

Abstract
Hymeglusin (1233A; F244; L-659-699) is established as a specific beta lactone inhibitor of eukaryotic hydroxymethylglutaryl-CoA synthase (HMGCS). Inhibition results from formation of a thioester adduct to the active site cysteine. In contrast, hymeglusin’s effects on bacterial HMG-CoA synthase, mvaS, have been minimally characterized. Hymeglusin blocks growth of Enterococcus faecalis. After removal of inhibitor from culture media, a growth curve inflection point at 3.1 hr is observed (versus 0.7 hr for uninhibited control). Upon hymeglusin inactivation of purified E. faecalis mvaS, the thioester adduct is more stable than that measured for human HMGCS. Hydroxylamine cleaves the thioester adduct; substantial enzyme activity is restored at a rate that is 8-fold faster for human HMGCS than for mvaS. Structural results explain these differences in enzyme-inhibitor thioester adduct stability/solvent accessibility. The E. faecalis mvaS-hymeglusin co-crystal structure (1.95 Å) reveals virtually complete occlusion of bound inhibitor in a narrow tunnel that is largely occluded from bulk solvent. In contrast, eukaryotic (Brassica juncea) HMGCS binds hymeglusin in a more solvent exposed cavity.
Hymeglusin [(2E,4E,7R)-11-[(2R,3R)-3-(hydroxymethyl)-4-oxooxetan-2-yl]-3,5,7-trimethylundeca-2,4-dienoic acid] was chemically characterized by Aldridge et al. (1) and described as an antibiotic but its biological properties were not reported. Tomoda et al. (2) subsequently demonstrated hymeglusin’s antimicrobial activity against several fungi and bacteria.
Hymeglusin structure
After Miziorko and Behnke (3, 4) identified 3-chloropropionyl-CoA as a mechanism based inactivator of animal HMG-CoA1 synthase (EC 4.1.3.5), hymeglusin was identified as the first natural product that specifically inhibits this cholesterogenic enzyme (5, 6). This led to the expanded effort in characterizing hymeglusin inhibition of eukaryotic cholesterol biosynthesis (6, 7) and of eukaryotic HMG-CoA synthase (8). Both 3-chloropropionyl-CoA and hymeglusin target the active site cysteine involved in forming the two reaction intermediates, which are thioester adducts of enzyme to acetyl or HMG-CoA groups (Schemes 1, 2).
Scheme 1.

HMG-CoA synthase (mvaS) reaction
Scheme 2.

Reaction of hymeglusin with active site cysteine of HMGCoA synthase (mvaS)
The confirmation of the active site function of the cysteine targeted by these inactivators was provided by the demonstration (9) that alanine or serine substitution of this residue produced an enzyme that was devoid of catalytic activity and unable to form the covalent reaction intermediates. More recently, Pojer et al. (10) reported the structure of a Brassica juncea HMG-CoA synthase adduct with hymeglusin. These results showed that the inhibitor’s extended aliphatic chain bound in a funnel shaped cavity of this plant enzyme and confirmed thioester formation between the inhibitor’s beta lactone and the active site cysteine.
While hymeglusin was originally described as an antibiotic, little is known about its interaction with mvaS, the bacterial HMG-CoA synthase. This report characterizes the inhibitor’s effect on Enterococcus faecalis and on isolated E. faecalis mvaS. Differences between persistence of the inhibition observed for bacterial cells in light of analogous observations documented for animal cells (7) prompted comparison of the enzyme-inactivator adducts for E. faecalis and human enzymes. The results suggested possible differences in the hymeglusin binding sites. Crystallization of hymeglusin-inactivated mvaS allowed a test of this hypothesis. The complementary biochemical and structural results of these studies are presented.
EXPERIMENTAL PROCEDURES
Materials
Acetyl-CoA and acetoacetyl-CoA were synthesized using acetic anhydride and diketene, respectively, according to the procedure of Simon and Shemin (11). Hymeglusin (L-659-699) was a generous gift from M.D. Greenspan (Merck Research Laboratories). Hydroxylamine hydrochloride was purchased from Eastman Laboratory Chemicals. Other biological and chemical products used in these studies were reagent grade materials purchased from Fisher Scientific or Sigma-Aldrich.
Cloning, overexpression, and purification of E. faecalis mvaS
A gene fragment encoding the entire mvaS open reading frame (residues 1-383) was amplified from E. faecalis genomic DNA by PCR, digested with BamHI and NotI endonucleases, and subcloned into the corresponding restriction sites of the prokaryotic overexpression vector pT7HMT (12). This vector directs IPTG-inducible expression of an N-terminally hexahistidine-tagged form of the mvaS protein, which can be digested from the affinity tag following treatment with TEV protease. Following confirmation of the mvaS sequence, the plasmid described above was transformed into E. coli BL21(DE3) cells. Selection of positive transformants, as well as bacterial growth and induction protein overexpression were carried out according to previously published methods (13).
Soluble, tagged E. faecalis mvaS was isolated from 1 L of induced E. coli cells through a combination of affinity and ion-exchange chromatographies. Briefly, the cells were resuspended, homogenized by microfluidization, and a soluble extract was prepared by high-speed centrifugation as described by Barta et al. (13). The tagged mvaS enzyme was then recovered from this supernatant using a Ni2+-NTA Sepharose column (GE Biosciences), again as previously described (12). Upon elution from the affinity column, recombinant TEV protease was used to digest the mvaS enzyme away from its affinity tag (12); however, the sequence GSTGS remains at the enzyme N terminus as an artifact of the subcloning procedure. Following buffer exchange into 20 mM Tris (pH 8.0), final purification to apparent homogeneity was achieved by Resource Q anion-exchange chromatography (GE Biosciences). The purified mvaS was concentrated to 5 mg/ml, buffer exchanged into 10 mM Tris (pH 7.5), 50 mM NaCl, and stored at 4 ºC for further use.
Inhibition of E. faecalis culture growth by hymeglusin
Two samples (10 mL) of sterile LB culture media, containing either 0 or 25 μM hymeglusin, were inoculated with 200 μL of an overnight culture of E. faecalis. Samples were incubated with shaking for 3 hours at 37 ºC. A 3 mL aliquot of each culture was centrifuged at 3000g for 5 minutes to pellet bacteria before resuspension in either 3 ml of fresh LB or fresh LB containing 25 μM hymeglusin. At 30 minute intervals, absorbance of each culture was measured at 600 nm.
Kinetic characterization of hymeglusin inhibition of E. faecalis mvaS
Enzyme activity was measured at 412 nm by the DTNB method of Skaff and Miziorko (14). Purified mvaS (48 nM) was incubated with hymeglusin (75–600 nM) in 100 mM Tris-Cl (pH 8.0). The reaction was performed at 18 ºC to allow measurement of activity at an adequate number of time points while maintaining elevated concentration ratios of hymeglusin/enzyme. At the specified time points, 400 μM acetyl-CoA (~Km level) was added to the incubation mix to acetylate free enzyme and protect against further formation of any hymeglusin adduct. Acetoacetyl-CoA (7 μM) was then added to initiate measurement of enzyme activity, which was performed in the presence of 0.2 mM DTNB. The data, which indicated time dependent loss of activity, were fit to semi-log plots of % residual activity versus time using a linear model and Microsoft Excel; correlation coefficients ranged from 0.970 to 0.995. Nonlinear regression fits (GraphPad Prism 4) of the data indicated a kinact = 3.5±0.6 min−1 and KI = 700±18.5 nM.
Recovery of HMGCS and mvaS activity from hymeglusin inhibition
Purified human HMGCS (14) or E. faecalis mvaS samples (9 μM) were incubated with 20 μM hymeglusin at room temperature in 0.1 M Tris-Cl (pH 8.0) for one hour. After this incubation period, each protein sample retained less than 10 percent residual activity as determined by the DTNB assay method (0.1 M Tris (pH 8.0) with 0.2 mM DTNB, 400 μM AcCoA and 7 μM AcAcCoA). Each mixture was then passed through a G50 centrifugal column equilibrated with 0.1 M Tris (pH 8.0) to remove any residual unbound hymeglusin and A280 was measured to confirm the comparability of the protein concentration of each sample. 1 mM neutralized hydroxylamine was added to each sample. Activity assays were performed at the indicated incubation time points over a one hour period following the hydroxylamine additions.
Crystallization, diffraction data collection, structure determination, refinement, and analysis
E. faecalis mvaS was crystallized by vapor diffusion of hanging drops at 20 ºC. In the current protocol, 1 μl of protein solution (5 mg/ml) was mixed with 1 μl of reservoir solution (0.1 M Bis-Tris (pH 6.3–6.5), 0.2 M NaCl, and 23% (w/v) polyethylene glycol 3350) that had been previously diluted with an equal volume of ddH2O. Block-shaped crystals appeared after 2 days and continued to grow in size over the course of 1–2 weeks. Crystals were harvested and flash cooled in a cryoprotectant solution of reservoir buffer where the polyethylene glycol 3350 concentration had been increased to 30% (w/v). Co-crystallization was achieved by adding hymeglusin (final concentration of 0.5 mM) to a small volume of purified mvaS protein, prior to establishing crystallization drops as described above.
Monochromatic X-ray diffraction data were collected from single crystals at −173 ºC using beamline 22-BM of the Advanced Photon Source, Argonne National Laboratory (Table 1). Upon completing data collection, the individual reflections were indexed, integrated, merged, and scaled using the HKL2000 software package (15). Initial phase information was obtained for both structures presented here by molecular replacement using phenix.automr (16). Chain A of a previously determined ligand-free E. faecalis mvaS structure (PDB code 1X9E) was used as a search model (17). The highest scoring solution for this crystal system contained an mvaS tetramer in the asymmetric unit; this corresponded to a Matthews coefficient of 2.21 Å3/Da and a solvent content of 44.23%.
Table 1.
Diffraction Data Collection and Structure Refinement Statistics
| Data Collectiona | ||
| Crystal | Ef mvaS | Ef mvaS + hymeglusin |
| Beamline | APS 22-BM | APS 22-BM |
| Wavelength (Å) | 1.000 | 1.000 |
| Space Group | P21 | P21 |
| Cell Dimensions (Å, °) | a=109.53 | a=108.33 |
| b=56.28 | b=52.76 | |
| c=123.89 | c=117.58 | |
| β=100.02 | β=99.51 | |
| Resolution (Å) | 50.00–1.60 | 50.00–1.95 |
| Reflections (unique) | 1,306,891 (193,904) | 445,318 (92,792) |
| Completeness (%) | 98.5 (95.8) | 96.8 (87.3) |
| Redundancy (fold) | 6.7 | 4.8 |
| <I>/<σI> | 22.2 (3.6) | 11.3 (2.6) |
| Rmerge (%)b | 6.2 (45.6) | 12.7 (42.1) |
| Refinement | ||
| RCSB Accession Code | 3V4N | 3V4X |
| Protein Molecules/AU | 4 | 4 |
| Rwork/Rfree (%)c | 14.1/19.4 | 17.1/21.8 |
| Number of Atoms | ||
| Protein | 11,975 | 11,862 |
| Ligand | n/a | 92 |
| Solvent | 1,494 | 1087 |
| RMSD | ||
| Bond Length (Å) | 0.003 | 0.010 |
| Bond Angle (°) | 0.72 | 1.25 |
| B factor (Å2) | ||
| Protein | 27.7 | 19.74 |
| Ligand | n/a | 32.34 |
| Solvent | 35.9 | 24.95 |
| Coordinate Error (Å) | 0.400 | 0.200 |
| Phase Error (°) | 20.3 | 21.8 |
| Ramachandran Plot (%) | ||
| Favored | 97.7 | 97.4 |
| Allowed | 2.3 | 2.6 |
| Outliers | 0.0 | 0.0 |
Numbers in parentheses are for the highest-resolution shell.
Rmerge = ΣhΣi|Ii(h)−<I(h)>|/ ΣhΣiIi(h), where Ii(h) is the ith measurement of reflection h and <I(h)> is a weighted mean of all measurements of h.
R=Σh|Fobs(h)−Fcalc(h)|/ ΣhΣFobs|. Rcryst and Rfree were calculated from the working and test reflection sets, respectively. The test set constituted 10% of the total reflections not used in refinement.
Structure refinement was carried out using additional protocols as implemented within the PHENIX suite (16). First, the initial molecular replacement model was subjected to parallel automated rebuilding using phenix.autobuild (16). This partially refined model was then improved by individual coordinate and atomic-displacement factor refinement using phenix.refine (16), which also allowed calculation of both 2Fo-Fc and Fo-Fc difference electron density maps. These maps were used to iteratively improve the model by manual rebuilding in Coot (18), followed by additional rounds of refinement as described above. Ordered solvent molecules were added according to the default criteria of phenix.refine, and validated by manual inspection in Coot prior to model completion. A complete list of refinement statistics can be found in Table 1.
Ligand Fitting
A model for the hymeglusin ligand was generated using the PRODRG server (19), and the corresponding molecular restraint files were generated with phenix.elbow (16). Examination of the initial Fo-Fc difference maps indicated unmodeled, contiguous density that corresponded to ordered hymeglusin at high occupancy within the active site of all four copies of mvaS within the asymmetric unit. Phenix.ligandfit (16) was then used to place and model a single hymeglusin molecule in each active site. Positional and atomic displacement factor refinement of the hymeglusin-bound mvaS structure was carried out as described above, with the exception that individual atom occupancy refinement was also used to model the hymeglusin bound at each mvaS active site independently.
RESULTS
Hymeglusin inhibition of E. faecalis growth and recovery in the absence of inhibitor
The genetic disruption of mvaS and several other genes encoding mevalonate pathway enzymes has been demonstrated to block growth of Streptococcus pneumoniae (20). In order to more specifically test of the consequences of mvaS inhibition and the consequent restriction of downstream polyisoprenoid metabolites on E. faecalis growth, cells were incubated at 37 °C in LB media either supplemented with 25 μM hymeglusin or without inhibitor. Cells maintained in the presence of inhibitor exhibited no growth over the 5 hour course of the experiment. In the absence of inhibitor, the cells reached a growth curve inflection point at 0.7 hours (Fig. 1). A control sample of cells exhibiting no growth were pelleted and then resuspended in fresh LB media in the absence of inhibitor. These cells reached a growth curve inflection point at 3.1 hours (Fig. 1; Table 2). The observations indicate that the putative adduct of hymeglusin to mvaS can slowly be hydrolyzed to restore enzyme activity and the pool of essential polyisoprenoid metabolites. In contrast with these observations for E. faecalis, animal cells require only 1 hour to exhibit a 50% rebound from inhibition (7). Such observations would be compatible with the hypothesis of different hydrolysis rates for inhibitor adducts to animal HMG-CoA synthase versus bacterial mvaS.
Figure 1.
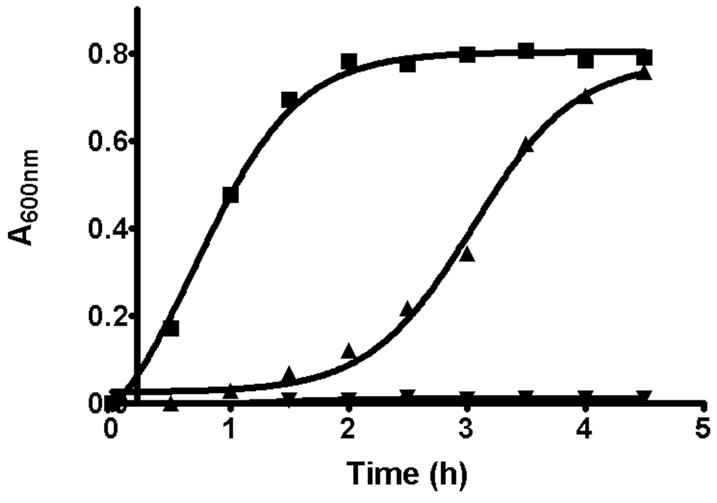
Recovery of E. faecalis cellular growth after removal of hymeglusin. E. faecalis cells were grown in LB media for 3 hours at 37°C in the presence, or absence, of 25 μM hymeglusin. Cells were pelleted then resuspended in fresh LB media and again incubated at 37°C. Absorbance was measured at 600 nm. The traces correspond to: LB prior to cell pelleting followed by post-resuspension growth in LB (■); hymeglusin/LB followed by post-resuspension LB (▲); hymeglusin/LB followed by post-resuspension hymeglusin/LB (▼). The individual time points are averages of data from three experiments performed in parallel. Curves represent nonlinear regression fits of the data to a sigmoidal equation (GraphPad Prism 4). Inflection times and error estimates are provided in Table 2.
Table 2.
Hymeglusin Inhibition of E. FAECALIS and Human HMG-CoA synthasea
| Sample/Enzyme | Time to growth curve inflection point (hours) | Inactivation Kinetic Parameters | Rate of activity restoration by 1 mM hydroxylamine (mUnits activity recovered/minute) |
|---|---|---|---|
| E. faecalis cells | 0.71±0.03 | ||
| E. faecalis cells +hymeglusin | 3.05±0.04 | ||
| E. faecalis mvaS | Nonlinear fit method: | 0.0012±0.0002 | |
| KI = 700±18.5 nM | |||
| kinact = 3.5±0.6 min−1 | |||
| kinact/KI= 0.0050±0.0016 nM−1min−1 | |||
| Graphical method: | |||
| KI = 606 nM | |||
| kinact = 2.75 min−1 | |||
| kinact/KI=0.0045 nM−1min−1 | |||
| Human HMGCSb | KI = 53.7 nM | 0.0097±0.0001 | |
| kinact = 1.06 min−1 | |||
| kinact/KI = 0.0197 nM−1min−1 |
Time dependent hymeglusin inactivation of E. faecalis mvaS
Ambiguities in the nature of hymeglusin inhibition of animal HMG-CoA synthase (6, 7) were resolved by the results of Rokosz et al. (8). Work with purified recombinant human HMG-CoA synthase clearly indicated time dependent inactivation. In contrast, the nature of hymeglusin inhibition of bacterial mvaS has not been studied in detail. A basic kinetic characterization of purified E. faecalis mvaS (Vm = 10 U/mg (corresponding to kcat = 6.9 s−1); Km AcCoA = 350 μM; Km AcAcCoA = 10 μM) has been reported (21) but the scope of that report did not include work on mvaS inhibitors or inactivators. To directly test the efficacy of hymeglusin as an inhibitor, homogeneous E. faecalis mvaS was incubated with this compound at 18 °C with concentrations ranging from 75 to 600 nM. In each sample, a time dependent loss of activity was observed (Fig. 2), with >95 % inhibition achieved at extended incubation times. These results suggest hymeglusin inactivation of the enzyme by formation of a covalent adduct between inhibitor and protein, as observed with animal enzyme. Inactivation follows first order kinetics and the double reciprocal replot of apparent kinact values as a function of hymeglusin concentration indicates saturable binding and provides estimates of a KI value = 606 nM and kinact = 2.75 min−1 (inset, Fig. 2; Table 2). Nonlinear regression analysis of the primary data indicates a KI value = 700±18.5 nM and kinact = 3.5±0.6 min−1 (Table 2). A partition ratio of 11.7±0.6 characaterizes mvaS inactivation by hymeglusin. In comparison, hymeglusin inhibition of human HMG-CoA synthase is characterized by a KI value of 53.7 nM and kinact of 1.06 min−1 (8).
Figure 2.

Time-dependent inactivation of E. faecalis HMG-CoA synthase/mvaS by hymeglusin. Hymeglusin was incubated with HMGCS/mvaS protein for the indicated times prior to addition of acetyl-CoA and DTNB. After recording a baseline level of 412 nm absorbance, acetoacetyl-CoA was added to initiate the activity assay. Reaction mixtures contained: Tris-Cl, 100 mM (pH 8.0); DTNB (0.2 mM); Ac-CoA (400 μM); acetoacetyl-CoA (7 μM); enzyme (2 μg/ml). A semi-log plot of percent HMGCS activity versus time is shown. Concentrations of hymeglusin used were 75 nM (■), 150 nM (◆), 225 nM (▲), 300 nM (□) and 600 nM (◇). Progress curves were fit to a linear model with R2 values of greater than 0.97. Inset: Replot of the reciprocal of apparent kobserved values (derived from t ½ values of data sets depicted in the main figure) versus the reciprocal of hymeglusin concentration. Intercepts on the x and y axes were used to determine KI = 606 nM and kinact= 2.75 min−1, respectively. Nonlinear regression fits to the primary data were also performed (GraphPad Prism 4) and indicate KI=700±18.5 nM and kinact= 3.5±0.6 min−1 (Table 2).
Recovery of activity of hymeglusin inactivated mvaS and human HMG-CoA synthase upon treatment with hydroxylamine
Neutralized hydroxylamine disrupts the thioester adducts of the HMG-CoA synthase reaction intermediates in which in the acyl groups of acetyl-CoA or HMG-CoA are linked to the active site cysteine (22). In hymeglusin inactivated E. faecalis mvaS or human HMG-CoA synthase, any thioester adduct formed between the beta lactone inhibitor and the active site cysteine should also be disrupted if accessible to an aqueous solution of hydroxylamine. Upon release of the resulting hydroxamic acid derivative of hymeglusin from the active site cavity, enzyme activity could, in principle, be restored. This possibility was tested by incubation of hymeglusin inactivated bacterial and human enzymes with 1 mM hydroxylamine. Substantial activity was restored at a rate for the human enzyme (0.0097±0.0001 mU/min) that was 8-fold higher than measured for the E. faecalis enzyme (0.0012±0.0002 mU/min) (Table 2). These results are compatible with the hypothesis that, for E. faecalis mvaS, the enzyme-S-hymeglusin thioester adduct is less accessible to hydroxylamine in aqueous solvent than the corresponding adduct formed using human HMG-CoA synthase.
Crystallographic analysis of hymeglusin bound to mvaS
To further test this hypothesis, attempts were made to determine the structure of hymeglusin-mvaS adduct, which would provide additional information on solvent access to the active site cysteine while the hymeglusin aliphatic chain occupies the active site cavity. Previous studies have established crystallization conditions for E. faecalis mvaS, which were used at that time to determine its structure both free and bound to the substrate acetoacetyl-CoA (17). Although initial attempts to reproduce these crystals with the enzyme preparation described here were unsuccessful, an alternative crystal form was identified (Experimental Procedures). This novel crystal form was characterized by a change to a lower-symmetry space group (P21 from I222) but with an increase in apparent resolution (1.6 Å from 2.4 Å) for the unbound enzyme. Using these crystals, a structure of mvaS was solved and refined to Rwork and Rfree values of 14.1 and 19.4%, respectively. This structure consists of four crystallographically unique copies of the enzyme within the asymmetric unit (Supplemental Fig. 1 and Table 1). By contrast, the previously determined structure of the same enzyme contained two polypeptides within its asymmetric unit (17).
Diffraction quality samples were also obtained from E. faecalis mvaS that had been briefly incubated with an equimolar concentration of hymeglusin immediately prior to establishment of the crystallization drops. These crystals of hymeglusin-bound mvaS displayed only minor changes in cell constants when compared to the unbound enzyme, and diffracted synchrotron X-rays to 1.95 Å limiting resolution (Table 1). After collecting a nearly complete data set, the corresponding structure was solved by molecular replacement and refined to Rwork and Rfree values of 17.1 and 21.8%, respectively (Fig. 3 and Table 1). Examination of both 2Fo-Fc and Fo-Fc difference electron density maps following initial rounds of refinement revealed the presence of largely contiguous, unmodeled density within the active site cavity for all four crystallographically unique copies of mvaS present within the asymmetric unit (Supplemental Fig. 2). The extended nature of this density was consistent with the branched, aliphatic tail group of hymeglusin. Furthermore, it was contiguous with the active site Cys111 side chain in all four copies; this was indicative of a covalent adduct that would be anticipated following hymeglusin modification of mvaS.
Figure 3.

1.95 Å structure of E. faecalis mvaS covalently modified by hymeglusin. Crystals of E. faecalis mvaS that had been briefly incubated with hymeglusin were produced as described in methods and used to collect monochromatic diffraction data. Following structure solution by molecular replacement, electron density maps were calculated that allowed modeling of bound ligand in each of the four crystallographically independent active sites. (A) Stereogram of the mvaS active site for chain A. Protein backbone is depicted in cartoon format (purple), while the sidechains of Glu79, Tyr143, and His233 are shown as cyan sticks. The catalytic Cys111 is colored green, and carbon atoms of covalently-bound hymeglusin are colored yellow. 2Fo-Fc map (blue mesh at 1.1σ contour) of the refined structure corresponding to the Cys111 thiolate-hymeglusin adduct is also shown. (B) Overlay of hymeglusin molecules as modeled in chains A-D, colored blue, green, orange, and red, respectively. The position of C8, which forms a covalent bond with the thiolate sulfur derived from Cys111, is indicated with an arrow. Additional information for all four active sites can be found in Supplemental Figures 2–4.
The readily interpretable nature of this unmodeled density allowed accurate placement of a single hymeglusin ligand in each active site (Fig. 3A). Comparison of the hymeglusin conformation in all four active sites revealed that the ligand bound in essentially identical orientations with respect to mvaS (Fig. 3B and Supplemental Figs. 2–3). In particular, superposition of all four hymeglusin chains within the refined structure yielded an average overall RMSD value of 0.72 Å across the entire ligand molecule. Interestingly, whereas the regions of the hymeglusin molecules involved in the covalent linkages to the Cys111 thiolates superimpose quite well with one another, areas of greater conformational variability between the ligands bound at different mvaS active sites are found in the distal regions of the inhibitor. This is most likely because this region of hymeglusin is quasi-linear and aliphatic in nature, and (aside from the terminal carboxylate) lacks hydrogen bond donors/acceptors or other noteworthy structural features that could significantly restrict its conformation toward the entrance of the mvaS catalytic cleft.
The tube-like nature of the Ef mvaS active site channel allows for nearly the entire hymeglusin molecule to be buried within the cavity upon binding. On average, 381.4 Å2 of surface area are buried through forming the enzyme/inhibitor interaction. This corresponds to approximately 84% of the total surface area available within the hymeglusin molecule. Despite the fact that a large portion of hymeglusin is aliphatic, two key polar interactions between its various functional groups and mvaS side chains stand out (Supplemental Fig. 4). These are hydrogen bonds between the side chain Nε2 of His233 and the O6 alcohol group of hymeglusin (2.6 Å distance), and the side chain carboxylate of Glu79 and the O5 alcohol of the inhibitor (2.9 Å distance). Previous kinetic and structural studies have consistently suggested that Glu79 functions as a general acid/base during catalysis (17, 22–24). His233 has also been ascribed a role as a general acid/base during the condensation partial reaction of HMG-CoA synthesis (24); however it is also worth noting this residue contributes to binding of the second substrate, AcAc-CoA, by hydrogen bonding to its acetoacetyl group (22–24). Similar interactions have also been described for His247 and Glu48 of the Brassica juncea HMG-CoA synthase in its hymeglusin-bound state (10). This further emphasizes the highly conserved nature of these specific side chains to HMG-CoA synthase function across diverse evolutionary lineages.
Differences in active site accessibility between the eukaryotic and prokaryotic synthases explain the altered stability of their hymeglusin adducts
While Ef mvaS and the human and plant synthases share only approximately 27 and 28% identity, respectively, their overall folds are quite similar (Fig. 4A) and all active site residues with clearly defined function are conserved. This conservation of mechanism is reflected in the analogous pairs of interactions formed between key enzyme side chains and the polar groups of the inhibitor hymeglusin, as mentioned above. Nevertheless, the conserved nature of the active site residues provided no insight regarding the differences in kinetic inactivation or hydroxylamine reactivation of mvaS versus its eukaryotic counterpart (Table 2). This suggested that regions at the active site periphery instead may be responsible for these differences.
Figure 4.

Comparison of hymeglusin-bound structures of eukaryotic HMG-CoA synthase and bacterial mvaS. (A) The crystal structure of hymeglusin-bound Brassica juncea HMG-CoA synthase (PDB code 2F9A, shown in orange cartoon) was superimposed with that of hymeglusin-bound E. faecalis mvaS (PDB code 3V4X, shown in purple cartoon). Positions of the respective catalytic cysteine residues are highlighted with green color. (B) Brassica juncea HMG-CoA synthase shown as a molecular surface in an identical orientation to panel A. Bound hymeglusin is shown as a yellow ball-and-stick. The positions of landmark residues that define the boundaries of the active site entrance are indicated and highlighted in purple. (C) Enterococcus faecalis mvaS shown as a molecular surface in an identical orientation to panel A. Bound hymeglusin is shown as a yellow ball-and-stick. The positions of landmark residues that define the boundaries of the active site entrance are indicated and highlighted in orange.
Indeed, an immediate and striking difference between the Ef mvaS and eukaryotic (B. juncea) HMG-CoA synthase involves the nature of the entrances to their respective active site cavities (Fig. 4). The plant enzyme is characterized by a relatively wide cavity that is reminiscent of a triangular or pyramidal-shaped funnel, with the bound hymeglusin laying flat against one its internal faces and extending nearly 16 Å into the catalytic core. For the purposes of illustration, the Cβ of Lys256 serves as a convenient landmark for defining boundaries of the active site funnel, since it lies only 3.2 Å from the hymeglusin carboxylate. Using this as a guide, the approximate dimensions of the funnel opening are 15.6 Å (Lys256 to Cγ1 of Val204), 17.2 Å (Lys256 to Cβ of Lys34), and 20.2 Å (Lys256 to Cβ of Pro155). It is particularly notable that the catalytic cysteine (Cys117), which participates in the thioester bond that covalently traps the inhibitor in place, has otherwise unrestricted access to bulk solvent via the unoccupied side of this funnel.
In contrast, the Ef mvaS entrance is far more restricted and resembles a tunnel or tube. In this case, the backbone O of Gly148 can be used as a landmark as it lies only 4.8 Å from the hymeglusin carboxylate. While this tunnel extends nearly 16 Å into the catalytic core, its opening is only 9.4 Å in diameter (as measured from Gly148 to the side chain amide of Asn202). Because of this, covalently linked hymeglusin appears capable of quite effectively occluding access of bulk solvent to the Ef mvaS catalytic Cys111. Thus, consideration of these architectural differences at the respective active-site peripheries provides a structural explanation for the ~8-fold slower activity recovery upon hydroxylamine treatment for mvaS relative to the eukaryotic synthase (Table 2).
DISCUSSION
Hymeglusin has been recognized and described as a potent, covalent inhibitor of HMG-CoA synthases from bacterial, plant, and animal origin. Yet while inhibition of human HMG-CoA synthase by this fungal polyketide has been studied in some detail (8), relatively little information is available regarding the specific nature of hymeglusin’s effects on the bacterial synthase. This prompted us to conduct the current study, which investigated the kinetic parameters of mvaS inactivation by hymeglusin both in live Enterococcus faecalis cells and in vitro. Despite a common mode of inactivation (i.e. covalent modification), we observed significant differences in a number of biochemical parameters between the human and bacterial synthases (Table 2). The human synthase is far more sensitive to inhibition by hymeglusin, as demonstrated by its 10-fold lower KI value relative to mvaS. (53.7 nM versus 700 nM, respectively). This differential affinity influences the inactivation efficiency (kinact/KI), which is higher for human HMG-CoA synthase than for E. faecalis mvaS ( 0.0197 versus 0.0050, respectively; Table 2). However significant differences between both the formation and stability of hymeglusin-synthase adducts were seen. Hymeglusin-mediated inactivation manifests itself at a faster rate in bacterial mvaS relative to human HMG-CoA synthase. Inactivation is also more transient (shorter in duration) for the human enzyme than for its bacterial counterpart. Apparent differences in duration of inactivation are in accord with the approximately 8-fold slower activity recovery rate following hydroxylamine treatment for inhibited E. faecalis mvaS when compared to human HMG-CoA synthase. This strongly suggested that differences in active site solvent accessibility contributed to the inactivation differences between the human and bacterial enzymes.
We obtained further insight into the basis for hymeglusin inhibition of E. faecalis mvaS through structural studies of the covalently modified enzyme. In doing so, a novel crystal form of E. faecalis mvaS was identified that provided improved diffraction limits when compared to that previously reported for the same enzyme (17). Interestingly, the unit cell parameters for the crystal system described here match exceedingly well those reported by Campobasso and colleagues for S. aureus mvaS (which shares ~60% identity) when bound to its substrate acetoacetyl-CoA (23). Comparison of the hymeglusin-bound E. faecalis mvaS structure presented here to those of either the E. faecalis (17) or S. aureus (23) enzymes bound to various CoA derivatives reveals that the aliphatic tail of hymeglusin occupies a largely identical region within the enzymes’ relatively long, active site tunnel as does the pantothenic acid moiety of CoA in these closely related structures (Figure 5). That said it is noteworthy that the hymeglusin tail is completely extended, and not kinked as in the distal, reactive region of CoA. This elongated conformation allows the covalent inhibitor to effectively bypass the hydrophobic crevice formed by Tyr205, Met239, and Tyr306 of mvaS, which itself accommodates the reactive cysteine/thiol-containing functionality of the coenzyme. Intriguingly, this stands in contrast to what is observed for the B. juncea HMG-CoA synthase when bound to hymeglusin, acetyl-CoA, or HMG-CoA (10). Here, the aliphatic hymeglusin chain actually follows, rather than bypasses, the kink found in the distal region of the CoA (Figure 6). In doing so, the terminal region of the hymeglusin chain binds to an opposing face of the funnel-shaped entrance to the enzyme’s active site than is seen for either CoA derivative. Such a dramatic difference in ligand-binding mode is not possible within the bacterial enzyme, as its tunnel-shaped active site entrance imposes significant structural restraints on any inhibitors/substrates that occupy this region. One important consequence of these architectural differences among HMG-CoA synthases across various kingdoms of life is that the active sites of the eukaryotic forms appear to have relatively free access to bulk solvent, whereas their bacterial counterparts are more restricted. While the physiological relevance of this difference is not clear at this time, it does provide a rational basis for the notably altered kinetic inactivation parameters reported here (Figures 1, 2, and Table 2) and elsewhere (8). Furthermore, it also strongly suggests that inhibitors which are highly selective for the bacterial enzymes over their human homolog can be discovered, optimized, and developed.
Figure 5.

Comparison of E. faecalis mvaS structures bound to either acetoacetate and CoA or hymeglusin. (A) Chain B from the structure of mvaS bound to acetoacetate and CoA (PDB code 1YSL). The polypeptide is shown in purple cartoon while the ligands are shown in ball and stick format. The catalytic cysteine residue is drawn in green. (B) Chain A from the structure of mvaS bound to hymeglusin (PDB code 3V4X). (C) Superposition of the CoA and hymeglusin structures as they lie within the mvaS active site. Note that the alphatic tail of hymeglusin largely occupies the same position as the pantothenic acid moiety of CoA. The positions of acetoacetate (AcAc), Coenzyme-A (CoA), and the C8 carbon of hymeglusin (C8) are indicated with arrows.
Figure 6.

Comparison of available ligand-bound structures for E. faecalis and S. aureus mvaS and B. juncea HMG-CoA synthases. (A) Superposition of enzyme monomers from E. faecalis mvaS bound to hymeglusin (PDB code 2V4X) and acetyl-CoA (PDB code 1YSL, chain B), S. aureus mvaS bound to acetoacetyl-CoA (PDB code 1TXT, chain A), and B. juncea HMG-CoA synthase bound to hymeglusin (PDB code 2F9A), acetyl-CoA (PDB code 2FA3), and HMG-CoA (PDB code 2FA0). The E. faecalis and S. aureus structures are shown in purple and blue, respectively, while the B. juncea structures are drawn in orange. The location of the helical insertion domain that is only found in the eukaryotic enzyme is highlighted with a dashed green circle. The positions of the bound ligands/inhibitors are shown in ball and stick format. Note that each of these ligands/inhibitors adopts the same positional conformation, except for hymeglusin when bound to B. juncea HMG-CoA synthase. Instead, this inhibitor binds to an opposing face of the active site funnel when compared to the CoA derivatives. (B) Magnified views of the positions of each of the ligands/inhibitors from panel B as they lie within the superimposed enzyme active sites. All CoA-derived ligands are colored with their carbon atoms in yellow, while hymeglusin as found in the E. faecalis and B. juncea enzymes is colored with purple and orange carbon atoms, respectively.
In reporting the eukaryotic HMGCS-hymeglusin adduct structure (10), it was noted that the active site glutamate hydrogen bonds to the C2 hydroxyl of the ring opened inactivator. Based on such structural observations, additional function for the catalytic glutamate in catalysis of thioester hydrolysis was proposed. Such a hypothesis can be discounted based on results of direct functional tests of the thioester hydrolysis partial reaction (22). Alanine substitution of the catalytic glutamate produces a mutant enzyme that is unimpaired in catalysis of this partial reaction. Neither Vm nor Km values are substantially different from wild type enzyme values. In contrast, however, this mutant exhibits more than a five orders of magnitude diminution of the overall reaction (Scheme 1) in comparison with wild type enzyme. This confirms previous functional assignment of this active site-resident glutamate in the condensation partial reaction (22, 24)
In addition to hymeglusin’s ability to inactivate both prokaryotic and eukaryotic HMG-CoA synthase, we recently demonstrated the ability of epoxide containing molecules to inhibit eukaryotic enzyme (25). Cerulenin is an antibiotic that inhibits beta ketoacyl acyl carrier protein synthases, the bacterial fatty acid condensing enzymes (26). Its epoxide moiety reacts with the active site cysteine to form a stable thioether adduct. These enzymes as well as HMG-CoA synthases are members of the family of initial condensation enzymes and they exhibit considerable structural and functional homology. On this basis, it seemed reasonable to test cerulenin against HMG-CoA synthase; the human enzyme proves to be sensitive to this inhibitor, displaying time dependent inactivation (25). S. aureus mvaS also exhibits time dependent inactivation by cerulenin (Misra and Miziorko, unpublished results). Ceestatin, a novel epoxide containing compound, inhibits hepatitis C virus replication via a mechanism attributed to its demonstrated time dependent inactivation of eukaryotic (human) HMG-CoA synthase (25). While these epoxide inhibitors are effective, in vitro, at higher concentrations than required for hymeglusin, the thioether adducts that presumably form upon active site cysteine modification are not labile under the same physiological conditions that permit thioester adduct hydrolysis. This is particularly relevant in terms of potential anti-infective applications of hymeglusin and related compounds, since data presented here show that hymeglusin’s effect on live bacterial cells is reversible on the order of hours (Figure 1 and Table 2). Thus, in any future work on design or optimization of HMG-CoA synthase inhibitors, the chemical nature of the protein-inhibitor adduct should receive consideration. In this regard, the structural information available on enzyme-inhibitor complexes along with the characterized differences in kinetic inactivation parameters presented herein should prove to be a valuable asset moving forward.
Supplementary Material
Acknowledgments
Dr. Michael Greenspan (Merck) generously provided the hymeglusin used in these studies. Dr. Ila Misra (Medical College of Wisconsin) determined that hymeglusin is a time dependent inactivator of Staphylococcus aureus mvaS. The authors acknowledge generous technical assistance of Drs. Rod Salazar, Andy Howard, and John Chrzas during X-ray diffraction data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) beamlines at the Advanced Photon Source, Argonne National Laboratory. A list of supporting member institutions may be found at www.ser-cat.org/members.html.
Footnotes
Abbreviations used are: HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; Ax-CoA, acetyl-CoA; AcAc-CoA, acetoacetyl-CoA; F244 (or 1233A), hymeglusin; mvaS, prokaryotic HMG-CoA synthase; HMGCS, eukaryotic HMG-CoA synthase; DTNB, dithiobisnitrobenzoic acid; Ef, Enterococcus faecalis; LB, Luria-Bertani medium.
SUPPORTING INFORMATION AVAILABLE
Four supplemental figures and the corresponding figure legends accompany this manuscript. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Aldridge DC, Giles D, Turner WB. Antibiotic 1233A: a fungal beta-lactone. J Chem Soc Perkin. 1971;123:3888–3891. doi: 10.1039/j39710003888. [DOI] [PubMed] [Google Scholar]
- 2.Tomoda H, Kumagai H, Takahashi Y, Tanaka Y, Iwai Y, Omura S. F-244 (1233A), a specific inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A synthase: taxonomy of producing strain, fermentation, isolation and biological properties. J Antibiot (Tokyo) 1988;41:247–249. doi: 10.7164/antibiotics.41.247. [DOI] [PubMed] [Google Scholar]
- 3.Miziorko HM, Behnke CE. Active-site-directed inhibition of 3-hydroxy-3-methylglutaryl coenzyme A synthase by 3-chloropropionyl coenzyme A. Biochemistry. 1985;24:3174–3179. doi: 10.1021/bi00334a015. [DOI] [PubMed] [Google Scholar]
- 4.Miziorko HM, Behnke CE. Amino acid sequence of an active site peptide of avian liver mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase. J Biol Chem. 1985;260:13513–13516. [PubMed] [Google Scholar]
- 5.Omura S, Tomoda H, Kumagai H, Greenspan MD, Yodkovitz JB, Chen JS, Alberts AW, Martin I, Mochales S, Monaghan RL, et al. Potent inhibitory effect of antibiotic 1233A on cholesterol biosynthesis which specifically blocks 3-hydroxy-3-methylglutaryl coenzyme A synthase. J Antibiot (Tokyo) 1987;40:1356–1357. doi: 10.7164/antibiotics.40.1356. [DOI] [PubMed] [Google Scholar]
- 6.Greenspan MD, Yudkovitz JB, Lo CY, Chen JS, Alberts AW, Hunt VM, Chang MN, Yang SS, Thompson KL, Chiang YC, et al. Inhibition of hydroxymethylglutaryl-coenzyme A synthase by L-659,699. Proc Natl Acad Sci U S A. 1987;84:7488–7492. doi: 10.1073/pnas.84.21.7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenspan MD, Bull HG, Yudkovitz JB, Hanf DP, Alberts AW. Inhibition of 3-hydroxy-3-methylglutaryl-CoA synthase and cholesterol biosynthesis by beta-lactone inhibitors and binding of these inhibitors to the enzyme. Biochem J. 1993;289(Pt 3):889–895. doi: 10.1042/bj2890889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rokosz LL, Boulton DA, Butkiewicz EA, Sanyal G, Cueto MA, Lachance PA, Hermes JD. Human cytoplasmic 3-hydroxy-3-methylglutaryl coenzyme A synthase: expression, purification, and characterization of recombinant wild-type and Cys129 mutant enzymes. Arch Biochem Biophys. 1994;312:1–13. doi: 10.1006/abbi.1994.1273. [DOI] [PubMed] [Google Scholar]
- 9.Misra I, Narasimhan C, Miziorko HM. Characterization of a Recumbinant Cholesterogenic Isozyme and Demonstration of the Requirement for a Sulfhydryl Functionality in the Formation of the Acetyl-Enzyme Reaction Intermediate. J Biol Chem. 1993;268:12129–12136. [PubMed] [Google Scholar]
- 10.Pojer F, Ferrer JL, Richard SB, Nagegowda DA, Chye ML, Bach TJ, Noel JP. Structural basis for the design of potent and species-specific inhibitors of 3-hydroxy-3-methylglutaryl CoA synthases. Proc Natl Acad Sci U S A. 2006;103:11491–11496. doi: 10.1073/pnas.0604935103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon EJ, DS The preparation of S-succinyl Coenzyme A. J Am Chem Soc. 1953;73:2520. [Google Scholar]
- 12.Geisbrecht BV, Bouyain S, Pop M. An Optimized System for the Expression and Purification of Secreted Bacterial Proteins. Prot Expr Purif. 2006;46:23–32. doi: 10.1016/j.pep.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Barta ML, Skaff DA, McWhorter WJ, Herdendorf TJ, Miziorko HM, Geisbrecht BV. Crystal structures of Staphylococcus epidermidis mevalonate diphosphate decarboxylase bound to inhibitory analogs reveal new insight into substrate binding and catalysis. J Biol Chem. 2011;286:23900–23910. doi: 10.1074/jbc.M111.242016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skaff DA, Miziorko HM. A visible wavelength spectrophotometric assay suitable for high-throughput screening of 3-hydroxy-3-methylglutaryl-CoA synthase. Anal Biochem. 2010;396:96–102. doi: 10.1016/j.ab.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 15.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 16.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty MW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Cryst D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steussy CN, Vartia AA, Burgner JW, Sutherlin A, Rodwell VW, Stauffacher CV. X-ray Crystal Structures of HMG-CoA Synthase from Enterococcus faecalis and a Complex with Its Second Substrate/Inhibitor Acetoacetyl-CoA. Biochemistry. 2005;44:14256–14267. doi: 10.1021/bi051487x. [DOI] [PubMed] [Google Scholar]
- 18.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and Development of Coot. Acta Cryst D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuettelkopf AW, van Aalten DMF. PRODRG - a Tool for High-throughput Crystallography of Protein-Ligand Complexes. Acta Cryst D. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 20.Wilding EI, Kim DY, Bryant AP, Gwynn MN, Lunsford RD, McDevitt D, Myers JE, Jr, Rosenberg M, Sylvester D, Stauffacher CV, Rodwell VW. Essentiality, expression, and characterization of the class II 3-hydroxy-3-methylglutaryl coenzyme A reductase of Staphylococcus aureus. J Bacteriol. 2000;182:5147–5152. doi: 10.1128/jb.182.18.5147-5152.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutherlin A, Hedl M, Sanchez-Neri B, Burgner JW, 2nd, Stauffacher CV, Rodwell VW. Enterococcus faecalis 3-hydroxy-3-methylglutaryl coenzyme A synthase, an enzyme of isopentenyl diphosphate biosynthesis. J Bacteriol. 2002;184:4065–4070. doi: 10.1128/JB.184.15.4065-4070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chun KY, Vinarov DA, Zajicek J, Miziorko HM. 3-Hydroxy-3-methylglutaryl-CoA synthase. A role for glutamate 95 in general acid/base catalysis of C-C bond formation. J Biol Chem. 2000;275:17946–17953. doi: 10.1074/jbc.M909725199. [DOI] [PubMed] [Google Scholar]
- 23.Campobasso N, Patel M, Wilding IE, Kallender H, Rosenberg M, Gwynn MN. Staphylococcus aureus 3-hydroxy-3-methylglutaryl-CoA synthase: crystal structure and mechanism. J Biol Chem. 2004;279:44883–44888. doi: 10.1074/jbc.M407882200. [DOI] [PubMed] [Google Scholar]
- 24.Theisen MJ, Misra I, Saadat D, Campobasso N, Miziorko HM, Harrison DH. 3-hydroxy-3-methylglutaryl-CoA synthase intermediate complex observed in "real-time". Proc Natl Acad Sci U S A. 2004;101:16442–16447. doi: 10.1073/pnas.0405809101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng LF, Schaefer EA, Maloof N, Skaff A, Berical A, Belon CA, Heck JA, Lin W, Frick DN, Allen TM, Miziorko HM, Schreiber SL, Chung RT. Ceestatin, a novel small molecule inhibitor of hepatitis C virus replication, inhibits 3-hydroxy-3-methylglutaryl-coenzyme A synthase. J Infect Dis. 2011;204:609–616. doi: 10.1093/infdis/jir303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price AC, Choi KH, Heath RJ, Li Z, White SW, Rock CO. Inhibition of beta-ketoacyl-acyl carrier protein synthases by thiolactomycin and cerulenin. Structure and mechanism. J Biol Chem. 2001;276:6551–6559. doi: 10.1074/jbc.M007101200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.