

Abstract
While p73 overexpression has been associated with increased apoptosis in cancer tissues, p73 overexpressing tumors appear to be of high grade malignancy. Why this putative tumor suppressor is overexpressed in cancer cells and what the function of overexpressed p73 is in breast cancers are critical questions to be addressed. By investigating the effect of p53 inactivation on p73 expression, we found that both protein and mRNA levels of TAp73 were increased in MCF-7/p53siRNA cells, MCF-7/p53mt135 cells and HCT-116/p53−/− cells, as compared to wild type control, suggesting that p53 inactivation by various forms upregulates p73. We showed that p53 knockdown induced p73 was mainly regulated at the transcriptional level. However, although p53 has a putative binding site in the TAp73 promoter, deletion of this binding site did not affect p53 knockdown mediated activation of TAp73 promoter. Chromatin immuno-precipitation (ChIP) data demonstrated that loss of p53 results in enhanced occupancy of E2F-1 in the TAp73 promoter. The responsive sequence of p53 inactivation mediated p73 upregulation was mapped to the proximal promoter region of the TAp73 gene. To test the role of E2F-1 in p53 inactivation mediated regulation of p73 transcription, we found that p53 knockdown enhanced E2F-1 dependent p73 transcription, and mutations in E2F-1 binding sites in the TAp73 promoter abrogated p53 knockdown mediated activation of TAp73 promoter. Moreover, we demonstrated that p21 is a mediator of p53-E2F crosstalk in the regulation of p73 transcription. We concluded that p53 knockdown/inactivation may upregulate TAp73 expression through E2F-1 mediated transcriptional regulation. p53 inactivation mediated upregulation of p73 suggests an intrinsic rescuing mechanism in response to p53 mutation/inactivation. These findings support further analysis of the correlation between p53 status and p73 expression and its prognostic/predictive significance in human cancers.
Introduction
Loss of p53 function, including genetic mutation and functional inactivation, has been associated with carcinogenesis and therapeutic resistance [1], [2]. However, some tumor cells with mutant p53 remain sensitive to DNA damage. Recent advances suggest that regulation of p73, a new p53 family member, may play a role in this process. Nevertheless, how p73 is regulated to compensate p53 function in p53 inactivated cells remains unclear.
The p73 gene has two separate sets of promoters that result in the transactivating (TA) and ΔN classes of products known as TAp73 and ΔNp73 [3]. Alternative splicing at the carboxyl terminus of both TAp73 and ΔNp73 yields further complexity via p73 isoforms, which include p73 α, β, γ, δ, ε, ζ, and η [4], [5]. Among them, p73 α and β are the major TA isoforms. Similar to p53, TAp73s are able to transactivate some p53 target genes and induce apoptosis and growth arrest. Functional p73 has been linked to chemosensitivity [6]. More importantly, p73 is able to induce apoptosis/growth arrest in cells with mutant p53 [7]. These findings not only support p73 as a tumor suppressor but also underscore the significance of p73 in p53-independent anti-tumor mechanisms. In contrast to TAp73, ΔNp73s have opposing activity and function as oncoproteins by inactivating p53 and TAp73 through the formation of heterodimers with these tumor suppressors [3].
Although in vitro studies suggest that p73 (which indicates TAp73 if not otherwise specified) is a putative tumor suppressor, the role of p73 in cancer development remains to be established. Different from p53, p73 mutation in cancer tissues is rare. Interestingly, overexpression of wild type p73 is frequently detected in different types of human cancers, including breast, lung, prostate, bladder and other types of cancers [8], [9], [10]. In breast cancers, increased p73 protein and mRNA levels have been detected in approximately one third of the cases that have been studied [11]. While p73 overexpression has been associated with increased apoptosis in cancer tissues, p73 overexpressing tumors appear to be of high grade malignancy [12]. Why this putative tumor suppressor is overexpressed in cancer cells and what the function of overexpressed p73 is in breast cancers are critical questions to be addressed. Among the factors that may contribute to p73 overexpression, interactions between p53 and p73 have been targeted by many investigators. It was reported that increased p73 mRNA levels was correlated with p53 mutation detected using mutant p53 specific antibody (clone 1801) [8]. Other studies using different tissues also suggest that p73 overexpression/alteration was linked to defective p53, as examined by loss of heterozygous (LOH) and immunohistochemistry [13]. However, DNA sequencing of the p53 gene from 8 cases of breast cancers overexpressing p73 in a separate report failed to show a similar correlation, which might be due to limited case number and no microdissection of the samples [11]. Therefore, correlation between p53 status and the expression of individual major isoforms of p73, and its clinical relevance in breast cancers require further studies.
Available results suggest that p53 and p73 interact in multiple ways. On the one hand, some p53 mutants, such as R175H and R248W, can bind to TAp73 and inactivate its function [14]. On the other, overexpression of ΔNp73 is able to bind to and inactivate p53 and TAp73, thereby functioning as a dominant negative inhibitor [3]. In addition, upregulation of p53 induces ΔNp73 transcription and expression, suggesting a feedback network between p53 and p73 [15]. It was also reported that overexpression of p53 or p73 induced p73 transcription in certain cell lines [16]. If only based on the above findings, one would conclude that p53 mutation simultaneously inactivates both p53 and p73 systems; and the role of p73 in p53 independent apoptosis in p53 mutant cells would be largely excluded. In fact, p73 is commonly overexpressed in cancer cells and is functional in many cell lines with mutant p53 [7]. This indicates that there might be additional mechanisms that coordinate p53 inactivation and p73 activation, such that p73 is upregulated/activated to rescue some, if not all, of p53 function.
In this study, we investigated the communication between p53 mutation/inactivation and p73 expression/activation. Using several p53 inactivation models we demonstrated that inactivation of p53 upregulated TAp73 expression. The underlying mechanisms involve E2F-1 mediated regulation of TAp73 transcription modulated by p21 activity.
Materials and Methods
Cell culture and transfection
MCF-7 cells were maintained in DMEM/F12 medium (Sigma, MO) supplemented with 10% fetal bovine serum (FBS). HCT-116 and HCT116p53−/− cells were a gift from Dr. Bert Vogelstein (The Johns Hopkins University). MCF-7 cell line expressing p53SiRNA was established by transfecting MCF-7 cells with p53/SiRNA plasmid (Imagenex, CA), followed by G418 selection. G418 resistant clones were pooled for further characterization. For transient transfection, cells were inoculated 24 hours before transfection. The transfection was performed using FuGENE6 (Roche, IN) according to manufacturer's protocol.
Plasmid construction
Luciferase reporter plasmids containing full-length p73 promoter (p73PF, −2713/+77) as well as promoter deletion mutants, p73NruI (−1210/+77), p73SacI (−883/+77), p73PstI (−299/+77) and p73PvuII (−220/+77) were constructed as in a previous report [17]. p73pvuII-E2F-1–155/−132 double mutant was generated in a previous study [18]. To construct the luciferase reporter vector containing full length p73 promoter that lacks p53-binding site, the 61-bp fragment containing putative p53 binding sequence was eliminated by use of following primers: Δ61F: 5′ GCC ATG AAG ATG TGC GAG T 3′ and Δ61R: 5′ GAA GTT CAT GGC CGC CGC CTG CCG C 3′. The resulting PCR product was cloned into pGVB2. This construct lacking p53 binding site was designated as p73-PFΔ61. pcDNA3-p21 plasmid was a gift of Dr. Todd Sladek.
Electrophoretic Mobility Shift Assay
Sixty-one oligonucleotides corresponding to wild type and mutant p53-binding sequences were synthesized and radiolabeled with [γ-32P] dATP by using T4 polynucleotide kinase. DNA binding reactions were performed in 20 µl containing 10 µg of protein from nuclear lysates of MCF-7, MCF-7/SiRNA, HCT116 or HCT116/p53−/− cells, 4 µl of 5× binding buffer (50 mM Tris pH 8.0, 750 mM KCl, 2.5 mM EDTA, 0.5% Triton-X 100, 62.5% glycerol (v/v), 1 mM DTT), 2 µl of polydIdC, and 50000 cpm of radiolabeled probe per sample. Twenty nanograms of unlabeled probes corresponding to wild type or mutant oligonucleotides were used for competition. Antibody supershift was performed using 3 µg of anti-p53 antibody. Binding reactions were incubated at room temperature for 30 minutes and then resolved on 4% PAGE at room temperature (5–6 hours at 70v). Gels were dried and exposed to film at −80°C.
Luciferase Reporter Assays
Cells were cultured at 1×105 cells/well in 12-well tissue culture plates 24 hours prior to transfection. The reporter plasmid was cotransfected with pSV-β-gal and appropriate plasmids, such as pcDNA3, pcDNA3/p53, pcDNA3/E2F-1 or pcDNA3/p21, using FuGENE 6 by following the standard protocol. Cells were harvested 40 hours post transfection and luciferase activity was measured using Luciferase Reporter Assay System (Roche, MN) according to the manufacturer's instructions on a luminometer. Results were normalized with β-gal activity and presented as fold increase over control luciferase activity. All experiments were performed in triplicates.
Western Blotting
Fifty µg of protein lysate was loaded onto each lane of a gel. Proteins were separated with 12% SDS-PAGE and transferred to PVDF membrane. The membrane was probed with a specific primary antibody at appropriate dilutions, followed by washing and probing with a corresponding secondary antibody. The specific protein band was visualized by autoradiography using an ECL kit (GE Health Care). Antibodies against p53, E2F-1, p21 and Actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody against p73 (AB3) was purchased from Calbiochem.
RNA Extraction and RT-PCR
Total RNA was isolated from control and treated cells using RNeasy Mini Kit (QIAGEN, CA). First strand cDNA synthesis was performed using SuperscriptIII™ First Strand synthesis system (Invitrogen). The primer sequences used were as follows: TAp73 forward (FW): 5′-GCA CCA CGT TTG AGC ACC TCT-3′; TAp73 reverse (RV): 5′-GCA GAT TGA ACT GGG CCA TGA-3′; DNp73 FW: 5′-CAA ACG GCC CGC ATG TTC CC-3′; DNp73 RV: 5′-TTG AAC TGG GCC GTG GCG AG-3′; Actin FW: 5′-GCA CCA CAC CTT CTA CAA TGA GC-3′ Actin RV: 5′-GAC GTA GCA CAG CTT CTC CTT AAT G-3′; PCR reactions were performed in a 50 µl reaction volume for 30–35 cycles. Twenty microliters of PCR product was electrophoresed on 1.8% agarose gel and visualized by Ethidium Bromide staining. Quantitative detection of TA and DNp73 mRNA levels was performed with real time RT-PCR using a Bio-Rad CFX96 Touch™ Real-Time PCR Detection System.
ChIP Assays
ChIP assays were carried out with a ChIP assay kit from the Upstate Biotechnology, Inc. (NY). MCF-7/control and MCF-7/sip53 cells were grown on 100 mm plates to 90% confluence in DMEM/F12 medium containing 10% FBS before collection. The cells were cross-linked with 1% formaldehyde (final concentration) by direct adding to the cell medium for 10 minutes at 37°C, followed by washing with cold PBS containing 1× protease inhibitor cocktail. The cells were then scraped, centrifuged for 5 minutes at 4°C, 4000 rpm, and lysed by incubation with 1 mL of l× SDS lysis buffer for 10 minutes on ice. The lysates were subjected to 15 cycles of sonication (15 sec pulse/2 min incubation for each cycle) at 50% duty cycle using an Out Control intensity of 2 (VirSonic 475) followed by centrifugation at 4°C, 16000 rpm for 10 minutes. Three hundred micrograms of supernatant protein were diluted 10-fold with ChIP dilution buffer and immunocleared with 70 µl of Protein A Agarose/Salmon Sperm DNA (50% Slurry) for 1 hour at 4°C with agitation. Collected agarose beads were saved as control IgG. Immunoprecipitation was performed overnight at 4°C with E2F-1 antibody (SC-20, Santa Cruz Biotechnology Inc.) at 3 µg/mL of precleared supernatant. Immunocomplexes were extracted by adding 65 µl of Protein A Agarose/Salmon Sperm DNA (50% Slurry) for 1.5 hour at 4°C followed by gentle centrifugation (1000 rpm, 1 minute, 4°C). Precipitates were washed sequentially with 1 mL of low-salt washing buffer, high-salt washing buffer, LiCl washing buffer, and were washed twice with 1 mL of TE buffer and extracted twice with 250 µl of freshly made elution buffer (1% SDS, 0.1 M NaHCO3). The pooled eluates in a total volume of 500 µl were mixed with 20 µL of 5 M NaCl, heated at 65°C for 10 hours to reverse the formaldehyde crosslinking. DNA fragments were then purified by phenol/chloroform and precipited with ethanol.
Five µL from the 30 µL DNA extraction was amplified by PCR with the following pairs of primers for the proximal promoter region of the TAp73 gene: Forward: 5′-GCCCATATAACCCGCCTA-3′; and Reverse: 5′-CCCTGGGCCTCCTACCTG-3′. The PCR conditions were: initial 10 minutes of denaturing at 94°C followed by 35 cycles of denaturing for 30 seconds at 94°C, annealing for 30 seconds at 59°C, and elongating for 45 seconds at 72°C; the final extension took place at 72°C for 5 minutes. Equal volumes of each PCR sample were subjected to electrophoresis in a 2% agarose gel, followed by gel documentation.
Results
p53 knockdown/mutation induces p73 upregulation in cancer cells
To investigate the specific role of p53 in the regulation of p73, we established a stable MCF-7 subline in which wild type p53 was knocked down using p53-targeting siRNA (MCF-7/p53siRNA). As shown in Fig. 1A, p53 knockdown was highly efficient, as indicated by the striking difference in p53 and p21 levels between control and MCF-7/p53siRNA cells in response to DNA damaging agent doxorubicin. We next examined p73 expression in the paired cell lines and found that p73 protein levels were significantly increased in MCF-7/p53siRNA cells (Fig. 1B), suggesting that loss of p53 may upregulate p73 expression. This hypothesis was supported by the increase of p73 levels in HCT116/p53−/− cells, as compared to the wild type control (Fig. 1C). To exclude the possibility of clonal selection in the stable cell lines and to demonstrate that p53 mutation may also induce p73 upregulation, MCF-7 cells were transiently transfected with control vector, p53siRNA, mutant p53 (mtp53) (G135A) and wild type p53 (wtp53), followed by p73 detection. We found that transfection of p53siRNA resulted in decreased p53 but increased p73 levels (Fig. 1D), which was consistent with the data from the stable cell lines. In the cells transfected with mtp53 (G135A), protein levels of both p53 and p73 were increased. Because dimer formation between mutant and wild type p53 increases p53 stability and inactivates wtp53 [19], increased p73 levels in these cells suggest that p53 inactivation by point mutation may also induce p73 expression. Interestingly, p73 protein levels in the cells transfected with wtp53 were modestly increased, instead of decrease. This may suggest the complexity of p53–p73 interaction, which will be discussed later. Taken together, these data demonstrated that p53 inactivation, either by knockdown/knockout or by mutation, upregulates p73 protein levels in these cells.
Figure 1.
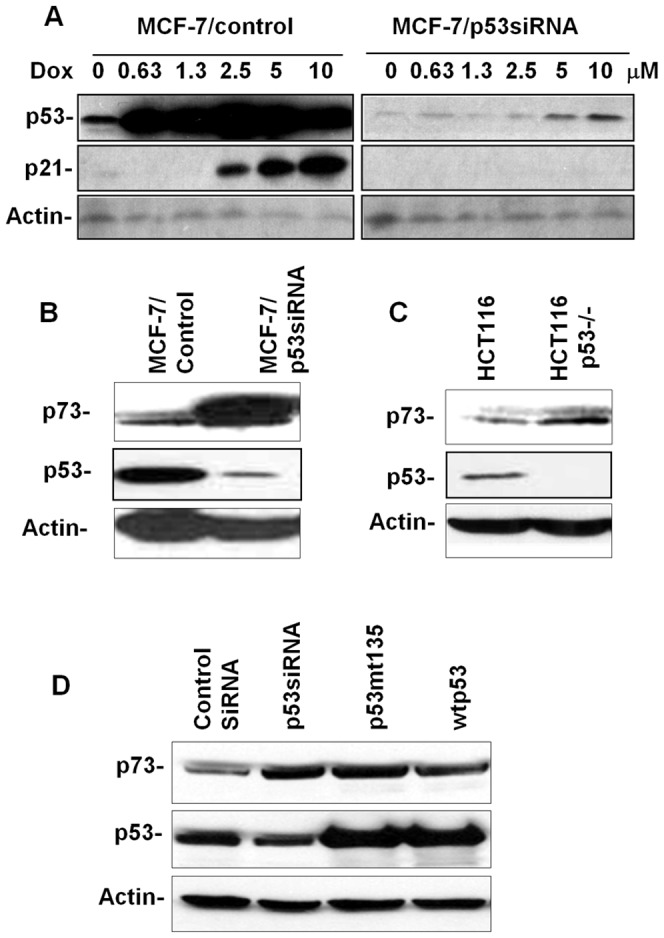
A. p53 knockdown in MCF-7 cells. A stable MCF-7 subline was established by transfection pGeneSupressor/p53siRNA followed by G418 selection. Control and MCF-7/p53siRNA cells were treated with doxorubicin (Dox) at indicated concentrations for 20 h. Protein levels of p53, p21 and actin were detected by Western blot. B & C. Upregulation of p73 β in MCF-7 (B) and HCT-116 (C) cells with p53 knockdown/knockout. Protein levels of p53, p73 β and Actin in control and p53 knockdown/knockout cells were detected with Western blot. D. Regulation of p73 by p53 in transient transfection system. MCF-7 cells were transiently transfected with control vector, pGeneSupressor/p53siRNA, pCMV/mtp53 (135G/A) and pCMV/wtp53, respectively. Protein levels of p53, p73 β and Actin were detected using Western blot.
p53 inactivation mediated upregulation of p73 is primarily regulated at the transcriptional levels
To test whether p53 inactivation mediated p73 upregulation occurs at transcriptional and/or translational levels, we examined the mRNA levels and protein half-life of TAp73 in cells with different p53 statuses. Using cycloheximide blockage assay we found that TAp73 protein degradation was similar between MCF-7/control and MCF-7/p53siRNA cells (data not shown). In contrast, RT-PCR amplification of p73 mRNA from both MCF-7 and HCT116 paired cell lines indicated that TAp73 mRNA levels were significantly upregulated in the cells with p53 inactivation (Fig. 2A).
Figure 2. p53 inactivation mediated upregulation of p73 is primarily regulated at the transcriptional level.

A & B. p53 inactivation upregulates TAp73 but not DNp73 mRNA levels. Relative mRNA levels of TAp73 (A) and DNp73 (B) in control cells (with wtp53) and p53 knockdown/knockout (p53KD/KO) MCF-7 and HCT-116 cells were detected with real-time RT-PCR using TAp73 and DNp73 specific primers. Specific mRNA levels were normalized with β-Actin mRNA. C & D. Loss of p53 induces the activation of p73 promoter in stable MCF-7 and HCT-116 sublines. Cells of each subline were co-transfected with luciferase reporter construct encoding full length (−2713 to +77) p73 promoter (p73-PF), pSV-β-gal. Luciferase activity was measured 40 hours after transfection. The experiments were performed at least three times in triplicates. E. Transfaction of p53 siRNA and mutant p53 induces p73 promoter activity in MCF-7 cells: MCF-7 cells were co-transfected with reporter construct encoding full length p73 promoter (p73-PF), pSV-β-gal, and vectors of control siRNA, p53siRNA, wild type p53 (wtp53) or mutant p53 (mtp53). Luciferase activity was determined as described above. (* p<0.01).
It was previously shown that p53 induces the expression of its antagonist DNp73 in H1299, Saos2 and LS174T cells, which forms an autoregulatory feedback loop [15]. Therefore, we also examined the transcription of DNp73 in these two paired cell lines. As shown in Fig. 2B, DNp73 mRNA levels were insignificantly increased in MCF-7/p53siRNA cells but remarkably decreased in HCT-116/p53−/− cells, as compared to respective controls. These data suggest that the impact p53 inactivation on TAp73 and DNp73 transcription was different. We focused on the regulation of TAp73 in this study.
We next characterized p53 inactivation mediated regulation of TAp73 transcription using reporter gene assay. The reporter construct used was the luciferase gene driven by a promoter fragment of the TAp73 gene spanning from −2713 to +77 of the starting codon (p73PF) [17]. Results from two paired cell lines, MCF-7 control vs. MCF-7-p53siRNA and HCT116 control vs. HCT116p53−/−, showed that p53 knockdown/knockout in both cell lines strongly activated the TAp73 promoter (Fig. 2 C &D), which supported the transcriptional regulation of p73 by p53 inactivation. By testing the activation of the TAp73 promoter in a transient transfection system, we found TAp73 promoter activity was increased in MCF-7 cells transfected with p53siRNA or mtp53 (G135A), and it was decreased in response to wtp53 transfection (Fig. 2E). Taken together, these data indicate that p53 inactivation mediated upregulation of p73 is primarily regulated at the transcriptional level.
p53 inactivation mediated upregulation of p73 is independent of p53 binding to the TAp73 promoter
Since p53 is a transcription factor, it is necessary to address whether p53 inactivation mediated upregulation of TAp73 is DNA binding dependent. Previously, it was reported that the TAp73 promoter contains a putative p53 responsive element (p53RE) between −2634 and −2574. Reporter assays based on a chimeric promoter containing the p53RE suggested that it may function as an enhancer [16]. However, reports from later studies showed that co-transfection of p53 did not activate the TAp73 promoter [6], [15], [20]. To determine whether this p53RE plays any roles in p53 inactivation modulated p73 regulation, we examined DNA binding activity of p53 to this putative binding site using electrophoretic mobility shift assay (EMSA). The labeled probe, which was a 61 bp oligonucleotide fragment (−2634 to −2574) containing the putative binding site, was incubated with nuclear protein lysate extracted from MCF-7, HCT116 or HCT116p53−/− cells in the presence/absence of competitive oligonucleotides. As shown in Fig. 3A, the probe formed a weak complex with the nuclear protein extract from both wild type MCF-7 and wild type HCT116 cells, but not with the p53−/− extract nor in the presence of competitive wild type oligomers. This signifies that p53 was able to bind to the putative binding site. To determine whether this DNA binding activity plays a role in p53 inactivation mediated upregulation of p73, we deleted this putative p53 binding sequence in the full-length p73 promoter (p73PF) to create a reporter construct named p73PFΔ61. By transfecting p73PF or p73PFΔ61 into control and p53siRNA expressing MCF-7 cells, we found that deletion of putative p53 binding sequence from the full-length p73 promoter did not affect p53 knockdown-mediated increase in the p73 promoter activity (Fig. 3B). These results suggest that p53 binding site in the p73 promoter is not involved in p53 knockdown-mediated upregulation of p73 transcription, although p53 has some affinity to this site.
Figure 3. p53 inactivation mediated upregulation of p73 is independent of p53's binding to the p73 promoter.
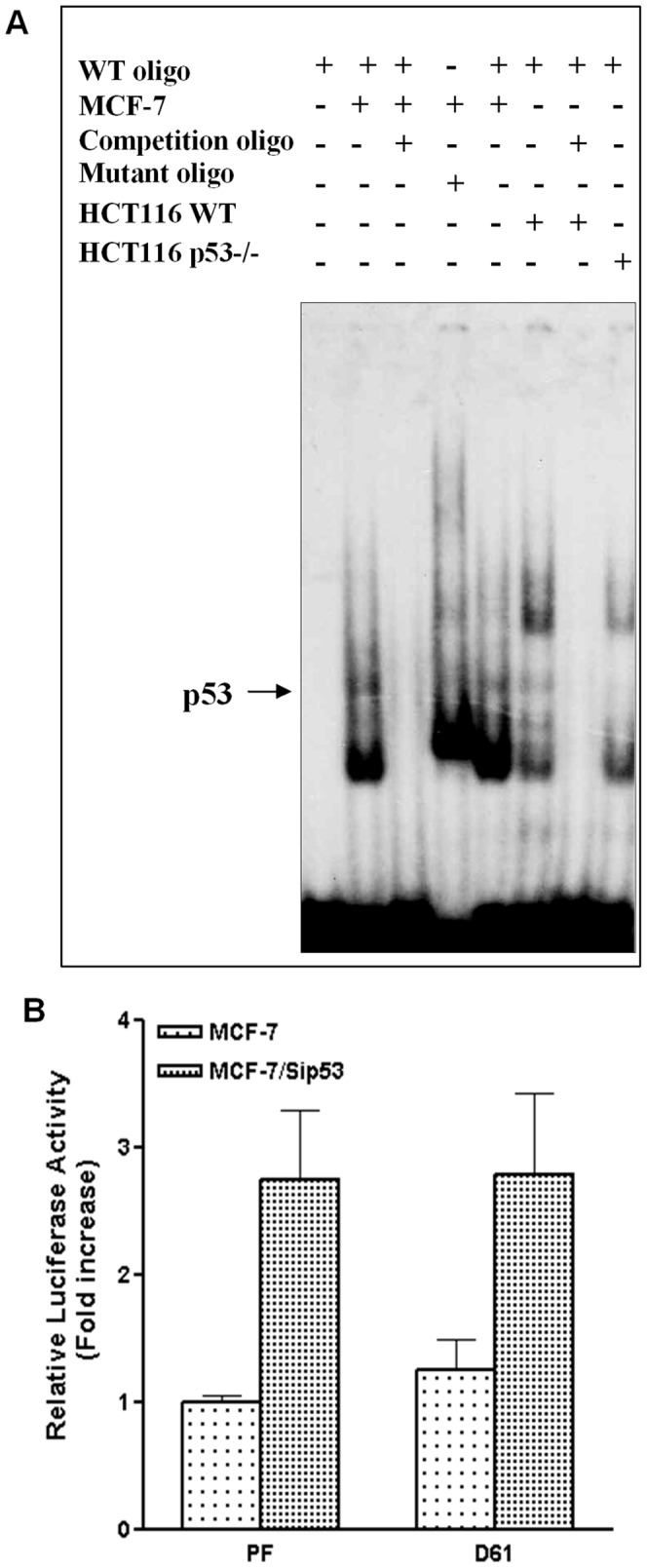
A. p53 binds to the p73 promoter. EMSA was performed using 32P-labeled 61 bp oligonucleotide (−2634 to −2574) containing putative p53-binding sequence in the p73 promoter and nuclear extracts of MCF-7 control and MCF-7/p53siRNA cells. Control and p53 knockout (p53−/−) HCT116 cells were used as control. Arrow indicates p53 bound to the p73 promoter sequence. B. Deletion of p53 binding site in the p73 promoter did not abrogate p53-knockdown mediated upregulation of p73 promoter activity. MCF-7 cells were transfected with the luciferase reporter constructs encoding full length p73 promoter, p73PF (PF) or the promoter lacking 61 bp p53 binding sequence, p73-PFΔ61 (D61) in the presence or absence of p53 knockdown with siRNA.
The responsive sequence of p53 inactivation mediated transcriptional activation of p73 is mapped to the proximal promoter region of the TAp73 promoter
To identify the minimal promoter region that is required for p53 inactivation-mediated enhancement of p73 transcription, we transfected a series of reporter constructs with various deletions in the TAp73 promoter region (Fig. 4A) into MCF-7/control and MCF-7/p53siRNA cells, respectively. Luciferase analysis revealed that p53 inactivation was able to enhance the promoter activity of each construct (Fig. 4B). The response of the shortest fragment (p73-pvuII, −220 to +71) was even greater than that of the full length promoter (p73PF). These results suggest that this short fragment upstream of the start codon was sufficient for p53-knockdown-mediated upregulation of p73 transcription.
Figure 4. Mapping of the DNA sequence that is responsible for p53 inactivation mediated p73 upregulation.
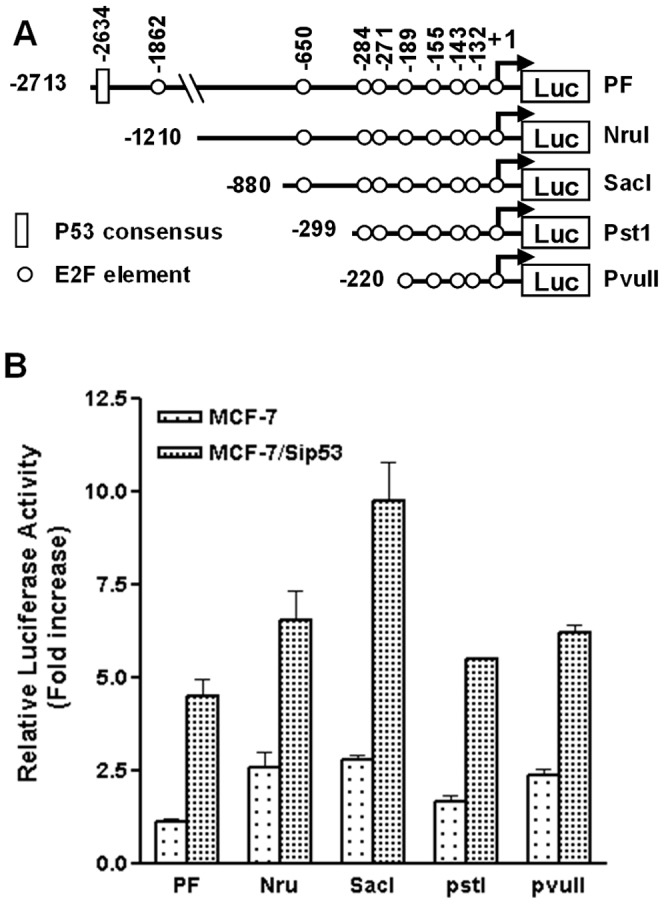
A. Reporter constructs of TAp73 promoter with a serious of deletions from the 5′ end. Putative binding sites for E2F and p53 are depicted. The drawing is not proportional to the actual size. B. MCF-7 cells were co-transfected with luciferase reporter constructs with different lengths of p73 promoter, pSV-β-Gal, and vectors of control siRNA or p53siRNA. Luciferase activity was measured 40 hours after transfection, which was followed by β-galactosidase normalization.
p53 inactivation upregulates p73 transcription through E2F-1
Since we had not found any potential p53 binding site in the first 220 bp region of the TAp73 promoter, we speculated that p53 inactivation might regulate p73 transcription through indirect mechanisms. Because E2F-1 is a major transcription factor that regulates TAp73 transcription and there are multiple E2F binding sites in the proximal promoter region [18], [21], [22], we reasoned that modulation of E2F-1 activity might play a role in p53 inactivation mediated upregulation of p73 transcription. To test our hypothesis, we co-transfected p73PF reporter gene with pcDNA3/E2F-1 or control vector into MCF-7/control and MCF-7/p53siRNA cells, respectively. We found that overexpression of E2F-1 drastically enhanced the activation of the TAp73 promoter, especially in MCF-7/p53siRNA cells (Fig. 5A). Consistent with the reporter assays, results from RT-PCR detection of TAp73 mRNA in the cells with the same transfection settings also showed that transcription of TAp73 was significantly boosted in MCF-7/p53siRNA cells (Fig. 5B).
Figure 5. Critical role E2F-1 in p53 knockdown-mediated upregulation of p73 transcription.
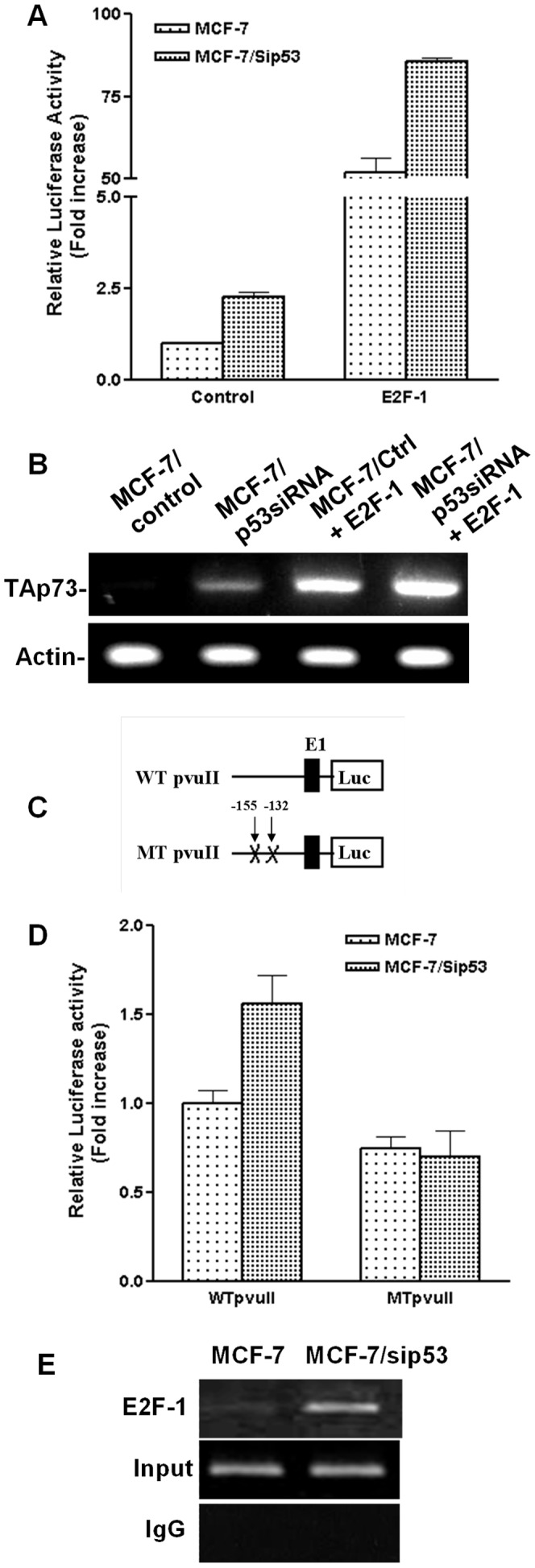
A. Additive effect of E2F-1 overexpression on p53 knockdown mediated increase in p73 promoter activity. MCF-7 cells were co-transfected with p73-PF/luciferase, pSV-β-Gal, and control or p53siRNA plasmids either in presence or in absence of E2F-1. Luciferase and β-galactosidase activity was determined 40 hours post-transfection. B. TAp73 mRNA levels in MCF-7 cells transfected with control vector, p53siRNA and/or E2F-1 cDNA. MCF-7 cells were transiently transfected with either control siRNA or p53-specific siRNA either in the presence or in the absence of E2F-1. RT-PCR analysis was performed using gene-specific primers. C & D. E2F-1 binding is required for p53 knockdown mediated increase in p73 promoter activity. MCF-7 cells were co-transfected with control or p53siRNA vector and reporter construct encoding wild type or mutant p73 promoter (p73PVUII, −220 to +71), in addition to pSV-β-Gal plasmid. The mutant PVUII promoter fragment contains mutant E2F-1 binding sites at −155 and −132 (C). Luciferase and β-gal activity was determined 40 hours post- transfection (D). E. Occupancy of the E2F responsive element in the TAp73 promoter by E2F-1 is enhanced in MCF-7/p53siRNA cells. Detected with chromatin immunoprecipitation (ChIP) assay, DNA fragment of the TAp73 promoter was amplified from the complexes immunoprecipitated with E2F-1 antibody from the paired cell lines. Input row were the DNA fragment amplified from the extracts before immunoprecipitation. In the control immunoglobulin G (IgG) reaction, PCR was done in the eluates from beads collected after preclearing of these extracts with normal rabbit serum.
To further demonstrate the specific role of E2F-1 in p53 inactivation regulated p73 transcription, we tested whether mutation of E2F binding sites abolishes p53 inactivation mediated upregulation of p73 transcription. It has been shown that, although there are multiple putative E2F binding sites in the proximal region of the TAp73 promoter, simultaneous mutation of two major E2F binding sites, −132 and −155, reduces the promoter activity by more than 90% [18]. We therefore transfected reporter constructs with wild type p73pvUII (wtp73pvuII) or p73pvUII with double mutations at −132/−155 (mtp73pvuII) (Fig. 5C) into MCF-7/control and MCF-7/p53siRNA cells, respectively. As shown in Fig. 5D, in contrast to induction in the cells transfected with wtp73pvuII, there was no upregulation of luciferase activity in MCF-7/p53siRNA cells transfected with mutant reporter construct (mtp73pvuII).
To support the reporter assay data, we performed chromatin immunoprecipitation (ChIP) assay to determine the effect of p53 inactivation on E2F-1 binding to the p73 promoter. As shown in Fig. 5E, PCR reaction targeting the p73 promoter region amplified a greater amount of DNA from the E2F-1 immunoprecipitates derived from MCF-7/p53siRNA cells, indicating that loss of p53 resulted in enhanced occupancy of E2F-1 in the TAp73 promoter (Fig. 5E). These results demonstrate that modulation of E2F activity plays a critical role in p53 inactivation mediated upregulation of p73 transcription.
p21 is a mediator of p53-E2F crosstalk in the regulation of p73 transcription
To bridge the gap between p53 and E2F-1 interactions in the regulation of p73 transcription, we tested the role of p21 in this crosstalk. p21 is a cyclin-dependent kinase inhibitor and a major transcriptional target of p53 [23]. It is known that p21 blocks retinoblastoma protein (pRb) phosphorylation by inhibition of cyclin/CDK complexes [24], [25], which constrains the transcriptional activity of E2F. To determine the role of p21 in p53 inactivation mediated upregulation of p73, we co-transfected p73PF reporter gene with pcDNA3/p21 or control vector into MCF-7/control and MCF-7/p53siRNA cells, respectively. As shown in Fig. 6A, transfection of p21 cDNA reversed the increase of p73 promoter activity by p53siRNA. Consistently, in contrast to p53siRNA induced upregulation, overexpression of wtp53 inhibited p73 promoter activation, especially in the presence of E2F-1 overexpression (Fig. 6B). We further demonstrated that overexpression of p21 abolished p53siRNA induced activation of TAp73 promoter in the absence or presence of E2F-1 overexpression, in a pattern similar to wtp53 overexpression (Fig. 6C). Taken together, these data suggest that downregulation of p21 and the consequent activation of E2F-1 activity are critical mediators of p53 knockdown-induced upregulation of p73 transcription.
Figure 6. p21 is a mediator of p53 inactivation induced upregulation of p73 transcription.
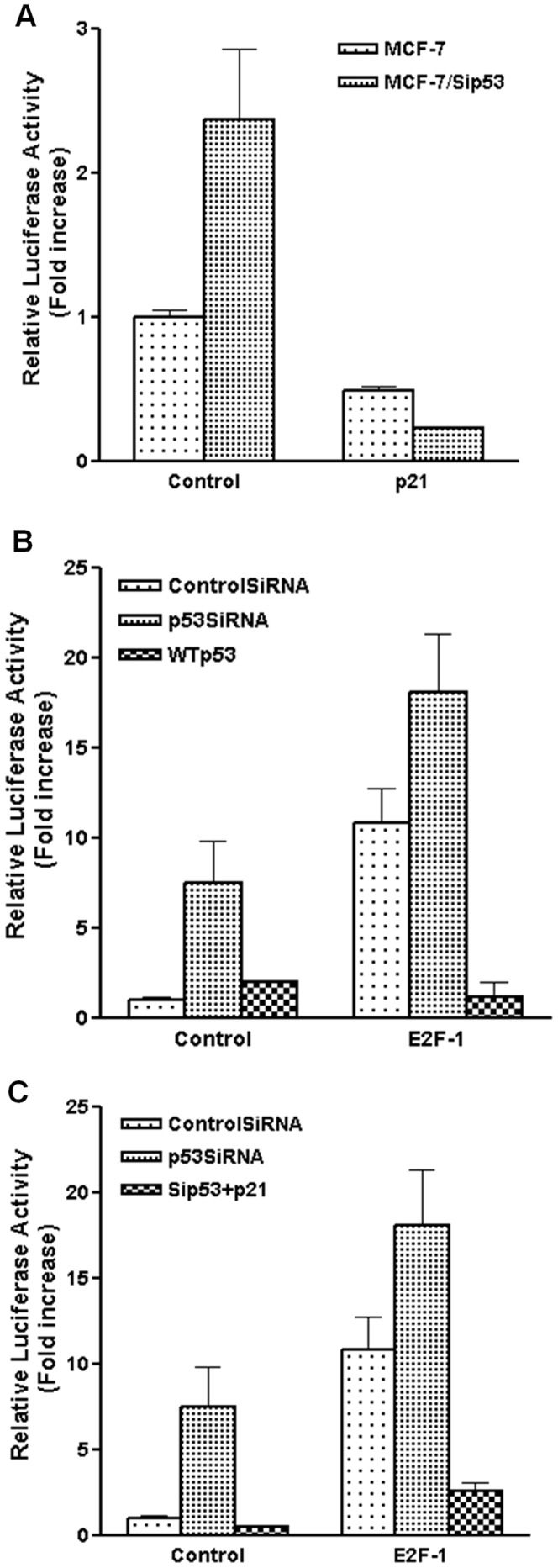
A. Overexpression of p21 abolishes p73 upregulation in control and MCF-7/p53siRNA cells. MCF-7/control and MCF-7/p53siRNA cells were cotransfected with p73-PF/pSV-β-Gal and control vector or pcDNA3/p21. Cell lysate was collected for luciferase assay 40 hours post-transfection. All the experiments were performed at least three times in triplicates. B. Overexpression of wtp53 reverses p53siRNA induced p73 transcription in the presence or absence of E2F-1 overexpression. p73-PF/pSV-β-Gal and pcDNA3/E2F-1 or control vector were cotransfected with the plasmids encoding control siRNA, p53siRNA or wtp53 into MCF-7 cells. Luciferase activity was determined as described above. C. Overexpression of p21 abrogates p53siRNA induced p73 transcription in the presence or absence of E2F-1 overexpression. p73-PF/pSV-β-Gal and pcDNA3/E2F-1 or control vector were cotransfected with the plasmids encoding control siRNA, p53siRNA or pcDNA3/p21 into MCF-7 cells. Luciferase activity was determined as described above.
Discussion
As a p53 family member and a critical molecule involved in apoptosis, cell cycle regulation and differentiation, p73 is regulated at multiple levels. It is known that p73 expression is regulated by multiple splicing, alternative promoters, and alternative initiation of translation [26], [27]. Methylation of p73 promoter has also been detected in human tumors [28], [29]. In response to cellular stresses, such as DNA damage, p73 transcription and activity can be regulated by phosphorylation, acetylation, and interacting with other cellular factors [30], [31], [32]. In particular, the interaction between p53 and p73 has been a critical issue in the understanding of the role of p73 in tumor development and therapeutic responses [33], [34]. Our data, which demonstrate p53 inactivation mediated upregulation of TAp73 in cancer cells, reveal a novel mechanism that regulates p53–p73 interactions.
Overexpression of wtp73 has been frequently detected in human cancers [8], [9], [10]. Immunohistochemical (IHC) data suggest a correlation between p73 overexpression and p53 mutation [8], [13]. Although it was difficult to discern suppressor (TAp73) from oncogenic (DNp73) isoforms in IHC tests, examination of specific isoforms of p73 mRNA indicated that both suppressor and oncogenic p73 were significantly upregulated in tumor tissues [35]. Our preliminary data also showed a correlation between p53 (mAb1801) staining, a surrogate marker of p53 mutation and p73 expression (data not shown). However, while overexpression of DNp73 was linked to wtp53 activation [35], the mechanism of TAp73 overexpression in cancer tissues remains poorly understood. We approached this critical issue by examination of p73 expression in paired cell lines with specific knockdown/knockout of wtp53. We found that inactivation of p53 in MCF-7 and HCT-116 cells resulted in increased expression of TAp73, but not DNp73, which suggests that p53 inactivation-mediated-upregulation of p73 may contribute to the p73 overexpression detected in human cancers. In addition to p53 knockdown and knockout, we found that inactivation of p53 by overexpression of mtp53 (G135A) led to p73 upregulation, indicating that p53 inactivation by means other than deletion/knockdown also induce p73 expression. However, because there are numerous p53 mutations and these mutations may have various functional outcomes [36], [37], the effect of specific p53 mutations on p73 transcription requires further analysis. Nevertheless, our data support a correlation between loss of p53 function and increase of p73 transcription.
We showed that p53 inactivation mediated upregulation of p73 was primarily regulated at the transcriptional level, and this activity was mapped to the proximal region of the p73 promoter. Previously, it was reported that there was a potential p53 binding site at the distal region (∼−2634 to−2574) of p73 promoter and expression of wtp53 in SAOS cells induced p73 expression [16]. In our tests, we found that the radio-labeled probe that contained the potential p53 binding site was able to form a weak complex with p53 from wild type MCF-7 and HCT-116 cells. However, results from reporter gene assays with series deletion of p73 promoter indicated that p53 inactivation was able to activate the luciferase reporter construct that only contained the proximal region p73 promoter (−220 to +71). These data suggest that p53 inactivation upregulated p73 transcription, which was independent of p53 binding. Increased activation of the full length p73 promoter (p73PF), which includes the p53 binding site, by p53 inactivation suggest that, at least in the cell lines tested, DNA binding independent regulation overrides DNA binding effect in p53–p73 communication.
Based on our evidence showing that p53 inactivation upregulates p73 transcription, one would expect a decrease of p73 expression in the cells transfected with wtp53. However, p73 protein levels in these cells were not decreased or even modestly increased (Fig. 1D). This might be due to the involvement of other mechanisms in the case of wtp53 overexpression, which are not symmetrical to p53 knockdown or knockout. Consistently, previous reports by different authors have shown that the TAp73 promoter is not modulated by p53 overexpression in co-transfection experiments [6], [15], [20]. In context with the transcriptional data in Fig. 2E, which shows no increase in TAp73 promoter activation upon wtp53 transfection, the results suggest that increased p73 protein levels in the presence of p53 overexpression or transfection-associated stress might induce modified protein stability and/or additional mechanisms. Given the complex nature of p53–p73 communication, differentiation of these mechanisms in p53–p73 crosstalk requires further investigation.
Our data further demonstrate that modulation of E2F-1 regulated transcription of p73 is responsible for upregulated p73 by p53 inactivation. It is known that E2F is a powerful transcription factor that regulates a number of molecules involved in cell cycle progression and apoptosis, including TAp73 [38]. E2F activity is tightly regulated by phosphorylation status of pRb, a well known tumor suppressor [38]. Hence, our results not only address how p53 inactivation upregulates p73 transcription in a DNA binding independent manner but also underscores the role of pRb/E2F-1 pathway in the regulation of p73 expression.
Based on previous reports, p53-E2F interaction may regulate E2F target genes in different ways. For the regulation of cdc2, an E2F target gene, it was reported that p53 represses the cdc2 promoter by inducing p21. Increased p21 inhibits cyclin-dependent kinase activity that enhances the binding of p130 and E2F4, which together bind to and repress the cdc2 promoter [39]. It was also reported that p53 represses survivin expression by interfering with E2F mediated transcription through forming E2F-1/p53 complex in a DNA binding dependent manner [40]. Because we found no p53 binding site in the p73PVUII fragment, and mutation of two major E2F sites or overexpression of p21 was sufficient to abolish p53 mediated upregulation of p73, it is likely that decreased p21 in p53 inactivated cells may enhance E2F mediated transcription of p73. However, because p53 may also affect E2F activity through other factors, such as p300 [41], it is possible that p53 inactivation modulates E2F mediated p73 transcription by additional mechanisms. This may partially explain our observation that p53 knockdown/knockout mediated upregulation of p73 is more evident than p53 overexpression mediated inhibition of p73.
Association between p53 and p73 status and its impact on clinical outcomes are complex issues. This is complicated by the existence of functionally opposite p73 isoforms and redundant alterations. In context with previous reports, it appears that p53 may communicate with p73 by multiple means. These include dimers between certain p53 mutants and p73 that downregulates p73 activity [33], [42]; a potential interaction between p53 and the p73 promoter at the distal region [16]; p53 mediated induction of oncogenic DNp73; and our finding that p53 inactivation upregulates p73 transcription. Different from the others, p53 inactivation mediated upregulation of TAp73 transcription could be a mechanism that explains the compensatory role of p73 in p53 mutant or inactivated cells. However, how this regulation contributes to the regulation of p53 independent apoptosis and tumor suppression requires further investigation. The outcomes would be determined by the balance between oncogenic and tumor suppressor p73 isoforms. In this context, our data showed that p53 inactivation induced remarkable decrease of DNp73 in HCT-116 cells but not MCF-7 cells, which is consistent with previous report that p53 activation induces expression of DNp73 in some cancer cells. Nevertheless, since p53 and E2F status might be also associated with the expression of other isoforms, such as ΔEx2/3p73, a comprehensive analysis of p53 inactivation regulation of other p73 isoforms/variants will be followed.
Taken together, our results reveal that p53 inactivation may upregulate TAp73 expression through E2F-1 mediated transcriptional regulation, which involves modulated p21 activity. These findings support further molecular analysis of the correlation between p53 status and TAp73 expression in human cancers, as suggested by the IHC data [8], [13]. Studies on the functional impact of p73 upregulation on apoptosis and tumor suppression in cancer cells lacking wtp53 will advance our understanding of this complex issue. Moreover, given the role of E2F-1 in the regulation of p73 transcription and p53–p73 communication, cancer therapeutics targeting E2F-p73 axis might be of great potential. Future studies on the effects of specific p53 mutation/inactivation variations on the expression of different p73 isoforms, and characterization of physical interaction between E2F-1 and p53 in the regulation of p73 transcription will advance our understanding of p53–p73 communication in tumor suppression and therapeutic responses.
Acknowledgments
We are grateful to Dr. Bert Vogelstein for the paired HCT-116 cells lines and Dr. Todd Sladek for the p21 plasmid. Special thanks are also given to Ms. X Cao for her assistance with manuscript preparation.
Funding Statement
This work was supported in part by a Seed Grant to Dr. XH Yang from the University of Oklahoma Cancer Center Institutional Research Grant from the American Cancer Society ASC-IRG #IRG-05-066-01 (http://www.cancer.org/), and by a Research Scholar Grant from American Cancer Society (RSG-08-138-01-CNE, http://www.cancer.org/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88: 323–331. [DOI] [PubMed] [Google Scholar]
- 2. Gudkov AV, Komarova EA (2003) The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer 3: 117–129. [DOI] [PubMed] [Google Scholar]
- 3. Grob TJ, Novak U, Maisse C, Barcaroli D, Luthi AU, et al. (2001) Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ 8: 1213–1223. [DOI] [PubMed] [Google Scholar]
- 4. De Laurenzi V, Costanzo A, Barcaroli D, Terrinoni A, Falco M, et al. (1998) Two new p73 splice variants, gamma and delta, with different transcriptional activity. J Exp Med 188: 1763–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Laurenzi VD, Catani MV, Terrinoni A, Corazzari M, Melino G, et al. (1999) Additional complexity in p73: induction by mitogens in lymphoid cells and identification of two new splicing variants epsilon and zeta. Cell Death Differ 6: 389–390. [DOI] [PubMed] [Google Scholar]
- 6. Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, et al. (2003) Chemosensitivity linked to p73 function. Cancer Cell 3: 403–410. [DOI] [PubMed] [Google Scholar]
- 7. Willis AC, Pipes T, Zhu J, Chen X (2003) p73 can suppress the proliferation of cells that express mutant p53. Oncogene 22: 5481–5495. [DOI] [PubMed] [Google Scholar]
- 8. Dominguez G, Silva JM, Silva J, Garcia JM, Sanchez A, et al. (2001) Wild type p73 overexpression and high-grade malignancy in breast cancer. Breast Cancer Res Treat 66: 183–190. [DOI] [PubMed] [Google Scholar]
- 9. Guan M, Peng HX, Yu B, Lu Y (2003) p73 Overexpression and angiogenesis in human colorectal carcinoma. Jpn J Clin Oncol 33: 215–220. [DOI] [PubMed] [Google Scholar]
- 10. Yokomizo A, Mai M, Tindall DJ, Cheng L, Bostwick DG, et al. (1999) Overexpression of the wild type p73 gene in human bladder cancer. Oncogene 18: 1629–1633. [DOI] [PubMed] [Google Scholar]
- 11. Zaika AI, Kovalev S, Marchenko ND, Moll UM (1999) Overexpression of the wild type p73 gene in breast cancer tissues and cell lines. Cancer Res 59: 3257–3263. [PubMed] [Google Scholar]
- 12. Yamamoto T, Oda K, Kubota T, Miyazaki K, Takenouti Y, et al. (2002) Expression of p73 gene, cell proliferation and apoptosis in breast cancer: Immunohistochemical and clinicopathological study. Oncol Rep 9: 729–735. [PubMed] [Google Scholar]
- 13. Cai YC, Yang GY, Nie Y, Wang LD, Zhao X, et al. (2000) Molecular alterations of p73 in human esophageal squamous cell carcinomas: loss of heterozygosity occurs frequently; loss of imprinting and elevation of p73 expression may be related to defective p53. Carcinogenesis 21: 683–689. [DOI] [PubMed] [Google Scholar]
- 14. Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C (2001) A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol 21: 1874–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kartasheva NN, Contente A, Lenz-Stoppler C, Roth J, Dobbelstein M (2002) p53 induces the expression of its antagonist p73 Delta N, establishing an autoregulatory feedback loop. Oncogene 21: 4715–4727. [DOI] [PubMed] [Google Scholar]
- 16. Chen X, Zheng Y, Zhu J, Jiang J, Wang J (2001) p73 is transcriptionally regulated by DNA damage, p53, and p73. Oncogene 20: 769–774. [DOI] [PubMed] [Google Scholar]
- 17. Ding Y, Inoue T, Kamiyama J, Tamura Y, Ohtani-Fujita N, et al. (1999) Molecular cloning and functional characterization of the upstream promoter region of the human p73 gene. DNA Res 6: 347–351. [DOI] [PubMed] [Google Scholar]
- 18. Seelan RS, Irwin M, van der Stoop P, Qian C, Kaelin WG Jr, et al. (2002) The human p73 promoter: characterization and identification of functional E2F binding sites. Neoplasia 4: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sigal A, Rotter V (2000) Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res 60: 6788–6793. [PubMed] [Google Scholar]
- 20. Pediconi N, Ianari A, Costanzo A, Belloni L, Gallo R, et al. (2003) Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat Cell Biol 5: 552–558. [DOI] [PubMed] [Google Scholar]
- 21. Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, et al. (2000) Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 407: 645–648. [DOI] [PubMed] [Google Scholar]
- 22. Lissy NA, Davis PK, Irwin M, Kaelin WG, Dowdy SF (2000) A common E2F-1 and p73 pathway mediates cell death induced by TCR activation. Nature 407: 642–645. [DOI] [PubMed] [Google Scholar]
- 23. Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, et al. (1993) p21 is a universal inhibitor of cyclin kinases. Nature 366: 701–704. [DOI] [PubMed] [Google Scholar]
- 24. el-Deiry WS, Harper JW, O'Connor PM, Velculescu VE, Canman CE, et al. (1994) WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res 54: 1169–1174. [PubMed] [Google Scholar]
- 25. el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825. [DOI] [PubMed] [Google Scholar]
- 26. Ozaki T, Nakagawara A (2005) p73, a sophisticated p53 family member in the cancer world. Cancer Sci 96: 729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Melino G, De Laurenzi V, Vousden KH (2002) p73: Friend or foe in tumorigenesis. Nat Rev Cancer 2: 605–615. [DOI] [PubMed] [Google Scholar]
- 28. Kawano S, Miller CW, Gombart AF, Bartram CR, Matsuo Y, et al. (1999) Loss of p73 gene expression in leukemias/lymphomas due to hypermethylation. Blood 94: 1113–1120. [PubMed] [Google Scholar]
- 29. Corn PG, Kuerbitz SJ, van Noesel MM, Esteller M, Compitello N, et al. (1999) Transcriptional silencing of the p73 gene in acute lymphoblastic leukemia and Burkitt's lymphoma is associated with 5′ CpG island methylation. Cancer Res 59: 3352–3356. [PubMed] [Google Scholar]
- 30. Dulloo I, Sabapathy K (2005) Transactivation-dependent and -independent Regulation of p73 Stability. J Biol Chem 280: 28203–28214. [DOI] [PubMed] [Google Scholar]
- 31. Oberst A, Rossi M, Salomoni P, Pandolfi PP, Oren M, et al. (2005) Regulation of the p73 protein stability and degradation. Biochem Biophys Res Commun 331: 707–712. [DOI] [PubMed] [Google Scholar]
- 32. Tsai KK, Yuan ZM (2003) c-Abl stabilizes p73 by a phosphorylation-augmented interaction. Cancer Res 63: 3418–3424. [PubMed] [Google Scholar]
- 33. Strano S, Munarriz E, Rossi M, Cristofanelli B, Shaul Y, et al. (2000) Physical and functional interaction between p53 mutants and different isoforms of p73. J Biol Chem 275: 29503–29512. [DOI] [PubMed] [Google Scholar]
- 34. Murray-Zmijewski F, Lane DP, Bourdon JC (2006) p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ 13: 962–972. [DOI] [PubMed] [Google Scholar]
- 35. Dominguez G, Garcia JM, Pena C, Silva J, Garcia V, et al. (2006) DeltaTAp73 upregulation correlates with poor prognosis in human tumors: putative in vivo network involving p73 isoforms, p53, and E2F-1. J Clin Oncol 24: 805–815. [DOI] [PubMed] [Google Scholar]
- 36. Milner J (1995) DNA damage, p53 and anticancer therapies. Nat Med 1: 879–880. [DOI] [PubMed] [Google Scholar]
- 37. Cerutti P, Hussain P, Pourzand C, Aguilar F (1994) Mutagenesis of the H-ras protooncogene and the p53 tumor suppressor gene. Cancer Res 54: 1934s–1938s. [PubMed] [Google Scholar]
- 38. Polager S, Ginsberg D (2008) E2F - at the crossroads of life and death. Trends Cell Biol 18: 528–535. [DOI] [PubMed] [Google Scholar]
- 39. Taylor WR, Schonthal AH, Galante J, Stark GR (2001) p130/E2F4 binds to and represses the cdc2 promoter in response to p53. J Biol Chem 276: 1998–2006. [DOI] [PubMed] [Google Scholar]
- 40. Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M (2002) Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem 277: 3247–3257. [DOI] [PubMed] [Google Scholar]
- 41. Lee CW, Sorensen TS, Shikama N, La Thangue NB (1998) Functional interplay between p53 and E2F through co-activator p300. Oncogene 16: 2695–2710. [DOI] [PubMed] [Google Scholar]
- 42. Di Como CJ, Gaiddon C, Prives C (1999) p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol Cell Biol 19: 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]