

Abstract
As the sequencing of healthy and disease genomes becomes more commonplace, detailed annotation provides interpretation for individual variation responsible for normal and disease phenotypes. Current approaches focus on direct changes in protein coding genes, particularly nonsynonymous mutations that directly affect the gene product. However, most individual variation occurs outside of genes and, indeed, most markers generated from genome-wide association studies (GWAS) identify variants outside of coding segments. Identification of potential regulatory changes that perturb these sites will lead to a better localization of truly functional variants and interpretation of their effects. We have developed a novel approach and database, RegulomeDB, which guides interpretation of regulatory variants in the human genome. RegulomeDB includes high-throughput, experimental data sets from ENCODE and other sources, as well as computational predictions and manual annotations to identify putative regulatory potential and identify functional variants. These data sources are combined into a powerful tool that scores variants to help separate functional variants from a large pool and provides a small set of putative sites with testable hypotheses as to their function. We demonstrate the applicability of this tool to the annotation of noncoding variants from 69 full sequenced genomes as well as that of a personal genome, where thousands of functionally associated variants were identified. Moreover, we demonstrate a GWAS where the database is able to quickly identify the known associated functional variant and provide a hypothesis as to its function. Overall, we expect this approach and resource to be valuable for the annotation of human genome sequences.
The increasing number of sequenced human genomes is providing a catalog of the large number of individual variations present in the human genome (The International HapMap Consortium 2005, 2007; The 1000 Genomes Project Consortium 2010). Many of these variants are expected to be responsible for normal and disease phenotypes. Similarly, large, genome-wide association studies (GWAS) continue to map diseases to associated genomic regions from large cohorts of individuals (Hindorff et al. 2012). Initial interpretation of results generated by both of these approaches has been limited to DNA regions that cause disruption of gene function through coding sequence changes typically identified using an application such as PolyPhen-2 (Adzhubei et al. 2010). However, ∼95% of known variants within sequenced genomes and 88% of those variants from GWAS studies fall outside of coding regions and have been difficult to interpret (Hindorff et al. 2009).
Both large consortia and individual labs are generating a significant amount of regulatory information that is providing a better interpretation of the noncoding portions of the genome. The ENCODE Project, in particular, has mapped open chromatin and protein binding regions for large numbers of factors across many cell types, and many individual labs are generating similar types of data (The ENCODE Project Consortium 2012). These data can be utilized to provide a powerful resource to help demonstrate functional consequences of variants outside of genic regions. However, these results are often scattered in a variety of online resources, and their use to annotate personal genomes has not been previously described.
Here, we provide both a database, RegulomeDB, which integrates a large collection of regulatory information, and an approach that enables the functional assignment of regulatory information onto any set of variants derived from genomic sequencing or GWAS studies. These efforts demonstrate which variants have potential or demonstrated regulatory functions and through which mechanisms those functions might work. To generate these functional assignments, we make use of large sets of data including the following: (1) Manually curated regions that have been experimentally characterized to be involved in regulation; (2) ChIP-seq information for a variety of important regulatory factors across a diverse set of cell types; (3) chromatin state information across over 100 cell types; and (4) expression quantitative trait loci (eQTL) information allowing the association of distal sites with gene promoters.
In addition to the experimentally measured evidence, computational processing and machine learning can improve our predictive capabilities for regulatory variants. To further assist in functional annotation, we provide computational predictions through the following methods: (1) DNase footprinting to demonstrate exact protein binding sites; and (2) potential binding motif alterations of nucleotide variants.
To demonstrate the utility of our approach in annotating genomes for regulatory information, we describe the results from analysis of 69 fully sequenced genomes recently determined by Complete Genomics as well as one deeply sequenced genome from the HapMap project. Thousands of variant regions can be assigned to potential regulatory functions, and we demonstrate that there is likely as much variation affecting regulatory function as that affecting gene function. In addition, we are able to reproduce the results from a recent GWAS study showing a single nucleotide variant (SNV) as a likely causative variant in lupus (Adrianto et al. 2011). Furthermore, we are able to provide a testable hypothesis for this SNV by comparing the same region in our database. Overall, we demonstrate that much variation is likely to affect regulatory function and that our approach and RegulomeDB will be powerful tools for assigning information to genomic variants discovered in projects that sequence personal genomes and map the genetic basis of human disease.
Results
Our overall approach is to align variants with regulatory information from a variety of sources. To do this, we first developed a comprehensive database which can help assign functional roles to variants. We then use this database to identify functional variants in the genomes of 69 normal individuals and identify regulatory variants associated with a GWAS region. We further present an online tool which rapidly annotates and classifies variants as well as a queue system for annotating entire personal genomes.
Data sources for RegulomeDB
We first created an integrated database, RegulomeDB, which provides a flexible platform for the addition of various data sources. We currently include all available ENCODE transcription factor (TF) ChIP-seq, histone ChIP-seq, FAIRE, and DNase I hypersensitive site data (The ENCODE Project Consortium 2012). In addition, we have included transcription factor ChIP-seq data available from the NCBI Sequence Read Archive (Hollenhorst et al. 2009; Jolma et al. 2010; Verzi et al. 2010; Wei et al. 2010; Hu et al. 2011; Lo et al. 2011; Novershtern et al. 2011; Palii et al. 2011; Yu et al. 2011). We also include a large collection of eQTL (Myers et al. 2007; Stranger et al. 2007; Schadt et al. 2008; Veyrieras et al. 2008; Dimas et al. 2009; Gibbs et al. 2010; Montgomery et al. 2010; Pickrell et al. 2010; Zeller et al. 2010), dsQTL (Degner et al. 2012), and ChIP-exo (Rhee and Pugh 2011) data. A total of 962 experimental data sets are included, covering over 100 tissues and cells lines and representing nearly 60 million annotations (Table 1).
Table 1.
Database content
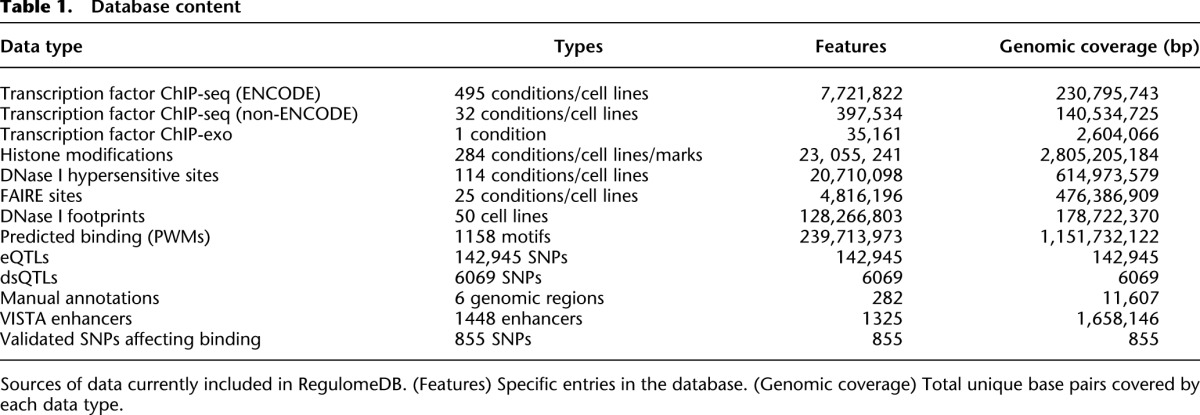
We extended the relevant biological assays with two computational predictors. First, we predict TF binding in a cell-type–specific manner by predicting DNase footprints from the DNase-seq data sets (Boyle et al. 2011; Pique-Regi et al. 2011). These results indicate a specific DNA–protein interaction event and, when combined with variant information, increase the confidence that a SNV in this region is functional. Secondly, we scanned the genome at a reasonable threshold for added positional weight matrices (PWMs) (Berger et al. 2006, 2008; Matys et al. 2006; Bryne et al. 2008; Badis et al. 2009; Scharer et al. 2009; Wei et al. 2010), which were further used to intersect with other functional data (see Methods). For these computational predictions of protein binding, 1158 motifs were considered, which resulted in over 365 million additional annotations in RegulomeDB.
We supplemented these high-throughput data sources through manual curation of literature sources. These provide valuable information from low-throughput but high-quality assays to aid in assigning function to SNVs. As an initial release, RegulomeDB contains manual curation from 97 papers focused on six loci, resulting in 188 genomic annotations. We have also included 1448 validated enhancer regions from the VISTA Enhancer Browser (Visel et al. 2007) and 855 SNVs shown to directly affect NFKB and RNA Pol 2 binding in lymphoblastoid cells (Kasowski et al. 2010; Karczewski et al. 2011).
We refer to the January 1, 2012 release of information with ∼425 million entries as RegulomeDB v1.0. RegulomeDB will continue to be improved through continual manual curation as well as the automated monitoring of high-throughput data sources.
Variant classification system
Although the catalog of information is important, its interpretation is even more so. To assist comparison among annotations, we developed a heuristic scoring system based on functional confidence of a variant. The scoring system represents with increasing confidence that a variant lies in a functional location and likely results in a functional consequence (i.e., alteration of TF binding and a gene regulatory effect) (Table 2). As such, we consider variants that are known eQTLs for genes, and thus have been shown to be associated with expression, as most likely to be significant and label these variants as Category 1. However, 58% of eQTL SNVs are not associated with any other functional annotation and are labeled Category 6. Within Category 1, subcategories indicate additional annotations from the most confident (1a, which has TF binding, a motif for that TF, and a DNase footprint) to the least confident (1f, which has only TF binding or a DNase peak).
Table 2.
RegulomeDB variant classification scheme
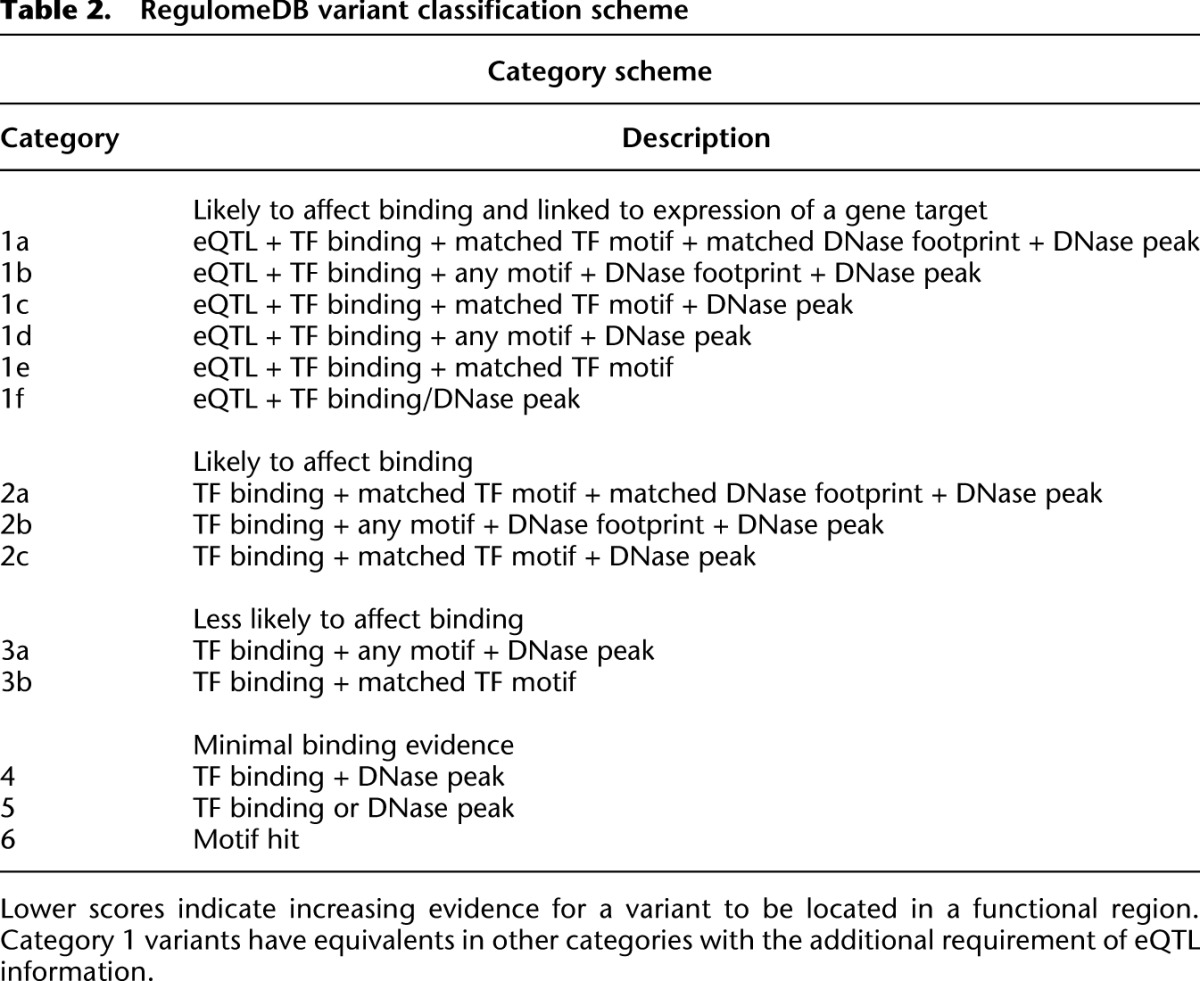
The additional categories represent analogous annotations to Category 1 but without eQTL data and, thus, no known direct effect on binding. Category 2(a–c) demonstrates direct evidence of binding through ChIP-seq and DNase with either a matched PWM to the ChIP-seq factor or a DNase footprint. Category 3(a–b) is considered less confident in affecting binding due to a more incomplete set of evidence. These sites have ChIP-seq evidence and either a motif that matches the ChIP-seq data but no DNase evidence, or DNase evidence and any other motif. Finally, Categories 4–6 lack evidence of the variant actually disrupting the site of binding. These include DNase and ChIP-seq evidence (Category 4), DNase or ChIP-seq evidence (Category 5), or any single annotation not in the above categories (Category 6).
As a verification of the heuristic system, we compared the enrichment of SNVs associated with a GWAS (Supplemental Table S1; Hindorff et al. 2009). In all cases, SNVs in perfect linkage with GWAS lead SNVs are significantly enriched for the heuristic categories, and the level of this enrichment increases in tandem with higher-confidence categories. Detailed analysis of the application of RegulomeDB to GWAS and the enrichment of GWAS SNVs in functional regions is available as an additional ENCODE companion paper (Schaub et al. 2012).
Regulatory variation across 69 individuals
Access to the deep whole genome sequences of 69 individuals from Complete Genomics (Drmanac et al. 2010) allows us to investigate the average frequency of variants that intersect functional DNA elements. These genomes collectively contain 19,124,349 SNVs and 3,040,170 insertion/deletions. The average individual contains 3,870,827 variants, of which ∼56.23% fall within any of our annotations (excluding histones, which alone cover 99.5% of the SNVs, standard deviation = 0.005). This count is substantially higher than those SNVs that affect protein coding sequences alone (∼9%). As expected, variants intersect multiple regulatory annotations. An example is shown in Figure 1; this variant falls within several regulatory elements, resides in several genomes, and has been shown to directly affect binding of NFKB (Kasowski et al. 2010).
Figure 1.

A SNV (rs9261424) overlapping many regulatory features. (A) This SNV falls within peak regions for many ChIP-seq factors as well as DNase-seq peaks from multiple cell lines. (B) The same SNV overlaps a motif match to the NFKB motif and has been shown to alter binding. The signal tracks represent ChIP-seq peaks of NFKB at the SNV site for three individuals: homozygous to reference allele (G), heterozygous, and homozygous to alternate allele (C) (Kasowski et al. 2010).
The incidence of variants falling into each RegulomeDB category is remarkably stable across individuals particularly when comparing heterozygous and homozygous variant calls. On average, 11.20% of variants fall into ChIP-seq peaks, 21.53% into DNase peaks, 1.29% into eQTLs, 6.15% into footprints, and 39.25% into PWM matches (Supplemental Table S2). Of significance is the difference in rates of annotations for heterozygous and homozygous variant calls (Fig. 2A). In the cases of PWMs, DNase HS sites, FAIRE regions, histone modifications, DNase footprints, and TF ChIP-seq data, the rate of homozygous and heterozygous variants compared to the total count of homozygous/heterozygous variants in each is consistently and significantly different in that homozygous variants in these regulatory annotations are significantly less than those of heterozygous variants (paired, two-tailed t-test; P < 5 × 10−15) (Supplemental Table S2). A genomic background would expect equal fractions of homozygous and heterozygous variants to be covered by regulatory features. Thus, these results indicate a purifying selection is occurring in the RegulomeDB annotated regions.
Figure 2.

Incidence of SNVs in features and categories. Average percent count of SNVs in each genomic feature (A) and in each RegulomeDB category (B). Although the differences between homozygous and heterozygous SNV counts are small, they are nevertheless significant (P < 5 × 10−15). Actual SNV count in features (C) and categories for the cell line GM12878 (D).
The SNV rate within the variant classification system is also stable (standard deviations < 0.004). On average, 0.55% of variants are Category 1, 1.48% Category 2, 1.16% Category 3, 4.44% Category 4, 17.96% Category 5, and 30.67% Category 6 (Fig. 2B; Supplemental Table S3). As expected, the significant enrichment of the rate of heterozygous and homozygous variants in RegulomeDB features is also evident in the enrichment of variants in the score categories of 2–6 (paired, two-tailed t-test; P < 5 × 10−15) (Supplemental Table S3). However, Category 1 variants show an opposite bias, likely due to the lack of rare variants in eQTL studies.
Although several cases of common variants associated with complex diseases have been identified, there is a growing interest in the effect of rare variants on common diseases (Manolio et al. 2009). Susceptibility to many complex diseases could be attributed to multiple rare variants rather than a single causative polymorphism. To capture these rare variants, it is necessary to sequence entire genomes rather than using array-based genotyping. The April 2010 release from the 1000 Genomes Project contains genotype data from 629 individuals worldwide (The 1000 Genomes Project Consortium 2010). We use this information to assign global MAFs (minor allele frequency) to the variants annotated in each of the categories annotated by RegulomeDB. The variants are classified as “Rare” if the MAF is <1%, “Less Common” if the MAF is between 1 and 5%, and “Common” if the MAF is >5%.
Interestingly, the proportions of rare (∼20%), less common (∼30%), and common (∼50%) variants show little difference across each category other than Category 1. Category 1, which requires eQTL information, has very few rare variants due to the bias in selection of candidates on the arrays used (Supplemental Table S4).
Regulatory annotation of an individual genome
We next determined whether our database can perform a detailed analysis of the personal genome of a sequenced individual. We examined the cell line GM12878, a European female, which has been deeply sequenced as part of the 1000 Genomes Project and is included in the Complete Genomics samples. This individual is also a Tier 1 cell line of the ENCODE Project allowing for verification of the annotation of its genome. A diploid map of this individual's genome has recently been generated, and it has been mapped with allele-specific expression and allele-specific binding (Rozowsky et al. 2011).
In order to obtain an overview of the distribution of regulatory variants, we annotated the SNVs of GM12878 from Complete Genomics with RegulomeDB (Fig. 2C,D) and mapped them onto genes and noncoding regions using the GENCODE v7 gene information (available from UCSC Table Browser) (Harrow et al. 2012). We find that a majority of variants are noncoding, with only 77,214 (2.1%) falling within exons. The coding variants can be further classified as those likely to cause damaging mutations in genes as predicted using PolyPhen-2 (Fig. 3; Supplemental Fig. S1; Adzhubei et al. 2010). In total, 2457 coding variants (0.07%) are predicted to cause damaging mutations in genes (PolyPhen-2 prediction of probably or possibly damaging). Of these, 626 (0.01%) are homozygous and likely to affect the gene product of both alleles.
Figure 3.

Protein coding and noncoding SNVs can be classified as potentially functional by Polyphen-2 and RegulomeDB, respectively. Heterozygous, damaging coding SNVs can act in conjunction with a heterozygous regulatory SNV on the opposite allele to create a compound heterozygote and loss of function on both alleles (one regulatory, the other coding).
In contrast to functional coding variants, RegulomeDB allows us to focus on noncoding variants that are likely to directly affect binding (Category 1 and 2), resulting in 70,216 SNVs (18,842 [0.51%] and 51,374 [1.47%] in Categories 1 and 2, respectively). Of these, 25,654 (0.69%) are homozygous and likely to affect gene expression of both alleles. Therefore, many variants are predicted to affect regulatory elements in noncoding regions, and this figure is more than 30 times higher than those that are predicted to disrupt gene function using current methods. However, these regulatory variants can only be associated with altered expression levels of genes and may not be as disruptive as the damaging variants in coding regions which may alter or destroy gene products.
By combining heterozygous damaging variants in the coding regions with associated heterozygous regulatory variants on the opposite allele (matched to genes by being an eQTL), we find 667 SNVs in possible instances of compound heterozygotes which directly affect the function of 93 genes. In these situations, binding site variants could inactivate gene expression in one allele, and a damaging mutation in the other allele might effectively completely disrupt gene function, thereby inactivating gene expression by regulatory/coding region compound heterozygosity. Furthermore, we find that, of the 40 genes in GM12878 that have heteroallelic expression and potential damaging mutations, half also have associated compound heterozygous mutations in a regulatory region (Supplemental Table S5).
Of these compound heterozygotes with allele specific expression, rs10895991 is of particular interest as it contains a damaging SNV on the preferentially expressed maternal allele in the CAPN1 gene and also contains two SNVs which are eQTLs for this gene and score as Category 1a (likely to affect binding and linked to expression of a gene target). The CAPN1 gene codes for a calcium-sensitive cysteine protease present in brain synapses, the inhibition of which has been shown to be protective against Alzheimer's disease (Trinchese et al. 2008). Thus, the CAPN1 gene is likely to be a hypomorph in this individual relative to others. This demonstrates the power of combining regulatory information with coding information in annotating personal genomes.
Application to GWAS
Classifying variants based on the above criteria is also highly informative to genome-wide association studies. We demonstrate this by repeating the search for a causative SNV for systemic lupus erythematosus in a 500-KB region around the TNFAIP3 gene (Adrianto et al. 2011).
In the initial 500-KB region, there are ∼2604 SNVs present at >1% MAF (dbSNP132), of which 109 are classified by RegulomeDB as having a potentially functional consequence. Using an association test on 113 SNVs in the tested European and Asian populations, we are able to identify 28 SNVs in association with the disease in common between Europeans and Asians. Of these SNVs, our approach classifies three as having potential functional consequence—each of which provides an easily testable hypothesis.
Furthermore, the study authors further reduced the size of the risk haplotype to a 16.3-kb region through use of LD structure and conditional association analysis, which resulted in eight SNVs only one of which is assigned as putatively functional by RegulomeDB. This SNV is the same one that the study authors conclude to be the most likely functional polymorphism.
The supporting evidence for this likely functional SNV (rs117480515) is detailed in Figure 4A. A set of immune-associated proteins are shown by ChIP-seq to bind regions overlapping this SNV: NFKB, BCL11A, BCLAF1, EBF1, MEF2A, and MEF2C (Fig. 4B,C). However, there is only one putative binding site (based on PWMs) overlapping this SNV and that belongs to the BCL family, indicating that BCL binding is disrupted by this polymorphism. In fact, the actual TT>A polymorphism decreases the information content match to the BCL consensus site by 3.24 bits and moves it below our PWM call threshold. The study authors demonstrate a decrease in NFKB binding with the polymorphism and conclude that this variant is likely to influence TNFAIP3 expression by decreasing factor binding in response to pro-inflammatory signals. However, in our analysis, any NFKB binding sites are intact, and we find it likely that the actual cause of the binding disruption is due to a BCL motif disruption. It is possible that BCL binding assists NFKB binding at this genomic location.
Figure 4.

TNFAIP3-associated SNV. (A) RegulomeDB results for rs117480515 which is likely a functional variant associated with systemic lupus erythematosus. (B) This SNV was the most likely to be functional in the associated region but might be missed in a standard study because it lies >20 kb downstream from its target. (C) An enlargement of the region around rs117480515 (red line) shows the overlap with a large number of functional elements (NFKB, purple; BCL, light blue; and DNase, green) as well as the motif for BCL.
Querying the database
Access to the database and scoring method is publically available via a web interface at http://RegulomeDB.org/. The integrated database is fully searchable using common variant formats (VCF, BED, GFF3, rsIDs) and through file upload of the same formats (Supplemental Fig. S2A). In addition, a user may input broad regions of interest which are converted to variants using dbSNP132 variants at >1% allele frequency. The initial results table provides a list of the coordinates of the variants, a dbSNP rsID (if it exists), a score assigned by our method, and links to external resources for each variant (Supplemental Fig. S2B). The list is sorted by our classification scheme, with the SNVs most likely to be functional listed first. This list of SNVs is also downloadable by the user for their own analysis.
In addition, by clicking on a score, the SNV summary page is displayed which provides all evidence of function associated with a SNV (Supplemental Fig. S2C). This display includes six major categories: Protein Binding, Motifs, Chromatin Structure, eQTLs, Histone Modifications, and Related Data (which includes gene information and other manual annotations). Each of these categories provides detailed information about the transcription factor, cell line, and a literature source of the information to provide the user with direct access for addressing their hypothesis.
Discussion
We present here an approach using an integrated database to quickly generate prioritized hypotheses for the function of variants affecting both coding and noncoding regions in a genome by combining a large array of data sources into a single, integrated database. In particular, we include extensive information on annotated and computed regulatory elements in the human genome. Access to this novel approach via a simple and straightforward interface allows for easy query submission, and the scoring system provides for instant classification of significant variants. In addition, the SNV summary page will allow a user to quickly form a hypothesis as to the true functional consequence of a variant.
Recently, Ward and Kellis (2012) published the HaploReg database which aims to provide a similar annotation by providing an intersect of SNVs with chromatin state (Ernst and Kellis 2010). Our database provides additional information well beyond this by prioritizing SNVs within general regulatory regions based on specific TF, chromatin, eQTL, and PWM information. Furthermore, we allow for a query of personal SNPs which account for a large proportion of variation in the population.
We have shown the utility of RegulomeDB by providing two types of examples of common experiments using variant information. First, we demonstrate that personal genomes can be annotated with SNVs of functional consequence and allow for individual genomic interpretation. This example highlights a potential damaging allele that might merit further investigation in the individual. In many cases, the interpretation needs to be combined with damaging alleles in coding sequences. Given the large number of personal genomes sequences that are expected in the foreseeable future, a comprehensive analysis of both coding and regulatory information will be essential for clinical and phenotypic genome interpretation (Chen et al. 2012). Second, we showed that a GWAS study can derive a valid hypothesis in a straightforward manner. We compare a literature case and show that RegulomeDB rapidly generates the same result and suggests the specific cause of the decrease of binding of a transcription factor.
While our examples deal with single nucleotide variants only, the database can also be used to annotate insertions and deletions. Using the online resource, these data would be inserted in the same manner as SNVs and are not distinguished as being of a different category. However, because the database is searched for a feature that overlaps the region that contains the insertion or deletion, the same information will be returned as to potential binding sites being disrupted by the insertion/deletion. Integration of all types of variants (SNVs, Indels, and SVs) with all types of noncoding and coding variants will provide detailed functional interpretation of personal and nonpersonal genomes.
The scoring system of our approach is a simple heuristic which can be improved over time as more functional SNVs are validated. Our analysis method and database are centered around a likely disruption of a protein–DNA interaction and/or expression. Therefore, a majority of our confidence in scoring comes from TF ChIP-seq data, particularly when a TF matches a PWM in the same context. We also derive confidence from SNVs overlapping eQTLs which have been shown to alter expression. It is likely that there are additional sources of data that reinforce each other in a different manner which should be explored. However, as shown when comparing to GWAS-lead and linkage SNVs, the scoring system provides significant enrichment concurrent with better category scores. As such, we believe that the database and scoring scheme provide the best current system for annotating and prioritizing variants.
Furthermore, our system still represents an early functional annotation of the genome. Once enough data are available, it will be possible to match annotations from specific tissues allowing for even more detailed hypotheses. Also, with the limited number of conditions that are presently covered, it is very difficult to score a gain of a regulatory site. In this scenario, we might show a new PWM, but there will be no additional functional data, resulting in a poor score for the site. Nonetheless, the database and approach have significant value in their present form. As additional functional data are collected from a variety of sources, these limitations will diminish. A collective community goal is to have sufficient information such that functional information is available on every base in the human genome at a level to be predictive of molecular and phenotypic outcomes.
Methods
Data from ENCODE
For this analysis, we only use ENCODE data from the January 2011 freeze. ChIP-seq peaks determined using SPP (Kharchenko et al. 2008) at a loose threshold and a cutoff was selected based on the IDR method (A Kundaje, Q Li, B Brown, J Rozowsky, A Harmanci, S Wilder, S Batzoglou, I Dunham, M Gerstein, E Birney, et al., in prep.). DNase-seq data were based on “peak” calls from both Duke University and the University of Washington (this excludes hotspots).
Data from other sources
The FASTQ files for ChIP-seq assays were downloaded from NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) with searching key word: “chip-seq transcription factor human.” Data sets were frozen by July 25, 2011. Experiments that were overlapped with the ENCODE Project were removed to avoid redundancy. All the FASTQ files were mapped to hg19 by BWA with the default setting. Only uniquely mapping reads (with q > 0) were used for the peaks calling. For ChIP-seq with replicates, the peaks were called by SPP-IDR pipeline with FDR less than 0.01 (Kharchenko et al. 2008). For ChIP-seq without replicates, peaks were called by MACS with parameter –p equal to 1 × 10−7 (Zhang et al. 2008).
PWM matching
All positional weight matrices from TRANSFAC (Matys et al. 2006), JASPAR CORE (Bryne et al. 2008), and UniPROBE (Berger et al. 2006, 2008; Badis et al. 2009; Newburger and Bulyk 2009; Scharer et al. 2009; Wei et al. 2010) were used to call motif matches in the genome. The PWMs were scored with a 1% added pseudocount and a uniform background. The TFM P-value program was used to determine a threshold at 4 × 10−8 for each PWM (Touzet and Varré 2007). All possible kmers that are above this threshold are aligned to the genome using Bowtie to give a final motif mapping (Langmead et al. 2009).
Footprinting
PWMs combined with all ENCODE DNase-seq data sets were run through CENTIPEDE to generate footprint calls (Pique-Regi et al. 2011). In addition to the DNase-seq data, the PWM score and conservation score (average phastCons score across the aligned bases) were provided to CENTIPEDE. Only those footprints with a posterior probability ≥ 0.99 were retained.
Detection of damaging SNVs
Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) was run on all exonic SNVs from GM12878 to determine their likelihood of causing a damaging mutation (Adzhubei et al. 2010). Mutations that were both damaging and heterozygous were examined to determine if they formed compound heterozygous damaging mutations with RegulomeDB SNVs.
Identification of literature for manual annotation
Literature relevant to the genes were identified by querying PubMed using the approved HGNC gene symbol, name, and alias, including a set of noncoding regions (introns OR promoter OR UTR OR miRNA OR insulator OR enhancer OR silencer) and restricted to abstracts that have been indexed to the MeSH term “human.” The full texts of these papers were downloaded via PubGet (http://pubget.com/) and EndNote (http://www.endnote.com/) and converted into plain text using pdf2text (http://www.foolabs.com/xpdf/home.html).
The full text of these articles was searched for word stems “bind” and “muta” in a single paragraph. The pdf2text conversion software keeps paragraphs together as a single line. Therefore, both words did not need to exist in a single sentence. The word stem “bind” was chosen because it can represent DNA binding or RNA binding activities independent of an assay, while the word stem “muta” (for mutated or mutant or mutagenesis) indicates that studies were performed to assess whether that nucleotide or region is necessary and sufficient for activity.
Manual curation of nucleotide regions
Nucleotide sequences from publications that demonstrated their function were mapped to the H. sapiens hg19 assembly using BLAST. The coordinates of these sequences were curated with the following data types when available: mutant sequence or sequence variant, experimental method, cell line or tissue, regulatory factors, and effect on gene expression. References for each manual annotation are available in the database as a link from overlapping SNVs.
Data access
The RegulomeDB database is accessible at http://www.regulomedb.org/. All ENCODE data are available through the ENCODE portal at http://encodeproject.org/. Complete Genomics data are available from http://www.completegenomics.com/ using Complete Genomics assembly software version 1.10.0.17.
Acknowledgments
This work was supported by the ENCODE Consortium under Grant No. NIH 5U54 HG 004558 and by the Beta Cell Consortium under Grant No. VUMC 38204.
Footnotes
[Supplemental material is available for this article.]
Article and supplemental material are at http://www.genome.org/cgi/doi/10.1101/gr.137323.112.
Freely available online through the Genome Research Open Access option.
References
- The 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, Bates JS, Hu Y, Kelly JA, Kaufman KM, et al. 2011. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet 43: 253–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. 2009. Diversity and complexity in DNA recognition by transcription factors. Science 324: 1720–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW III, Bulyk ML 2006. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol 24: 1429–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Peña-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET, et al. 2008. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133: 1266–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AP, Song L, Lee B-K, London D, Keefe D, Birney E, Iyer VR, Crawford GE, Furey TS 2011. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res 21: 456–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryne JC, Valen E, Tang M-HE, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A 2008. JASPAR, the open access database of transcription factor-binding profiles: New content and tools in the 2008 update. Nucleic Acids Res 36: D102–D106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HYK, Chen R, Miriami E, Karczewski KJ, Hariharan M, Dewey FE, et al. 2012. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148: 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Pai AA, Pique-Regi R, Veyrieras J-B, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. 2012. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482: 390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Arcelus MG, Sekowska M, et al. 2009. Common regulatory variation impacts gene expression in a cell type–dependent manner. Science 325: 1246–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2012. An integrated encyclopedia of DNA elements in the human genome. Nature (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M 2010. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol 28: 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai S-L, Arepalli S, Dillman A, Rafferty IP, Troncoso J, et al. 2010. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet 6: e1000952 doi: 10.1371/journal.pgen.1000952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res (this issue). doi: 10.1101/gr.135350.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA 2009. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci 106: 9362–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, MacArthur J, Wise A, Junkins H, Hall P, Klemm A, Manolio T 2012. A catalog of published genome-wide association studies http://www.genome.gov/gwastudies/
- Hollenhorst PC, Chandler KJ, Poulsen RL, Johnson WE, Speck NA, Graves BJ 2009. DNA specificity determinants associate with distinct transcription factor functions. PLoS Genet 5: e1000778 doi: 10.1371/journal.pgen.1000778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Schones DE, Cui K, Ybarra R, Northrup D, Tang Q, Gattinoni L, Restifo NP, Huang S, Zhao K 2011. Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome Res 21: 1650–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium 2005. A haplotype map of the human genome. Nature 437: 1299–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449: 851–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolma A, Kivioja T, Toivonen J, Cheng L, Wei G, Enge M, Taipale M, Vaquerizas JM, Yan J, Sillanpää MJ, et al. 2010. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res 20: 861–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Tatonetti NP, Landt SG, Yang X, Slifer T, Altman RB, Snyder M 2011. Cooperative transcription factor associations discovered using regulatory variation. Proc Natl Acad Sci 108: 13353–13358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, Habegger L, Rozowsky J, Shi M, Urban AE, et al. 2010. Variation in transcription factor binding among humans. Science 328: 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, Park PJ 2008. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26: 1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo KA, Bauchmann MK, Baumann AP, Donahue CJ, Thiede MA, Hayes LS, des Etages SAG, Fraenkel E 2011. Genome-wide profiling of H3K56 acetylation and transcription factor binding sites in human adipocytes. PLoS ONE 6: e19778 doi: 10.1371/journal.pone.0019778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. 2009. Finding the missing heritability of complex diseases. Nature 461: 747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. 2006. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34: D108–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, Guigo R, Dermitzakis ET 2010. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464: 773–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L, Kaleem M, Leung D, Bryden L, Nath P, et al. 2007. A survey of genetic human cortical gene expression. Nat Genet 39: 1494–1499 [DOI] [PubMed] [Google Scholar]
- Newburger DE, Bulyk ML 2009. UniPROBE: An online database of protein binding microarray data on protein–DNA interactions. Nucleic Acids Res 37: D77–D82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, Habib N, Yosef N, Chang CY, Shay T, et al. 2011. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144: 296–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palii CG, Perez-Iratxeta C, Yao Z, Cao Y, Dai F, Davison J, Atkins H, Allan D, Dilworth FJ, Gentleman R, et al. 2011. Differential genomic targeting of the transcription factor TAL1 in alternate haematopoietic lineages. EMBO J 30: 494–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras J-B, Stephens M, Gilad Y, Pritchard JK 2010. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464: 768–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique-Regi R, Degner JF, Pai AA, Gaffney DJ, Gilad Y, Pritchard JK 2011. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res 21: 447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF 2011. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147: 1408–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, et al. 2011. AlleleSeq: Analysis of allele-specific expression and binding in a network framework. Mol Syst Biol 7: 522 doi: 10.1038/msb.2011.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. 2008. Mapping the genetic architecture of gene expression in human liver. PLoS Biol 6: e107 doi: 10.1371/journal.pbio.0060107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer CD, McCabe CD, Ali-Seyed M, Berger MF, Bulyk ML, Moreno CS 2009. Genome-wide promoter analysis of the SOX4 transcriptional network in prostate cancer cells. Cancer Res 69: 709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M 2012. Linking disease associations with regulatory information in the human genome. Genome Res (this issue). doi: 10.1101/gr.136127.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, et al. 2007. Population genomics of human gene expression. Nat Genet 39: 1217–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touzet H, Varré J-S 2007. Efficient and accurate P-value computation for Position Weight Matrices. Algorithms Mol Biol 2: 15 doi: 10.1186/1748-7188-2-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchese F, Fa' M, Liu S, Zhang H, Hidalgo A, Schmidt SD, Yamaguchi H, Yoshii N, Mathews PM, Nixon RA, et al. 2008. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest 118: 2796–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi MP, Shin H, He HH, Sulahian R, Meyer CA, Montgomery RK, Fleet JC, Brown M, Liu XS, Shivdasani RA 2010. Differentiation-specific histone modifications reveal dynamic chromatin interactions and partners for the intestinal transcription factor CDX2. Dev Cell 19: 713–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyrieras J-B, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, Stephens M, Pritchard JK 2008. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet 4: e1000214 doi: 10.1371/journal.pgen.10002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA 2007. VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res 35: D88–D92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD, Kellis M 2012. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 40: D930–D934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G-H, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, et al. 2010. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J 29: 2147–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Cui K, Jothi R, Zhao D-M, Jing X, Zhao K, Xue H-H 2011. GABP controls a critical transcription regulatory module that is essential for maintenance and differentiation of hematopoietic stem/progenitor cells. Blood 117: 2166–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, Maouche S, Germain M, Lackner K, Rossmann H, et al. 2010. Genetics and beyond—the transcriptome of human monocytes and disease susceptibility. PLoS ONE 5: e10693 doi: 10.1371/journal.pone.0010693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137 doi: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]