

Abstract
Vigabatrin (VGB) is a commonly prescribed antiepileptic drug designed to inhibit GABA-transaminase, effectively halting seizures. Unfortunately, VGB treatment is also associated with the highest frequencies of peripheral visual field constriction of any of the antiepileptic drugs and the mechanisms that lead to these visual field defects are uncertain. Recent studies have demonstrated light exposure exacerbates vigabatrin-induced retinal toxicity. We further assessed this relationship by examining the effects of vigabatrin treatment on the retinal structures of mice with genetically altered photoreception. In keeping with previous studies, we detected increased toxicity in mice exposed to continuous light. To study whether cone or rod photoreceptor function was involved in the pathway to toxicity, we tested mice with mutations in the cone-specific Gnat2 or rod-specific Pde6g genes, and found the mutations significantly reduced VGB toxicity. Our results confirm light is a significant enhancer of vigabatrin toxicity and that a portion of this is mediated, directly or indirectly, by phototransduction signaling in rod and cone photoreceptors.
Introduction
Vigabatrin (VGB) is an antiepileptic drug that inhibits γ–aminobutyric acid (GABA) transaminase (T), an enzyme that degrades GABA. Vigabatrin’s blockade of GABA-T activity elevates free cellular GABA activity in astrocytes [1]. As GABA is a major inhibitory neurotransmitter in the brain, VGB treatment greatly reduces seizure frequency, and many patients report being seizure-free on the medication [2]. However, a well-known side effect of this drug is retinal toxicity, which causes irreversible peripheral vision loss in 30% to 50% of adult patients [3], varying degrees of retinal atrophy, and decreased visual function in children with infantile spasms [4], [5]. As awareness of this side effect has grown, it has become common to monitor patients’ vision with visual field examinations and electroretinograms (ERG). Recently, Moskowitz et al. used electroretinography to identify worsening vision among pediatric patients taking the drug and concluded that ERG testing alone may be insufficient for this purpose [6]. In addition to the ERG, sweep visual evoked potential (VEP) testing has shown promise as a tool for monitoring pediatric patients taking vigabatrin [4], [7]. Nevertheless, the mechanism of the drug’s toxicity is still poorly understood, leading to a gap in our knowledge of how to prevent its vision loss side effect in patients.
Several studies have produced leads to a possible explanation for this phenomenon, showing a correlation between vigabatrin-induced damage and phototoxicity. Duboc et al. found that the retinal sections of rats treated with VGB at a dose of 250 mg/kg/day for 45 days began showing lesions, but only when the rats were maintained in a lighted room; their counterparts that remained in darkness showed no lesions [8]. Similarly, retinal damage was observed even with mice exposed to the normal daylight cycle [9]. Yukitoshi Izumi et al. found that the retinas of mice injected with VGB and maintained in a darkroom for 20 hours after injection remained normal, with no histologic retinal damage, which would suggest light plays the definitive role in VGB toxicity. While the significance of lesions is still unclear, maintaining animals in darkness during VGB treatment may eliminate the lesions completely from the albino mouse retina [10], [11], [12]. The results of these studies support the hypothesis that light plays a critical role in VGB retinal toxicity.
Our present study attempts to confirm and extend these findings, by testing the independent role of rod and cone phototransduction in VGB retinal toxicity. We examined retinal abnormalities resulting from taking the medication in mice with mutations in Gnat2 and Pde6g and showed the dependence of VGB toxicity on light. Comparisons of fold formation among the various mutant mice were used to elucidate light’s role in the mechanism of VGB retinal toxicity.
Materials and Methods
Mouse Lines and Husbandry
Ethics statement
Mice were used in accordance with the Statement for the Use of Animals issued by the Association for Research in Vision and Ophthalmology, as well as the Policy for the Use of Animals in Neuroscience Research of the Society for Neuroscience. Albino mice were used for these experiments, as they exhibit increased photoreceptor sensitivity to light compared to pigmented strains [13]. The albino MF1 strain (Harlan Laboratories) served as wild-type controls. Albino Gnat2 mice (Jackson Laboratory) carry a mutation that extinguishes important functional domains of cone α-transducin [14], [15]. Albino W70A mice carry a null mutation in Pde6g and a transgene expressing a mutant PDE6g protein with a W70A substitution. They have a desensitized photoresponse, as PDE6γ is required for rod hyperpolarization [13]. The methods for creating a W70A mouse strain have been detailed elsewhere [16], [17]. Animals were housed individually and kept on a light–dark cycle (12 hour–12 hour) before the experiment. Food and water were available ad libitum.
Determining the Incidence of Folding in Mouse Retina
Mice were weighed to determine the volume of drug to be injected. VGB (Sabril®) was dissolved in sterile PBS and was delivered by intraperitoneal (i.p.) injection at doses ranging from 400 to 3000 mg/kg. Injections were performed when mice were between three and six weeks of age. Cages were placed in a light-proof room, maintained in a room with a 12 hour on/off light cycle, or kept under constant light exposure (5000 lux) for a predetermined period between 6–48 hours. Mice were then euthanized; eyes were enucleated and fixed in ½ Karnovsky’s fixative for 24 hours. The eyes were then embedded in paraffin and sectioned at a thickness of five microns. Approximately 10 to 12 sections were collected containing the optic nerve heads. Retinal sections were stained with hematoxylin and eosin (H&E) and were examined by light microscopy.
Determining Folding Orientation and Size
Mice were exposed for 24 hours to VGB at 1500 mg/kg and kept under constant light (5000 lux). Before enucleation, a small scar was introduced to the cornea at the 12 o’clock position (Fig. 1A). This scar was used to orient the globe within a paraffin block with the assistance of a magnifying glass. An equal number of eyes from VGB treated mice were sectioned along the 12 to 6 o’clock and 9 to 3 o’clock axes. Retinas taken from VGB-treated mice were serial sectioned starting from the optic nerve head, proceeding at 5 micron sections for an additional 60 sections to collect ∼300 microns of retina (Fig. 2A). These sections were collected on 7.5 cm×5 cm slides.
Figure 1. Retinal folding in two different orientations.

Retinas used in this experiment are from mice exposed for 24 hours to 1500 mg/kg of VGB and kept under constant light (A). Mouse illustration shows the orientation of the section planes with respect to the position of the eye globe relative to the body. A corneal scar was introduced with a cautery pen at the 12 o’clock position. This scar, visible under a magnifying lens, can used to orient the eye globe during embedding so that the plane of section is known. (B–D) Examples of retinal folds detected along the 12–6 o’clock plane. (E–H) Retinal folds along the 9–3 o’clock plane. Scale bar: 100 µm.
Figure 2. Retinal folding collected in 60 sections.

Schematic of a mouse eye (A), showing the cutting pattern of the serial sections displayed from B-M. Five-micron sections were collected at regular intervals starting at the optic nerve head and continuing outwards relative the center. Approximately 60 sections were collected; every fifth section is shown here. Arrowheads indicate the position of a fold seen in all 60 sections. Retinas used in this experiment are from mice exposed for 24 hours to 1500 mg VGB/kg and exposed to a constant light source (B-M). Scale bar: 200 µm.
Statistical Analysis
A statistical analysis of the results was performed using the Statistical Package for the Social Sciences. The Chi-squared 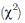 test was used to compare the incidence of the fold formation in Gnat2 and Pde6g-W70A mice with MF1 mice. A p-value of p<0.05 was taken as a criterion for statistical significance.
test was used to compare the incidence of the fold formation in Gnat2 and Pde6g-W70A mice with MF1 mice. A p-value of p<0.05 was taken as a criterion for statistical significance.
The lengths of folds were measured and expressed as a percentage comparing fold length to the total length between the optic nerve and the ciliary body. We tested only in folding-positive mice (mice that displayed folds) (Fig. 3). The slides were serial sectioned and slides displaying the lengthiest folds were selected for measurement. Group results were expressed as the means of individual percentages ± SEM. Statistical comparisons between mutant mouse groups were performed by t-test.
Figure 3. Folding coverage percentage in different mutated mice.
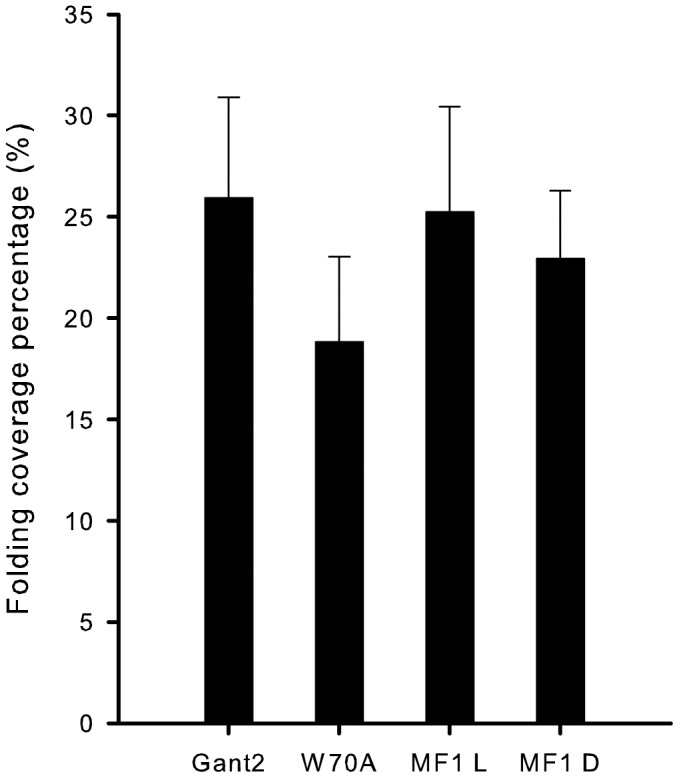
Folding coverage percentages measured in the slides of folding-positive mice were compared between groups of mutant mice by t-test. No significant differences were found between each group. (t = 0.09, p>0.05 for Gnat2 and MF1-light; t = 0.56, p>0.05 for W70A and MF1-light; t = 0.71, p>0.05 for Gnat2 and W70A). (MF1L: MF1 mice kept in light; MF1D: MF1mice kept in dark).
Results
In keeping with previous studies, in which single doses of up to 1000 mg/kg have been administered to test toxicity effects in the mouse retina [9], [18], we used an expanded VGB dosage range, from 400 mg/kg to 3000 mg/kg. These higher doses, together with a light intensity of 5000 lux, were expected to result in elevated VGB levels, thus significant decreases in GABA transaminase activity and significant increases in GABA levels as soon as 2 hours after intraperitoneal injection [9], [18], [19]. Our pilot experiment using MF1 mice involved a range of VGB doses (400, 600, 800, and 1000 mg/kg). Examination of H&E stained retinas from this series demonstrated abnormal retinal structure in two of the four retinas at the 1000 mg/kg dose. This effect was restricted to the outer nuclear layer, in which a portion of the ONL folded back on itself (Fig. 4). The retinas from lower VGB doses appeared normal. Next we tested higher VGB doses ranging from 1000 to 3000 mg/kg under various light exposure durations and conditions (Table 1). Doses greater than 2000 mg/kg were found potentially lethal, but lower doses were not associated with death 24 hours after injection. Only non-lethal doses of 1500 mg/kg were used in the mice examined for retinal folding. Examination of retinal sections in pilot experiments demonstrated ONL folding in 5 of 35 cases.
Figure 4. Retinal folding after VGB treatment.
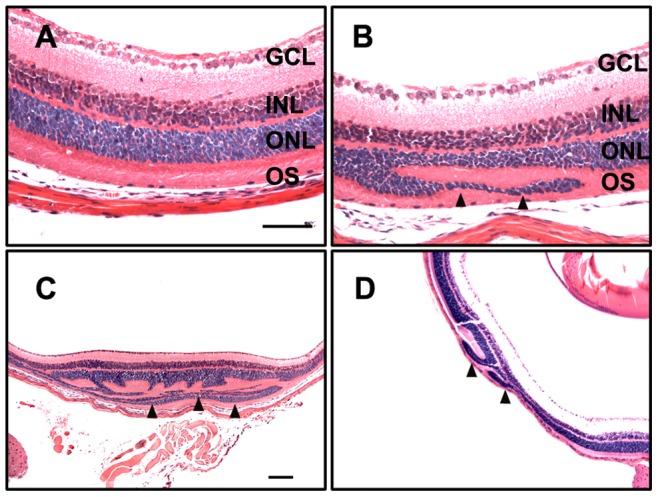
Retinas taken from mice injected with saline (A) or VGB at 1000 mg/kg (B-D). The ONL appears to “fold” over itself (arrowheads) and includes the OS layer. The appearance and extent of the folds can vary; compare with B, C, and D. GCL: ganglion cell layer; INL: inner nuclear layer; ONL: outer nuclear layer. Scale bars: 100 µm.
Table 1. Pilot study under various conditions.
| VGB Dose (mg/kg) | Light | Exposure (hours) | Mouse | Total eyes | Negative | Positive | Comment |
| 1000 | cycle | 24 | 2 | 4 | 4 | 0 | |
| 1000 | light | 24 | 4 | 8 | 8 | 0 | |
| 1000 | light | 12 | 1 | 2 | 2 | 0 | |
| 1000 | light | 6 | 1 | 2 | 2 | 0 | |
| 1000 | dark | 12 | 1 | 2 | 2 | 0 | |
| 1000 | dark | 6 | 1 | 2 | 2 | 0 | |
| 1500 | cycle | 24 | 2 | 4 | 3 | 1 | |
| 1500 | light | 24 | 2 | 4 | 3 | 1 | |
| 2000 | cycle | 24 | 2 | 4 | 3 | 1 | 1 died overnight |
| 2000 | light | 24 | 2 | 4 | 4 | 0 | 1 died overnight |
| 2000 | cycle | 12 | 1 | 2 | 2 | 0 | |
| 2000 | light | 12 | 1 | 2 | 2 | 0 | |
| 3000 | cycle | 24 | 1 | 2 | 2 | 0 | 1 died overnight |
| 3000 | light | 24 | 1 | 2 | 2 | 0 | 1 died overnight |
| 3000 | light | 12 | 2 | 4 | 3 | 1 | |
| 3000 | dark | 12 | 1 | 2 | 1 | 1 |
Since both the frequency of VGB-induced folding and the numbers of mice tested for each variable were low, we selected a single condition and increased the number of mice in an attempt to establish a meaningful folding frequency. Therefore, we treated 10 mice at a 1500 mg/kg dose, exposing these mice to constant light (5000 lux) for 24 hours (Table 2). The same number of control mice were injected with saline, exposed to constant light, and harvested at 24 hours. In this experiment, we found VGB-associated folding occurred at a rate of 35% with 7 of the 20 mouse retinas displaying folding. We observed one instance of folding in the control untreated animals, which may have been due to light exposure alone (Table 2, Fig. 5J).
Table 2. Incidence of VGB-associated folding at the 1500 mg/kg dose.
| VGB Dose (mg/kg) | Exposure (hours) | Light (lux) | Mouse | Total eyes | Negative | Positive | Frequency |
| NO DRUG | 24 | 5000 | MF1 | 16 | 15 | 1 | 6% |
| 1500 | 24 | 5000 | MF1 | 20 | 13 | 7 | 35% |
| 1500 | 48 | 5000 | MF1 | 24 | 11 | 13 | 54% |
Figure 5. Examples of retinal sections under various conditions in different mutant mice.

Panels show retinas taken from the experiments described in Tables 2 and 3. The frequencies of folding are indicated in the tables. The experimental conditions for each sample are indicated in each panel. Panels B, D, F, H, J, and L show folds (*). Panels A, C, E, G, I, and K show retinas with no detectable folds; samples are shown at the same magnification. Scale bar: 100 µm.
To determine whether the rate of detection was influenced by folds’ small size or region-specificity, we performed orientation and serial sectioning experiments. For the orientation experiment, retinas were sectioned along either the 12 to 6 o’clock or 9 to 3 o’clock axes, with a roughly equal number of eyes with folds occurring in each group (Fig. 1). For the sectioning experiments, four retinas were sectioned completely through 300 microns, starting at the edge of the optic nerve head and continuing outwards away from the center (Fig. 2A). One of the four retinas in this test condition exhibited a fold, which was detectable in all 60 sections (Fig. 2B–M). The results suggest the orientation and the size of folds were not the reasons for low incidences of folding; the folds’ locations were independent of both the eye globe orientations and the planes of section.
Next, we tested whether extending the time of light exposure between drug injection and analysis would increase the frequency of fold detection. We injected 12 mice with 1500 mg/kg VGB and maintained them under constant light exposure (5000 lux) for 48 hours. Under these new conditions, we found that the incidence of retinal folding increased from 35% to 54% (Table 2). We next examined whether light exposure could influence vigabatrin toxicity. Mice given the standard 1500 mg/kg VGB dose but kept for 48 hours in a light-proof room showed a reduced folding incidence of 30% (dark), as compared to the control of 54% (light) (Table 3). These results suggest prolonged light exposure exacerbates VGB-induced retinal toxicity.
Table 3. The incidence of retinal folding in different mice.
| VGB Dose (mg/kg) | Exposure (hours) | Light (lux) | Mouse | Total eyes | Negative | Positive | Frequency |
| 1500 * | 48 | 5000 | MF1 | 24 | 11 | 13 | 54% |
| 1500 | 48 | Dark | MF1 | 23 | 16 | 7 | 30% |
| 1500 | 48 | 5000 | Gnat2 | 38 | 29 | 9 | 24% |
| 1500 | 48 | 5000 | W70A | 48 | 38 | 10 | 21% |
Data from the 48-hour exposure under 5000 lux light experiment, shown in Table 2, is shown again here for comparison.
To extend these findings, we next tested if genetic determinants of cone or rod phototransduction play a role in VGB toxicity by analyzing VGB-treated Gnat2 and Pde6g-W70A transgenic mice (Table 3, Fig. 5J, H, K, L). Similarly to the dark-exposed mice, Gnat2 mutant mice, which completely lack α-transducin function in cone phototransduction [15], showed a reduced folding incidence (24%) compared to their counterparts the MF1 wild-type mice (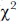 = 5.97, p<0.05). Likewise, the incidence of folding in Pde6g-W70A transgenic mice, which have abnormal rod phototransduction activity, was significantly reduced relative to controls (= 8.18, p<0.01). Figure 3 summarizes the folding coverage percentages among the folding-positive groups of mice. Comparisons of the four mouse groups by t-tests revealed no significant differences in folding coverage percentages among the folding-positive mice belonging to all four groups (Fig. 3) (t = 0.09, p>0.05 for Gnat2 and MF1-light; t = 0.56, p>0.05 for W70A and MF1-light; t = 0.71, p>0.05 for Gnat2 and W70A).
= 5.97, p<0.05). Likewise, the incidence of folding in Pde6g-W70A transgenic mice, which have abnormal rod phototransduction activity, was significantly reduced relative to controls (= 8.18, p<0.01). Figure 3 summarizes the folding coverage percentages among the folding-positive groups of mice. Comparisons of the four mouse groups by t-tests revealed no significant differences in folding coverage percentages among the folding-positive mice belonging to all four groups (Fig. 3) (t = 0.09, p>0.05 for Gnat2 and MF1-light; t = 0.56, p>0.05 for W70A and MF1-light; t = 0.71, p>0.05 for Gnat2 and W70A).
Discussion
Vigabatrin can induce structural changes in the retina. The drug has been demonstrated to cause movement of photoreceptor nuclei toward the retinal pigment epithelium in several studies [8], [19]. These previous studies are consistent with our observation of folds in the outer nuclear layer of the retina. Although the formation of folds is closely related to vigabatrin dose [21] and duration of light exposure [9], [11], we observed that genetic mutations could also influence fold efficiency. In this study, we tested mice that carried mutations that affected rod or cone photoreceptor signaling and observed decreased folding formation.
Light has been thought to be an important factor in influencing VGB toxicity. In this study, the folding efficiency of MF1 mice kept in the dark was significantly lower than that of the light-exposed group. One mouse developed retinal folding after sustained light exposure, even without VGB injection. These results are consistent with the findings of Yukitoshi et al. [9], who reported that light is necessary for acute VGB retinotoxicity. Photoreceptor displacement was also observed in previous studies [11], [20]. Our research extended these findings: mice maintained in light conditions for 48 hours displayed increased folding incidence compared with those maintained in light conditions for 24 hours. These results suggest drug-induced retinal toxicity will occur in lighting conditions, but will be inhibited when exposure to light is limited.
VGB-elicited photoreceptor degeneration has been reported to be light-dependent by several studies [9], [11], [12]. Our results (Table 3) showed that engineered mice carrying mutations in phototransduction genes interfering with light transduction showed a decreased incidence of folding formation when given VGB. These results suggest light-induced PDE activation in rod or cone photoreceptors contributes to vigabatrin toxicity.
Considering this light dependence, we speculate rod or cone phototransduction pathway genes thus play an important role in vigabatrin toxicity. Phototransduction begins at photoreceptor outer segments [22]; activation of rhodopsin in rods and cones by light occurs, followed by stimulation of G-protein-coupled receptors that activate phosphodiesterase (PDE) [23]. Our study utilized W70A-strain and Gnat2 mutant mice, which carry phototransduction signaling defects in their rods or cones. The W70A mice used in this study carried a point mutation in the gene encoding for the γ-subunit of rod cGMP phosphodiesterase (PDE6γ). The Gnat2 mutant mice have extinguished important functional domains of the G-protein α-subunit [19], which has been shown to interact with all cone opsins and phosphodiesterase α-subunits [25] and to play an important role in transmitting visual signals [24]. Our findings are consistent with the interpretation that vigabatrin-induced retinal toxicity is dependent on phototransduction signaling. Interestingly, once folds had formed, their relative size and coverage did not differ among different groups of mice (Fig. 3). More investigation is needed to establish how the visual pathway is subject to interference from vigabatrin, and how these folds form in response to the toxicity.
Connecting some hypotheses which have been developed in the past about the mechanisms of VGB-induced retinal toxicity, we would consider the possibility that the drug may cause retinal toxicity through improper depolarization of the cells. In the retina, GABA is an established inhibitory neurotransmitter associated with several different types of cells [26]. Three different groups of GABA receptors are distributed in retina, termed GABAA, GABAB, and GABAC. GABAA receptors are distributed in all neurons except for rod photoreceptors, while GABAC receptors gate the Cl-channels located in cone photoreceptors and bipolar cells. GABAC receptors are more sensitive to GABA than are GABAA receptors. The highest concentration of these receptors is on axon terminals of rod bipolar cells [27], where they form a positive feedback loop with Ecl, increasing the sensitivity of the mechanism that controls release of GABA and leads to cells’ depolarization. VGB elevates GABA concentration, keeping cells depolarized for longer periods of time.
In the normal phototransduction pathway, light strikes the photoreceptor, activating PDE6, diminishing the supply of cGMP, hyperpolarizing the photoreceptor cell, and causing less glutamate to be released. Then ON bipolar cells become depolarized [23]. Excessive or prolonged depolarization of these cells induces a potentially cytotoxic calcium influx [28]. The mice with mutations in the Gnat2 or Pde6g genes would have reduced photoreceptor hyperpolarization, leading to reduced rates of ON bipolar cells’ depolarization. We speculate that the mutant mice are protected from retinal toxicity in this manner. The precise relationship between fold formation and cells’ depolarization is unclear but presumably involves additional mechanisms.
In summary, this study reports on VGB-elicited retinal toxicity in mice with different phototransduction mutations that have not been previously described, and gives insight into the mechanisms of light-mediated retinal toxicity. Our findings suggest that phototransduction is a significant enhancer of vigabatrin toxicity. More experiments should be done to confirm these results before they can be applied to patients who are treated with VGB.
Funding Statement
This work was funded by the National Institutes of Health grants 5P30CA013696, R01EY018213, P30EY019007, the Department of Defense grant DOD-TS080017-W81XWH- 09-1-0575, and the Foundation Fighting Blindness. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Yee JM, Agulian S, Kocsis JD (1997) Vigabatrin enhances promoted release of GABA in neonatal rat optic nerve. Epilepsy Res 29: 195–200. [DOI] [PubMed] [Google Scholar]
- 2. Livingston JH, Beaumont D, Arzimanoglou A, Aicardi J (1989) Vigabatrin in the treatment of epilepsy in children. Br J Clin Pharmacol 27: 109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waterhouse EJ, Mims KN, Gowda SN (2009) Treatment of refractory complex partial seizures: role of vigabatrin. Neuropsychiatr Dis Treat 5: 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Durbin S, Mirabella G, Buncic JR, Westall CA (2009) Reduced Grating Acuity Associated with Retinal Toxicity in Children with Infantile Spasms on Vigabatrin Therapy. Vis Sci 50: 4011–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buncic JR, Westall CA, Panton CM, Munn JR, MacKeen LD, et al. (2004) Characteristic retinal atrophy with secondary “inverse” optic atrophy identifies vigabatrin toxicity in children. Ophthalmology 111(10): 1935–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moskowitz A, Hansen RM, Eklund SE, Fulton AB (2012) Electroretinographic (ERG) responses in pediatric patients using vigabatrin. Doc Ophthalmol 124: 197–209. [DOI] [PubMed] [Google Scholar]
- 7. Van der Torren K, Graniewski-Wijnands HS, Polak BC (2002) Visual field and electrophysiological abnormalities due to vigabatrin. Doc Ophthalmol 104: 181–188. [DOI] [PubMed] [Google Scholar]
- 8. Duboc A, Hanoteau N, Simonutti M, Rudolf G, Nehlig A, et al. (2004) Vigabatrin, the GABA-Transaminase Inhibitor, Damages Cone Photoreceptors in Rats. Ann Neurol 55: 695–705. [DOI] [PubMed] [Google Scholar]
- 9. Izumi Y, Ishikawa M, Benz AM, Izumi M, Zorumski CF, et al. (2004) Acute Vigabatrin Retinotoxicity in Albino Rats Depends on Light But Not GABA. Epilepsia 45: 1043–1048. [DOI] [PubMed] [Google Scholar]
- 10. Gibson JP, Yarrington JT, Loudy DE, Gerbig CG, Hurst GH, et al. (1990) Chronic toxicity studies with vigabatrin, a GABA-transaminase inhibitor. Toxicol Pathol 18: 225–238. [DOI] [PubMed] [Google Scholar]
- 11. Jammoul F, Wang Q, Nabbout R, Coriat C, Duboc A, et al. (2009) Taurine deficiency is a cause of vigabatrin-induced retinal phototoxicity. Ann Neurol 65: 98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y, Wang QP, Ye W (2009) Neuronal Plasticity Induced by Vigabatrin Can Be Suppressed by Darkness in the Mouse Retina. China J Clin Neurosci 5: 457–462. [Google Scholar]
- 13. Lyubarsky AL, Daniele LL, Pugh EN (2004) From candelas to photoisomerizations in the mouse eye by rhodopsin bleaching in situ and the light-rearing dependence of the major components of the mouse ERG. Vision Res 44: 3235–3251. [DOI] [PubMed] [Google Scholar]
- 14. Allen AE, Brown TM, Lucas RJ (2011) A distinct contribution of short-wavelength- sensitive cones to light-evoked activity in the mouse pretectal olivary nucleus. J Neurosci 31(46): 16833–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang B, Dacey MS, Hawes NL, Hitchcock PF, Milam AH, et al. (2006) Cone Photoreceptor Function Loss-3, a Novel Mouse Model of Achromatopsia Due to a Mutation in Gnat2 . Invest Ophthalmol Vis Sci 47(11): 5017–21. [DOI] [PubMed] [Google Scholar]
- 16. Tsang SH, Gouras P, Yamashita CK, Kjeldbye H, Fisher J, et al. (1996) Retinal degeneration in mice lacking the gamma subunit of the rod cGMP phosphodiesterase. Science. 272: 1026–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsang SH, Burns ME, Calvert PD, Gouras P, Baylor DA, et al. (1998) Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science 282: 117–121. [DOI] [PubMed] [Google Scholar]
- 18. Sills GJ, Butler W, Forrest G, Ratnaraj N, Patsalos PN, et al. (2003) Vigabatrin, but not Gabapentin or Topiramate, Produces Concentration-related Effects on Enzymes and Intermediates of the GABA Shunt in Rat Brain and Retina. Epilepsia 44(7): 886–892. [DOI] [PubMed] [Google Scholar]
- 19. Sills GJ, Patsalos PN, Butler E, Forrest G, Ratnaraj N, et al. (2001) Visual field constriction: accumulation of vigabatrin but not tiagabine in the retina. Neurology 57(2): 196–200. [DOI] [PubMed] [Google Scholar]
- 20. Wang QP, Jammoul F, Duboc A, Gong J, Simonutti M, et al. (2008) Treatment of epilepsy: the GABA-transaminase inhibitor, vigabatrin, induces neuronal plasticity in the mouse retina. Eur J Neurosci 27: 2177–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kjellström U, Bruun A, Ghosh F, Andréasson S, Ponjavic V (2009) Dose-related changes in retinal function and PKC-alpha expression in rabbits on vigabatrin medication: Effect of vigabatrin in the rabbit eye. Graefes Arch Clin Exp Ophthalmol 247(8): 1057–67. [DOI] [PubMed] [Google Scholar]
- 22. Arshavsky VY, Burns ME (2012) Photoreceptor signaling: supporting vision across a wide range of light intensities. J Biol Chem 287(3): 1620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsang SH, Tsui I, Chou CL, Zernant J, Haamer E, et al. (2008) A novel mutation and phenotypes in phosphodiesterase 6 deficiency. Am J Ophthalmol 146: 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen CK, Woodruff ML, Chen FS, Shim H, Cilluffo MC, et al. (2010) Replacing the rod with the cone transducing α subunit decreases sensitivity and accelerates response decay. J Physiol 588(17): 3231–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Y, Arshavsky VY, Ruoho AE (1996) Interaction sites of the COOH-terminal region of the gamma subunit of cGMP phosphodiesterase with the GTP-bound alpha subunit of transducin. J Biol Chem 271(43): 26900–7. [DOI] [PubMed] [Google Scholar]
- 26. Mustafa BA (1995) Diversity of GABA receptors in the vertebrate outer retina. Trends Neurosci 18(3): 118–200. [DOI] [PubMed] [Google Scholar]
- 27. Staley KJ, Soldo BL, Proctor WR (1995) Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 269: 977–981. [DOI] [PubMed] [Google Scholar]
- 28. Kinirons P, Cavalleri GL, Shahwan A, Wood NW, Goldstein DB, et al. (2006) Examining the role of common genetic variation in the gamma2 subunit of the GABA(A) receptor in epilepsy using tagging SNPs. Epilepsy Res 70: 229–38. [DOI] [PubMed] [Google Scholar]