

Abstract
PPARGC1A is a transcriptional coactivator that binds to and coactivates a variety of transcription factors (TFs) to regulate the expression of target genes. PPARGC1A plays a pivotal role in regulating energy metabolism and has been implicated in several human diseases, most notably type II diabetes. Previous studies have focused on the interplay between PPARGC1A and individual TFs, but little is known about how PPARGC1A combines with all of its partners across the genome to regulate transcriptional dynamics. In this study, we describe a core PPARGC1A transcriptional regulatory network operating in HepG2 cells treated with forskolin. We first mapped the genome-wide binding sites of PPARGC1A using chromatin-IP followed by high-throughput sequencing (ChIP-seq) and uncovered overrepresented DNA sequence motifs corresponding to known and novel PPARGC1A network partners. We then profiled six of these site-specific TF partners using ChIP-seq and examined their network connectivity and combinatorial binding patterns with PPARGC1A. Our analysis revealed extensive overlap of targets including a novel link between PPARGC1A and HSF1, a TF regulating the conserved heat shock response pathway that is misregulated in diabetes. Importantly, we found that different combinations of TFs bound to distinct functional sets of genes, thereby helping to reveal the combinatorial regulatory code for metabolic and other cellular processes. In addition, the different TFs often bound near the promoters and coding regions of each other's genes suggesting an intricate network of interdependent regulation. Overall, our study provides an important framework for understanding the systems-level control of metabolic gene expression in humans.
PPARGC1A (formerly known as PGC-1α), encoded by the PPARGC1A gene, is a powerful transcriptional coactivator that has been implicated in a wide array of human diseases including type II diabetes, cardiovascular disease, Huntington's disease, and other diet-related diseases (Muller et al. 2003; Handschin et al. 2005; Soyal et al. 2006; Sihag et al. 2009; Weydt et al. 2009). PPARGC1A is activated in response to a number of environmental stresses and coordinates tissue-specific programs of gene regulation that most prominently affect the expression of genes involved in energy metabolism and mitochondrial function and biogenesis (Lin et al. 2005). PPARGC1A belongs to the PGC1 family of coactivators, along with the highly related protein PPARGC1B and the more distantly related PPRC1 (also known as PRC), that regulate transcription by docking to transcription factors (TFs) at the promoters of their target genes (Andersson and Scarpulla 2001; Meirhaeghe et al. 2003; Lin et al. 2005). PPARGC1A contains an N-terminal activation domain and additional regulatory domains that can recruit chromatin-modifying proteins such as histone acetyltransferases as well as components of the mediator complex (Lin et al. 2005). PPARGC1A is a highly versatile protein that can interact with most members of the nuclear receptor (NR) family of TFs as well as several non-NR TFs (Finck and Kelly 2006). This feature enables PPARGC1A to appropriately respond to diverse signaling pathways and exert precise control in regulating distinct sets of target genes.
Numerous studies have identified an important role for PPARGC1A in maintaining blood glucose levels by up-regulating genes involved in gluconeogenesis and the beta-oxidation of fatty acids in the liver (Yoon et al. 2001; Lin et al. 2004). PPARGC1A expression is activated in response to cAMP and glucocorticoid signaling in fasted mice (Yoon et al. 2001). In diabetic mice, it is aberrantly up-regulated in the absence of fasting, a factor that contributes to elevated hepatic glucose production and hyperglycemia (Herzig et al. 2001). Single gene studies have identified HNF4A, NR3C1 (also known as GR), and FOXO1 as key TFs that are coactivated by PPARGC1A in order to regulate downstream gluconeogenic target genes including phosphoenolpyruvate carboxykinase (PCK1) and glucose 6 phosphatase (G6PC) (Puigserver et al. 2003; Rhee et al. 2003). PPARGC1A also interacts with peroxisome proliferator-activated receptor alpha (PPARA) and estrogen-related receptor alpha (ESRRA) to regulate the expression of genes involved in fatty acid oxidation and oxidative phosphorylation in hepatocytes (Vega et al. 2000; Mootha et al. 2004). Although previous work has identified many TFs that are coregulated by PPARGC1A, it is likely that important TF partners remain to be identified. In addition, the complete repertoire of PPARGC1A target genes is unknown. Uncovering how PPARGC1A cooperates with a multitude of TFs across the genome and how it functions within the context of transcriptional regulatory networks that coordinate numerous biological pathways will deepen our understanding of the biological roles of PPARGC1A.
Recent advances in high-throughput DNA sequencing technology have made it possible to rapidly and efficiently study the genome-wide activity of TFs, coregulators, and other regulatory proteins (Farnham 2009). In the present study, we sought to use next-generation DNA sequencing to investigate the global PPARGC1A regulatory circuitry by mapping the genome-wide occupancy of PPARGC1A and several of its network partners in a human hepatocarcinoma cell line (HepG2) under conditions that activate PPARGC1A. HepG2 cells have been a widely used model cell line for the study of metabolic pathways in humans (Javitt 1990). To help build this circuitry, we utilized a two-step approach in which the genomic binding sites of PPARGC1A were first analyzed in an unbiased manner to discover DNA sequence motifs corresponding to potential novel regulatory partners. Next, ChIP-seq analysis of several known and novel partners was performed in order to determine the combinatorial binding patterns that these factors form with each other and with PPARGC1A and the likely gene targets regulated by each combination of factors.
Results
Genome-wide identification of PPARGC1A binding sites by ChIP-seq
In order to determine the genome-wide binding sites of PPARGC1A, we performed chromatin immunoprecipitation with a PPARGC1A-specific antibody in human HepG2 cells. The cells were treated with forskolin to stimulate the cAMP signaling pathway that activates PPARGC1A; this is the same pathway that is normally activated by glucagon in the fasted state (Yoon et al. 2001). PPARGC1A-associated DNA was isolated by immunoprecipitation, and processed and subjected to high-throughput DNA sequencing using the Illumina Genome Analyzer II platform. As a control, input DNA isolated under the same conditions was also sequenced. The short sequence tags (27 bp) were then mapped to the human genome. Using two biological replicate experiments, a total of 13.6 and 11.8 million mapped sequence tags were obtained for PPARGC1A and input DNA, respectively (Table 1). We generated signal maps for PPARGC1A and input DNA by extending the sequence tags in the 3′ direction to 200 bp (the average length of DNA fragments in the sequenced samples), and counting the number of overlapping tags at each position in the genome; 1886 PPARGC1A binding regions with significantly higher signals in the PPARGC1A signal map relative to input DNA were identified using the PeakSeq algorithm (Rozowsky et al. 2009) (1% false discovery rate [FDR] threshold; Supplemental Table S1 and Supplemental Data set S1). As shown in Figure 1A, the average signal at PPARGC1A peak positions was highly elevated relative to both the surrounding genomic regions (±2 kb from the peak) and the same positions in the input DNA sample. In addition, the genomic regions occupied by PPARGC1A exhibited strong evolutionary sequence conservation as demonstrated by their high average phastCons conservation scores (Siepel et al. 2005) relative to the surrounding genomic regions (Fig. 1B). Thus, these sites are likely to serve a functionally important role as regulatory elements in vivo. We tested 20 putative PPARGC1A binding sites using ChIP followed by quantitative PCR (ChIP-qPCR) and validated enrichment at 18 of these sites, indicating that our PPARGC1A binding data set is highly specific (Supplemental Fig. S1; Supplemental Table S3).
Table 1.
Summary of Illumina tag sequencing data and PeakSeq scoring results
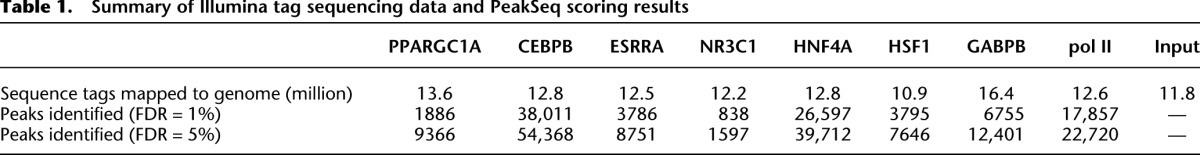
Figure 1.

Genome-wide occupancy of PPARGC1A determined by ChIP-seq. (A) Plot of the mean PPARGC1A ChIP enrichment signal (blue) in the region spanning ±2 kb from the signal peak across all 1886 PPARGC1A-occupied regions. (Gray) Mean signals from input DNA in the same regions. (B) Plot of the mean phastCons conservation scores in the region spanning ±2 kb from the signal peak across all PPARGC1A-occupied regions. The phastCons conservation score (ranging from 0 to 1) represents the probability that a base is in a conserved element (Siepel et al. 2005). (C) Pie chart displaying the distribution of PPARGC1A binding sites relative to UCSC Known Genes transcript annotations (Hsu et al. 2006). Each PPARGC1A binding site was mapped to one of four annotation categories in the following order of preference: TSS (peak within 5 kb), 3′-end (peak within 5 kb), intragenic, intergenic. The total number of sites mapping to each category is displayed within the corresponding portion of the pie chart. (D) Distribution of all 715 TSS-associated PPARGC1A binding sites relative to the position of the nearest TSS. As demonstrated in the transcript diagram below the plot, binding positions are plotted with respect to the orientation of the associated transcript; negative positions indicate binding in the upstream region and positive positions indicate binding in the downstream region.
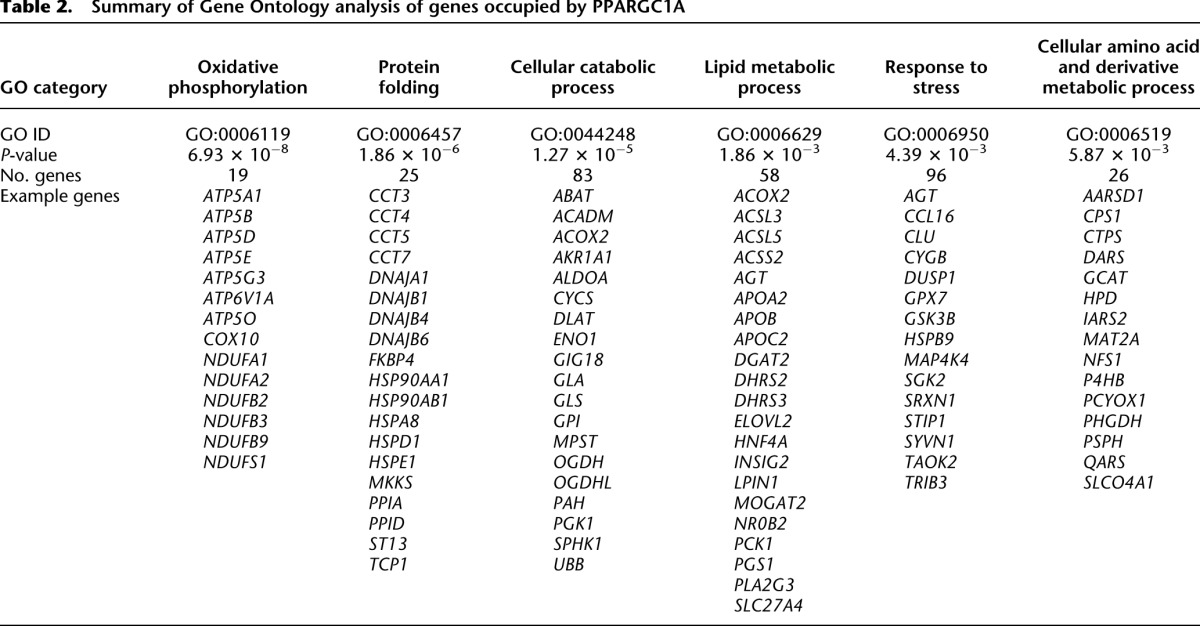
We next examined the distribution of PPARGC1A peaks relative to transcripts in the UCSC Known Genes track (Fig. 1C; Supplemental Table S2; Hsu et al. 2006). PPARGC1A binding sites occurred most commonly within promoter regions, defined as ±5 kb from a transcriptional start site (TSS). Binding within promoters was observed for 715 PPARGC1A peaks, comprising 37.9% of all PPARGC1A binding sites (Fig. 1C). These sites were predominantly located in the upstream region within 1 kb of the TSS (Fig. 1D). Among the remaining sites, 630 (33.4%) were located in intergenic regions, 411 (21.8%) in intragenic regions, and 130 (6.9%) within 5 kb of a 3′-end (Fig. 1C). These findings suggest that, in addition to promoter regions, a large proportion of PPARGC1A binding events occur at distal regulatory elements that may correspond with enhancer regions. Importantly, PPARGC1A peaks were found adjacent to numerous previously identified target genes, including PCK1, ALAS1, ACADM, ATP5B, CYP7A1, CYP8B1, TRIB3, CYCS, ESRRA, and SOD2, further supporting the validity of our binding data set (Supplemental Table S2). Gene ontology (GO) analysis (The Gene Ontology Consortium 2000) of PPARGC1A-occupied genes revealed enrichment of several functional categories that have been associated with PPARGC1A, such as oxidative phosphorylation and lipid metabolism, in addition to novel categories such as protein folding (Supplemental Table S4). Six representative GO categories and examples of PPARGC1A-occupied genes are listed in Table 2.
Table 2.
Summary of Gene Ontology analysis of genes occupied by PPARGC1A
Enriched motifs at PPARGC1A-occupied sites suggest novel regulatory partners
Numerous studies have demonstrated that PPARGC1A interacts with DNA-binding TFs in order to operate as a transcriptional coactivator (Lin et al. 2005; Soyal et al. 2006). We hypothesized, therefore, that analysis of the DNA sequences underlying PPARGC1A peaks identified by ChIP-seq would reveal known and novel regulatory partners of PPARGC1A based on the enrichment of their consensus DNA binding motifs. To identify enriched sequence motifs in an unbiased manner, we examined the DNA sequences (±100 bp from the signal peak) corresponding to the top 250 PPARGC1A binding sites using the program MEME (Bailey and Elkan 1994). MEME analysis identified three significantly enriched DNA sequence motifs (Fig. 2A; Supplemental Table S5), each corresponding to a known TF motif in the TRANSFAC database (Matys et al. 2006). Surprisingly, the motif exhibiting the highest statistical significance matched the heat shock element (HSE) that serves as the recognition site for the heat shock TF, HSF1, a TF that has not been known previously to function with PPARGC1A. The core HSE contains three inverted repeats of the consensus sequence nGAAn, as depicted in the sequence logo in Figure 2A; however, some HSEs are known to extend beyond this core by one or more additional nGAAn repeats. We observed the HSE motif at 344 PPARGC1A peaks (18.2%) and ∼20% of these contain more than three copies of the nGAAn motif (Supplemental Fig. S2). The second MEME motif corresponded to a NR half-site strongly resembling the consensus binding motif of estrogen-related receptor alpha (ESRRA). ESRRA is one of the principal TFs that cooperate with PPARGC1A to regulate the expression of genes involved in mitochondrial biogenesis and oxidative phosphorylation in cardiac and skeletal muscle and in the liver (Huss et al. 2002; Mootha et al. 2004; Charest-Marcotte et al. 2010). In addition, expression of the ESRRA gene is strongly induced by PPARGC1A (Schreiber et al. 2003; Laganiere et al. 2004; Mootha et al. 2004). Thus, the unbiased recovery of this motif supports the validity of our PPARGC1A ChIP-seq data set. The third motif identified by MEME analysis matched the consensus motif for CCAAT/enhancer-binding protein beta (CEBPB) and its related proteins (Osada et al. 1996). Cooperation between CEBPB and PPARGC1A has not been demonstrated previously; however, several studies have suggested a link between these factors (see Discussion). The ESRRA and CEBPB motifs were present in 403 and 338 PPARGC1A peaks, respectively (Fig. 2B). In total, 917 out of 1886 PPARGC1A peaks (48.6%) contained at least one of the three motifs.
Figure 2.

Discovery of enriched sequence motifs in PPARGC1A-occupied regions. (A) (Left panel) The top three DNA sequence motifs identified in an unbiased analysis of PPARGC1A-occupied regions using MEME. The MEME E-value, a measure of statistical significance (Bailey and Elkan 1994), is displayed above each motif. (Right panel) The most similar motif in the TRANSFAC database is displayed for each MEME motif (Matys et al. 2006). Comparison with the TRANSFAC database was performed using Tomtom (Gupta et al. 2007). The motif accession number and name from TRANSFAC and P-value from Tomtom are listed above each TRANSFAC motif. Motif logos were generated using the WebLogo tool (http://weblogo.berkeley.edu/). (B) The genomic DNA sequences of all PPARGC1A-occupied regions (±100 bp from each peak) were scanned for matches to each of the three motifs identified by MEME analysis. Colored wedges within each pie chart represent the fraction of all PPARGC1A binding sites that contain at least one match to the indicated DNA sequence motif. The number of regions in which a match is present or absent is indicated. Motif matches were identified using FIMO with a similarity P-value threshold of 0.01 (Bailey et al. 2009). (C) The mean phastCons conservation score (Siepel et al. 2005) across all motif-containing PPARGC1A binding sites is plotted in a 200-bp window centered on the start position of the motif. (Black boxes) Position of the corresponding motif. (Open circles) Points in each plot that correspond to base pairs constituting the motif. (D) PPARGC1A binds in close proximity to HSEs in a subset of occupied regions. A PPARGC1A binding site in the promoter of the HSP90AA1 gene is shown as an example. The genomic position of the HSE in the HSP90AA1 promoter is indicated as a red box beneath the PPARGC1A signal map. The sequence of the HSE, containing six inverted repeats of the consensus sequence nGAAn (Hickey et al. 1989; Mathur et al. 1994), is displayed in the expanded view. (X-axis) Chromosomal positions. Gene structure is shown to scale above the signal plot. (Arrow) Genomic coordinate of the PPARGC1A signal peak.
To determine whether the motifs enriched at PPARGC1A binding sites are evolutionarily conserved, we aligned all the occurrences of each motif and calculated the average phastCons conservation score at each position in addition to the surrounding genomic regions in a 200-bp window (±100 bp from the motif start position) (Fig. 2C). All three motifs exhibited an increase in average conservation relative to the genomic background (Fig. 2C). The HSE motif displayed the highest average conservation; in addition, we noted that PPARGC1A signal peaks tended to be particularly high at sites containing an HSE, and PPARGC1A peaks were frequently located in very close proximity to these elements. An example of PPARGC1A occupancy at an HSE within the promoter of the known HSF1 target gene, HSP90AA1 (Hickey et al. 1989), is displayed in Figure 2D.
ChIP-seq reveals genome-wide patterns of TF binding and colocalization of PPARGC1A and its network partners
Our initial ChIP-seq studies suggested that PPARGC1A functions with known and novel partners in HepG2 cells. To better understand how PPARGC1A and its network partners cooperate to regulate their target genes across the genome in response to metabolic signals, we performed ChIP-seq experiments to map the genome-wide occupancy of the three regulators identified in the unbiased motif analysis—HSF1, ESRRA, and CEBPB. To place these factors within a wider context, we also selected three additional TFs that have been shown to function with PPARGC1A, namely hepatocyte nuclear factor 4 alpha (HNF4A), glucocorticoid receptor (NR3C1), and GA-binding protein (GABP, also known as nuclear respiratory factor 2, NRF2) (Knutti et al. 2000; Rhee et al. 2003; Mootha et al. 2004). DNA binding motifs for both HNF4A and GR were found to be enriched when PPARGC1A-occupied sites were analyzed using the complete set of motifs in the TRANSFAC database with the program TFM-explorer (Defrance and Touzet 2006). HNF4A and NR3C1 are NRs that bind to PPARGC1A directly through LXXLL motifs in the PPARGC1A N terminus (Rodgers et al. 2008). GABP, an ETS-family TF consisting of alpha and beta subunits, is coactivated by PPARGC1A at mitochondrial genes in muscle (Mootha et al. 2004). In addition to these six factors, we also determined the genome-wide occupancy of the unphosphorylated form of RNA polymerase II, a marker of transcription initiation. Biological replicate experiments were performed for each TF. We obtained approximately a total of 10.9–16.4 million mapped sequence tags. Using an FDR threshold of 1% and the program PeakSeq, high-confidence TF binding sites were obtained (Table 1).
The binding results of the six site-specific TFs and RNA pol II in HepG2 cells treated with forskolin are listed in Table 1. The number of binding sites was highest for CEBPB and HNF4A (38,011 and 26,597, respectively), suggesting more general roles for these factors in maintaining hepatocyte gene expression. ESRRA, GABP, and HSF1 each localized to an intermediate number of sites (3786, 6755, and 3795, respectively). NR3C1 occupied 838 sites, likely a small number relative to the full complement of target sites that are bound in the presence of a glucocorticoid ligand such as dexamethasone (Reddy et al. 2009). ChIP-qPCR experiments directed against selected targets of CEBPB and HSF1 confirmed that the majority (∼95%) of binding sites identified by ChIP-seq are bound in vivo (Supplemental Fig. S1; Supplemental Table S3). In addition, the binding sites of all six TFs exhibited elevated sequence conservation based on their average phastCons scores (Supplemental Fig. S3). Our results are consistent with several previous studies that have examined the genomic binding of several of these factors in various cell types (Wallerman et al. 2009; Charest-Marcotte et al. 2010; Schmidt et al. 2010). For example, Schmidt and colleagues recently identified a similar number of HNF4A binding sites (∼27,800) in primary human hepatocytes (Schmidt et al. 2010). Peak signals tended to be higher overall for the six TFs than for PPARGC1A, likely due to their direct binding to DNA compared with the indirect binding of PPARGC1A. For example, although similar numbers of sequence reads were obtained for each factor, only 12 PPARGC1A peaks contained >100 overlapping sequence tags, compared with 5673 peaks for CEBPB and 2271 for GABP (Supplemental Table S1; Supplemental Data set S1). It is important to note that some of the variation we observed among these factors in binding site frequency and peak signal may be due to technical limitation of ChIP, such as differences in antibody efficiency.
To examine the overlap among all seven regulators, we first identified genomic regions that contain clusters of adjacent binding sites by extending each peak ±200 bp and merging regions that overlapped. This analysis revealed 16,712 genomic regions that are occupied by two or more regulators (Supplemental Table S6). We refer to these regions as multiple regulatory factor binding regions (multi-RFBRs). We next determined the number of multi-RFBRs co-occupied by each distinct pair of regulators (Fig. 3A). The patterns of overlap at multi-RFBRs suggested that these different factors co-occur with distinct preferences. For example, although ESRRA and HSF1 occupy nearly identical numbers of multi-RFBRs (3193 and 3198, respectively), ESRRA is much more frequently found at multi-RFBRs that contain HNF4A (2806 ESRRA-HNF4A multi-RFBRs vs. 2077 HSF1-HNF4A multi-RFBRs) (Fig. 3A). To determine which groups of regulators associate most strongly throughout the genome, we next clustered all seven regulators based on the similarity of their colocalization patterns (Fig. 3B). For this analysis, we computed a colocalization score (odds ratio from a Fisher's exact test) for each pair of regulators based on the observed versus expected number of co-occupied multi-RFBRs. As depicted in the heatmap in Figure 3B, two groups emerged with high levels of association—Group 1, consisting of PPARGC1A, ESRRA, and HSF1, and Group 2 consisting of GABP and NR3C1. The strong association observed among the regulators in each of these groups indicates that they share an especially high number of binding sites and potentially function in a modular fashion to regulate the expression of their target genes.
Figure 3.

Genome-wide patterns of binding and colocalization of PPARGC1A and its network partners. (A) Pairwise colocalization of transcriptional regulators within multi-RFBRs. Numbers in each cell represent the number of multi-RFBRs that are co-occupied by the corresponding pair of regulators. The first cell in each column indicates the total number of multi-RFBRs bound by each regulator. Cells are colored by the fraction of all 16,712 multi-RFBRs identified in the genome. Factors are ordered by decreasing number of multi-RFBRs bound. (B) Heatmap depicting the clustering of transcriptional regulators based on the similarity of their colocalization patterns. Colors in the heatmap indicate the strength of association between each pair of regulators (odds ratio from a Fisher's exact test; see Methods). Two groups of regulators exhibiting highly similar colocalization patterns based on the results of clustering are outlined in blue boxes. (C) Distribution of transcriptional regulator binding sites relative to UCSC Known Genes transcript annotations (Hsu et al. 2006). Factors are ordered by increasing proportion of TSS-adjacent binding sites. (D) Bar graph depicting the fraction of binding sites within 5 kb of an RNA pol II peak for each regulator.
Comparison of the binding sites of each factor with transcripts in the UCSC Known Genes track also revealed similarities and differences in their binding properties (Fig. 3C). For example, binding outside of promoter regions was quite prevalent for CEBPB and HNF4A (68.0% and 63.3% of sites, respectively), similar to our results with PPARGC1A (Fig. 3C). In contrast, GABP and NR3C1 were much more common at promoters (72.0% and 91.3% of sites, respectively) and most similar in their binding distribution to RNA pol II (Fig. 3C). To examine the relationship of each factor with RNA pol II in more detail, we also determined the fraction of sites that was adjacent to an RNA pol II binding site (Fig. 3D). These results were consistent with the patterns we observed relative to TSSs, indicating that the subset of promoter-proximal binding sites of each factor is associated with sites of transcription initiation in the genome (Fig. 3D).
Correlation of binding with gene expression suggests that many of the binding sites are functional
To determine whether these factors are associated with genes that are differentially expressed in the fasted state, we measured gene expression using Illumina BeadArrays before and after 6 h treatment with forskolin. Genes were ranked by fold-induction and compared with the binding site information for the different factors. As shown in the heatmap in Figure 4, PPARGC1A and its network partners are strongly associated with both the top and bottom of the ranked gene list, indicating that the binding of these factors may promote the up- or down-regulation of their target genes. The biased representation of target genes at the top and bottom of the ranked list was statistically significant for each factor (P < 10−3) based on the results of gene set enrichment analysis (Subramanian et al. 2005), with the exception of NR3C1, which was significantly associated only with the top ranking genes (Fig. 4).
Figure 4.

Regulator occupancy corresponds with gene expression changes in forskolin-treated HepG2 cells. (Left panel) Replicate gene expression measurements from Illumina BeadArrays were used to rank 18,000 genes by fold induction after 6 h treatment with forskolin. (Right panel) The distribution of regulator-occupied genes within the ranked list is depicted as a heatmap. The ranked list was divided into 18 rank groups containing 1000 genes each and genes containing 5′-proximal, 3′-proximal, or intragenic binding sites were identified for each factor. Within each rank group, the fraction of genes that are occupied by a given factor was compared with the fraction expected from a random distribution. Colors in the heatmap represent the level of over- or under-representation of regulator-occupied genes. Association of regulator-occupied genes with both the top and bottom of the ranked list was determined to be statistically significant (P < 10−3) for CEBPB, HNF4A, GABP, HSF1, PPARGC1A, and ESRRA using gene set enrichment analysis (Subramanian et al. 2005). NR3C1-occupied genes were significantly associated with the top of the ranked list only (P < 10−3).
PPARGC1A functions with different combinations of TFs at distinct types of gene targets
In order to examine the combinatorial binding patterns of PPARGC1A with each of its network partners, we next analyzed the subset of interactions involving PPARGC1A in more detail. We first grouped the entire set of PPARGC1A binding sites based on the number of regulators bound (Supplemental Table S8); these interactions are visualized as a heatmap in Figure 5A. Notably, 1741 PPARGC1A binding sites (92.3%) are located in multi-RFBRs, the highest proportion among all of the regulators that we examined. This finding is consistent with the requirement of PPARGC1A to localize to its genomic targets through TFs that directly bind DNA; it likely also reflects the fact that factors were chosen based on association with PPARGC1A. PPARGC1A also likely interacts with TFs not included in the present study at the remaining 145 sites located outside of multi-RFBRs. Another intriguing finding is that PPARGC1A binding sites are typically co-occupied by more than one additional TF. Indeed the two largest groups of PPARGC1A-bound multi-RFBRs contain two to three additional TFs (Fig. 5A). The top three unique regulator combinations within each group are displayed in Figure 5B. Overall, the most prevalent combination was PPARGC1A–HNF4A–CEBPB, observed at 350 multi-RFBRs (Fig. 5B). Other examples of frequently observed combinations are PPARGC1A–HNF4A–CEBPB–HSF1 (188 multi-RFBRs) and PPARGC1A–HNF4A–CEBPB–ESRRA (160 multi-RFBRs) (Fig. 5B).
Figure 5.

Recruitment of PPARGC1A to distinct clusters of TF binding sites. (A) Combinatorial binding patterns of PPARGC1A with its network partners displayed as a heatmap. PPARGC1A binding sites were grouped based on the number of bound factors, as indicated to the left of each group. Sites containing two or more factors correspond to PPARGC1A-bound multi-RFBRs. Within each group, binding sites were sorted by decreasing intensity of PPARGC1A binding, based on the number of sequence tags mapping to multi-RFBRs or within 200 bp of the PPARGC1A peak for sites bound by PPARGC1A only. Colors from yellow to red reflect increasing tag count. (Gray) Sites that are not bound by a given TF. TFs are ordered from left to right by decreasing colocalization with PPARGC1A. RNA pol II binding is included for reference in the last column. (B) Predominant regulator combinations at PPARGC1A-bound multi-RFBRs. PPARGC1A-bound multi-RFBRs were grouped based on the number of bound factors as in A. For each group, the total number of multi-RFBRs is listed, along with the top three distinct regulator combinations and the number of times each combination is observed. (Colored boxes) Binding by the respective regulator. (C) Differential enrichment of GO categories among genes occupied by PPARGC1A and each of its network partners. Relative fold enrichment is displayed as a heatmap with columns clustered by similarity. Representative GO categories are shown; the complete set of differentially enriched GO categories is listed in Supplemental Table S9. NR3C1 is excluded due to the small number of targets overlapping with PPARGC1A.
One challenge in genomics has been to understand how different combinations of regulators work together to target specific sets of genes involved in distinct biological pathways (Blais and Dynlacht 2005). To explore this problem in the context of PPARGC1A and its network partners, we identified the set of genes occupied by each distinct PPARGC1A–TF pair and functionally categorized these genes using GO (The Gene Ontology Consortium 2000). We then determined the level of over- or under-representation of each GO category for each PPARGC1A–TF pair and used this information to cluster GO categories based on the similarity of their enrichment profiles (Supplemental Table S9). Representative differentially enriched GO categories are displayed in the heatmap in Figure 5C. Our results suggest that several important pathways are preferentially regulated by distinct TF combinations. For example, genes involved in mitochondrial metabolism, including GO categories such as oxidative phosphorylation and the respiratory electron transport chain, were preferentially occupied by both the regulator combinations of PPARGC1A–ESRRA and PPARGC1A–GABP, in agreement with previous studies of these TFs (Mootha et al. 2004; Charest-Marcotte et al. 2010). In contrast, genes involved in lipid catabolism were enriched among genes occupied by PPARGC1A–ESRRA but under-represented among those occupied by PPARGC1A–GABP (Fig. 5C). Genes involved in protein folding and related categories were enriched among PPARGC1A–HSF1-occupied genes (Fig. 5C; Supplemental Table S9). Surprisingly, the protein folding categories were also enriched among PPARGC1A–GABP-occupied genes, suggesting that these three factors work together to regulate protein folding and other pathways associated with the heat shock response. Lipid metabolic pathways were slightly over-represented among genes occupied by PPARGC1A in combination with HNF4A or CEBPB (Fig. 5C). PPARGC1A–HNF4A- and PPARGC1A–CEBPB-occupied genes exhibited less distinct patterns of differential enrichment, presumably reflecting the broad binding of these TFs to the genome (Supplemental Table S9). Overall, these results indicate that distinct combinations of TFs bind distinct gene targets.
Transcriptional regulatory circuitry of PPARGC1A and its network partners
We next sought to explore the regulatory circuitry formed by PPARGC1A and its network partners by investigating their binding relationships with each other and with other TFs. We first examined the genes encoding each transcriptional regulator and determined which regulators were bound. As shown for the PPARGC1A gene in Figure 6A, we frequently observed binding of different combinations of regulators not only at promoter regions, but also at multiple sites within the gene body and adjacent to the 3′-end. Thus, we considered any such binding event to be a potential regulatory connection and used this information to construct a transcriptional regulatory network. In this network, “nodes” represent each transcriptional regulator and “edges” connecting nodes represent binding of one regulator to the gene encoding another regulator. As diagrammed in Figure 6B, the seven regulators form a highly connected network suggesting complex patterns of interdependent regulation. Various network motifs (the smallest units of network structure) are abundant in this network, including feed-forward loops, multi-component loops, and multi-input motifs (Blais and Dynlacht 2005). One particularly striking feature of this network is that every transcriptional regulator exhibits autoregulation, a motif that characterizes master regulators of important cellular pathways in various cell types (Boyer et al. 2005; Odom et al. 2006; Reed et al. 2008). Notably, an autoregulatory interaction occurs in intron 2 of the PPARGC1A gene (Fig. 6A), a region in which single nucleotide polymorphisms (SNPs) have been associated with the age at onset of Huntington's disease (Taherzadeh-Fard et al. 2009; Weydt et al. 2009).
Figure 6.

Transcriptional regulatory circuitry of PPARGC1A and its network partners. (A) Regulator binding at the PPARGC1A locus. Signal maps are displayed for four factors that occupy PPARGC1A. Significant peaks (q < 0.01) are indicated by colored lines under each signal map. (X-axis) Chromosomal positions. Gene structure is shown to scale below the signal maps. (B) Transcriptional regulatory network diagram displaying interactions among CEBPB, ESRRA, GABP, NR3C1, HNF4A, HSF1, and PPARGC1A. (Arrows) Direct binding of one regulator to the 5′-proximal, 3′-proximal, or intragenic region of the gene encoding another regulator (or the same regulator in the case of autoregulatory loops). (C) Regulatory hierarchy among the seven regulators depicted in B. Factors are ranked first by the number of incoming network connections (“kin”) then by the number of outgoing network connections (“kout”) (Borneman et al. 2006). (D) Expanded transcriptional regulatory network. Additional TFs were added to the core network shown in B, represented here by the circle in the center of the diagram, if they contained four or more incoming network connections. The number of incoming connections is indicated by arrow types according to the legend. Fifteen representative TFs are shown; the complete list of 155 TFs in the expanded network and their bound regulators is available in Supplemental Table S10.
The factors in this network also exhibit a hierarchical relationship in terms of the number of transcriptional regulators that they occupy, represented by the number of outgoing edges of each node (Fig. 6C). CEBPB and HNF4A, for example, are highly connected nodes, forming direct regulatory connections with all seven regulators that we examined. These findings indicate that CEBPB and HNF4A can influence the expression of a large assortment of genes both by direct interactions and by interactions mediated by multiple downstream regulators, and are consistent with the observed roles of HNF4A and CEBP family members in specifying and maintaining the hepatocyte transcriptional program (Lekstrom-Himes and Xanthopoulos 1998; Odom et al. 2004; Kyrmizi et al. 2006). ESRRA, GABP, HSF1, and PPARGC1A, on the other hand, occupy lower positions in the hierarchy, as each of these factors binds to a more limited subset of three to four regulators, suggesting that they influence the expression of genes that are involved in more focused biological pathways. One basic property of transcriptional regulatory networks is that edges are directional; thus, each node can contain differing numbers of incoming and outgoing connections (Barabasi and Oltvai 2004; Borneman et al. 2006). In the network formed by PPARGC1A and its partner TFs, both ESRRA and HNF4A contain six incoming connections, compared with only three to four for the remaining regulators (Fig. 6C). These findings indicate that the ESRRA and HNF4A genes are subject to greater regulatory input and potentially responsive to a wider array of biological stimuli. Previous work in yeast suggests that TFs that are downstream targets of many regulators can serve as “target hubs” in regulatory networks whose activity represents the combined output of multiple upstream signals (Borneman et al. 2006; Zhu et al. 2007). These factors often serve as master regulators of important cellular pathways (Borneman et al. 2006; Zhu et al. 2007).
To identify additional regulators that might form important connections with the seven factors in our “core” network, we expanded our network by including target genes encoding other transcriptional regulators that were occupied by at least four factors (Fig. 6D; Supplemental Table S10). The expanded network contains 155 additional regulators (Supplemental Table S10), many of which have demonstrated roles in critical cellular processes related to the function of PPARGC1A. Indeed, several of these regulators are known targets of PPARGC1A coactivation, including RXRA, RXRB, YY1, PPARD, and NR1H3 (also known as LXRA) (Fig. 6D; Delerive et al. 2002; Oberkofler et al. 2003; Wang et al. 2003; Cunningham et al. 2007). Intriguingly, YY1, one of the TFs with the most connections to the core network, has been shown to bind directly to the PPARGC1A promoter in skeletal muscle, and YY1 DNA binding motifs are highly enriched in the promoters of mitochondrial genes regulated by PPARGC1A (Cunningham et al. 2007). Another factor in the expanded network, NR0B2 (also known as SHP), negatively regulates the expression of PPARGC1A in brown adipocytes (Wang et al. 2005). The presence of regulators such as YY1 and NR0B2 and other TFs in our expanded network not only implicates these factors as players in the PPARGC1A regulatory network in the liver, but also suggests that a broad transcriptional network is influenced by PPARGC1A and its associated factors.
Discussion
In this study, we have utilized a genomic approach to identify potential regulatory partners of the transcriptional coactivator PPARGC1A. Using ChIP-seq, we mapped PPARGC1A binding sites across the genome in human hepatocytes under conditions simulating the fasted state. MEME analysis of the underlying sequences at these sites revealed an enrichment of consensus motifs corresponding to known TFs, suggesting PPARGC1A coactivation or cooperation with these factors in the liver. Of the three motifs discovered in an unbiased manner, two motifs, corresponding to HSF1 and CEBPB, represent novel associations with PPARGC1A. Subsequent mapping of these TFs revealed a high degree of overlap throughout the genome. Although direct binding of PPARGC1A with these TFs may account for the strong motif enrichment and target overlap that we observed, it is also possible that these factors interact indirectly within larger multi-protein complexes that form at regulatory elements in the genome. Regardless, PPARGC1A mediates its activity by working with a number of other TFs. Our selection of additional TFs for ChIP-seq analysis was guided by searches for known consensus motifs, knowledge of PPARGC1A binding partners from the literature, expression in HepG2 cells, and the availability of suitable antibodies for ChIP-seq.
The highly significant enrichment of conserved HSEs, elevated PPARGC1A peak signals at these sites, and strong tendency of PPARGC1A and HSF1 to associate within multi-RFBRs are particularly striking and may have important implications for understanding the role of PPARGC1A in normal metabolic function and human disease. HSF1 regulates the expression of chaperone proteins that aid in the repair of protein damage in response to cellular stress (Hooper 2009). Like PPARGC1A, HSF1 and its downstream chaperones have been implicated in the pathogenesis of type II diabetes. For example, the HSF1 target gene HSP72 exhibits reduced levels in the skeletal muscle of patients with insulin resistance and diabetes (Kurucz et al. 2002; Bruce et al. 2003; Chung et al. 2008). Furthermore, studies in diabetic non-human primates suggest that HSF1 and heat shock protein deficiency in the liver contributes to insulin resistance (Kavanagh et al. 2009). Intriguingly, the diet-responsive NAD-dependent deacetylase SIRT1, a critical regulator of PPARGC1A activity in the fasted liver (Rodgers et al. 2005), was recently shown to interact with and deacetylate HSF1 (Westerheide et al. 2009). Our results suggest that PPARGC1A functions closely with HSF1 and that HSF1 activity may be linked to dietary conditions such as fasting and calorie restriction. Our results also suggest novel combinatorial relationships between HSF1 and the other TFs that we examined. For example, HSF1 and ESRRA (along with PPARGC1A) were found to be strongly associated when we clustered TFs based on their pairwise colocalization patterns (Fig. 3B). HSF1 and ESRRA were also found to participate in a multicomponent loop in which each factor binds to the gene encoding the other (Fig. 6B). In another example, we found that genes involved in the response to unfolded protein were enriched in the binding data sets of both HSF1 and GABP (Fig. 5C). Although PPARGC1A and HSF1 appear to bind in close proximity, we have so far been unable to detect a stable complex by co-immunoprecipitation of the native proteins in HepG2 cells. One possibility is that they may only form a stable complex in the presence of DNA, a phenomenon that we have previously observed in yeast (Borneman et al. 2006). Alternatively, they may exist in separate complexes that localize to a common set of regulatory regions.
The finding of CEBPB motif enrichment in our PPARGC1A binding data set is also intriguing since several previous studies have identified links in the expression and regulation of these two factors. For example, CEBPB activity was found to be highly elevated in the livers of PPARGC1A knockout mice (Lin et al. 2004) and CEBPB has been shown to regulate the expression of the PPARGC1A gene by binding to a cAMP response element in the PPARGC1A promoter (Karamanlidis et al. 2007; Wang et al. 2008). In addition, C/EBP-family proteins participate in a number of cellular pathways that are also regulated by PPARGC1A, such as gluconeogenesis (Roesler 2001).
Our findings demonstrate that PPARGC1A and its network partners localize to the genome in a combinatorial manner with distinct preferences for genes involved in different functional pathways. The genomic binding sites of these factors typically occur in clusters (i.e., multi-RFBRs) that are found at both TSS proximal and distal regulatory regions. Indeed, PPARGC1A typically co-occupies sites containing two to three additional TFs and the majority of PPARGC1A binding sites are located outside of promoter regions. The manner in which PPARGC1A associates with TFs at multi-RFBRs is likely to be complex. For example, a single PPARGC1A protein may interact with several TFs simultaneously in a protein complex, an arrangement that would be facilitated by the multiple interfaces for protein–protein interactions contained on PPARGC1A (Rodgers et al. 2008). Alternatively, PPARGC1A may physically interact with one or more TFs independently; such an arrangement may occur when the DNA binding sites of these TFs are not located in close proximity. Overall our mapping experiments suggest a complex code in which distinct combinations of factors work together to activate distinct types of gene targets. The mapping of seven key factors involved in metabolism is likely to be a valuable resource in elucidating the metabolic transcriptional code. Given the importance of transcriptional regulatory networks in diabetes, our work is likely to form an important foundation for understanding basic mechanisms of human disease.
Methods
Cell culture
HepG2 cells were cultured in DMEM supplemented with 10% FBS and 100 U/mL penicillin/streptomycin. For ChIP-seq experiments reflective of the low glucose state, cells were cultured for 12 h in DMEM with 0.5% BSA then treated for 6 h with forskolin (1 μM) in low-glucose DMEM containing 1 mM pyruvate.
ChIP and ChIP-seq
ChIP was performed as described using two to three independent biological replicate experiments (Reed et al. 2008). Antibodies used were rabbit anti-PGC1 (sc-13067), rabbit anti-C/EBP β (sc-150), rabbit anti-ERRα (sc-66882), rabbit anti-GABP-α (sc-22810), rabbit anti-GR (sc-1002), rabbit anti-HNF-4α (sc-6556), and rabbit anti-HSF1 (sc-9144) from Santa Cruz Biotechnology, and mouse anti-POLR2A (RNA pol II) (MMS-126R) from Covance. ChIP and input DNA samples were prepared in parallel and sequenced on an Illumina Genome Analyzer II according to the manufacturer's protocol (Illumina).
Data analysis
Raw images generated from Illumina Genome Analyzer II runs were analyzed using Illumina's recommended software pipeline to generate lists of base-called sequence tags. Sequence tags (27 bp) were mapped to the human genome (hg18/NCBI Build 36.1) downloaded from the UCSC Genome Browser (http://genome.ucsc.edu/) (Kent et al. 2002) using the program ELAND. Sequence tags from independent replicates were combined into a single data set, then extended in the 3′ direction to 200 bp and converted to signal map files representing the integer count of mapped tags overlapping at each genomic position. The signal maps were scored using the program PeakSeq (Rozowsky et al. 2009) in order to identify factor binding sites. Statistical significance was calculated using a binomial test followed by the Benjamini-Hochberg correction for multiple hypothesis testing to yield a q-value for each candidate region. High confidence bound regions were selected with a q-value cutoff of 0.01, corresponding to an overall FDR of 1%. A q-value cutoff of 0.05 was also used to identify a set of lower confidence regions.
Target validation
To validate binding targets quantitative PCR with site-specific primers was performed in duplicate on a BioRad MyiQ real-time PCR cycler with BioRad iQ SYBR Green supermix. Primers for ChIP-qPCR were designed for 20–23 genomic binding sites selected to represent the distribution of peak intensities for PPARGC1A, CEBPB, and HSF1, and for a negative control unbound region. Normalized Ct (δCt) values for each sample were calculated by subtracting the Ct value obtained using input DNA from the Ct value obtained using ChIP DNA (δCt = CtChIP – Ctinput). Fold enrichment was then calculated using the formula 2−(δCt[target] – δCt[control]). For the purpose of estimating specificity, targets with mean qPCR fold enrichment >2 were considered enriched (Supplemental Fig. S1). Primer sequences are listed in Supplemental Table S3.
Motif analysis
The motif discovery program MEME (Bailey and Elkan 1994) was used to identify candidate protein–DNA interaction motifs in an unbiased manner. Input to the MEME program consisted of 200-bp sequences centered at the peak position for the top 250 bound regions (ranked by q-value). The results of MEME analysis are in Supplemental Table S5. No additional motifs were identified beyond the three listed in Figure 2A. The top-scoring motifs from MEME were queried against the TRANSFAC database (Matys et al. 2006) using Tomtom (Gupta et al. 2007) to identify the best matching known motifs. Locations of matches to MEME motifs were determined using the program FIMO (Bailey et al. 2009) with a similarity P-value threshold of 10−4. The mammalian phastCons conservation scores (Siepel et al. 2005) at motif matches within PPARGC1A-bound regions were obtained from the UCSC Genome Browser (Karolchik et al. 2008). Enrichment of known motifs was determined using the program TFM-explorer (Defrance and Touzet 2006).
Functional classification
Statistical analysis of the enrichment of GO categories (The Gene Ontology Consortium 2000) was performed using the GOstats package in R (Falcon and Gentleman 2007). To identify differentially enriched GO categories among genes occupied by each PPARGC1A–TF pair, categories exhibiting significant enrichment (P < 0.01, hypergeometric test) in at least one PPARGC1A–TF pair and containing at least 20 member genes were identified. Next, an enrichment score was determined by calculating the ratio of observed/expected genes for each category and retaining categories with a standard deviation of at least 0.5 across all PPARGC1A–TF pairs (Supplemental Table S9). Enrichment scores of example categories in Figure 5C were log-transformed, mean-centered, and hierarchically clustered using Cluster 3.0 (de Hoon et al. 2004) and visualized using Java Treeview (Saldanha 2004).
Gene expression analysis
Total RNA from 5 × 106 forskolin-treated and untreated HepG2 cells was collected using TRIzol-containing buffer (Invitrogen) and further purified using a Qiagen RNeasy kit. Purified total RNA was submitted to the Microarray Resource at the Yale W.M. Keck Foundation Biotechnology Resource Laboratory for labeling and hybridization to Illumina HumanRef-8 Expression BeadChips. Raw intensities were log2-transformed and quantile-normalized, and probe sequences were remapped to Entrez GeneIDs using the lumi package in R (Du et al. 2008). The complete expression data set is summarized in Supplemental Table S7.
Multi-RFBR identification and clustering
Multi-RFBRs were identified by combining the peaks of all seven regulators into one data set, extending these peaks ±200 bp, and merging regions that overlapped. This analysis identified 57,436 unique regions, of which 16,712 were occupied by more than one factor and designated multi-RFBRs (Supplemental Table S6). The remaining 40,724 regions consisted almost entirely of single binding sites for CEBPB (24,123 regions) and HNF4A (12,533 regions). To cluster regulators based on the similarity of their colocalization patterns, the observed and expected numbers of multi-RFBRs co-occupied by each pair of regulators were compared using a Fisher's exact test in order to derive an odds ratio representing the strength of association of each pair. Hierarchical clustering was then performed on the 7 × 7 matrix of odds ratios using the “heatmap” function in R with default parameters.
Data access
Sequence reads, quality scores, alignment coordinates, and PeakSeq results for factors in this study have been submitted to the Encyclopedia of DNA elements (ENCODE) project (The ENCODE Project Consortium 2007), and are available for download or visualization as genome browser tracks at the UCSC ENCODE Data Coordination Center website (http://genome.ucsc.edu/ENCODE/). Sequence reads have been submitted to the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRA050697.
Acknowledgments
This work was supported by grants from the NHGRI (F31HG004299).
Footnotes
[Supplemental material is available for this article.]
Article and supplemental material are at http://www.genome.org/cgi/doi/10.1101/gr.127761.111.
Freely available online through the Genome Research Open Access option.
References
- Andersson U, Scarpulla RC 2001. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol Cell Biol 21: 3738–3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS 2009. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi AL, Oltvai ZN 2004. Network biology: Understanding the cell's functional organization. Nat Rev Genet 5: 101–113 [DOI] [PubMed] [Google Scholar]
- Blais A, Dynlacht BD 2005. Constructing transcriptional regulatory networks. Genes Dev 19: 1499–1511 [DOI] [PubMed] [Google Scholar]
- Borneman AR, Leigh-Bell JA, Yu H, Bertone P, Gerstein M, Snyder M 2006. Target hub proteins serve as master regulators of development in yeast. Genes Dev 20: 435–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. 2005. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122: 947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce CR, Carey AL, Hawley JA, Febbraio MA 2003. Intramuscular heat shock protein 72 and heme oxygenase-1 mRNA are reduced in patients with type 2 diabetes: Evidence that insulin resistance is associated with a disturbed antioxidant defense mechanism. Diabetes 52: 2338–2345 [DOI] [PubMed] [Google Scholar]
- Charest-Marcotte A, Dufour CR, Wilson BJ, Tremblay AM, Eichner LJ, Arlow DH, Mootha VK, Giguere V 2010. The homeobox protein Prox1 is a negative modulator of ERRα/PGC-1α bioenergetic functions. Genes Dev 24: 537–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Nguyen AK, Henstridge DC, Holmes AG, Chan MH, Mesa JL, Lancaster GI, Southgate RJ, Bruce CR, Duffy SJ, et al. 2008. HSP72 protects against obesity-induced insulin resistance. Proc Natl Acad Sci 105: 1739–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P 2007. mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature 450: 736–740 [DOI] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, Miyano S 2004. Open source clustering software. Bioinformatics 20: 1453–1454 [DOI] [PubMed] [Google Scholar]
- Defrance M, Touzet H 2006. Predicting transcription factor binding sites using local over-representation and comparative genomics. BMC Bioinformatics 7: 396 doi: 10.1186/1471-2105-7-396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delerive P, Wu Y, Burris TP, Chin WW, Suen CS 2002. PGC-1 functions as a transcriptional coactivator for the retinoid X receptors. J Biol Chem 277: 3913–3917 [DOI] [PubMed] [Google Scholar]
- Du P, Kibbe WA, Lin SM 2008. lumi: A pipeline for processing Illumina microarray. Bioinformatics 24: 1547–1548 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R 2007. Using GOstats to test gene lists for GO term association. Bioinformatics 23: 257–258 [DOI] [PubMed] [Google Scholar]
- Farnham PJ 2009. Insights from genomic profiling of transcription factors. Nat Rev Genet 10: 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Kelly DP 2006. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J Clin Invest 116: 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium 2000. Gene ontology: Tool for the unification of biology. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS 2007. Quantifying similarity between motifs. Genome Biol 8: R24 doi: 10.1186/gb-2007-8-2-r24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C, Lin J, Rhee J, Peyer AK, Chin S, Wu PH, Meyer UA, Spiegelman BM 2005. Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1α. Cell 122: 505–515 [DOI] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, et al. 2001. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413: 179–183 [DOI] [PubMed] [Google Scholar]
- Hickey E, Brandon SE, Smale G, Lloyd D, Weber LA 1989. Sequence and regulation of a gene encoding a human 89-kilodalton heat shock protein. Mol Cell Biol 9: 2615–2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper PL 2009. Inflammation, heat shock proteins, and type 2 diabetes. Cell Stress Chaperones 14: 113–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D 2006. The UCSC known genes. Bioinformatics 22: 1036–1046 [DOI] [PubMed] [Google Scholar]
- Huss JM, Kopp RP, Kelly DP 2002. Peroxisome proliferator-activated receptor coactivator-1α (PGC-1α) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-α and -γ. Identification of novel leucine-rich interaction motif within PGC-1α. J Biol Chem 277: 40265–40274 [DOI] [PubMed] [Google Scholar]
- Javitt N 1990. Hep G2 cells as a resource for metabolic studies: Lipoprotein, cholesterol, and bile acids. FASEB J 4: 161–168 [DOI] [PubMed] [Google Scholar]
- Karamanlidis G, Karamitri A, Docherty K, Hazlerigg DG, Lomax MA 2007. C/EBPβ reprograms white 3T3-L1 preadipocytes to a Brown adipocyte pattern of gene expression. J Biol Chem 282: 24660–24669 [DOI] [PubMed] [Google Scholar]
- Karolchik D, Kuhn RM, Baertsch R, Barber GP, Clawson H, Diekhans M, Giardine B, Harte RA, Hinrichs AS, Hsu F, et al. 2008. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res 36: D773–D779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh K, Zhang L, Wagner JD 2009. Tissue-specific regulation and expression of heat shock proteins in type 2 diabetic monkeys. Cell Stress Chaperones 14: 291–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The human genome browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutti D, Kaul A, Kralli A 2000. A tissue-specific coactivator of steroid receptors, identified in a functional genetic screen. Mol Cell Biol 20: 2411–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurucz I, Morva A, Vaag A, Eriksson KF, Huang X, Groop L, Koranyi L 2002. Decreased expression of heat shock protein 72 in skeletal muscle of patients with type 2 diabetes correlates with insulin resistance. Diabetes 51: 1102–1109 [DOI] [PubMed] [Google Scholar]
- Kyrmizi I, Hatzis P, Katrakili N, Tronche F, Gonzalez FJ, Talianidis I 2006. Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes Dev 20: 2293–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganiere J, Tremblay GB, Dufour CR, Giroux S, Rousseau F, Giguere V 2004. A polymorphic autoregulatory hormone response element in the human estrogen-related receptor α (ERRα) promoter dictates peroxisome proliferator-activated receptor γ coactivator-1α control of ERRα expression. J Biol Chem 279: 18504–18510 [DOI] [PubMed] [Google Scholar]
- Lekstrom-Himes J, Xanthopoulos KG 1998. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J Biol Chem 273: 28545–28548 [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, et al. 2004. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 119: 121–135 [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM 2005. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab 1: 361–370 [DOI] [PubMed] [Google Scholar]
- Mathur SK, Sistonen L, Brown IR, Murphy SP, Sarge KD, Morimoto RI 1994. Deficient induction of human hsp70 heat shock gene transcription in Y79 retinoblastoma cells despite activation of heat shock factor 1. Proc Natl Acad Sci 91: 8695–8699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. 2006. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34: D108–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirhaeghe A, Crowley V, Lenaghan C, Lelliott C, Green K, Stewart A, Hart K, Schinner S, Sethi JK, Yeo G, et al. 2003. Characterization of the human, mouse and rat PGC1 β (peroxisome-proliferator-activated receptor-γ co-activator 1 β) gene in vitro and in vivo. Biochem J 373: 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, et al. 2004. Errα and Gabpa/b specify PGC-1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci 101: 6570–6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller YL, Bogardus C, Pedersen O, Baier L 2003. A Gly482Ser missense mutation in the peroxisome proliferator-activated receptor γ coactivator-1 is associated with altered lipid oxidation and early insulin secretion in Pima Indians. Diabetes 52: 895–898 [DOI] [PubMed] [Google Scholar]
- Oberkofler H, Schraml E, Krempler F, Patsch W 2003. Potentiation of liver X receptor transcriptional activity by peroxisome-proliferator-activated receptor γ co-activator 1 α. Biochem J 371: 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA, Gifford DK, et al. 2004. Control of pancreas and liver gene expression by HNF transcription factors. Science 303: 1378–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom DT, Dowell RD, Jacobsen ES, Nekludova L, Rolfe PA, Danford TW, Gifford DK, Fraenkel E, Bell GI, Young RA 2006. Core transcriptional regulatory circuitry in human hepatocytes. Mol Syst Biol 2: 2006.0017 doi: 10.1038/msb4100059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada S, Yamamoto H, Nishihara T, Imagawa M 1996. DNA binding specificity of the CCAAT/enhancer-binding protein transcription factor family. J Biol Chem 271: 3891–3896 [DOI] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, et al. 2003. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 423: 550–555 [DOI] [PubMed] [Google Scholar]
- Reddy PH, Mao P, Manczak M 2009. Mitochondrial structural and functional dynamics in Huntington's disease. Brain Res Brain Res Rev 61: 33–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M 2008. Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet 4: e1000133 doi: 10.1371/journal.pgen.1000133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan M, Gonzalez FJ, Spiegelman BM 2003. Regulation of hepatic fasting response by PPARγ coactivator-1α (PGC-1): Requirement for hepatocyte nuclear factor 4α in gluconeogenesis. Proc Natl Acad Sci 100: 4012–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P 2005. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434: 113–118 [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P 2008. Metabolic adaptations through the PGC-1 α and SIRT1 pathways. FEBS Lett 582: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesler WJ 2001. The role of C/EBP in nutrient and hormonal regulation of gene expression. Annu Rev Nutr 21: 141–165 [DOI] [PubMed] [Google Scholar]
- Rozowsky J, Euskirchen G, Auerbach RK, Zhang ZD, Gibson T, Bjornson R, Carriero N, Snyder M, Gerstein MB 2009. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat Biotechnol 27: 66–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ 2004. Java Treeview—extensible visualization of microarray data. Bioinformatics 20: 3246–3248 [DOI] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, et al. 2010. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328: 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A 2003. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor α (ERRα). J Biol Chem 278: 9013–9018 [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ 2009. PGC-1α and ERRα target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol 46: 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyal S, Krempler F, Oberkofler H, Patsch W 2006. PGC-1α: A potent transcriptional cofactor involved in the pathogenesis of type 2 diabetes. Diabetologia 49: 1477–1488 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taherzadeh-Fard E, Saft C, Andrich J, Wieczorek S, Arning L 2009. PGC-1α as modifier of onset age in Huntington disease. Mol Neurodegener 4: 10 doi: 10.1186/1750-1326-4-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Huss JM, Kelly DP 2000. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 20: 1868–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallerman O, Motallebipour M, Enroth S, Patra K, Bysani MS, Komorowski J, Wadelius C 2009. Molecular interactions between HNF4a, FOXA2 and GABP identified at regulatory DNA elements through ChIP-sequencing. Nucleic Acids Res 37: 7498–7508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM 2003. Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell 113: 159–170 [DOI] [PubMed] [Google Scholar]
- Wang L, Liu J, Saha P, Huang J, Chan L, Spiegelman B, Moore DD 2005. The orphan nuclear receptor SHP regulates PGC-1α expression and energy production in brown adipocytes. Cell Metab 2: 227–238 [DOI] [PubMed] [Google Scholar]
- Wang CE, Tydlacka S, Orr AL, Yang SH, Graham RK, Hayden MR, Li S, Chan AW, Li XJ 2008. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum Mol Genet 17: 2738–2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Anckar J, Stevens SM Jr, Sistonen L, Morimoto RI 2009. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323: 1063–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Soyal SM, Gellera C, Didonato S, Weidinger C, Oberkofler H, Landwehrmeyer GB, Patsch W 2009. The gene coding for PGC-1α modifies age at onset in Huntington's Disease. Mol Neurodegener 4: 3 doi: 10.1186/1750-1326-4-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, et al. 2001. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413: 131–138 [DOI] [PubMed] [Google Scholar]
- Zhu X, Gerstein M, Snyder M 2007. Getting connected: Analysis and principles of biological networks. Genes Dev 21: 1010–1024 [DOI] [PubMed] [Google Scholar]