

Abstract
Gene regulatory programs in distinct cell types are maintained in large part through the cell-type–specific binding of transcription factors (TFs). The determinants of TF binding include direct DNA sequence preferences, DNA sequence preferences of cofactors, and the local cell-dependent chromatin context. To explore the contribution of DNA sequence signal, histone modifications, and DNase accessibility to cell-type–specific binding, we analyzed 286 ChIP-seq experiments performed by the ENCODE Consortium. This analysis included experiments for 67 transcriptional regulators, 15 of which were profiled in both the GM12878 (lymphoblastoid) and K562 (erythroleukemic) human hematopoietic cell lines. To model TF-bound regions, we trained support vector machines (SVMs) that use flexible k-mer patterns to capture DNA sequence signals more accurately than traditional motif approaches. In addition, we trained SVM spatial chromatin signatures to model local histone modifications and DNase accessibility, obtaining significantly more accurate TF occupancy predictions than simpler approaches. Consistent with previous studies, we find that DNase accessibility can explain cell-line–specific binding for many factors. However, we also find that of the 10 factors with prominent cell-type–specific binding patterns, four display distinct cell-type–specific DNA sequence preferences according to our models. Moreover, for two factors we identify cell-specific binding sites that are accessible in both cell types but bound only in one. For these sites, cell-type–specific sequence models, rather than DNase accessibility, are better able to explain differential binding. Our results suggest that using a single motif for each TF and filtering for chromatin accessible loci is not always sufficient to accurately account for cell-type–specific binding profiles.
Multicellular organisms require mechanisms for maintenance of cell-type–specific gene expression programs. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) now enables genome-wide localization of transcription factors (TFs) and other regulators that orchestrate these programs. However, a key limitation is that each ChIP-seq assay only captures the binding profile in a single cell type. Therefore, accurately modeling the DNA sequence preferences of TFs and predicting their genomic binding sites continue to be key problems in regulatory genomics. The ENCODE Consortium and other groups have undertaken large-scale efforts to profile the genome-wide binding sites of numerous TFs across multiple human cell lines using ChIP-seq (The ENCODE Project Consortium 2011). The ENCODE project also provides genome-wide data on chromatin state in many of the same cell lines, including ChIP-seq profiling of histone modifications and DNase-seq assays to profile chromatin accessibility (Ernst et al. 2011; Thurman et al. 2012). We use this wealth of data to systematically explore the determinants of differential TF binding across cell types and further elucidate the underlying TF recognition code.
Cell-type–specific usage of regulatory elements is frequently associated with one or more chromatin alterations. These include histone modifications (Barrera et al. 2008; Heintzman et al. 2009), DNA methylation status (Deaton et al. 2011; Wiench et al. 2011), and accessibility of regulatory elements as measured by DNase sensitivity (Boyle et al. 2011; Thomas et al. 2011). Additionally, chromatin conformation and DNA looping can also influence TF occupancy in a cell-type–specific manner (Gheldof et al. 2010). The question of whether TFs have cell-type–specific DNA binding site sequences has been less systematically studied. Recent analyses of TF ChIP-seq profiles across multiple cell types have typically used motif discovery approaches, searching for proximal cofactors that may establish a favorable chromatin context or act as recruitment factors (e.g., Heinz et al. 2010). These motifs are usually represented as single position-specific scoring matrices (PSSMs) and discovered through enrichment analysis using tools such as MEME (Bailey and Elkan 1994). Indeed, most computational methods for learning sequence preferences focus on finding motifs one cell type at a time and modeling regulatory sequences as cis-regulatory modules composed of multiple PSSMs (Bailey and Noble 2003; Zhou and Wong 2004; Sinha et al. 2006, 2008).
Recent work for predicting in vivo binding of a TF in a given cell type has combined information on the chromatin state with DNA binding motif scanning or discovery. In particular, chromatin marks (Heintzman et al. 2007; Whitington et al. 2009) and DNase-hypersensitive regions (Hesselberth et al. 2009) have been used as filters for PSSM motif hits to predict cell-type–specific gene expression. Several integrative methods have combined PSSMs and chromatin data in probabilistic models (Ernst et al. 2010; Won et al. 2010; Pique-Regi et al. 2011) or identified chromatin states representative of promoters and enhancers that are enriched for known TF binding sites (Ernst and Kellis 2010).
We present a novel discriminative framework for learning DNA sequence and chromatin signals that predict cell-type–specific TF binding. First we investigated the rules governing TF binding irrespective of cell type. While PSSMs are compact and interpretable, they can underfit ChIP-seq data by failing to capture subtle but detectable sequence signals, including direct DNA-binding preferences of the TF, cofactor binding sequences, accessibility signals, and other discriminative sequence features. Furthermore, there has been limited work on extracting more general motif models, such as the use of boosting with PSSMs (Hong et al. 2005) or feature motif models (Sharon et al. 2008). Therefore, we used discriminative sequence models based on support vector machines (SVMs) and flexible k-mer patterns. This approach allowed us to better leverage ChIP-seq data to learn in vivo DNA sequence features that more accurately discriminate between TF ChIP-seq peaks and nonpeaks than traditional motif discovery methods that find enriched PSSMs or k-mers. We also explored what chromatin state information is most predictive of TF binding by training discriminative spatial chromatin models using histone modification ChIP-seq or DNase-seq data (Fig. 1A). We found that spatial SVM DNase models are more accurate than standard methods based on combining chromatin marks or DNase read counts for identifying TF-occupied regions. Finally, we found that a simple combination of sequence and chromatin models strongly improved accuracy over using either model alone and, for most TFs, enabled “transfer learning” of a binding model trained in one cell type to make accurate predictions in a second cell type.
Figure 1.

Schematic of models to predict transcription factor occupancy from sequence and chromatin. (A) We developed DNA sequence and chromatin models based on flexible k-mer patterns and spatial organization of histone modifications and DNase accessibility. The models were trained to discriminate between regulatory ChIP-seq peaks and flanking regions within a single cell type using a support vector machine. (B) To study cell-type–specific DNA sequence preferences, we simultaneously train on binding site data from two cell types. This allowed us to jointly learn the cell-type–specific preferences (top and bottom).
We next used our framework to explore the determinants of cell-type–specific binding. Many TFs bound mostly similar genomic locations in the two hematopoietic cell lines; however, a handful bound dramatically different loci. We investigated the extent to which differentially bound sites could be explained by differences in chromatin context and/or sequence signal. We were able to maximize sensitivity to differential sequence signal using a multitask learning framework to simultaneously learn from both cell types (Fig. 1B). Intriguingly, JUND and YY1 both had cell-specific sequence models that were markedly different and that better explained differential binding than did DNase accessibility. In both cases, we found evidence that differential composition of the TF binding complex may explain the cell-type–specific sequence signal. For example, in the case of JUND, our analysis points to differential composition of the AP-1 heterodimeric complex.
Our results suggest a more complicated set of determinants of cell-type–specific binding than is currently implemented in analyses of high-throughput binding data. In particular, for some TFs, accurately predicting cell-type–specific binding cannot be achieved using a single motif coupled with a filter for DNase accessible loci. Rather, more flexible sequence models that can capture subtle TF sequence signals are required, and cell-dependent sequence preferences may be important for explaining cell-type–specific binding rather than chromatin accessibility alone.
Results
Discriminative sequence models better predict TF binding sites than motif discovery methods
TF binding sites typically contain factor-specific DNA sequences that enable binding. While some factors have a small set of canonical DNA sequences they typically bind, many transcriptional regulators can bind a broader range of DNA sequences. Moreover, TF-occupied regions may contain other discriminative DNA features, including cofactor sequences and subtle sequence signals for chromatin accessibility, that can be used to predict binding. We learned TF binding site sequence models from 238 TF ChIP-seq experiments generated by ENCODE, comprising 67 transcriptional regulators across two hematopoietic cell lines (GM12878, a lymphoblastoid cell line produced from blood of a HapMap donor by EBV transformation, and K562, an immortalized cell line generated from a patient with CML in blast crisis) and HeLa cells, with at least two replicates for all of the experiments (Supplemental Table S1). For each ChIP-seq experiment, we identified the top 1000 most significant peaks (see Methods) and took DNA regions of length 100 bp centered at these peaks as positive sequence examples and flanking 100-bp regions sampled 200 bp away as negative sequence examples. The proximity of the negative examples generated sequences with a similar background composition as the positive data (see Methods). Positive and negative sequence examples were evenly divided into training and held-out test sets. We trained sequence models on peaks versus flanks within a single ChIP-seq experiment and evaluated accuracy on held-out peaks/flanks from the same experiment.
We modeled TF-occupied DNA regions with string kernel SVMs. Specifically, we used the di-mismatch k-mer kernel, which we recently introduced for modeling in vitro TF-DNA binding preferences (Agius et al. 2010). Briefly, the kernel maps input sequences into a feature space indexed by a set of informative k-mers, where each feature is a weighted count of the number of times the corresponding k-mer occurs in the input sequence with up to m mismatches in the alphabet of dinucleotides (for parameter choices, see Methods) (Agius et al. 2010). To compare to traditional motif-discovery approaches, we also used the training data to estimate motif models using MDscan (Liu et al. 2002), cERMIT (Georgiev et al. 2010), DME (Smith et al. 2005), and Weeder (Pavesi et al. 2004). The first three methods learn PSSMs that can score sequences via a log likelihood ratio compared to a background model. Weeder simply identifies a set of overrepresented k-mers, which we convert to a scoring function by counting occurrences, allowing up to one mismatch (see Methods). We note that our problem is not the typical motif discovery task—where methods are evaluated based on their ability to find an enriched motif similar to a known motif in a database—but rather a prediction task, where each method must discriminate between ChIP-seq defined peaks and nearby nonpeak regions.
The statistical validity of all methods was evaluated by computing the area under the ROC curve (AUC score) on each test set. Figure 2A shows an example of the ROC curve comparing the di-mismatch SVM model against the motif discovery approaches for BCL11A in GM12878, and Figure 2B reports the mean AUC for each TF (across cell lines and replicates) of the SVM and motif methods across all ChIP-seq experiments. The SVM models had much higher accuracy than all the motif discovery approaches. Compared with the most accurate motif method, MDscan, SVM sequence models better capture the underlying sequence content of transcription binding sites for >90% of TFs (P < 1.3 × 10−11 by paired signed rank test), with a mean AUC improvement of 0.07.
Figure 2.

SVM sequence models better predict binding sites than traditional motif approaches. (A) The accuracy of our method is assessed by the area under the ROC curve, which provides a natural trade-off between false positives (x-axis) and sensitivity (y-axis). The ROC curve is shown for discriminating BCL11A ChIP-seq peaks from nonpeaks using four approaches: k-mer SVM, MDscan, cERMIT, and Weeder. (B) The accuracy (AUC) of k-mer SVM models (y-axis) is compared against motif-based algorithms (MDscan, cERMIT, DME, and Weeder; x-axis) for discriminating ChIP-seq peaks from flanking regions. We used training and test sets taken from the same experiment; only accuracy on the test set is shown. Results for transcription factors with multiple ChIP-seq experiments for replicates and cell types were averaged. The SVM models are significantly more accurate than each of the alternative methods (P-values inset and color-coded for each method). (C) The k-mer SVM model is able to learn degenerate motifs. We show the k-mer SVM scores (y-axis) versus the cERMIT motif score (x-axis) for binding sites of BCL11A in GM12878. Example binding sites that are detected by the SVM but receive low scores by the motif are enriched for a more degenerate motif instance, as found by MEME.
To give some intuition about why the SVM sequence models are able to better capture sequence content than single motifs, we compare the scores of the cERMIT PSSM to those of our di-mismatch model for BCL11A on binding sites in the lymphoblastoid cell line (Fig. 2C). We see that the SVM is able to detect true binding site sequences that receive low scores by the learned PSSM. If we select from the SVM-detected binding sites two groups based on high and low PSSM scores and feed these binding site sequences into MEME, we obtain two different versions of the motif, with only the high-PSSM scoring group matching the original cERMIT PSSM. The SVM is implicitly capturing this range of sequence preferences while still accurately discriminating between bound and unbound loci.
Not all of the transcriptional regulators assayed by ChIP-seq and included in our comparison are considered to be sequence-specific TFs. However, we found it interesting that nonetheless all the factors were partially predicted by underlying DNA sequence content, and the SVM models outperformed motif methods for these factors just as they did for classical sequence-specific TFs (Supplemental Fig. S2).We grouped these “nonsequence-specific” transcriptional regulators into those with low accuracy (AUC ≤ 0.6), including RDBP (NELF-E), SMARCB1 (INI1), POLR3, XRCC4, and WRNIP1 (WHIP); fair accuracy (0.6 < AUC < 0.75), including members of the POLR3 initiation complex (BDP1, BRF1, BRF2, SMARCA4 [BRG1]), members of the POLR2 complex (TBP, TAF1, GTF2B), POLR2 itself, and the histone methyltransferase SETDB1; and good accuracy (AUC ≥ 0.75), including EP300 (p300), POLR3A (RPC155), SMARCC1 (BAF155), SMARCC2 (BAF170), and RAD21. These results show that we do not need direct interaction with DNA via a DNA-binding domain to find a sequence signal. Moreover, both for sequence-specific TFs and these nonsequence-specific transcriptional regulators, our analysis suggests that the in vivo binding sequence signal is more of a continuum than a present/absent call.
Discriminative spatial models better capture TF chromatin signatures than read-count methods
We used discriminative training to learn chromatin signatures that predict TF occupied or unoccupied regions. Figure 3A shows the spatial organization of chromatin marks in 5000-bp windows centered at GABPA ChIP-seq peaks. In this figure, each row shows a single binding site, and each column shows the ChIP-seq read information in 100-bp bins from −2500 to 2500 bp relative to the binding sites. As can be seen, many chromatin marks are correlated with the TF binding sites and display different spatial patterns relative to the binding peaks; however, all marks show a depletion at the peak center, corresponding to a nucleosome-depleted region at the location of protein–DNA interaction.
Figure 3.

SVM spatial chromatin models better predict binding sites than simpler models. (A) The distribution of histone marks over 5000-bp windows centered at GABPA ChIP-seq peaks in K562 shows spatial organization of multiple correlated signals. (B) The accuracy of multiple chromatin models suggests that spatial signatures of DNase accessibility better predict binding sites than other methods. The cumulative distributions of prediction accuracy (AUC; x-axis) across a subset of ChIP-seq experiments are shown for multiple chromatin representations. Shown are an SVM model trained on all spatially binned histone marks (blue), which is more accurate than standard ranking based on best single mark read counts (black) or a logistic regression combination of read counts (red); similarly, an SVM model trained on spatially binned DNase-seq reads (brown) better describes binding sites than use of DNase bin counts (purple). Paired signed rank test P-values are shown. (C) Transcription factors that bind the core promoter, proximal to transcript start site, or distal to start site have distinctive spatial patterns of histone modifications. The four plots show spatial coordinates of the learned bin weights arranged along the x-axis, with the values of the weights shown on the y-axis. The bin weightings are averaged across subsets of core, proximal, and distal binding transcription factors. The valleys at the binding site suggest that spatial models are capturing predictive information regarding the differential spacing of nucleosome-depleted regions at core, proximal, and distal binding sites.
We used these multiple spatially binned histone modifications for training chromatin signatures of TF binding. That is, we used the chromatin data from the rows of Figure 3A as feature vectors to train SVM models to discriminate between the chromatin profiles at TF peaks versus nonpeaks. First, we trained a separate chromatin model for each ChIP-seq experiment. We measured the extent to which chromatin alone could predict binding, using the same training and test sets as used for learning the sequence models. We found that combining histone modifications using an SVM model was far more accurate than using read-counts of the best single modification, including H3K4me3 and H3K4me1, or combining modifications through logistic regression (Fig. 3B). Of practical interest, we found that an SVM trained on spatially binned DNase-seq data, used alone, was more accurate than the combination of histone modifications SVM model. Therefore, if the main task is to localize potential binding sites, the single experiment with greatest predictive value is DNase-seq, which is consistent with recent findings (Pique-Regi et al. 2011). We also noted that the single chromatin mark with greatest predictive value, when used as a spatial signature to localize TF binding sites, was H3K27ac.
Some insight into why the spatial information is so valuable can be gained by looking at the SVM chromatin model vectors. When we clustered the SVM model vectors for different TFs using standard hierarchical clustering, we found that the vectors clustered together based on the TF's genome-wide binding locations relative to genes. In particular, we observed three canonical patterns, one for generic TFs that bind the core promoter, one for TFs with ≥25% of peaks in the proximal promoters (within 2 kbp from genes), and one for TFs with distal binding locations (<25% proximal binding sites). Figure 3C shows the mean chromatin SVM w vectors for each of these three classes. These signatures are recognizing the nucleosome-depleted region centered at the binding peak to improve prediction.
Combining sequence and chromatin signatures improves binding site prediction and prediction in new cell lines
We observed that ChIP-seq occupancy for some TFs was well correlated with SVM sequence signal, while the occupancy of other TFs was better characterized by DNase accessibility (Fig. 4A). To quantify the importance of these two signals for predicting TF binding, we compared the accuracy of the DNase SVM and sequence SVM for different TFs (Fig. 4B; Supplemental Table S4). There are a number of outliers that tend to be much better specified by either chromatin accessibility or sequence signal. For instance, REST is known to act as a repressor and bind a long DNA sequence that provides high specificity (Johnson et al. 2007), so it is unsurprising that chromatin accessibility is not an ideal predictive signature. In contrast, factors such as PAX5 and EP300 are much better predicted by DNase than sequence signal, which is likely due to their ability to bind indirectly to enhancers and to highly degenerate sequences (Cobaleda et al. 2007; Visel et al. 2009).
Figure 4.

Combining chromatin and sequence models improves binding site prediction. (A) Binding sites for REST and PAX5 illustrate loci that have a high sequence signal or DNase accessibility, but not both. (B) Learning sequence models in a single cell type reveals that some TFs are better predicted by sequence signals (such as REST), whereas others are better predicted by DNA accessibility (such as EP300 and PAX5). The AUC was determined for each replicate in each cell type and then averaged. (C) When DNase accessibility information is added to k-mer SVM models, the combined model is more predictive of in vivo binding sites. The scatter plot compares the accuracy of a combination of sequence and DNase SVM signatures with that of the sequence model alone. Models were learned from one cell type and then used to predict binding sites in the same cell type (black) or a different cell type (red). Accuracy (AUC) for each TF was averaged across replicates and cell lines (same cell case) or only replicate experiments (transfer learning case). JUND is an outlier, where applying the sequence model across cell lines is significantly worse than applying it in the same cell line. POLR3 is poorly predicted in all settings and is not shown.
Next we asked whether combining sequence and chromatin signatures could significantly improve TF occupancy prediction. We found that a simple sum of normalized prediction scores from the sequence and DNase SVMs was more accurate than either model alone. Figure 4C shows that the combined model better described binding sites than the sequence-only SVM model for all but a handful of TFs when training and testing in the same cell type (black dots, P < 2.0 × 10−15, paired signed rank test). Improvements are also obtained when combining sequence with the histone signatures, although the average improvement is smaller (mean increase in AUC of 0.04 vs. 0.08 for DNase signatures).
We also wanted to transfer TF binding predictions to a new cell type where there is chromatin data but no TF binding data. Figure 4C also shows that using a sequence model and DNase model trained on one cell line gives good generalization to the other cell line—where for predictions, we used chromatin data collected in the new cell type—and improved over using sequence only in almost all cases (red dots, P < 8.3 × 10−3, paired signed rank test; mean AUC improvement of 0.05). For many TFs, within-cell-type accuracy (i.e., train and test sites belong to the same cell type) and the between-cell-types accuracy (i.e., train on binding sites from one cell type and test on the other) is comparable. A notable exception is JUND, where the sequence-only model accuracy was much poorer when trained in one cell line and tested in the other. Even the combined JUND sequence and DNase model showed a small reduction in accuracy for the between-cell-types task compared with the within-cell-type task.
TFs can display strong cell-type–specific binding patterns
We next wanted to better understand and quantify cell-type–specific binding. We first noted that some TFs had high ChIP-seq signal in one cell line, but very little in the other (Fig. 5A). To accomplish a genome-wide similarity measure of a TF's binding profiles across two cell lines, we determined the top 5000 ChIP-seq peaks in each cell line and quantile-normalized the log counts of reads per million aligned (RPM) mapping to these peak regions in each cell line. We then assessed the significance of the observed log read ratios, using an intensity-specific noise model for each TF based on replicate-to-replicate log RPM ratios within each cell type (see Methods). We say that a binding site is cell-type specific if the log RPM ratio between cell types has a significance of P < 0.01 based on the replicate noise model. For simplicity, we include only binding sites that consistently satisfy this P-value threshold for both pairs of GM12878 versus K562 replicate experiments (see Methods).
Figure 5.

Cell-type–specific transcription factor binding is measured by ChIP-seq and correlated with differential gene expression. (A) ChIP-seq of USF1 reveals sites that are bound in both cell lines (left), only GM12878 (middle), or only K562 (right). Units are reads per million aligned (RPM). (B) We find cell-type–specific binding sites by measuring replicate-to-replicate noise and comparing it to cell-to-cell variation. Replicate and cell-specific binding are shown for REST, MAX, and JUND. The top row of scatterplots shows the ChIP-seq read counts [in RPM, scaled by log(x + 1)] for the top 5000 peaks in two replicate experiments in the same cell type (GM12878). The bottom row of scatterplots shows the log ChIP-seq read counts in GM12878 versus K562 for the union of the top 5000 peaks in each cell line. In these plots, each point is a binding site, and the x- and y-axes show the log read counts aligning to the site in the respective replicates (top row) or cell types (bottom row). (C) We find that the most proximal genes near cell-type–specific binding sites are differentially expressed between cell types. The cumulative distribution of log expression level changes are shown. Expression is estimated by RNA-seq in units of reads per thousand nucleotides of transcript per million reads aligned (RPKM).
Figure 5B shows the replicate versus replicate log read count scatterplot within a single cell type (GM12878) for the top 5000 ChIP-seq peaks in this cell type for the TFs REST, MAX, and JUND (top row) and the corresponding scatterplots between cell types (K562 vs. GM12878, bottom row). The top row shows that replicate-to-replicate noise varies considerably for different TFs. Specifically, we see that lower intensity binding is subject to greater variance, suggesting an adaptive noise model. In the bottom row, the boundary of the shaded “funnel” corresponds to the P < 0.01 significance threshold based on replicate-to-replicate noise, and the points outside the funnel are the cell-type–specific binding sites.
In fact, for all three TFs shown in Figure 5B, a large fraction of the top 5000 binding sites across the two cell types display cell-to-cell log read ratios that place them outside the funnel (36.1%, 32.0%, and 31.9% for REST, MAX, and JUND, respectively). However, it is clear that in the case of REST, most of the binding sites with more reads in GM12878 actually have low read counts in both cell lines. In contrast, JUND has a large number of cell-type–specific binding sites that have high read counts in one cell line and low read counts in the other. To reflect this difference, we use the term cell-type exclusive to describe binding sites that are cell-type specific (outside the funnel) but are also not bound, based on a RPM cut-off of 1, in the other cell type. By this measure, JUND has a much larger proportion of cell-type–exclusive binding sites (24.9%) compared with REST (7.4%), with MAX falling in between (18.3%). Complete lists of the fraction of cell-type–specific and cell-type–exclusive binding sites for the 10 TFs for which high-quality replicate experiments were available are provided in Supplemental Table S6.
We note that cell-type–specific binding sites, as identified by our statistical procedure, are correlated with expression of nearby genes. When we examined the expression levels as measured by RNA-seq of genes proximal to cell-type–specific binding sites, we found that these genes were significantly differentially expressed in GM12878 versus K562 based on their cumulative distribution of log expression changes relative to all expressed genes and genes bound in both cell lines (Fig. 5C).
A subset of TFs display distinct cell-type DNA sequence specificity
We next wished to learn if TFs can display cell-type–specific sequence preferences. To maximize sensitivity to independent sequence preferences, we learned cell-type–specific k-mer SVM sequence models for each TF using multitask learning, and examined cases where these sequence models were significantly different. Multitask learning is an attractive framework for simultaneously training GM12878- and K562-specific sequence models while also learning what is shared in both GM12878 and K562 binding sites. Specifically, we jointly learn two models, namely, the GM12878-specific model w0 + wGM and the K562-specific model w0 + wK, where the common sequence signal in both cell lines is given by w0. The model vectors w0, wGM, and wK are learned simultaneously and over the same k-mer feature space. We also confirmed that the cell-type–specific models learned through multitask joint training were more accurate than models individually trained on cell-type–exclusive sites from a single cell line (Supplemental Fig. S3).
We then asked how well differential DNase accessibility and differential sequence scores correlate with cell-type–specific binding. The cell-type–specific sequence scores are determined by (wGM − wK) · x, where x is the vector of k-mer features for a particular binding site and wGM and wK are the cell-type–specific k-mer SVMs. We first examined the differential DNase accessibility and differential SVM sequence scores as a function of K562 versus GM12878 log read count scatterplots. Figure 6A shows differential DNase read counts for USF1 (top) and YY1 (bottom), where bins are colored red if the binding sites inside the bin are more DNase accessible in K562 and blue if they are more accessible in GM12878. Figure 6B shows differential k-mer SVM sequence scores, also for USF1 and YY1. These TFs both display cell-type–specific binding patterns (fraction of cell-type–specific peaks about the top 5000 peaks in both cell types is 28.4% for USF1 and 38.9% for YY1). However, for USF1, only the differential DNase accessibility correlates with differential occupancy, while for YY1, the differential sequence scores correlate more strongly with differential binding. It is worth noting that the examples used to train the SVM models have been removed in these analyses.
Figure 6.

Cell-type–specific TF binding is associated with differential DNase accessibility, sequence signal, or both. (A) Differential DNase accessibility (color) is shown for K562 versus GM12878 with respect to cell-type–specific binding (x-axis for GM12878; y-axis for K562). Each point represents a single binding site, and if there are a sufficient number of points in a region, their value is averaged and appears as a square. DNase accessibility, as measured by read-counts, for USF1 (top) correlates with cell-specific binding. This contrasts with YY1 (bottom), where DNase accessibility is evenly distributed across cell-type–specific and nonspecific peaks. (B) Differential sequence preference (color) is shown for K562 versus GM12878. k-mer SVM models are learned from K562 and GM12878 binding sites, and their differential scores are shown by color gradient. For YY1, but not USF1, we see that the differential k-mer SVM scores distinguish cell-type–specific binding sites. (C) Binding sites with differential TF occupancy also have differential DNase accessibility. Each line represents a TF that has been assayed in GM12878 and K562. The x-axis plots a ranking from the most K562-specific binding site to the most GM12878-specific binding sites, based on cell-to-cell log read count ratios, while the y-axis shows the difference in DNase-accessibility ranks in GM12878 and K562. The line plot is smoothed using the mean over a window of 500 binding sites. (D) For the same TFs, we plot the difference in K562- and GM12878-specific k-mer SVM score ranks (y-axis) as a function of the ranking of cell-to-cell log read count ratios, from the most K562-specific binding site to the most GM12878-specific binding sites. The line plot is smoothed using the mean over a window of 500 binding sites.
To see the correlation between DNase and cell-type–specific binding more clearly, Figure 6C shows the difference in ranks of DNase log ChIP-seq read counts in GM12878 versus K562 as a function of the ranking from most K562-specific binding site to most GM12878-specific binding sites, as measured by K562-vs-GM12878 log read ratios for the TFs GABPA, USF1, YY1, and JUND. In all cases, there is some correlation between differential ChIP-seq occupancy and differential DNase accessibility between cell types. The correlation is particularly strong for JUND and USF1 but only marginal for YY1. Next, we computed similar line plots but calculated the difference in ranks of K562- versus GM12878-specific SVM sequence scores and a function of the ranking of binding sites K562-vs-GM12878 log read ratios (Fig. 6D). Here it is clear that the differential sequence signal strongly correlates with differential binding for YY1 and JUND, while the differential sequence signal for USF1 does not correlate with its differential occupancy.
Cell-type–specific sequence preferences can be explained by cell-type–specific heteromeric complexes
Since cell-specific sequence models for several TFs correlate strongly with their cell-type–specific binding, we further examined the sequence differences between these models to gain clues about the mechanism underlying cell-dependent sequence signals. A simple visualization of the k-mer information used to train the cell-type–specific models is suggestive: When we clustered the rows and columns of the k-mer feature matrix for the cell-type–exclusive binding site examples of either YY1 or JUND, we identified blocks of co-occurring k-mers that were strongly enriched either in the K562-exclusive sites or the GM12878-exclusive sites (Supplemental Fig. S4). To examine cell-type–specific information for these two TFs more carefully, we considered GM12878- and K562-exclusive binding sites that were not used in training the sequence models and plotted the Z-transformed k-mer SVM scores for the K562-specific model (using model vector w0 + wK) against the k-mer SVM scores for the GM12878-specific model (using model vector w0 + wGM), as shown in Figure 7, A and B. We identified examples that received a Z-transformed SVM discriminant score >1.5 for at least one of the models. We also required that the difference between the scores for the cell-type–specific models be above 1.
Figure 7.

Cell-type–specific sequence models for JUND and YY1 capture different primary motifs. (A) Cell-type–exclusive JUND sites were scored by the GM12878- and K562-specific k-mer SVM models (x- and y-axes, respectively) to identify groups of differentially scored sites. Sites with high K562 and low GM12878 scores (red) and high GM12878 and low K562 scores (blue) were used as input to MEME to produce the different sequence motifs shown with significance P < 10−77. (B) Same as previous panel but using cell-type–exclusive YY1 binding sites with significance P < 10−21.
This procedure identified a set of binding sites that were exclusively bound in one cell type or the other and also differentially scored by the two cell-type–specific SVM sequence models. We used these binding site sequences as input to the MEME algorithm and show the top extracted motifs (Fig. 7A,B). The K562 and GM12878 motifs for JUND and YY1 that are derived from this procedure are quite interesting and suggest mechanisms for cell-type–specific binding.
The extracted JUND motifs represent heterodimer motifs with different spacing. The K562 motif is an octamer, where the two 3-mer motifs are separated by a spacer of 2 nucleotides (nt). The GM12878 motif is a heptamer, where the spacer separating the dimeric motifs is a single nucleotide. This difference suggests a cell-type–dependent change in composition of the AP-1 heterodimeric complex (van Dam and Castellazzi 2001). To look for additional evidence in support of this hypothesis, we examined expression levels of a set of AP-1 cofactors in GM12878 and K562 as measured by RNA-seq (Supplemental Fig. S5). We indeed found that BATF, which forms a heterodimer with JUND and acts to negatively regulate AP-1 targets (Echlina et al. 2000), is more highly expressed in GM12878 than K562. Potentially, a greater proportion of JUND-BATF heterodimers in GM12878 relative to K562 may explain the distinct cell-type–specific sequence preferences of JUND.
We also find that YY1 binds to a longer motif in K562 that includes at least four additional specificity nucleotides around the core ATGGC motif (as shown in Fig. 7B). In contrast, sites exclusively bound in GM12878 contain motifs with two different nucleotides around the core YY1 motif. When we searched for the K562 motif (exact AAGATGGCGG k-mer matches, one mismatch allowed), we found it in 70% of K562-specific and only 13% of GM12878-specific sites (odds ratio [OR] of 15, P < 1.1 × 10−35). Similarly, we found the GM12878 motif (AATGGCT) in 36% of K562-specific sites and 66% of GM12878-specific sites (OR = 3.6, P < 1.8 × 10−10). We did not find any significant secondary motifs that were present in more than 10% of cell-type–specific binding sites (as determined by MEME). Previously, a similar longer YY1 motif was characterized through a dimeric YY1-DNA crystal structure, where the first zinc finger of YY1 makes base contact to the extended nucleotide (Houbaviy et al. 1996). Interestingly, mutation of the first zinc finger of YY1 also ablates all sequence specificity outside of the core ATGGC motif (Kim and Kim 2009). We next examined whether any known cofactors of YY1 are significantly differentially expressed in the two cell types (Supplemental Fig. S5). We found that MNDA, which is known to increase YY1 DNA-binding affinity in vitro but does not have any independent sequence specificity in vitro (Xie et al. 1998), is highly up-regulated in GM12878. Meanwhile, the cofactor CtBP, whose presence in the nucleus is required for YY1 binding in Drosophila (Srinivasan and Atchison 2004), is highly expressed in both cell lines but is expressed higher in GM12878. These previous findings, in combination with our high-resolution sequence models, suggest that allosteric alterations in one or multiple binding domains, possibly through cofactor interaction or post-translational modification, may be capable of altering the genome-wide DNA-binding preferences of YY1.
Cell-type–specific sequence signal, rather than chromatin accessibility, explains cell-type–exclusive sites for JUND and YY1
To ask whether cell-type sequence signal influences whether a locus is bound in a given cell type, we returned to the cell-type–exclusive binding sites in GM12878 and K562 for three TFs—USF1, YY1, and JUND—and searched for loci that were chromatin accessible in both cell types, even though these loci were bound in only one cell type. Figure 8A shows for each of these three TFs the number of binding sites across both cell types, the number of K562- and GM12878-exclusive binding sites, and the number of cell-type–exclusive binding sites that are also DNase-accessible in GM12878. The top heatmap in Figure 8B shows all the cell-type–exclusive binding sites for USF1. For this example, cell-type–specific DNase accessibility almost perfectly correlates with cell-type–specific binding, shown by the ChIP-seq read signals, and the GM12878 and K562 sequence scores are well-correlated with each other and appear to provide no additional discriminative information about differential binding. In contrast, in the middle and bottom heatmaps of Figure 8B, we focus on the cell-type–exclusive binding sites that are also DNase accessible in GM12878 for YY1 and JUND. In these examples, a subset of the cell-type–exclusive binding sites are DNase accessible in both cell types but are only bound in K562. For this subset of sites for YY1 and JUND, the K562-specific SVM sequence scores clearly correlate with binding, while the GM12878 SVM assigns low scores. Therefore, for these TFs, cell-type–specific sequence models can explain cell-type–specific binding for loci that are DNase accessible in both cell types.
Figure 8.

Cell-type–specific sequence models can predict cell-type–specific binding at loci that are DNase accessible in both cell lines. (A) The number of binding sites, cell-type–exclusive binding sites, and exclusive binding sites that are DNase accessible in GM12878. (B) Cell-type–exclusive binding sites can be explained by cell-type–specific sequence preferences when a binding site is accessible in both cell lines. Cell-type–exclusive binding sites for USF1, YY1, and JUND are shown. For USF1, all GM12878- and K562-exclusive binding sites are shown, and DNase accessibility is able to explain cell-type–exclusive binding. In contrast, for JUND and YY1, there are cell-type–exclusive binding sites in GM12878 and K562 that are DNase accessible in both cell lines, and only these examples are plotted in the middle and bottom heatmaps. For these examples, the cell-type–specific SVM sequence scores can explain the cell-type–specific binding. (C) AUC values for the task of discriminating between GM12878-exclusive peaks and K562-exclusive peaks by differential DNase reads (x-axis) or by cell-type–specific SVM sequence scores. For the SVM models, the GM12878- and K562-specific models were each used to discriminate between GM12878- and K562-exclusive binding sites, and the mean AUC over both models was reported. Binding site sequences used in training the models were held out of test sets for this evaluation. For most TFs, the cell-type–exclusive binding sites are well-predicted by differential DNase accessibility (I, IV). For REST, DNase is not predictive in general and the SVM models are consistent between the two cell lines (II). For JUND and YY1 (III), DNase is not predictive of cell-type–exclusive binding, as many sites are DNase accessible in both cell lines; however, the cell-type–specific peaks tend to have different underlying k-mer sequences, enabling accurate discrimination by cell-type–specific SVM sequence models.
In order to better quantify the predictive value of cell-specific sequence preferences versus chromatin accessibility in this setting, we evaluated the ability of differential DNase accessibility and cell-type–specific SVM sequence models to discriminate between GM12878- and K562-exclusive binding sites. First, we scored each TF's cell-type–exclusive binding sites by the difference in log DNase reads between the two cell types and used the AUC to measure how well this score discriminated between GM12878- and K562-exclusive binding sites. Next, for each TF, we used the previously trained GM12878- and K562-specific SVM sequence models to discriminate between exclusive sites for each cell type, and we averaged the AUCs for each model to report a single SVM sequence AUC score. Results for this discrimination task are reported in Figure 8C. Binding site sequences used in training the SVM sequence models were held out of test sets for this analysis.
We found that for most TFs, the cell-type–exclusive binding sites are well-discriminated solely by differential DNase accessibility (region I in Fig. 8C). For JUND and YY1 (region III), differential DNase is not predictive of cell-type–exclusive binding due to many sites that are DNase accessible in both cell lines, whereas the cell-type–specific SVM models do accurately discriminate between loci bound exclusively in GM12878 and K562 (as described above). REST (region II) has consistent SVM models between cell types, but differential DNase also poorly discriminates between its cell-type–exclusive binding sites, likely due to its enrichment in repressed regions of the genome. Interestingly, SRF and FOS (c-Fos) are partially predicted by sequence signal in addition to differential DNase accessibility (region IV). We hypothesized that this may be a consequence of differential proximal cofactors pre-establishing differential DNase accessible sites in the cell lines, as has been previously suggested (e.g., Heinz et al. 2010). We found that an ETS motif (exact k-mer CCGGA) is weakly more prevalent in GM12878 SRF sites (OR = 2, P < 0.012), suggesting that an ETS family member may differentially prime sites that are subsequently bound by SRF. In contrast, the accuracy difference for FOS reflects a depletion of the primary motif (TGAGTCA, allowing one mismatch) in GM12878 sites (OR = 0.23, P < 0.0014). While unknown cofactors may supply additional sequence specificity, MEME did not yield any significant motifs for >10% of the sites. It is also possible that the depletion may be an artifact of higher false-positive rate in the GM12878 ChIP-seq experiments.
Discussion
We have presented a framework for modeling protein–DNA binding sites across multiple cell types. In using discriminative sequence models based on k-mer patterns to capture the cell-type–specific sequence preferences, we are proposing that TF ChIP-seq binding profiles contain much richer and more subtle sequence information than can be captured by a single motif. Indeed, we have seen that our k-mer–based SVM models can capture a range of high and low information content binding sites that would need to be described by several slightly different PSSMs. In our framework, we find that DNA sequence signal is quantitative and varies across regulators; rather than finding sequence-specific and nonspecific factors, we find a continuum of sequence specificity and SVM sequence model performance across the factors in our study.
While the cell-type specificity of a gene expression program may be largely maintained through chromatin state, TF complex composition may also control cell-type–specific expression. In particular, differential TF binding between cell lines can be predicted by cell-type–specific primary sequence preferences, not just differences in chromatin accessibility. While differential binding due to complex composition has been observed previously at specific loci (van Dam and Castellazzi 2001; Saccani et al. 2003; Leung et al. 2004; So et al. 2007), we present the first genome-wide assessment of how these complexes may contribute to cell-type specificity. We also note that the sequence preference differences we find are alterations in binding of the primary factor, not the binding of proximal cofactors. Naturally, proximal cofactors can also influence binding through recruitment and chromatin reorganization (e.g., Mullen et al. 2011); however, the factors in our analysis that are significantly enriched in cell-type–specific proximal binding sites of cofactors are better predicted by DNase accessibility.
These observations lead us to believe that TFs should not be treated as isolated or static, but should be considered in the context of their heteromeric complexes, their post-translational modifications, the specific nucleotide sequence they are binding, and other allosteric alterations, which are all likely to play a role in DNA binding preferences. These factors also naturally introduce competition into complex formation, as has been explicitly described for the NFKB complex (Saccani et al. 2003). Furthermore, different k-mer patterns not only may recruit different complexes but may actually allosterically alter the protein function (Meijsing et al. 2009). Given the massive data sets being generated by the ENCODE Consortium and other large-scale efforts, there is an excellent opportunity to learn richer sequence representations to more fully understand the sequence information that specifies TF binding and activity.
Methods
ChIP-seq processing
We obtained raw reads (fastq files) from the ENCODE section of the UCSC Genome Browser (http://genome.ucsc.edu/ENCODE/). We aligned reads to the hg19 reference genome allowing one mismatch and keeping only uniquely aligned reads.
TF binding sites were determined by peak calling using the SPP (Kharchenko et al. 2008) R package. We estimated DNA fragment size using the cross-strand correlation measure and estimated FDR q-values using DNA input controls. In all our analyses, we examined peaks with q < 0.2 or the top n peaks, where n is given in the main text. The number of reads mapping to peaks was normalized by RPM.
DNase accessibility (from the Stamatoyannopoulos ENCODE laboratory) and histone modification occupancy (from the Berstein ENCODE laboratory) were estimated using overlapping read counts. Histone modification ChIP-seq reads were strand-extended by 150 bp, and the middle 75 bp was used to estimate overlaps. Occupancy profiles were then binned at 100-bp resolution. We note that data from an alternative DNase assay (Boyle et al. 2011) gave highly correlated, though not identical, results (Supplemental Fig. S6).
Learning DNA sequence models
To learn sequence models, we selected bound (positive) and unbound (negative) genomic regions. To select bound examples, we extracted the 100-nt DNA sequence centered at ChIP-seq peaks. Unbound examples were selected 200 bp from the flanking regions of the ChIP-seq peaks. This flanking region is significantly more difficult to predict correctly than random genomic regions (data not shown).
For the SVM models, we transformed the positive and negative examples into features by computing the dinucleotide mismatch k-mer features of the 100-nt regions (as previously described by Agius et al. 2010). This feature representation is the set of weighted counts of selected k-mers allowing for m mismatches in a dinucleotide alphabet. Each example is represented as the number of weighted inexact matches with respect to a feature set of the 1000 maximally discriminative individual k-mers. We then trained support vector classifiers using a slightly modified version of the LIBSVM package (Chang and Lin 2001). We use parameters k = 8, m = 2 as these settings are able to capture shorter and longer motifs.
We also assessed the accuracy of PSSM motif discovery methods (cERMIT, DME, and MDscan) and a k-mer enrichment method (Weeder). DME and MDscan requires specification of motif length, which we set to 6, 8, 10, 12, 14, and 16. DME uses flanking sequences to explicitly discriminate whereas MDscan uses the flanking sequences to construct a Markov model background. cERMIT requires a ranking of sequences, which we determined from the ChIP-seq read counts, and corresponding negative examples were ranked after all positive sequences and in the same order as their flanking positive example. By default, we obtain five motifs for each motif width for MDscan and DME. cERMIT returns 10 PSSMs of variable widths. For cERMIT, MDscan, and DME we selected the best performing PSSM motif. We also used Weeder to find the 50 most enriched 6-, 8-, and 10-bp motifs (allowing one mismatch) using human genome-wide k-mer frequencies as the background.
When scoring new sequences by PSSMs, we computed the log ratio of the PSSM with respect to human background nucleotide frequency. We then scanned the entire 100-nt region with the PSSM and used the maximum value as the PSSM score. To apply Weeder k-mers to unseen 100-nt regions, we counted the total number of occurrences of the enriched k-mers allowing for one mismatch (weighting k-mers by a linear regression model reduced accuracy; data not shown). Applying the di-mismatch SVMs to new sequences required calculation of the 1000 k-mer di-mismatch counts, weighted by the w vector learned during SVM training.
Learning TF chromatin models
We examined 5-kbp regions and determined overlapping read counts binned at 100 bp (described above, shown in Fig. 3A). This resulted in 50-dimensional vectors for each chromatin-related experiment. To train an SVM classifier, we selected positive examples centered at TF peaks. Negative examples were sampled from ±200, 500, 1000 bp away from the peak site, so that each positive example generated six negative examples. The closely sampled negatives allowed us to test if it is possible to learn high-resolution predictors.
We compared the spatial SVM against several simpler methods. When examining a single histone modification or DNase accessibility, we compared against total read counts mapping to various window locations and sizes centered at the peak and flanking negative regions. Specifically, we examined single windows of sizes 100 bp and 200 bp across all possible bins, any two windows of size 100 bp, and symmetric windows around the center of the example taken at all possible radii. We report results for the most-accurate set of windows.
When examining a set of histone modifications (with or without DNase), we compared our spatial SVMs against a logistic regression combination of the histone marks. Prior to regression, we reduced the dimensionality of the examples by extracting only the middle 1000 bp, which greatly increased the hold-out accuracy of the regression (data not shown).
Identifying cell-type–specific binding sites
To identify cell-type–specific binding sites, we identified peaks in both of two experimental replicates in each of the two cell lines. To identify the total set of peaks across replicates and cell lines, we performed peak calling on each cell line and replicate independently (as described above) and examined the top 5000 peaks (or peaks with q-value <0.2 if less than 5000). We matched overlapping peaks (within 100-bp radius) to ensure that peaks were not double counted and also included peaks occurring in only one cell line. This resulted in a total of 5000–10,000 peaks across the two cell lines. Next, the number of strand-extended reads whose starting position fell within a 200-bp window of each peak was determined (in RPM) was quantile-normalized.
We then estimated a background noise model from the replicates to determine significance of cell specificity. The noise model was derived from the replicate 1 RPM values x and replicate 2 RPM values y. We computed the geometric average between the two replicates a = log(x)/2 + log(y)/2 and the log ratio of RPM m = log(x/y). We then estimated the standard deviation θ(a) of the log ratio at a given geometric average a and fit this to an exponential noise model 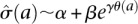 . The parameters α, β, γ were fit by nonlinear least squares, and we used
. The parameters α, β, γ were fit by nonlinear least squares, and we used 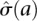 as the estimated standard deviation of the log ratio at a given RPM level. A normal distribution with zero-mean and standard deviation
as the estimated standard deviation of the log ratio at a given RPM level. A normal distribution with zero-mean and standard deviation 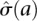 was used to estimate P-values for cell-to-cell variation in RPM counts at a given RPM.
was used to estimate P-values for cell-to-cell variation in RPM counts at a given RPM.
We considered two types of cell-type–specific binding. The first only required a binding site differential RPM to pass a P < 0.01 significance threshold. The second required that the differential RPM be significant at P < 0.01 and that the cell line considered unbound had less than 1 RPM. We call the first definition “cell-type specific” and the second “cell-type exclusive.” For each definition, we enforce the conservative stance that the differentially bound locus must be consistently differentially bound in both of the replicate comparisons. That is, the differential binding must be consistent in the two comparisons: (1) replicate 1 of the first cell line compared with replicate 1 of the second cell line; and (2) replicate 2 of the first cell line compared with replicate 2 of the second cell line.
Learning cell-type–specific sequence preferences
We used a regularized multitask learning framework (Evgeniou et al. 2005) to learn cell-type–specific sequence preferences. In this approach, weight vectors wi, i = 1,2 are learned collectively for each individual task, and a weight vector w0 is learned on the collection of features for all tasks; after training, the model vector w0 + wi is used as the model vector for classification task i. Formally, in the SVM constrained optimization problem, the objective function to be minimized is
where the parameter λ trades off between the common and specific tasks; a soft margin constraint is introduced for each training example of each task, namely:
where 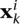 is the kth training example for task i,
is the kth training example for task i, 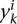 is its binary label, and
is its binary label, and 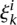 is the corresponding slack variable. We found that taking λ = 1 gave good performance across TFs, and we assume this parameter value in the formulation below.
is the corresponding slack variable. We found that taking λ = 1 gave good performance across TFs, and we assume this parameter value in the formulation below.
Multitask SVM learning can be reduced to a standard SVM problem as follows. If FGM and FK represent the feature matrices for the individual tasks of learning binding preferences for GM12878 and K562, that is, the matrix of di-mismatch k-mer counts for the training sequences (rows) with respect to the k-mers (columns), then the multitask feature matrix is defined to be
and a weight vector (w0, wGM, wK) is learned using a regular SVM. The set of training sequences for FGM and FK were sampled from cell-type–exclusive binding site examples. To prevent any biases, we maintained the same set of k-mers for both cell-type feature maps, namely, the union of the best 1000 k-mers for each cell type.
Data access
The code, training, and test sequences for learning sequence models are available at http://cbio.mskcc.org/leslielab/TFcelltype/.
Acknowledgments
We thank Anshul Kundaje from the Human ENCODE Consortium for extensive discussions and assistance with the ENCODE ChIP-seq data sets. This work was supported by NIH U54 award HG004695 (W.S.N.) and NIH R01 award HG006798 (C.L.).
Footnotes
[Supplemental material is available for this article.]
Article and supplemental material are at http://www.genome.org/cgi/doi/10.1101/gr.127712.111.
Freely available online through the Genome Research Open Access option.
References
- Agius P, Arvey A, Chen W, Noble WS, Leslie C 2010. High resolution models of transcription factor-DNA affinities improve in vitro and in vivo binding predictions. PLoS Comput Biol 6: e1000916 doi: 10.1371/journal.pcbi.1000916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings of ISMB 2: 28–36 [PubMed] [Google Scholar]
- Bailey TL, Noble WS 2003. Searching for statistically significant regulatory modules. Bioinformatics 19: ii16–ii25 [DOI] [PubMed] [Google Scholar]
- Barrera LO, Li Z, Smith AD, Arden KC, Cavenee WK, Zhang MQ, Green RD, Ren B 2008. Genome-wide mapping and analysis of active promoters in mouse embryonic stem cells and adult organs. Genome Res 18: 46–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AP, Song L, Lee B-K, London D, Keefe D, Birney E, Iyer VR, Crawford GE, Furey TS 2011. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res 21: 456–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-C, Lin C-J 2001. LIBSVM: a library for support vector machines. http://www.csie.ntu.edu.tw/∼cjlin/libsvm
- Cobaleda C, Schebesta A, Delogu A, Busslinger M 2007. Pax5: The guardian of B cell identity and function. Nat Immunol 8: 463–470 [DOI] [PubMed] [Google Scholar]
- Deaton AM, Webb S, Kerr ARW, Illingworth RS, Guy J, Andrews R, Bird A 2011. Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Genome Res 21: 1074–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echlina DR, Taeb H-J, Mitin N, Taparowsky EJ 2000. B-ATF functions as a negative regulator of AP-1 mediated transcription and blocks cellular transformation by Ras and Fos. Oncogene 19: 1752–1763 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2011. A user's guide to the encyclopedia of DNA Elements (ENCODE). PLoS Biol 9: e1001046 doi: 10.1371/journal.pbio.1001046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M 2010. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol 28: 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Plasterer HL, Simon I, Bar-Joseph Z 2010. Integrating multiple evidence sources to predict transcription factor binding in the human genome. Genome Res 20: 526–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evgeniou T, Micchelli CA, Pontil M 2005. Learning multiple tasks with kernel methods. J Mach Learn Res 6: 615–637 [Google Scholar]
- Georgiev S, Boyle A, Jayasurya K, Ding X, Mukherjee S, Ohler U 2010. Evidence-ranked motif identification. Genome Biol 11: R19 doi: 10.1186/gb-2010-11-2-r19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheldof N, Smith EM, Tabuchi TM, Koch CM, Dunham I, Stamatoyannopoulos JA, Dekker J 2010. Cell-type-specific long-range looping interactions identify distant regulatory elements of the CFTR gene. Nucleic Acids Res 38: 4325–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman N, Stuart R, Hon G, Fu Y, Ching C, Hawkins RD, Barrera L, Van Calcar S, Qu C, Ching K, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318 [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK, et al. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. 2009. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods 6: 283–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong P, Liu X, Zhou Q, Lu X, Liu J, Wong W 2005. A boosting approach for motif modeling using ChIP-chip data. Bioinformatics 21: 2636–2643 [DOI] [PubMed] [Google Scholar]
- Houbaviy HB, Usheva A, Shenk T, Burley SK 1996. Cocrystal structure of YY1 bound to the adeno-associated virus P5 initiator. Proc Natl Acad Sci 93: 13577–13582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502 [DOI] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, Park PJ 2008. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26: 1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim J 2009. YY1's longer DNA-binding motifs. Genomics 93: 152–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung TH, Hoffmann A, Baltimore D 2004. One nucleotide in a κB site can determine cofactor specificity for NF-κB dimers. Cell 118: 453–464 [DOI] [PubMed] [Google Scholar]
- Liu XS, Brutlag D, Liu J 2002. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat Biotechnol 20: 835–839 [DOI] [PubMed] [Google Scholar]
- Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR 2009. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 324: 407–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AC, Orlando DA, Newman JJ, Lovén J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, Dekoter RP, Young RA, et al. 2011. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 147: 565–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavesi G, Mereghetti P, Mauri G, Pesole G 2004. Weeder web: Discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Res 32: W199–W203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique-Regi R, Degner JF, Pai AA, Gaffney DJ, Gilad Y, Pritchard JK 2011. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res 21: 447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccani S, Pantano S, Natoli G 2003. Modulation of NF-κB activity by exchange of dimers. Mol Cell 11: 1563–1574 [DOI] [PubMed] [Google Scholar]
- Sharon E, Lubliner S, Segal E 2008. A feature-based approach to modeling protein-DNA interactions. PLoS Comput Biol 4: e1000154 doi: 10.1371/journal.pcbi.1000154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Liang Y, Siggia E 2006. Stubb: A program for discovery and analysis of cis-regulatory modules. Nucleic Acids Res 34: W555–W559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Adler AS, Field Y, Chang HY, Segal E 2008. Systematic functional characterization of cis-regulatory motifs in human core promoters. Genome Res 18: 477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD, Sumazin P, Zhang MQ 2005. Identifying tissue-selective transcription factor binding sites in vertebrate promoters. Proc Natl Acad Sci 102: 1560–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- So AY-LY, Chaivorapol C, Bolton EC, Li H, Yamamoto KR 2007. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet 3: e94 doi: 10.1371/journal.pgen.0030094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan L, Atchison ML 2004. YY1 DNA binding and PcG recruitment requires CtBP. Genes Dev 18: 2596–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Li XY, Sabo P, Sandstrom R, Thurman R, Canfield T, Giste E, Fisher W, Hammonds A, Celniker S, et al. 2011. Dynamic reprogramming of chromatin accessibility during Drosophila embryo development. Genome Biol 12: R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam H, Castellazzi M 2001. Distinct roles of Jun: Fos and Jun: ATF dimers in oncogenesis. Oncogene 20: 2453–2464 [DOI] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry Ml, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitington T, Perkins AC, Bailey TL 2009. High-throughput chromatin information enables accurate tissue-specific prediction of transcription factor binding sites. Nucleic Acids Res 37: 14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiench M, John S, Baek S, Johnson TA, Sung M-HH, Escobar T, Simmons CA, Pearce KH, Biddie SC, Sabo PJ, et al. 2011. DNA methylation status predicts cell type-specific enhancer activity. EMBO J 30: 3028–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won K-J, Ren B, Wang W 2010. Genome-wide prediction of transcription factor binding sites using an integrated model. Genome Biol 11: R7 doi: 10.1186/gb-2010-11-1-r7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Briggs JA, Briggs RC 1998. Human hematopoietic cell specific nuclear protein MNDA interacts with the multifunctional transcription factor YY1 and stimulates YY1 DNA binding. J Cell Biochem 70: 489–506 [PubMed] [Google Scholar]
- Zhou Q, Wong W 2004. CisModule: De novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc Natl Acad Sci 101: 12114–12119 [DOI] [PMC free article] [PubMed] [Google Scholar]