

Abstract
The characteristics and evolutionary forces acting on regulatory variation in humans remains elusive because of the difficulty in defining functionally important noncoding DNA. Here, we combine genome-scale maps of regulatory DNA marked by DNase I hypersensitive sites (DHSs) from 138 cell and tissue types with whole-genome sequences of 53 geographically diverse individuals in order to better delimit the patterns of regulatory variation in humans. We estimate that individuals likely harbor many more functionally important variants in regulatory DNA compared with protein-coding regions, although they are likely to have, on average, smaller effect sizes. Moreover, we demonstrate that there is significant heterogeneity in the level of functional constraint in regulatory DNA among different cell types. We also find marked variability in functional constraint among transcription factor motifs in regulatory DNA, with sequence motifs for major developmental regulators, such as HOX proteins, exhibiting levels of constraint comparable to protein-coding regions. Finally, we perform a genome-wide scan of recent positive selection and identify hundreds of novel substrates of adaptive regulatory evolution that are enriched for biologically interesting pathways such as melanogenesis and adipocytokine signaling. These data and results provide new insights into patterns of regulatory variation in individuals and populations and demonstrate that a large proportion of functionally important variation lies beyond the exome.
Protein-coding DNA constitutes ∼1.5% of the human genome, but ∼2.5%–15% is estimated to be functionally constrained (Mouse Genome Sequencing Consortium 2002; Lunter et al. 2006; Asthana et al. 2007; Meader et al. 2010; Ponting and Hardison 2011). Thus, a significant amount of functionally important DNA is located in noncoding regions, and genetic variation in such regions likely makes a significant contribution to phenotypic variation and disease susceptibility among individuals. For example, regulatory variation has been linked to the susceptibility of a wide variety of human diseases, including infectious, autoimmune, psychiatric, neoplastic, and neurodegenerative disorders (for review, see Skelly et al. 2009). In addition, regulatory variation is an important substrate for evolutionary change within and between species (King and Wilson 1975), and a number of examples in humans have been described of positive selection that are due to adaptive evolution of noncoding DNA (Bamshad et al. 2002; Hamblin et al. 2002; Bersaglieri et al. 2004; Tishkoff et al. 2007).
To date, the identification and interpretation of regulatory variation has been challenging because of the difficulty in accurately localizing functional noncoding elements that regulate transcription. Computational approaches for large-scale delineation of regulatory DNA have generally been disappointing. For instance, although sophisticated methods have been developed to identify cis-regulatory motifs, such as transcription factor binding sites and 3′ UTR elements (Hughes et al. 2000; Stormo 2000; Vavouri and Elgar 2005; Xie et al. 2005), it is often unclear how many of the predicted sites are functional. Furthermore, evolutionary-based methods (Siepel et al. 2005; Pollard et al. 2010) are a powerful approach for identifying conserved noncoding DNA that is likely to be functionally important, but only a fraction of such sites encode experimentally verifiable transcriptional control elements, while other data suggest that a large fraction of binding sites for specific regulatory factors is not constrained between species, in part due to lineage-specific use of regulatory elements (Dermitzakis and Clark 2002; The ENCODE Project Consortium 2007, 2012; Blow et al. 2010; Schmidt et al. 2010). Consequently, between-species conservation-based methods likely miss many functional elements.
Although computational and evolutionary-based methods play a critical role in understanding the biology of genomes and interpreting the consequences of putative regulatory variation, experimental methods are the most direct approach for assessing the functional significance of noncoding variation. To this end, large-scale experimental studies of noncoding DNA, harnessing new technologies, such as the ENCODE Project (The ENCODE Project Consortium 2007, 2012) are providing a detailed roadmap to the locations of regulatory DNA in the human genome.
A generic structural feature of animal regulatory DNA is extreme accessibility to nucleases in the context of intact nuclei (Gross and Garrard 1988), and hypersensitivity to the nonspecific endonuclease DNase I has been used for over 30 yr as a probe for regulatory DNA (Galas and Schmitz 1978). The binding of sequence-specific transcriptional regulators in place of canonical nucleosomes creates DNase I hypersensitive sites (DHSs). Nucleotide resolution analysis of DNase I cleavage patterns allows identification of the “footprints” of DNA-bound regulators (Galas and Schmitz 1978). In contrast to ChIP-chip and ChIP-seq, which probe the locations of regulatory sequences for a specific transcription factor, the nonspecificity of DNase I is a powerful feature that allows all DNA–protein interactions to be queried in a single experiment. Large-scale localization of in vivo DNase I cleavages using deep sequencing (Hesselberth et al. 2009) has enabled the creation of genome-scale maps of diverse functional noncoding elements marked by DHSs. For example, in the ENCODE Project, ∼3 million DHSs have now been mapped across 138 cell types (Thurman et al. 2012). In addition, genomic DNase I footprinting of 41 cell diverse cell types has resulted in the localization of 8.4 million DNase I footprints (Neph et al. 2012b).
Here, we describe a comprehensive analysis into patterns of genetic variation in regulatory DNA marked by DHSs and DNase I footprints (The ENCODE Project Consortium 2012). By analyzing whole-genome sequence data, we are able to directly compare characteristics of regulatory and protein-coding variation and find that individuals harbor considerably more regulatory compared to protein-coding variants. Moreover, we demonstrate that significant heterogeneity of functional constraint exists across regulatory DNA between cell types and that regulatory DNA present in multiple broad categories of cell types is significantly more constrained. Finally, we quantify patterns of population structure in regulatory DNA and identify several hundred loci that contain signatures of local adaptation. In summary, these analyses represent the most comprehensive assessment of human regulatory variation described to date and have important implications for personal genomics, disease mapping studies, and human evolution.
Results and Discussion
Overview of DNase I and whole-genome sequence data
A schematic illustration of the classes of data used in our analyses is shown in Figure 1A. Within DHSs, DNase I “peaks” correspond to ∼150-bp regions of maximum hypersensitivity (Fig. 1A; see The ENCODE Project Consortium 2012). Embedded within peaks, are much smaller 6- to 20-bp DNase I footprints, which identify regions bound by regulatory factors (Fig. 1A). We also obtained publicly available whole-genome sequence data for 53 unrelated individuals that encompass five geographically diverse populations (Fig. 1B) from Complete Genomics. The average sequencing depth per individual was ∼40×. Variants were filtered for deviations from Hardy-Weinberg equilibrium, and partial genotype calls were set to missing data (see Methods). The high-coverage whole-genome sequence data are ideal for population genetics analyses as they are free from the confounding affects of ascertainment bias present in genotypes obtained from SNP chips (Tennessen et al. 2011).
Figure 1.

Overview of data used in the analyses. (A) Schematic of the DNase I data. Binding of regulatory proteins to DNA (blue rectangle) results in nucleosome (open circles) displacement and local chromatin remodeling, and these regions are susceptible to cleavage with the endonuclease DNase I. High-throughput sequencing of libraries made from digested nuclei reveals DNase I hypersensitive sites, detectable by increased depth of coverage. Peaks are defined as 150-bp windows centered on the area of maximum cleavage (The ENCODE Project Consortium 2012). Within hypersensitive sites, footprints of regulatory factor binding are observed as decreased cleavage. (B) Unrooted neighbor-joining tree of the 53 unrelated individuals colored by population. Abbreviations are described in Supplemental Table 2.
Pervasive regulatory variation across the human genome
Across all cell types, over 2.9 million DNase I peaks and 8.4 million DNase I footprints were identified across sampled cell types that collectively span 577 Mb and 156 Mb of sequence, respectively (18.7% and 5.1% of the genome for peaks and footprints, respectively). By use of the whole-genome sequence data, we observed 3.85 million, 1.01 million, and 0.15 million variants in DNase I peaks, DNase I footprints, and the exome, respectively. The large number of variants in peaks and footprints relative to exomes is a function of the total amount of sequence they encompass. For example, the number of variants per kilobase in peaks, footprints, and the exome is 6.7, 6.5, and 4.2, respectively.
To compare the number of putatively functional variants across peaks, footprints, and the exome we obtained GERP scores for each variant, which is a measure of evolutionary constraint with positive values indicating greater conservation (Cooper et al. 2005). Peaks and footprints not only have an overall larger number of variants relative to exomes but also manifest more high GERP variants compared with protein-coding regions (Fig. 2A), although the differences between categories becomes less dramatic. For example, at a threshold of GERP ≥ 3 (Cooper et al. 2005) 146,570, 61,933, and 36,935 variants are observed in peaks, footprints, and the exome, respectively. It is interesting to note that protein-coding DNA contains proportionally more putatively functional variation compared with noncoding DNA (i.e., 24.6%, 6.1%, and 3.8% of variants have a GERP ≥ 3 for exomes, footprints, and peaks, respectively), but the absolute number of functional variants in noncoding regions is larger because of the greater amount of genomic sequence they encompass. Thus, regulatory variation is pervasive across the human genome, and a substantial proportion of functional variation exists in noncoding DNA.
Figure 2.

Characteristics of regulatory variation among individuals. (A) Total number of variants in DNase I peaks, footprints, and the exome stratified by GERP score. (B) Distribution of the number of variants per individual in DNase I peaks, footprints, and the exome. (C) Distribution of the number of variants per individual with GERP ≥ 3 in DNase I peaks, footprints, and exomes.
Next, we investigated the distribution of putative regulatory and protein-coding variation across individuals. As expected, the average number of variants (±SD) per individual in peaks and footprints is dramatically higher than that found in the exome (741k ± 72k in peaks, 192k ± 18k in footprints, and 24.4k ± 2.2k in the exome) (Fig. 2B). A more interesting comparison, however, is the number of putatively functional regulatory and protein variants per individual. Therefore, we also determined the number of variants per individual with a GERP ≥ 3 in peaks, footprints, and the exome (Fig. 2C). On average, individuals contain 24.2k ± 2.3k, 10.1k ± 0.92k, and 4.7k ± 0.40k high GERP variants in peaks, footprints, and the exome, respectively (Fig. 2C). Although evolutionary constraint is not a perfect proxy for function, these results suggest that individuals possess more regulatory versus protein-coding variants. Assuming the probability that a variant is functional is the same between coding and noncoding DNA for a given GERP value, we estimate that individuals contain up to five times as many regulatory compared with protein-coding variants. This assumption, however, is dubious (McVicker et al. 2009), and more definitive inferences on the proportion of functional variants in noncoding versus coding DNA will ultimately require further experimental data. In addition, it is interesting to note that, as expected, the average number of variants per individual in peaks and footprints is significantly higher for individuals of African ancestry compared to non-Africans (859k vs. 710k in Africans and non-Africans, respectively; P < 9.95 × 10−10) (Fig. 2B).
Patterns of nucleotide diversity in regulatory DNA sequence motifs
The unique scope of the data sets analyzed here allows us for the first time to systematically investigate genomic patterns of variation in DNA sequence motifs. To this end, we scanned DNase I footprints for 732 known motifs (see Methods), and for each motif, we calculated nucleotide diversity, π, averaged across all instances of the motif in these regions. To facilitate interpretation of motif diversity, we also calculated π for fourfold synonymous sites, a proxy for neutrally evolving DNA, and protein-coding sequences. As shown in Figure 3A, average diversity varies by over sevenfold across known regulatory motifs, ranging from 2.67 × 10−4 to 2.0 × 10−3. Approximately 60% of motifs have average diversities significantly lower than fourfold synonymous sites (Fig. 3A), indicative of purifying selection.
Figure 3.

Significant variation of diversity between 732 cis-regulatory motifs. (A) For each motif, average diversity is plotted as a black circle, and 95% confidence intervals obtained by bootstrapping are shown as gray lines. The light blue and yellow rectangles denote the 95% confidence intervals of diversity in fourfold synonymous sites (FFSs) and the exome, respectively. (Red vertical lines) Motifs that belong to the indicated class of transcription factor. (Black vertical lines) Motifs where at least 50% of all instances of that motif contain a CpG dinucleotide. (B) Normalized diversity in motifs versus non-normalized diversity. Motifs with a CpG (defined as above) are plotted in red. (Dashed line) Best fit for non-CpG motifs (r = 0.70, P < 10−16).
Figure 3A also highlights motif diversity for several important classes of transcriptional regulators. For example, HOX-, POU-, and FOX-domain factors are heavily enriched in developmental regulators and controllers of cellular differentiation. Motifs for transcription factors belonging to these classes are markedly shifted toward lower diversity, and motifs for several individual factors exhibit levels of diversity that are reduced beyond that of protein-coding sequences (Fig. 3A). In contrast, diversity in motifs for tandem zinc finger transcription factors, which comprise the largest and most diverse class of human transcription factors, is distributed relatively evenly across the diversity spectrum (Fig. 3A). Members of this group include core regulatory factors such as CTCF and YY1, developmental regulators such as PRDM1 and ZIC3, and numerous chromatin repressors such as RREB1, REST, and the KRAB-ZNF family of proteins. Because many of the canonical motifs for these factors contain one or more CG dinucleotides, we hypothesized that the increased average diversity for these factors might be a consequence of higher mutation rates at CpG sites. To explore this hypothesis, we identified factors for which >50% of the motif instances in regulatory DNA contained CpGs, which revealed that the ubiquitous presence of CpG sites is a common characteristic of motifs with high levels of diversity (Fig. 3A).
To more systematically control for mutation rate heterogeneity, we also calculated π normalized for divergence (see Methods) for each motif. As shown in Figure 3B, normalized diversity has the most dramatic effects on motifs that contain CpGs, highlighting the potentially large contribution that mutation rate has on observed levels of π. The effect of normalization on non-CpG motifs is more modest, and normalized and unnormalized diversity levels among these motifs are strongly correlated (r = 0.70; P < 10−16). Nonetheless, these data demonstrate that heterogeneity in both selective constraint and mutation rate likely contribute to the differences in diversity observed among motifs. In the following, we will focus on normalized π to mitigate variation in mutation rate among sequences being compared.
Heterogeneity of functional constraint across cell types
We next tested the hypothesis that levels of functional constraint acting on regulatory DNA varied across cell types. To this end, we calculated normalized π averaged across all DNase I peaks for each of the 138 cell lines. We found marked differences in normalized diversity between cell lines (P < 10−4) (Fig. 4A), which ranged from a low of 5.52 × 10−4 in primary hepatocytes to a high of 6.15 × 10−4 in the immortalized B-lymphoblastoid cell line GM12864. The majority of cell types exhibited average levels of normalized diversity that are within the range of fourfold degenerate sites (Fig. 4A). Note, as we are averaging over many megabases of sequence in each cell type, this does not mean that specific sites, such as motifs embedded within peaks, are evolving neutrally. Six cell types (retinal pigment epithelial, neuroblastoma, primary liver, skeletal muscle myoblast, umbilical vein endothelial, and prostate adenocarcinoma cells, corresponding to cell lines HRPEpiC, SK-N-SH, Hepatocyte, Hsmm, Huvec, and LNCaP, respectively) exhibited average levels of normalized diversity that are significantly lower (ranging from P = 0.024 to P < 10−4) than fourfold degenerate sites, indicative of stronger functional constraint.
Figure 4.

Heterogeneity of polymorphism across cell types. (A) Distribution of normalized nucleotide diversity (black points) across DNase I peaks in 138 cell types. Vertical bars around peaks indicate 95% confidence intervals obtained by bootstrapping. (Blue rectangle) 95% confidence interval for normalized nucleotide diversity in fourfold degenerate sites. (B) Venn diagram showing the amount of shared and unique sequence for DNase I peaks among normal/primary, malignant, and iPS/ES cell types. The barplot on the left shows average normalized diversity for several categories of peaks in the Venn diagram. Shared all and shared two denote peaks shared among all three categories and between any two categories, respectively. N, M, and SC denotes peaks specific to normal/primary, malignant, and stem cell (iPS/ES) cell types, respectively.
It is important to note that the magnitude of reduced diversity in these six cell types is much less than that observed for protein-coding genes. Specifically, normalized diversity across the exome is 4.04 × 10−4, a reduction of 31.2% compared with fourfold degenerate sites. The stronger signature of purifying selection on exomic sequence relative to regulatory regions defined by DNase I hypersensitivity is likely attributable to both the higher proportion of functionally important variants in protein-coding versus noncoding DNA and that, on average, mutations in exonic sequences are more deleterious than mutations in regulatory regions. Indeed, numerous studies have found that regulatory mutations tend to be mildly deleterious (Asthana et al. 2007; Chen et al. 2007; Ronald and Akey 2007).
We next investigated differences in normalized diversity between “core” DHS and DHS found in only one category of cell types. To this end, all of the cell types can be grouped into one of three categories: normal/primary, iPS/ES, and malignant. To minimize potential contributions from experimental noise, we focused on a subset of 92 cell types with high-quality DNase I data in which >40% all sequence tags map within DHSs (equivalent to average signal-to-noise of ∼100:1) (Thurman et al. 2012) and calculated normalized π in DNase I peaks that are shared and unique to each cell type category (Fig. 4B). Eight percent of peaks are found in all three categories, whereas 6.4%, 31.1%, and 28.2% of peaks are unique to iPS/ES, malignant, and normal/primary cell types, respectively (Fig. 4B). Overall, there is significant variation (P < 10−4) in normalized diversity among peaks shared between cell type categories versus those found in a single category (Fig. 4B). In particular, DNase I peaks shared by two or three categories of cell types exhibit the lowest levels of normalized diversity (Fig. 4B), consistent with stronger selective constraint. Conversely, peaks found in only one cell category have significantly higher normalized diversity than shared peaks, (Fig. 4B). These results suggest that the “core” set of DHSs, present in more than one cell type category, is subject to stronger purifying selection because they are necessary for proper transcriptional programs in multiple cell types.
Evidence for ectopic activation of DHSs in malignant cell types
Many cancers are characterized by disruptions in chromatin maintenance pathways (Wang et al. 2007; Morin et al. 2010; Jiao et al. 2011). Additionally, many immortalized cells express different complements or ratios of transcriptional factors than are found in normal differentiated cells (Zaidi et al. 2007). These observations suggest that immortalized and malignant cell lines may experience increased “ectopic” activation of DHSs. To explore the potential ectopic activation of DHSs in malignant and immortalized cell types further, we calculated the proportion of DNase I peaks that are present in only one cell line, as noncanonical chromatin remodeling would be expected to result in an excess of cell type–restricted DHSs. Again, we used the same 92 cell types as described above.
We found that 54% of peaks specific to normal/primary cells are present in a single cell type. In contrast, 81% of malignant-specific peaks and 86% of iPS/ES-specific peaks are present in a single cell line. However, these percentages are not directly comparable, because of sample size differences between categories (n = 58, 29, five for normal/primary, malignant, and iPS/ES, respectively). When we correct for the number of cell types per category (see Methods), we find that iPS/ES cells are not enriched for singleton DHSs compared to normal/primary cells (P = 0.236), whereas malignant cell types are significantly enriched (P < 10−4) for singleton DHSs compared to normal/primary cells (Fig. 5).These data raise the intriguing possibility that the DHSs found in malignant cells, though not increased significantly in number (data not shown), are enriched in elements resulting from ectopic cooperative transcription factor binding within neutrally evolving sequences.
Figure 5.

Malignant cell lines exhibit significantly more singleton DNase I peaks than normal cell lines. (Triangles) Observed proportion of singleton peaks. (Blue and green lines) Distribution (density histograms) of singleton peaks when randomly sampling 29 (blue) or five (green) cell types; this is the distribution of the number of singleton peaks we would expect if malignant or stem cells were similar to normal cells, respectively. Note the malignant category (blue) shows significantly more singleton peaks than expected given its sample size, but the stem cell category (green) falls within the expected range.
Signatures of positive selection
A large number of genome-wide scans for recent positive selection have been performed in humans (for review, see Akey 2009). Typically, these studies focus only on patterns of DNA sequence variation and are not informed by functional genomics data, although genome-wide analyses have been pursued on computationally predicted motifs. The large compendium of experimentally characterized regulatory regions provides a unique data set to interrogate for signatures of recent positive selection.
To this end, we performed a population genomics analysis to identify DNase I peaks that contain variants with large allele frequency differences between populations relative to the genome-at-large, which is a signature of geographically restricted selection (Akey et al. 2002; Akey et al. 2004). Specifically, we calculated locus-specific branch lengths (LSBLs) (Shriver et al. 2004) for variants in DNase I peaks in Africans, Asians, and Europeans. LSBL is a function of pairwise FST between populations (see Methods) and helps isolate the direction of allele frequency change (Shriver et al. 2004). To reduce the stochasticity inherent in summary statistics of population differentiation, we averaged LSBL across all variants in a peak. We excluded X-linked variants from our analysis due to its different effective population size.
The genome-wide distributions of population structure in DNase I peaks in the African, Asian, and European populations are shown in Figure 6A. We pursued two distinct approaches to interpret these data. First, to obtain general insights into the characteristics of DNase I peaks that exhibit large allele frequency differences between populations, we focused on peaks in the 1% tail of the empirical distribution of LSBLs in each population (Fig. 6A). Next, we identified all genes within 50 kb of these peaks (n = 3372, 3224, and 3099 such genes in Africans, Asians, and Europeans, respectively) and tested for enrichment of KEGG pathways. As shown in Table 1, this set of genes is significantly enriched for 15 KEGG pathways, seven of which are shared between two or more populations (including pathways related to cancer, axon guidance, and WNT signaling). Interestingly, the most significantly enriched pathway in Europeans is melanogenesis (Table 1), suggesting that in addition to protein-coding variants (Lamason et al. 2005), regulatory polymorphisms influencing pigmentation phenotypes have also been a target of recent positive selection. Moreover, our African sample is significantly enriched for chemokine and adipocytokine signaling pathways (Table 1), which is particularly interesting given the known differences in prevalence of insulin resistance and type 2 diabetes in individuals of African ancestry (Reimann et al. 2007).
Figure 6.

Genome-wide distribution of population structure in regulatory DNA. (A) Genome-wide distribution of locus-specific branch lengths (LSBLs) for Africans, Asians, and Europeans, respectively. Note that the valley of uniform LSBL on chromosome 17 in Europeans corresponds to the MAPT region that is segregating a large chromosomal inversion (Zody et al. 2008). (B) Distribution of the proportion of highly differentiated DNase I peaks found for different categories of cell types. (SC) Stem cells (iPS/ES); (I) immortalized; (M) malignant; (N) normal/primary cell types. (C) Distribution of African LSBL across intron 1 of VDR. (D) Distribution of European LSBL across intron 4 of FTO. In panels C and D, peaks are shown as red rectangles and exons as black rectangles.
Table 1.
Enriched KEGG pathways for genes within 50 kb of highly differentiated DNase I peaks

We also investigated the distribution of DNase I peaks that exhibit unusually large levels of population structure across cell types. To this end, we classified the 138 types into normal, immortalized, malignant, and pluripotent (iPS/ES) categories. The proportion of DNase I peaks that are in the 1% tail of the empirical distribution of LSBLs is significantly different across cell type categories (Kruskal-Wallis test, P = 3.2 × 10−12). Primary/normal cell lines had the highest proportion of differentiated peaks, whereas iPS/ES cell lines had the lowest proportion of differentiated peaks (Fig. 6B). The higher proportion of differentiated DNase I peaks in primary/normal cell lines is driven by a wide variety of cell types, including astrocytes (spinal cord [HA-sp], cerebellar [HA-c], and cortical [HA-h]), renal glomeral endothelial cells (HRGEC), and cardiac fibroblasts (HCFaa). Although these results are intriguing and offer preliminary insights into the types of tissues that contribute to fitness differences among individuals, more definitive inferences will require an even broader sampling of cell types.
Second, to develop a more refined list of putative targets of recent adaptive evolution, we focused on the most differentiated 1% of DHSs that also contain one or more highly differentiated variants with a GERP ≥ 3. In total, 323, 349, and 313 DHSs meet these criteria in Africans, Asians, and Europeans, respectively. We identified genes located within 50 kb of each of these peaks and identified 187, 174, and 179 genes in Africans, Asians, and Europeans, respectively (Supplemental Text 1). Notably, included in this set of peaks is the well-documented promoter variant in DARC that results in malaria resistance in African populations (Hamblin et al. 2002), which demonstrates the potential power of this data set to fine-scale map signatures of selection and identify selected alleles. Moreover, 61, 40, and 51 of these candidate selection genes in Africans, Asians, and Europeans, respectively, overlap previously reported signatures of selection collected by Akey (2009), which is significantly more than expected by chance (P < 10−6 for all populations). Thus, these observations suggest that the loci identified here are enriched for targets of recent positive selection. Particularly interesting examples of novel targets of selection include the vitamin D receptor (VDR) in Africans and the fat mass and obesity associated gene (FTO) in Europeans (Fig. 6C,D). The list of all candidate selection genes located within 50 kb of highly differentiated peaks is provided in Supplemental Text 1, which provides a powerful framework for more detailed analyses into recent adaptation of noncoding DNA in humans.
Conclusions
By synergistically integrating whole-genome sequences with genome-wide DNase I data, we provide new insights into the distribution and characteristics of human cis-regulatory variation in individuals and populations. Our results demonstrate that regulatory variation is pervasive throughout the genome, on average mildly deleterious, and individuals likely harbor more functionally important variants in noncoding compared with protein-coding DNA. The latter observation is important for disease mapping studies and suggests that a substantial proportion of disease alleles exist beyond the exome (Bamshad et al. 2011). Our results also suggest that ectopic activation of noncanonical cis-regulatory sequences contributes to the aberrant transcriptional changes that are observed in many cancers. Finally, we describe a large compendium of DHSs that exhibit unusually large levels of population structure, consistent with the action of geographically restricted selection. Genes adjacent to these highly differentiated regulatory sequences are enriched for a number of biologically interesting categories, such as signaling and disease related pathways. Although considerably more work is needed to elucidate the evolutionary history of these loci, they provide an important starting point for understanding how recent adaptive evolution has influenced regulatory networks.
Methods
DNase I data
We obtained DNase I peaks, footprints, and predicted motif locations from the ENCODE Project (The ENCODE Project Consortium 2012, http://genome.ucsc.edu/ENCODE/downloads.html). Peaks and footprints were empirically thresholded at a 1% false-discovery rate. For aggregate analyses over DHS across cell types, peak or footprint locations were merged across cell types using BEDOPS (http://code.google.com/p/bedops/) (Neph et al. 2012a). More information about the cell lines can be found in Supplemental Table 1 and http://genome.ucsc.edu/ENCODE/cellTypes.html.
Protein binding motif locations were generated genome-wide with FIMO motif scanning software (Bailey et al. 2009), version 4.6.1, using a P-value threshold of ≤1 × 10−5 threshold. Motif models were obtained from TRANSFAC (Matys et al. 2002), version 2011.1. For these analyses, all motifs were intersected with footprint data using BEDOPS.
Sequence data
We obtained whole-genome sequence data from 69 individuals that were sequenced to high coverage by Complete Genomics (http://www.completegenomics.com/sequence-data/download-data/). Among these 69 individuals, 54 are reported to be unrelated. To verify that these 54 individuals are unrelated, we performed relationship inference with KING (Manichaikul et al. 2010). Two Maasai individuals (NA21732 and NA21737) who were not reported as being related were found to be either siblings or parent–child. We removed NA21737 from further analyses as this individual had more missing data than NA21732. Thus, our final data set consists of 53 individuals from five populations (Supplemental Table 2). Genotype data were filtered to remove partial genotypes (i.e., where one allele is called and the other is reported as missing), by coverage (>20% of individuals must have calls), and by extreme departures from Hardy-Weinberg Equilibrium (P < 10−8, which corresponds to all individuals being heterozygous and therefore most likely a paralogous sequence variant). We defined fourfold degenerate sites using NCBI-called reading frames. We used the NimbleGen SeqCapEZ Exome, version 2.0, definition, downloaded from the NimbleGen website (http://www.nimblegen.com/products/seqcap/ez/v2/). Repeats were defined by RepeatMasker regions, obtained from the UCSC Genome Browser. A dinucleotide is conservatively called as CpG for NpG and CpN dinucleotides (where N = A,C,T, or G) in any of our 53 genomes, chimpanzee, orangutan, or rhesus macaque.
Statistical analyses
We calculated nucleotide diversity as: 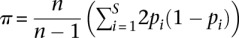 , where n is the number of chromosomes and pi is the frequency of the major allele for the ith segregating site, S. To obtain a per nucleotide estimate of π, we divided by the total number of bases considered for a particular analysis. Normalized diversity was calculated by dividing the per nucleotide π estimate by the estimated neutral mutation rate. For exonic sequence, we used the mutation rate calculated at fourfold synonymous sites, as this sequence is less likely to be influenced by selection compared to all synonymous sites. For DHSs, we expanded each region (peak or motif) by 1500 bp on either side and removed putatively selected sequence (footprints, exome, and peaks padded by 250 bp) from this region. All normalized π values were then multiplied by 2 × 10−8 to bring them into the range of non-normalized π values. Repeats were removed in all analyses. For π in DHS (normalized and non-normalized), exonic sequence was also removed. CpGs were removed in all normalized π calculations as described above.
, where n is the number of chromosomes and pi is the frequency of the major allele for the ith segregating site, S. To obtain a per nucleotide estimate of π, we divided by the total number of bases considered for a particular analysis. Normalized diversity was calculated by dividing the per nucleotide π estimate by the estimated neutral mutation rate. For exonic sequence, we used the mutation rate calculated at fourfold synonymous sites, as this sequence is less likely to be influenced by selection compared to all synonymous sites. For DHSs, we expanded each region (peak or motif) by 1500 bp on either side and removed putatively selected sequence (footprints, exome, and peaks padded by 250 bp) from this region. All normalized π values were then multiplied by 2 × 10−8 to bring them into the range of non-normalized π values. Repeats were removed in all analyses. For π in DHS (normalized and non-normalized), exonic sequence was also removed. CpGs were removed in all normalized π calculations as described above.
To evaluate the number of singleton peaks in malignant and iPS/ES cells relative to normal/primary cell lines, we performed a resampling procedure. Specifically, we randomly selected 1000 samples of five and 29 cell types from the 58 normal cell lines and, for each sample, calculated the proportion of singleton normal-specific DNase I peaks. Singleton peaks must occur in category-specific DHS; therefore, we calculate the percentage of category-specific DHS that is also singleton. This procedure generates an empirical distribution for the proportion of singleton peaks expected in a category with five or 29 cell types.
We calculated LSBLs as previously described (Shriver et al. 2004). In brief, pairwise FST between Africans (n = 16), Europeans (n = 13), and Asians (n = 8) was calculated as 1 − HS/HT, where HS and HT denote average subpopulation heterozygosity and total heterozygosity, respectively (Hartl and Clark 1997). These pairwise estimates of FST were then used to calculate LSBL for each population. For example, denote the FST between Africans and Europeans, Africans and Asians, and Europeans and Asians as dAB, dAC, dAB, respectively. The LSBL for Africans is (dAB + dAC − dBC)/2. LSBLs were calculated for all variants in peaks with 100% coverage (all individuals fully called), excluding the exome and repeats.
To test for enrichment of candidate selection genes in KEGG pathways, we used WebGestalt (Duncan et al. 2010; http://bioinfo.vanderbilt.edu/webgestalt). In these analyses, we used a background list of genes by identifying the closest gene to each DNase I peak, which recapitulates how genes were associated with the highly differentiated peaks. We used the false discovery rate (FDR) method of Benjamini and Hochberg (1995) to address multiple testing.
Acknowledgments
This work was supported in part by research grants from the NIH to J.M.A. (1R01GM076036 and R01GM078105) and J.A.S. (HG004592). We thank Ian Stanaway for help in obtaining estimates of fourfold synonymous sites, Louisa Jauregui for help with figures, and three anonymous reviewers for their comments.
Footnotes
[Supplemental material is available for this article.]
Article and supplemental material are at http://www.genome.org/cgi/doi/10.1101/gr.134890.111.
Freely available online through the Genome Research Open Access option.
References
- Akey JM 2009. Constructing genomic maps of positive selection in humans: Where do we go from here? Genome Res 19: 711–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD 2002. Interrogating a high-density SNP map for signatures of natural selection. Genome Res 12: 1805–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey JM, Eberle MA, Rieder MJ, Carlson CS, Shriver MD, Nickerson DA, Kruglyak L 2004. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol 2: e286 doi: 10.1371/journal.pbio.0020286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthana S, Noble WS, Kryukov G, Grant CE, Sunyaev S, Stamatoyannopoulos JA 2007. Widely distributed noncoding purifying selection in the human genome. Proc Natl Acad Sci 104: 12410–12415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS 2009. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Mummidi S, Gonzalez E, Ahuja SS, Dunn DM, Watkins WS, Wooding SP, Stone AC, Jorde LB, Weiss RB, et al. 2002. A strong signature of balancing selection in the 5′ cis-regulatory region of CCR5. Proc Natl Acad Sci 99: 10539–10544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12: 745–755 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300 [Google Scholar]
- Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg T, Schaffner SF, Drake JA, Rhodes M, Reich DE, Hirschhorn JN 2004. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet 74: 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2010. ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet 42: 806–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CTL, Wang JC, Cohen BA 2007. The strength of selection on ultraconserved elements in the human genome. Am J Hum Genet 80: 692–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, NISC Comparative Sequencing Program, Green ED, Batzoglou S, Sidow A 2005. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 15: 901–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET, Clark AG 2002. Evolution of transcription factor binding sites in Mammalian gene regulatory regions: Conservation and turnover. Mol Biol Evol 19: 1114–1121 [DOI] [PubMed] [Google Scholar]
- Duncan DT, Prodduturi N, Zhang B 2010. WebGestalt2: An updated and expanded version of the Web-based Gene Set Analysis Toolkit. BMC Bioinformatics (Suppl 4) 11: 1020053291 [Google Scholar]
- The ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2012. An integrated encyclopedia of DNA elements in the human genome. Nature (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galas DJ, Schmitz A 1978. DNaseI footprinting: A simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res 5: 3157–3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DS, Garrard WT 1988. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem 57: 159–197 [DOI] [PubMed] [Google Scholar]
- Hamblin MT, Thompson EE, Di Rienzo A 2002. Complex signatures of natural selection at the Duffy blood group locus. Am J Hum Genet 70: 369–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Clark AG 1997. Principles of population genetics, 3rd ed. Sinauer Associates, Sunderland, Massachusetts [Google Scholar]
- Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. 2009. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods 6: 283–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JD, Estep PW, Tavazoie S, Church GM 2000. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol 296: 1205–1214 [DOI] [PubMed] [Google Scholar]
- Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, et al. 2011. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331: 1199–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, Wilson AC 1975. Evolution at two levels in humans and chimpanzees. Science 188: 107–116 [DOI] [PubMed] [Google Scholar]
- Lamason RL, Mohideen MA, Mest JR, Wong AC, Norton HL, Aros MC, Jurynec MJ, Mao X, Humphreville VR, Humbert JE, et al. 2005. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 310: 1782–1786 [DOI] [PubMed] [Google Scholar]
- Lunter G, Ponting CP, Hein J 2006. Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput Biol 2: e5 doi: 10.1371/journal.pcbi.0020005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM 2010. Robust relationship inference in genome-wide association studies. Bioinformatics 26: 2867–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gössling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. 2002. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31: 374–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker G, Gordon D, Davis C, Green P 2009. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet 5: e1000471 doi: 10.1371/journal.pgen.1000471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meader S, Ponting CP, Lunter G 2010. Massive turnover of functional sequence in human and other mammalian genomes. Genome Res 20: 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. 2010. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42: 181–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S, et al. 2012a. BEDOPS: High performance genomic feature operations. Bioinformatics doi: 10.1093/bioinformatics/bts277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, Sandstrom R, Johnson AK, Maurano MT, et al. 2012b. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard K, Hubisz M, Rosenbloom K, Siepel A 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20: 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP, Hardison RC 2011. What fraction of the human genome is functional? Genome Res 21: 1769–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann M, Schutte AE, Schwarz PE 2007. Insulin resistance–the role of ethnicity: Evidence from Caucasian and African cohorts. Horm Metab Res 39: 853–857 [DOI] [PubMed] [Google Scholar]
- Ronald J, Akey JM 2007. The evolution of gene expression QTL in Saccharomyces cerevisiae. PLoS ONE 2: e678 doi: 10.1371/journal.pone.0000678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, et al. 2010. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328: 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver MD, Kennedy GC, Parra EJ, Lawson HA, Sonpar V, Huang J, Akey JM, Jones KW 2004. The genomic distribution of population substructure in four populations using 8,525 autosomal SNPs. Hum Genomics 1: 274–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 8: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly DA, Ronald J, Akey JM 2009. Inherited variation in gene expression. Annu Rev Genomics Hum Genet 10: 313–332 [DOI] [PubMed] [Google Scholar]
- Stormo GD 2000. DNA binding sites: Representation and discovery. Bioinformatics 16: 16–23 [DOI] [PubMed] [Google Scholar]
- Tennessen JA, O'Connor TD, Bamshad MJ, Akey JM 2011. The promise and limitations of population exomics for human evolution studies. Genome Biol 12: 127 doi: 10.1186/gb-2011-12-9-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, Silverman JS, Powell K, Mortensen HM, Hirbo JB, Osman M, et al. 2007. Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet 39: 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavouri T, Elgar G 2005. Prediction of cis-regulatory elements using binding site matrices - the successes, the failures and the reasons for both. Curr Opin Genet Dev 15: 395–402 [DOI] [PubMed] [Google Scholar]
- Wang GG, Allis CD, Chi P 2007. Chromatin remodeling and cancer, part I: Covalent histone modifications. Trends Mol Med 9: 363–372 [DOI] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M 2005. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434: 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SK, Young DW, Javed A, Pratap J, Montecino M, van Wijnen A, Lian JB, Stein JL, Stein GS 2007. Nuclear microenvironments in biological control and cancer. Nat Rev Cancer 7: 454–463 [DOI] [PubMed] [Google Scholar]
- Zody MC, Jiang Z, Fung HC, Antonacci F, Hillier LW, Cardone MF, Graves TA, Kidd JM, Cheng Z, Abouelleil A, et al. 2008. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet 40: 1076–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]