaerobic chamber. Suspended biofilm biomass was placed in a N2 gassed Hungate tube and pelleted via centrifugation at 2,000 x g at 4°C for 10 min. Planktonic biomass (10 ml) was collected from the CDC reactor and placed in a N2 gassed Hungate tube and pelleted as described. The collection tubes were pressurized with N2 and pellets were stored at -70°C until processed. Biofilm and planktonic pellets were suspended in 2 ml of Tris HCl buffer and pulse sonicated for 10 min. Supernatants were cleared by centrifugation at 9,400 x g for 30 min at 4°C followed by bubbling with N2 for 30 min to regenerate anoxic conditions. Supernatants were stored in N2 gassed Hungate tubes at -20°C until activity was measured (within 12 h after sonication).
Activity assays were done in a N2 flushed cuvette (10 min) that was sealed with a septum and components were added via a N2 flushed syringe. Initially, 0.6 ml of anoxic H20, 0.1 ml of 500 mM Tris HCl pH 8.0, 0.1 ml of 1 M β mercaptoethanol, and 0.05 ml of 100 mM methyl viologen were incubated for 1 min at room temperature in the cuvette. Next, 0.05 ml of crude sample was added and the mixture was incubated for 5 min at room temperature. Last, 0.1 ml of 1 M sodium formate was added to start the reaction and the OD578 was measured immediately after a quick inversion to mix the components. Readings were taken every 5 s for a total of 8 min. Activity was determined using the molar extinction coefficient for methyl viologen at OD578 = 9.8 mM-1 cm -1. Protein concentration of the crude extract was determined by the Bradford assay. Reduction of methyl viologen was minimal during initial incubation periods.
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
MEC carried out growth, biomass harvest, and activity assays, participated in array/protein data analysis, and drafted the manuscript. ZH carried out microarray hybridizations and data analysis. AMR carried out proteomic detection and data analysis. JMP and APA participated in data analysis and manuscript preparation. AM, JZ, JDK, and MWF participated in study design, study coordination, data analysis/interpretation, and manuscript preparation. All authors read and approved the final manuscript.
Supplementary Material
Confocal image of D. vulgaris biofilm stained with acridine orange.
{kind=link}
Comparison of biofilm transcriptomic and proteomic data when both samples were normalized to batch, exponential-phase planktonic cells. Genes of interest are highlighted.
{kind=link}
Whole genome transcript expression data for biofilm cells compared to planktonic, batch cells.
Whole genome transcript expression data for biofilm cells compared to planktonic, reactor cells.
Protein expression ratios between biofilm to batch, planktonic cells; biofilm to reactor, planktonic cells; or reactor, planktonic to batch, planktonic cells.
Expression and z values for 15 ribosomal protein genes. Expo1 and Expo2 represent exponential-phase cells and Stat1 through Stat4 represent stationary-phase cells. Blue and red colors denote level of expression for genes with a significant z score for up- and down-expression, respectively.
Analysis of enriched extracellular proteins from biofilms.
Contributor Information
Melinda E Clark, Email: MEW2FX@hscmail.mcc.virginia.edu.
Zhili He, Email: zhe@rccc.ou.edu.
Alyssa M Redding, Email: oxello@gmail.com.
Marcin P Joachimiak, Email: marcinjoachimiak@gmail.com.
Jay D Keasling, Email: jdkeasling@lbl.gov.
Jizhong Z Zhou, Email: jzhou@rccc.ou.edu.
Adam P Arkin, Email: aparkin@lbl.gov.
Aindrila Mukhopadhyay, Email: amukhopadhyay@lbl.gov.
Matthew W Fields, Email: matthew.fields@erc.montana.edu.
Acknowledgements
The research was supported by the U.S. Department of Energy, Office of Biological and Environmental Research, U. S. Department of Energy under Contract No. DE-AC02-05CH11231 via ENIGMA (Ecosystems and Networks Integrated with Genes and Molecular Assemblies). MWF would like to thank Peg Dirckx and Skye Saylor for graphic assistance.
References
- Dunne WM. Bacterial adhesion: seen any good biofilms lately? Clin Microbiol Rev. 2002;15:155–166. doi: 10.1128/CMR.15.2.155-166.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter R. Surfacing views of biofilm biology. Trends Microbiol. 2005;13:1–2. doi: 10.1016/j.tim.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Or D, Smets BF, Wraith JM, Dechesne A, Friedman SP. Physical constraints affecting bacterial habitats and activity in unsaturated porous media - a review. Adv Water Resources. 2007;30:1505–1527. doi: 10.1016/j.advwatres.2006.05.025. [DOI] [Google Scholar]
- Hamilton WA. Sulfate-reducing bacteria: Physiology determines their environmental impact. Geomicrobiol J. 1998;15:19–28. doi: 10.1080/01490459809378059. [DOI] [Google Scholar]
- Reardon CL, Cummings DE, Petzke LM, Kinsall BL, Watson DB, Peyton BM, Geesey GG. Composition and diversity of microbial communities recovered from surrogate minerals incubated in an acidic uranium-contaminated aquifer. Appl Environ Microbiol. 2004;70:6037–6046. doi: 10.1128/AEM.70.10.6037-6046.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfiffner SM, Cantu JM, Smithgalla A, Peacock AD, White DC, Moser DP, Onstott TC, van Heerdend E. Deep subsurface microbial biomass and community structure in Witwatersrand Basin mines. Geomicrobiol J. 2006;23:431–442. doi: 10.1080/01490450600875712. [DOI] [Google Scholar]
- Wanger G, Southam G, Onstott TC. Structural and chemical characterization of a natural fracture surface from 2.8 kilometers below land surface: Biofilms in the deep subsurface. Geomicrobiol J. 2006;23:443–452. doi: 10.1080/01490450600875746. [DOI] [Google Scholar]
- Beyenal H, Lewandowski Z. Dynamics of lead immobilization in sulfate reducing biofilms. Water Res. 2004;38:2726–2736. doi: 10.1016/j.watres.2004.03.023. [DOI] [PubMed] [Google Scholar]
- Beyenal H, Sani RK, Peyton BM, Dohnalkova AC, Amonette JE, Lewandowski Z. Uranium immobilization by sulfate-reducing biofilms. Environ Sci Technol. 2004;38:2067–2074. doi: 10.1021/es0348703. [DOI] [PubMed] [Google Scholar]
- Neal AL, Amonette JE, Peyton BM, Geesey GG. Uranium complexes formed at hematite surfaces colonized by sulfate-reducing bacteria. Environ Sci Technol. 2004;38:3019–3027. doi: 10.1021/es030648m. [DOI] [PubMed] [Google Scholar]
- Marsili E Beyenal H DiPalma L Meril C Dohnalkova AC Amonette JE Lewandowski Z Uranium removal by sulfate reducing biofilms in the presence of carbonates Water Sci Technol 20055249–55.16180408 [Google Scholar]
- Jan-Roblero J, Posadas A, de la Serna JZD, Garcia R, Hernandez-Rodriguez C. Phylogenetic characterization of bacterial consortia obtained of corroding gas pipelines in Mexico. World J Microbiol Biotechnol. 2008;24:1775–1784. doi: 10.1007/s11274-008-9674-x. [DOI] [Google Scholar]
- Beech IB, Sunner JA, Hiraoka K. Microbe-surface interactions in biofouling and biocorrosion processes. Inter Microbiol. 2005;8:157–168. [PubMed] [Google Scholar]
- Clark ME, Edelmann RE, Duley ML, Wall JD, Fields MW. Biofilm formation in Desulfovibrio vulgaris Hildenborough is dependent upon protein filaments. Environ Microbiol. 2007;9:2844–2854. doi: 10.1111/j.1462-2920.2007.01398.x. [DOI] [PubMed] [Google Scholar]
- Kuchma SL, O'Toole GA. Surface-induced and biofilm-induced changes in gene expression. Curr Opinion Biotechnol. 2000;11:429–433. doi: 10.1016/S0958-1669(00)00123-3. [DOI] [PubMed] [Google Scholar]
- Schembri MA, Kjaergaard K, Klemm P. Global gene expression in Escherichia coli biofilms. Mol Microbiol. 2003;48:253–267. doi: 10.1046/j.1365-2958.2003.03432.x. [DOI] [PubMed] [Google Scholar]
- Pysz MA, Conners SB, Montero CI, Shockley KR, Johnson MR, Ward DE, Kelly RA. Transcriptional analysis of biofilm formation processes in the anaerobic, hyperthermophilic bacterium Thermotoga maritima. Appl Environ Microbiol. 2004;70:6098–6112. doi: 10.1128/AEM.70.10.6098-6112.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite RD, Paccanaro A, Papakonstantinopoulou A, Hurst JM, Saqi M, Littler E, Curtis MS. Clustering of Pseudomonas aeruginosa transcriptomes from planktonic cultures, developing and mature biofilms reveals distinct expression profiles. BMC Genomics. 2006;7:162. doi: 10.1186/1471-2164-7-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemesh M, Tam A, Steinberg D. Differential gene expression profiling of Streptococcus mutans cultured under biofilm and planktonic conditions. Microbiol. 2007;153:1307–1317. doi: 10.1099/mic.0.2006/002030-0. [DOI] [PubMed] [Google Scholar]
- Sauer K, Camper AK. Characterization of phenotypic changes in Pseudomonas putida in response to surface-associated growth. J Bacteriol. 2001;183:6579–6589. doi: 10.1128/JB.183.22.6579-6589.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosthuizen MC, Steyn B, Theron J, Cosette P, Lindsay D, von Holy A, Brozel VS. Proteomic analysis reveals differential protein expression by Bacillus cereus during biofilm formation. Appl Environ Microbiol. 2002;68:2770–2780. doi: 10.1128/AEM.68.6.2770-2780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremoulet F, Duche O, Namane A, Martinie B, Labadie JC. Comparison of protein patterns of Listeria monocytogenes grown in biofilm or in planktonic mode by proteomic analysis. FEMS Microbiol Lett. 2002;210:25–31. doi: 10.1111/j.1574-6968.2002.tb11155.x. [DOI] [PubMed] [Google Scholar]
- Resch A, Leicht S, Saric M, Pasztor L, Jakob A, Gotz F, Nordheim A. Comparative proteome analysis of Staphylococcus aureus biofilm and planktonic cells and correlation with transcriptome profiling. Proteomics. 2006;6:1867–1877. doi: 10.1002/pmic.200500531. [DOI] [PubMed] [Google Scholar]
- O'Toole G, Kaplan HB, Kolter R. Biofilm formation as microbial development. Annu Rev Microbiol. 2000;54:49–79. doi: 10.1146/annurev.micro.54.1.49. [DOI] [PubMed] [Google Scholar]
- Stewart PS, Franklin MJ. Physiological heterogeneity in biofilms. Nat Rev Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- Wanner O, Gujer W. A multispecies biofilm model. Biotechnol Bioeng. 1986;28:314–328. doi: 10.1002/bit.260280304. [DOI] [PubMed] [Google Scholar]
- Nie L, Wu G, Zhang W. Correlation of mRNA expression and protein abundance affected by multiple sequence features related to translational efficiency in Desulfovibrio vulgaris: a quantitative analysis. Genetics. 2006;174:2229–2243. doi: 10.1534/genetics.106.065862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, Redding AM, Joachimiak MP, Arkin AP, Borglin SE, Dehal PS, Chakraborty R, Geller JT, Hazen TC, He Q, Joyner DC, Martin VJJ, Wall JD, Yang ZK, Zhou J, Keasling JD. Cell-wide responses to low-oxygen exposure in Desulfovibrio vulgaris Hildenborough. J Bacteriol. 2007;189:5996–6010. doi: 10.1128/JB.00368-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark ME, He Q, He Z, Huang KH, Alm EJ, Wan XF, Hazen TC, Arkin AP, Wall JD, Zhou JZ, Fields MW. Temporal transcriptomic analysis as Desulfovibrio vulgaris Hildenborough transitions into stationary phase during electron donor depletion. Appl Environ Microbiol. 2006;72:5578–5588. doi: 10.1128/AEM.00284-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Pitts B, Stewart PS, Camper AK, Yoon J. Comparison of the antimicrobial effects of chlorine, silver ion, and tobramycin on biofilm. Antimicro Agents Chemo. 2008;52:1446–1453. doi: 10.1128/AAC.00054-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin O, Hall EK, Kaiser C, Battin TJ, Richter A. Optimization of biomass composition explains microbial growth-stoichiometry relationships. Am Nat. 2011;177:E29–E42. doi: 10.1086/657684. [DOI] [PubMed] [Google Scholar]
- Beloin C, Ghigo JM. Finding gene-expression patterns in bacterial biofilms. Trends Microbiol. 2005;13:16–19. doi: 10.1016/j.tim.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Sebban-Kreuzer C, Blackledge M, Dolla A, Marion D, Guerlesquin F. Tyrosine 64 of cytochrome c553 is required for electron exchange with formate dehydrogenase in Desulfovibrio vulgaris Hildenborough. Biochem. 1998;37:8331–8340. doi: 10.1021/bi980142u. [DOI] [PubMed] [Google Scholar]
- Voordouw G. Carbon monoxide cycling by Desulfovibrio vulgaris Hildenborough. J Bacteriol. 2002;184:5903–5911. doi: 10.1128/JB.184.21.5903-5911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg JF, Seshadri R. et al. The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat Biotechnol. 2004;22:554–559. doi: 10.1038/nbt959. [DOI] [PubMed] [Google Scholar]
- Greene EA, Hubert C, Nemati M, Jenneman GE, Voordouw G. Nitrite reductase activity of sulphate-reducing bacteria prevents their inhibition by nitrate-reducing, sulphide-oxidizing bacteria. Environ Microbiol. 2003;5:607–617. doi: 10.1046/j.1462-2920.2003.00446.x. [DOI] [PubMed] [Google Scholar]
- He Q, Huang KH, He ZL, Alm EJ, Fields MW, Hazen TC, Arkin AP, Wall JD, Zhou JZ. Energetic consequences of nitrite stress in Desulfovibrio vulgaris Hildenborough, inferred from global transcriptional analysis. Appl Environ Microbiol. 2006;72:4370–4381. doi: 10.1128/AEM.02609-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WW, Culley DE, Nie L, Scholten JCM. Comparative transcriptome analysis of Desulfovibrio vulgaris grown in planktonic culture and mature biofilm on a steel surface. Appl Microbiol Biotechnol. 2007;76:447–457. doi: 10.1007/s00253-007-1014-9. [DOI] [PubMed] [Google Scholar]
- Hindre T, Bruggemann H, Buchrieser C, Hechard Y. Transcriptional profiling of Legionella pneumophila biofilm cells and the influence of iron on biofilm formation. Microbiol. 2008;154:30–41. doi: 10.1099/mic.0.2007/008698-0. [DOI] [PubMed] [Google Scholar]
- Kasprzak KS. Oxidative DNA and protein damage in metal-induced toxicity and carcinogenesis. Free Rad Biol Med. 2002;32:958–967. doi: 10.1016/S0891-5849(02)00809-2. [DOI] [PubMed] [Google Scholar]
- Banin E, Vasil ML, Greenberg EP. Iron and Pseudomonas aeruginosa biofilm formation. Proc Nat Acad Sci USA. 2005;102(31):11076–11081. doi: 10.1073/pnas.0504266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M, Cockayne A, Williams PH, Morrissey JA. Iron-responsive regulation of biofilm formation in Staphylococcus aureus involves fur-dependent and fur-independent mechanisms. J Bacteriol. 2005;187:8211–8215. doi: 10.1128/JB.187.23.8211-8215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L, Krogh A, Sonnhammer ELL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Colombatti A, Bonaldo P, Doliana R. Type A modules: interacting domains found in several non-fibrillar collagens and in other extracellular matrix proteins. Matrix. 1993;13:297–306. doi: 10.1016/s0934-8832(11)80025-9. [DOI] [PubMed] [Google Scholar]
- Ren DC, Bedzyk LA, Setlow P, Thomas SM, Ye RW, Wood TK. Gene expression in Bacillus subtilis surface biofilms with and without sporulation and the importance of yveR for biofilm maintenance. Biotechnol Bioeng. 2004;86:344–364. doi: 10.1002/bit.20053. [DOI] [PubMed] [Google Scholar]
- Domka J, Lee J, Wood TK. YliH (BssR) and YceP (BssS) regulate Escherichia coli K-12 biofilm formation by influencing cell signaling. Appl Environ Microbiol. 2006;72:2449–2459. doi: 10.1128/AEM.72.4.2449-2459.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMartino P, Fursy R, Bret L, Sundararaju B, Phillips RS. Indole can act as an extracellular signal to regulate biofilm formation of Escherichia coli and other indole-producing bacteria. Can J Microbiol. 2003;49:443–449. doi: 10.1139/w03-056. [DOI] [PubMed] [Google Scholar]
- Elias DA, Drury EC, Redding AM, Mukhopadyay A, Yen HCB, Fields MW, Hazen TC, Arkin AP, Keasling JD, Wall JD. Expression profiling of hypothetical genes in Desulfovibrio vulgaris leads to improved functional annotation. Nucleic Acids Res. 2009;37:2926–2939. doi: 10.1093/nar/gkp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabriere E, Vernède X, Guigliarelli B, Charon MH, Hatchikian, Fontecilla-Camps JC. Crystal structure of the free radical intermediate of pyruvate:ferredoxin oxidoreductase. Science. 2001;294:2559–2563. doi: 10.1126/science.1066198. [DOI] [PubMed] [Google Scholar]
- Caffrey SM, Park HS, Been J, Gordon P, Sensen CW, Voordouw G. Gene expression by the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough grown on an iron electrode under cathodic protection conditions. Appl Environ Microbiol. 2008;2008(74):2404–2413. doi: 10.1128/AEM.02469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcıa-Sanchez S, Aubert S, Iraqui I, Janbon G, Ghigo JM, d'Enfert C. Candida albicans biofilms: a developmental state associated with specific and stable gene expression patterns. Euk Cell. 2004;3:536–545. doi: 10.1128/EC.3.2.536-545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr Al, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- He Q, He Z, Joyner DC, Joachimiak MP, Price MN, Yang ZK, Yen HCB, Hemme CL, Chen W, Fields MW, Stahl DA, Keasling JD, Keller M, Arkin AP, Hazen TC, Wall JD, Zhou J. Impact of elevated nitrate on sulfate-reducing bacteria: a comparative study of Desulfovibrio vulgaris. Nature ISME J. 2010;4:1386–1397. doi: 10.1038/ismej.2010.59. [DOI] [PubMed] [Google Scholar]
- He Z, Wu L, Fields MW, Zhou JZ. Use of microarrays with different probe sizes for monitoring gene expression. Appl Environ Microbiol. 2005;71:5154–5162. doi: 10.1128/AEM.71.9.5154-5162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, He ZL, Alm EJ, Arkin AP, Baidoo EE, Borglin SC, Chen WQ, Hazen TC, He Q, Holman HY, Huang K, Huang R, Joyner DC, Katz N, Keller M, Oeller P, Redding A, Sun J, Wall J, Wei J, Yang ZM, Yen HC, Zhou JZ, Keasling JD. Salt stress in Desulfovibrio vulgaris Hildenborough: An integrated genomics approach. J Bacteriol. 2006;188:4068–4078. doi: 10.1128/JB.01921-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Huang KH, Alm EJ, Arkin AP. A novel method for accurate operon predictions in all sequenced prokaryotes. Nucleic Acids Res. 2005;33:880–892. doi: 10.1093/nar/gki232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding AM, Mukhopadhyay A, Keasling JD. Study of nitrate stress in Desulfovibrio vulgaris Hildenborough using iTRAQ proteomics. Brief Funct Genomic Proteomic. 2006;5:133–143. doi: 10.1093/bfgp/ell025. [DOI] [PubMed] [Google Scholar]
- Bhupathiraju VK, Hernandez M, Landfear D, Alvarez-Cohen L. Application of a tetrazolium dye as an indicator of viability in anaerobic bacteria. J Microbiol Methods. 1999;1999(37):231–243. doi: 10.1016/s0167-7012(99)00069-x. [DOI] [PubMed] [Google Scholar]
- Costa C, Teixeira M, LeGall J, Moura JJG, Moura I. Formate dehydrogenase from Desulfovibrio desulfuricans ATCC 27774: isolation and spectroscopic characterization of the active sites (heme, iron-sulfur centers and molybdenum) J Biol Inorganic Chem. 1997;2:198–208. doi: 10.1007/s007750050125. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Confocal image of D. vulgaris biofilm stained with acridine orange.
Comparison of biofilm transcriptomic and proteomic data when both samples were normalized to batch, exponential-phase planktonic cells. Genes of interest are highlighted.
Whole genome transcript expression data for biofilm cells compared to planktonic, batch cells.
Whole genome transcript expression data for biofilm cells compared to planktonic, reactor cells.
Protein expression ratios between biofilm to batch, planktonic cells; biofilm to reactor, planktonic cells; or reactor, planktonic to batch, planktonic cells.
Expression and z values for 15 ribosomal protein genes. Expo1 and Expo2 represent exponential-phase cells and Stat1 through Stat4 represent stationary-phase cells. Blue and red colors denote level of expression for genes with a significant z score for up- and down-expression, respectively.
Analysis of enriched extracellular proteins from biofilms.