

Synopsis
Airway smooth muscle (ASM) has long been recognized as the main cell type responsible for bronchial hyperresponsiveness. It has thus been considered as a target for bronchodilation. In asthma however, there is a complex relationship between ASM and inflammatory cells such as mast cells and T lymphocytes. Moreover, the increased ASM mass in the asthmatic airways is one of the key features of airway remodeling. This article aims to review the main concepts about the three possible roles of ASM in asthma including (i) contractile tone, (ii) inflammatory response and (iii) remodeling.
Keywords: Bronchodilators, Hyperresponsiveness, Inflammation, Remodeling, Smooth muscle
Introduction
Asthma is a chronic disease, characterized by the association of bronchial hyperresponsiveness (BHR), inflammation, and remodeling (1). Airway smooth muscle (ASM) has long been recognized as the main cell type responsible for bronchial contraction and BHR (2). It has thus been logically considered as the key target for bronchodilation. Regarding asthmatic inflammation, a high number of eosinophils infiltrate both bronchial epithelium and submucosa (3), but there is apparently no eosinophil infiltration of the ASM layer (4). However, other inflammatory cells such as mast cells (4) or T lymphocytes (5) have been shown to infiltrate the ASM suggesting a complex relationship between ASM cells and inflammatory cells and even a possible role for ASM to organize inflammation. Asthmatic bronchial remodeling is characterized by various structural changes including abnormal epithelium, sub-epithelial membrane thickening, alteration of the extracellular matrix (ECM) deposition, neoangiogenesis, mucus gland hypertrophy and increased ASM mass (6). Recent evidence suggests that the increased ASM mass is the key feature of bronchial remodeling in asthma since, on the one hand, it is associated with severe asthma phenotype (7), and, on the other hand, it is correlated with a decrease in lung function (5, 8). This article reviews the main concepts about the three possible roles of ASM in asthma including (i) contractile tone, (ii) inflammatory response and (iii) remodelling.
Airway smooth muscle shortening and relaxation
In normal airways, ASM cell contraction regulates airway caliber and bronchomotor tone. Using isolated bronchial preparations, studies have compared the isotonic length and/or force generation of tissues derived from asthmatics and non-asthmatics. Interestingly, not all studies demonstrated an increased force generation in asthmatic tissues when compared to control tissues (9). These conflicting data likely can be attributed to differences in the experimental and methodological approaches used (10). Several parameters, such as the degree of tissue elastance, smooth muscle mass, and knowledge of the optimal length, were found to be important factors when evaluating the force generating capacity of ASM preparations derived from asthmatics (9, 10).
Although conflicting data exist in studies comparing smooth muscle responsiveness between normal and asthmatic patients, a number of studies report that passive sensitization of human ASM with asthmatic serum induces a non-specific increase in smooth muscle responsiveness (11–14), demonstrating the existence of mediators in the serum of asthmatic patients that promote airway responsiveness. While the precise nature of these mediators remains incompletely defined, evidence suggests that TNF-α, IL-13 or IL-1β can induce BHR in both humans and animals. Cytokines also can “prime” ASM to become hyperresponsive to contractile agonists in vitro, supporting the concept that cytokines modulate agonist-induced ASM contractile function. Other pro-inflammatory mediators, such as lysophosphatidic acid, a bioactive lipid released from activated platelets, phospholipase A2 and leukotriene C4, also enhance ASM responsiveness in vitro to contractile agonists such as acetylcholine, methacholine and serotonin. Together, these studies suggest that pro-inflammatory mediators induce BHR by enhancing ASM contraction and/or altering ASM relaxation (see Figure 1). Understanding the mechanisms by which inflammatory mediators modulate ASM contractile reactivity may offer new insight into the molecular mechanisms that modulate BHR in asthma (reviewed in (15)).
Figure 1. Excitation-contraction coupling in airway smooth muscle.

Effects of pro-inflammatory cytokines and β2-adrenergic receptor agonists on excitation-contraction coupling in ASM cells. Contractile agonists activate receptors that influence intracellular signaling, affecting calcium homeostasis and sensitization as well as the function and expression of GPCRs and CD38. Inflammatory cytokines bind to receptors and modulate calcium homeostasis by increasing expression of CD38 and increasing Ca2+ release from the sarcoplasmic reticulum. Inflammatory cytokines such as IL-13, IL-1β and TNF-α also increase Rho kinase activity to modulate the calcium sensitization pathways. β2-adrenergic receptor agonists regulate calcium homeostasis and calcium sensitization by inhibiting RhoA activation, Ca2+ release from the sarcoplasmic reticulum, and actin-myosin crossbridging. cADPR, cyclic ADP ribose; CaM, calmodulin; DAG, diacylglycerol; GEF, guanine exchange factor; GPCR, G-protein–coupled receptor; IP3, inositol tri-phosphate; MLC, myosin light chain; MLCK, myosin light chain kinase; PIP2, phophatylinositol 4,5 biphosphate; PLC, phospholipase C; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; β2AR, β2-adrenergic receptor.
The level of intracellular calcium regulates, in part, ASM shortening. Activation of an ASM cell by an agonist induces a rapid rise in [Ca2+]i, associated with the release of intracellular calcium stores, to a peak level roughly tenfold higher than the resting level (100 nM to greater than 1 μM with maximum agonist stimulation). Following this peak, calcium levels fall but remain elevated provided that the excitatory stimulus remains present. The elevation in [Ca2+]i activates the calcium/calmodulin-sensitive myosin light chain kinase (MLCK), leading to phosphorylation of the regulatory myosin light chain (MLC20) at Serine 19. Phosphorylation of this residue by myosin ATPase activity initiates crossbridge cycling between myosin and actin. ATP binding, hydrolysis and ADP release continue as long as MLC20 is phosphorylated; dephosphorylation by the MLC phosphatase terminates crossbridge cycling and relaxes smooth muscle (reviewed in (16)).
Considering the central role of Ca2+ in regulating ASM contractile function, investigators postulate that alterations in Ca2+-regulatory mechanisms likely impair ASM contractility. Studies using cultured human tracheal or bronchial smooth muscle cells, as in vitro models of ASM responsiveness, convincingly demonstrated that Gq-protein coupled receptors (GPCR)-associated signaling in ASM can be modulated by a variety of inflammatory stimuli. Cytokines, such as TNF-α, augment agonist-induced ASM contractility by enhancing, in a non-specific manner, agonist-evoked Ca2+ transients (to bradykinin, carbachol) (15). The hypothesis that changes in GPCR-associated Ca2+ signaling represent an important mechanism underlying the development of BHR has also been supported by other studies. Tao and colleagues showed that ASM cells derived from hyperresponsive inbred rats have an augmented bradykinin-induced Ca2+ response when compared to ASM cells derived from normoresponsive rats (17). Deshpande and colleagues demonstrated that in addition to TNF-α, other cytokines including IL-1β and, in to a lesser degree, IFNγ augments Ca2+ responses induced by carbachol, bradykinin and thrombin (18). In a similar manner, IL-13, a Th2 type important mediator in allergic asthma (19), also non-specifically increased Ca2+ responses to agonists (20–23). Microarray technology used to study the modulation of gene expression of ASM by IL-13 revealed a diversity of potential molecular mechanisms influencing ASM responsiveness, including changes in cytoskeletal proteins, receptors or calcium regulators (24). Together, these data show that “pro-asthmatic” cytokines, in a non-specific manner, enhance GPCR-associated Ca2+ responses in ASM, a mechanism likely to affect ASM contractility.
Reports in C3H/HeJ, Balb/C and A/J mice revealed that differences in ASM contractility among species may not require changes in GPCR agonist-induced Ca2+ responses but rather involve changes in the Ca2+ sensitivity of the contractile apparatus (25). A possible mechanism involves the small monomeric G protein Rho that can augment ASM contractility by increasing levels of MLC phosphorylation via the Rho-activated kinase (ROCK) dependent suppression of MLC phosphatase (26, 27). Both RhoA and ROCK are activated by a variety of stimuli associated with the development of BHR including cytokines (28–31), sphingolipids (32–34), mechanical stress (35) and isoprostane (36). The RhoA/Rho kinase pathway regulates the expression of serum response factor-dependent smooth muscle specific genes in canine ASM cells (37), a mechanism that identifies the importance of the Rho-kinase pathway in maintaining a contractile phenotype recently described in bovine ASM tissues (38). Rho pathways modulate diverse cellular responses in ASM cells including the regulation of Ca2+ influx (39) and cell proliferation (40). Possibly, abnormal RhoA activity and/or expression will dramatically alter ASM contractility not only via the Ca2+ sensitization but also through the increased expression of Rho-dependent contractile proteins. A report using the Y-27632 inhibitor confirmed that the non-specific BHR as well as the specific allergen responsiveness induced by passive sensitization requires the activation of Rho-kinase (41).
Changes in ASM contractile properties play an important role in the development of BHR associated with chronic airway diseases such as asthma. In vitro studies support the concept that a variety of “pro-asthmatic” signals such as physical (repeated stretch) or chemical exposures (cytokines) drastically augment ASM contractile force by altering multiple key pathways: i) via the aberrant activation of contractile and/or impaired function of relaxant receptors (desensitization), ii) the alteration of Ca2+ regulatory signaling molecules (CD38, SERCA, Ca2+ channels), and iii) the activity of elements of the contractile apparatus through Rho-dependent pathways. Defining the inflammatory signals (factors and associated mechanisms) involved in the regulation of ASM responsiveness may represent a potential new target for the treatment of BHR. Pro-inflammatory mediators can modulate the density of contractile agonist receptors on ASM cells. TNF-α induced a dramatic decrease in muscarinic receptor density. In contrast, expression of the bradykinin B2 receptor was rapidly increased in ASM exposed to IL-1β or TNF-α by a prostanoid-dependent regulation of gene transcription and by the activation of the Ras/Raf/MEK pathway. Surprisingly, the β2-agonist fenoterol or the steroid methylprednisolone also increased expression of the histamine H1 and bradykinin B2 receptors, an effect that involved both increased gene expression and mRNA stability. This increase in H1 receptor expression was associated with an increase in the contractile response to histamine. Whether β2-agonists and steroids induce such effects in vivo remains unclear. These studies, however, suggest that current asthma therapy may also modulate BHR by altering contractile agonist receptor expression in ASM (reviewed in (15)).
In human ASM cells, contractile agonists bind GPCR and activate phospholipase C. The subsequent hydrolysis of phosphatidylinositol 4,5 bisphosphate into inositol trisphosphate and diacylglycerol ultimately results in an increase in [Ca2+]i (reviewed in (16)). Since most of the inflammatory agents do not evoke either a calcium response or phosphoinositide hydrolysis in human ASM, modulation of agonist-induced increases in [Ca2+]i by extracellular stimuli may be due to the modulation of downstream GPCR signaling. TNF-α increased the amount, as well as the activity, of G-proteins in several cell types including ASM (42, 43). The finding that TNF-α potentiated calcium mobilization in response to NaF (44), an agent that bypasses membrane receptors and directly activates G-proteins (45, 46), supports the notion that TNF-α may act directly at the level of G-proteins.
Studies now show that bradykinin-evoked phosphoinositide accumulation in human ASM is significantly enhanced by various cytokines such as TNF-α and IL-1β (44, 47–49). The effect of cytokines on agonist-evoked calcium responses seems to be stimulus-specific, however, since pretreatment of ASM cells with IL-1β diminished phosphoinositide metabolism induced by histamine (50). In addition to their effect on calcium signaling, cytokines may also modulate β2-adrenergic function. TNF-α as well as IL-1β also suppress isoproterenol-stimulated activation of adenylyl cyclase (51–53). A recent report showed that IL-13 is also able to impair ASM responsiveness to β2-adrenergic stimuli (54) showing that cytokines may promote BHR by impairing β2-adrenergic responsiveness in ASM cells.
Pro-inflammatory cytokines affect ASM contractility on many levels. Alterations in calcium homeostasis and sensitivity, as well as contractile agonist receptor expression and signal transduction pathways, have profound effects on airway hyperreactivity. The ability of anti-inflammatory therapies, such as corticosteroids, to modulate these effects are discussed later in this review (also reviewed in (15, 55)).
To date, the most effective therapeutic approaches in asthma are corticosteroids and β2-adrenergic receptor agonists, which abrogate airway inflammation and reverse bronchoconstriction respectively. Given the evidence that ASM cells secrete and express immunomodulatory proteins, investigators are now studying the cellular and molecular processes that regulate ASM synthetic function and examining the role of dexamethasone and β-agonists in modulating cytokine-induced synthetic responses and bronchodilation.
In asthma, β-agonists such as isoproterenol, albuterol, salmeterol and formoterol are therapeutic agents that promote bronchodilation by stimulating receptors coupled to Gs, and that, in turn, activate adenylyl cyclase, increase [cAMP]i and stimulate cAMP-dependent protein kinase (A-kinase) in ASM. In a similar manner, PGE2, which is produced in large quantities at sites of inflammation, also increases [cAMP]i in human ASM cells and is a potent and effective bronchodilator (56). Evidence suggests that [cAMP]i mobilizing agents in ASM cells also modulate cytokine-induced synthetic function. In TNF-α-stimulated ASM cells, both eotaxin and RANTES expression are effectively inhibited by isoproterenol, PGE2, dibutyl-[cAMP]i, or the phosphodiesterase inhibitors, rolipram and cilomast (57–59). TNF-α-induced IL-8 secretion was also inhibited by the combination of [cAMP]i mobilizing agents and corticosteroids (60). Similarly, sphingosine-1-phosphate, which activates a Gs protein coupled receptor and increases [cAMP]i, abrogated TNF-α-induced RANTES secretion in ASM cells (61).
In contrast to the effects of [cAMP]i on chemokine secretion, pharmacological agents that increase [cAMP]i also markedly stimulate secretion of IL-6 in human ASM cells (57). This appears to be due to effects on basal IL-6 promoter activity (62). Whether the secreted IL-6 modulates ASM cell function in an autocrine manner or alters leukocyte function in the submucosa remains unknown. Since studies show that overexpression of IL-6 decreases acetylcholine responsiveness in transgenic mice (63), the role of IL-6 in asthma may be that of an anti-inflammatory signal. More recently, investigators reported that cAMP limits secretion of GM-CSF by ASM cells. Cyclo-oxygenase inhibitors reduce PGE2 and enhance cytokine-induced secretion of GM-CSF (64, 65), while PDE type IV inhibitors reduce GM-CSF secretion in vitro and antigen-induced BHR in an animal model (64, 66). Taken together, current evidence suggests that some but not all pro-inflammatory functions in ASM cells are inhibited by [cAMP]i mobilizing agents.
Conflicting reports exist, however, concerning effects of increased [cAMP]i on lymphocyte adhesion and migration through cytokine-activated endothelial cells (67, 68). The controversy regarding the role of [cAMP]i in modulating cell adhesion likely reflects differences in the cytokines used to stimulate endothelial cells, or the temporal differences in the addition of the agonists used to increase [cAMP]i. Far less is known concerning [cAMP]i effects on smooth muscle-leukocyte adhesion. In human ASM cells, activation of [cAMP]i-dependent pathways inhibited, in part, both TNF-α-mediated induction of ICAM-1 and VCAM-1 expression and adhesion of activated T cells to ASM cells. Interestingly, the basal expression of ICAM-1 and VCAM-1, as well as the binding of activated T cells to unstimulated ASM, was resistant to increases in [cAMP]i (69). Together these studies suggest that cytokine-induced expression of cell adhesion molecules and T cell adhesion to ASM cells are modulated by changes in [cAMP]i.
Airway smooth muscle can also organize inflammation
There is plenty of evidence that, in asthma, a complex relationship involves ASM and inflammatory cells. Indeed, ASM produces a variety of chemotactic mediators and expresses different adhesion molecules (Table 1), which can participate to both the recruitment and the micro-localization of inflammatory cells within the ASM. Asthmatic ASM is infiltrated by both mast cells (4) and T lymphocytes (5) but apparently no eosinophil (4).
Table 1.
Airway smooth muscle production of chemotactic mediators and adhesion molecules
| Factors | Spontaneous | After stimulation | References |
|---|---|---|---|
| Chemokines | |||
| CCL2 (MCP-1) | − | + (IL-1β, TNF-α) | (146, 147) |
| CCL5 (RANTES) | − | + (IL-1β, TNF-α) | (147–149) |
| CCL11 (Eotaxin) | + | + (IL-1β, TNF-α) | (78, 150, 151) |
| CCL19 (MIP-3β) | + | (95) | |
| CXCL8 (IL-8) | − | + (IL-1α, IL-1β, TNF-α) | (78, 146, 152) |
| CXCL9 (Mig) | − | + (IFN-γ, IL-1β, TNF-α) | (78) |
| CXCL10 (IP-10) | − | + (IFN-γ, IL-1β, TNF-α) | (78) |
| CXCL12 (SDF-1α) | − | + (IFN-γ, IL-1β, TNF-α) | (78) |
| CX3CL1 (Fractalkine) | − | + (TNF-α) | (80) |
| Cytokines | |||
| SCF | + | (153) | |
| TGF-β1 | + | + (Angiotensin II) | (154) |
| IFN-γ | + | + (AS) | (155) |
| GM-CSF | + | + (AS, IL-1β, TNF-α) | (155–157) |
| L-2 | + | + (AS) | (155) |
| IL-5 | + | + (AS) | (155) |
| IL-6 | − | + (IL-1, TGF-β, TNF-α) | (149, 158) |
| IL-11 | − | + (IL-1, TGF-β, virus) | (158) |
| IL-12 | + | + (AS) | (155) |
| IL-33 | + | + (IFN-γ, TNF-α | (159) |
| Adhesion and co-stimulatory molecules | |||
| CD11a | + | (100) | |
| CD40 | + | + (TNF-α, IFN-γ) | (103) |
| CD40L | + | (103) | |
| CD44 | + | (99) | |
| CD80, CD86 | + | (100) | |
| ICAM-1 | + | + (TNF-α, IL-1β, IL-5, AS) | (99, 160, 161) |
| VCAM-1 | + | + (TNF-α, IL-1β) | (99, 160) |
Abbreviations: AS: asthmatic serum, GM-CSF: Granulocyte macrophage colony-stimulating factor, ICAM: intercellular adhesion molecule, IL: interleukin, IFN: interferon, SCF: stem cell factor, TGF: transforming growth factor, TNF: tumor necrosis factor, VCAM: vascular cell adhesion molecule.
The mast cell infiltration of the ASM layer, also called mast cell myositis (70) appeared to be a specific feature of asthma, as compared to that of patients suffering from eosinophilic bronchitis and healthy subjects (4, 71). Interestingly, the ASM mast cell infiltration is observed in various asthma phenotypes, including eosinophilic and non eosinophilic asthma (72), severe and non severe asthma (71, 73–75), but also atopic and non atopic asthma, even if the number of mast cells were significantly higher in the ASM of atopic asthmatics (76). Moreover, asthma treatments including inhaled corticosteroids did not change mast cell myositis (71, 73). The mechanism of such a myositis has been firstly related with the production of mast cell chemotactic factors by the ASM itself, through an auto-activation loop (77). Indeed, upon activation, mast cells release tryptase and pro-inflammatory cytokines, such as TNF-α, which stimulate the production of TGF-β1 and, to a lesser extent, SCF by ASM cells, which in turns, induce mast cell chemotaxis (77). Moreover, ASM promote mast cell chemotaxis through the secretion of a wide array of chemotactic factors, upon stimulation by Th1 (78), Th2 (78, 79) or pro-inflammatory cytokines (77, 80). ASM also produces functionally active CXCL10 (78), CXCL8 (79), CCL11 (79) and CX3CL1 (80), even if for CX3CL1, the additional presence of vasoactive intestinal peptide (VIP) is necessary (80). Taken together, mast cell migration is induced by the production of various mediators secreted by the ASM itself, which is closely related to the ASM inflammatory micro-environment.
Once present within the ASM bundle, mast cell can adhere to ASM. This adhesion has been initially reported to a cell - cell direct interaction involving an Ig superfamily member, i.e. CADM1 (cell adhesion molecule 1), previously known as TLSC-1 (tumor suppressor in lung cancer 1) (81, 82) (Figure 2). However, blocking CADM1 partially reduced the adhesion of mast cells to ASM, suggesting an alternative mechanism (81). Indeed, mast cell - ASM adherence also involved cell - extracellular matrix (ECM) - cell interaction through type I collagen, CD44 and CD51 (83) (Figure 2). This adhesion was improved under inflammatory conditions or using asthmatic ASM cells (83). These latter in vitro findings are in agreement with ex vivo ultrastructural analysis of asthmatic ASM using electron microscopy (84). Indeed, such analysis did not demonstrate any direct cell-cell contact between ASM and mast cells, but only close contacts without tight junction (84). The majority of mast cells infiltrating the asthmatic ASM bundles are typically of the MCTC phenotype, containing both tryptase and chymase (4, 85, 86). Interestingly, these mast cells infiltrate the ASM of both large and small airways, and exhibit marked features of chronic ongoing activation (74, 76). Such findings were also confirmed by ultrastructural analysis of asthmatic ASM using electron microscopy (84). However, little is known about the mechanisms by which mast cell activation may occur within the ASM layer (87). Mast cell degranulation may result from IgE-dependent activation, especially in atopic patients (76). However, IgE-independent mechanisms have also been evoked, following mast cell - ASM interaction through the complement C3a or SCF (82, 88–90), for instance, or, following bacterial or viral infection through Toll-like receptors (87).
Figure 2. Airway smooth muscle - mast cell interaction.
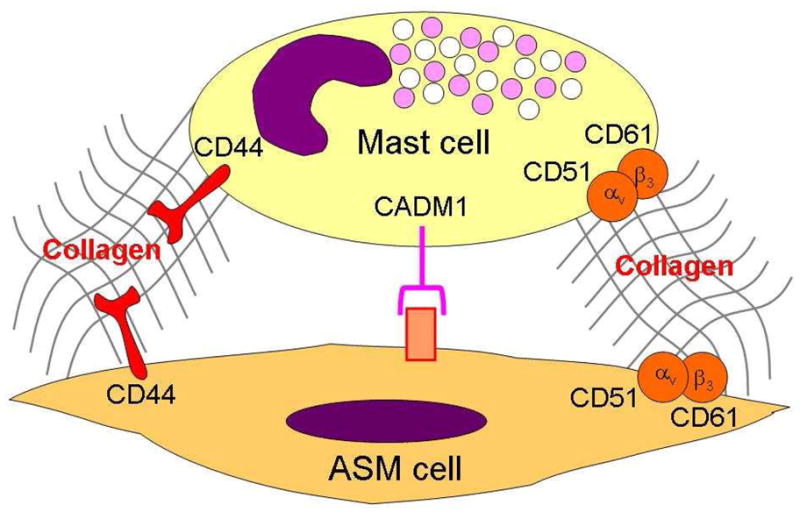
Airway smooth muscle (ASM) can adhere to mast cell through cell-cell direct interaction involving cell adhesion molecule 1 (CADM1) and through cell-extracellular matrix interaction involving type I collagen and both CD44 and CD51.
Taking into account the micro-localization of mast cells within the asthmatic ASM layer, the adherence of mast cells to the ASM, and, the features of mast cell activation within the ASM, it may be suggested a close functional relationship between these two cell types. On the one hand, mast cells are likely to alter functional and phenotypic properties of ASM cells. Indeed, mast cell-derived mediators may contribute to BHR (see part 1) and ASM remodeling (see part 3) (6). For instance, the major mast cell product tryptase induces ASM calcium increase (91) and non specific BHR to histamine in vitro (92) and in vivo (93). Tryptase also increases the production TGF-β1 by ASM cell (77), which promotes the differentiation of ASM cell towards a more contractile phenotype, characterized by both an increased expression of α-smooth muscle actin and an enhanced ASM contractility (88). The number of mast cells within the ASM layer is positively correlated with the degree of BHR (4, 71), and with the intensity of α-smooth muscle actin staining (88). Furthermore, mast cell myositis may also promote ASM remodeling. Indeed, mast cell-derived tryptase has been shown to stimulate ASM proliferation (94). Moreover, mast cell – derived CCL19 mediate ASM migration through the activation of ASM CCR7 (95). However, neither ASM proliferation (89, 96) nor ASM survival appeared to be modulated by co-cultured mast cells (89). In addition, CCL11/CCR3 mediated ASM cell migration was inhibited by mast cells (97). Furthermore, no correlation was found between the number of mast cells within the ASM and ASM mass, supporting the modest role of mast cells in ASM remodeling (76). On the other hand, ASM cells can alter mast cell functional and phenotypic properties. Indeed, ASM cells can promote mast cell survival and proliferation, through a mechanism involving a cooperative interaction between ASM membrane-bound SCF, soluble IL-6 and mast cell-CADM1 (82). ASM cell-derived ECM proteins may also promote mast cells differentiation towards a fibroblastoid phenotype, characterized by the expression of fibroblast markers and fibroblast-like morphology. This feature seems to be specific of mast cells within the ASM layer, since fibroblast markers were not expressed by mast cells within the submucosa (75).
Regarding T cell infiltration within the asthmatic ASM layer, only few studies have been performed. CD4+ T cell micro-localization was initially reported in an experimental asthma model within the ASM layer of OVA-sensitized rat as compared to non sensitized animals (98). These findings have been further confirmed in human asthmatics (5, 84) and are related to asthma severity (5). As compared to mast cell infiltration, the number of T cells seems to be lower within the ASM layer (4, 84). ASM cells are able to produce appropriate chemotactic factors for T cells, including CCL5 (RANTES) or CXCL10 (IP-10) (78). However, direct chemotactic properties of ASM to T cells remain to be demonstrated.
Close contacts between ASM and T cells have been shown in asthma (5, 84), suggesting cell - cell adherence between these two cell types. Lazaar and coworkers firstly demonstrated that activated T cells can adhere in vitro to resting ASM cells from non asthmatic patients and that such an adhesion was enhanced when ASM cells were primed with pro-inflammatory cytokines such as TNF-α (99). These findings were further confirmed independently (100). This adhesion involves on the one hand, CD44, ICAM-1 (Intercellular cell adhesion molecule 1), VCAM-1 (Vascular cell adhesion molecule 1) expressed by ASM cells, and, on the other hand, CD44, LFA-1 (Lymphocyte function-associated antigen 1) and VLA-4 (Very late antigen 4) expressed by T cells (99). More recently, non asthmatic ASM cells, pulsed to the superantigen staphylococcal enterotoxin A (SEA), have been shown to adhere to T cells by presenting the SEA via their MHC class II (101). Although ASM cells express MHC class II constitutively and under stimulation (100, 102), these cells are not classically considered as an antigen presenting cell. Consequently, these findings support an emerging role of ASM cell as an immunomodulatory cell. However, except for VCAM1, which forms clusters in the asthmatic ASM ex vivo suggesting VCAM1-mediated intercellular signaling, the role of the above molecules in adherence between T cell and ASM cell from asthmatic patients has not yet been considered. Two other ASM cell-surface molecules, CD40 (100, 103–105) and OX40 ligand (105–107), both expressed in asthmatic and non asthmatic ASM cells, have also been suggested in promoting ASM cell – T cell adherence. These co-stimulatory cell-surface molecules, members of the TNF superfamily, respectively bind to CD154 and OX40 on activated T cells (108, 109). However, their role in the adherence of T cells to asthmatic ASM remains to be investigated. By contrast, a possible role for mast cell chymase has been suggested since this mast cell protease is able to inhibit T cell adhesion to non asthmatic ASM cells in vitro (110).
Close interaction between T cells and ASM cells can stimulate a cross-talk between these two cell types, but little is known about the functional consequences of such interaction. On the one hand, T cells may alter functional properties of ASM cells. Firstly, T cells may alter ASM contractile phenotype, enhancing ASM contractility to acetylcholine and reducing its relaxation to isoproterenol in isolated rabbit bronchi (100). Moreover, T cells may also induce ASM remodeling, and more precisely ASM hyperplasia (5, 98, 99). Indeed, in an experimental rat asthma model, adoptively transferred CD4+ T cells from OVA-sensitized rats induced an increase in ASM mass, which was both related with an increased ASM proliferation and decreased apoptosis ex vivo (98). Such findings were confirmed in vitro only upon direct CD4+ T cells – ASM cells contact, highlighting the need for close cellular interaction between these two cell types (98). These observations are in agreement with a previous study demonstrating in vitro a role for T cell in ASM DNA synthesis and proliferation in ASM cells from non asthmatic patients (99). The role of T cells in driving ASM remodeling was later confirmed in human asthmatics (5). Moreover, the number of T cells infiltrating the asthmatic ASM correlated with ASM mass (5). Collectively, these findings suggest an emerging role of T cells in both ASM hyperresponsiveness and remodeling. On the other hand, ASM cells may also alter functional properties of T cells. Indeed, direct contact between CD4+ T cells and ASM cells also enhances T cell survival, thus possibly contributing to the perpetuation of bronchial inflammation (98). Cultured human non asthmatic ASM cells are able to present superantigens via their MHC class II molecules to resting CD4+ T cells (101), which induces CD4+ T cells activation, adherence between these cells and finally the release of IL-13, that, in turns, allow increasing the contractile response to acetylcholine of isolated rabbit bronchi (101).
Airway smooth muscle remodeling
The fact that the increased ASM mass in the asthmatic airway is one of the key features of the structural changes which constitute airway remodeling is well recognized (111). Moreover this increase has been attributed with a pivotal role in determining one of the key functional characteristics of the asthmatic airway namely airway hyperresponsiveness (112). The increased muscle mass is likely to be the most important abnormality responsible for the exaggerated response to bronchoconstricting stimuli in asthma, resulting in development of greater narrowing of the lumen. More recently there has been much investigation into the mechanisms underlying this increase. Whether hyperplasia, hypertrophy, or their combination is the major contributing process remains unclear, and it is possible that the changes in the muscle are not homogeneous throughout the airway. Ebina et al (113) described two patterns in lungs harvested post mortem – type I in which hyperplasia was present in the walls of large bronchi with no hypertrophy and type II in which hypertrophy was present in the whole airway and mild hyperplasia in the larger airways. Additional theories as to the reason for the increase in muscle bulk relate to an increase in ECM proteins deposited in and around the muscle bundles (see below) and migration of mesenchymal precursors from the peripheral blood into the lung (114).
Although the majority of research has focused recently on the mechanisms underlying hyperplasia, some interesting observations have been reported regarding hypertrophy and, specifically, that induced by stretch. This is important because bronchoconstriction itself constitutes a form of stretch or strain (115) and this is highly relevant to the asthmatic airway. Interestingly, the effect of mechanical stretch is to induce hypertrophy of human ASM and this appears to be mediated by the action of a microRNA (miRNA) specifically miR-26a (116). Stretch and/or overexpression of miR-26a induces human ASM hypertrophy via an effect on glycogen synthase kinase -3β. In these experiments, stretch also induced hyperplasia but the predominant effect was hypertrophy and miR26a did not affect proliferation.
A lot more attention has been focused on the underlying causes of hyperplasia which, in some studies, has been reported to be the sole pathology underlying the increase in muscle mass (117). This has been attributed to an upregulation of proproliferative pathways or, alternatively, a lack of endogenous braking mechanisms (Table 2). In 2001 (118), it was noted that ASM cells obtained from patients with asthma proliferated at a greater rate than those obtained from people without asthma and this finding was confirmed and extended by Trian et al (119) although this has not always been a consistent finding (89, 120). In the latter study, the increased growth was attributed to mitochondrial biogenesis and to dysfunctional calcium regulation (119). Further studies on calcium homeostasis have demonstrated that the expression and function of sarcoendoplasmic reticulum calcium ATPases, which play a role in the extrusion of calcium from the ASM cell, are downregulated (121, 122). With respect to hyperproliferative pathways, almost every signal transduction protein has been associated with exaggerated proliferation, including, but not limited to the MAP kinases ERK (123) and p38 (124), as well as PI3 kinases (125). Studies conducted using intact tissue, as opposed to cells from patients with asthma, have revealed increased immunohistochemical detection of a marker of proliferation – proliferative cell nuclear antigen, indicating that evidence for increased proliferation of ASM is not merely a phenomenon of cell culture (5, 126).
Table 2.
Modulation of human airway smooth muscle proliferation#
| Proproliferative | Antiproliferative |
|---|---|
| Growth factors | |
| EGF (162) | PGE2 (127, 163) |
| IGF (164) | heparin (163) |
| PDGF (165) | interleukin-13 (166) |
| FGF-2 (167) | VIP (168) |
| Plasma/Inflammatory cell mediators | |
| β-hexosaminidases (169) | Glucocorticoids ^ (128) |
| Elastase (170) | interleukin-4 (167) |
| α-thrombin (171) | beta agonists (127) |
| Tryptase (94) | |
| Sphingosine-1 phosphate (61) | |
| Contractile agents | |
| Endothelin-1* (162) | |
| Leukotriene D4* (172) | |
| Cysteinyl Leukotriene (173) | |
| Histamine (168) | |
| Extracellular matrix proteins | |
| Fibronectin (174) | |
| Collagen-1 (174) | |
Abbreviations: EGF: epidermal growth factor, IGF- insulin-like growth factor, PDGF: platelet derived growth factot, FGF-2: fibroblast growth factor-2,
a co-mitogen,
reviewed in Hirst et al., (175). VIP: vasoactive intestinal peptide,
nonasthmatic cells only
It is possible that internal mechanisms for limiting proliferation could be dysfunctional (Table 2). Levels of endogenous Prostaglandin E2, which is an inhibitor of mitogenesis (127), are decreased in cultures of asthma derived ASM, and this was also found to be the case for protein levels of the transcription factor C/EBPα- an inhibitor of proliferation (128). In addition, evidence that vitamin D inhibits ASM cell proliferation may suggest that low levels in vivo could contribute to unopposed growth (129).
It was originally thought that the structural changes in asthmatic airways that constitute airway remodeling developed as a result of a persistent inflammatory stimulus. This has been challenged by some recent reports regarding the mechanisms underlying remodeling. Bronchoconstriction induced by a non inflammatory stimulus such as methacholine, resulted in significant increases in parameters of remodeling to approximately the same extent as an allergen challenge (130). These changes were assessed by means of biopsies, and, although there was no actual measurement of ASM, the number of proliferating cells in the lamina propria was increased in each challenge. The exact nature of these cells was not explored but they could have been myocytes which had dedifferentiated into myofibroblasts and migrated into the submucosa in response to allergen challenge (131). Increases in sub basement membrane collagen and alterations in epithelial cell morphology – both hallmarks of airway remodeling- were a consistent feature observed in the study by Grainge (130) in response to both inflammatory (allergen) and non inflammatory (methacholine) challenges. Thus, it may be that the structural changes which constitute remodeling are not a consequence of chronic persistent inflammation, as was previously thought, but rather the two processes develop along separate parallel pathways- a hypothesis put forward by Martinez in 2007 (132).
Not only ASM cells per se but also their products – eg ECM proteins contribute to the area of the muscle bundle (133). The fractional area of the matrix is increased in the smooth muscle in cases of fatal asthma (134). The ECM provides a scaffold for support of the cells embedded in it. In addition, ECM proteins have profound biological effects on the smooth muscle – influencing proliferation, formation and release of growth factors, and matrix metalloproteinases (MMPs) which may cleave factors embedded in the matrix and release them to alter function. There is now accumulating evidence that remodeling in the asthmatic airway is associated with changes in the ECM proteins. When ASM cells are exposed to serum from asthmatic patients, increases in many ECM proteins are observed (135). Alterations in the ECM are a well recognized component of remodeling in the asthmatic airway, including enhanced deposition of collagens I, III and V, fibronectin, tenascin, hyaluronan, versican, laminin α2/β2, and perlecan (136–139), whereas decorin, collagen IV and elastin are decreased (140). ASM cells derived from patients with asthma produce a different profile of matrix proteins (141) including increased amounts of fibulin 1-C (142). The importance of these alterations in the matrix proteins lies in the fact that they can profoundly alter the properties of the ASM. Fibulin 1-C for example plays a role in vitro in the enhanced proliferation of asthmatic ASM. The components of the matrix may also modulate response to pharmacotherapy, conferring resistance to corticosteroids and β2 adrenoceptor agonists (143, 144). In intensive studies of tissue from asthma patients who have suffered a fatal attack, elastic fibers and fibronectin are increased and in this same cohort, changes in the MMPs were also noted, with increases in both MMP-9 and -12 detected immunohistochemically in the ASM of large airways (145). Whether these changes are consistent throughout the airways is unclear.
Conclusion
A better knowledge of the various roles of ASM in the pathophysiology of asthma is necessary to understand that ASM is not just a target for bronchodilation. Of course, changes in ASM contractile properties and/or impaired function of relaxant receptors play an important role in the development of BHR in asthma. Bronchodilators can also inhibit some but not all pro-inflammatory functions of ASM in vitro. Moreover, steroids are not able to reduce both mast cell myositis and increased proliferation of asthmatic ASM. This latter appears to be an important mechanism of ASM remodeling. Moreover, T cells may induce ASM hyperplasia and have been involved in BHR. All these findings confirm that the roles of ASM in the pathophysiology of asthma are complex and need further specific pharmaceutical developments.
Key Points.
A better knowledge of the various roles of ASM in the pathophysiology of asthma is necessary to understand that ASM is not just a target for bronchodilation.
Changes in ASM contractile properties and/or impaired function of relaxant receptors play an important role in the development of BHR in asthma.
The roles of ASM in the pathophysiology of asthma are complex and need further specific pharmaceutical developments.
Footnotes
Financial disclosures and conflicts of interest
Grants support for RAP (NIEHS ES013508, NIH HL097796), and PB (ANR N°2010 CESA 001 01 (2010-0145)).
PB has received fees for speaking or consulting from Novartis, Glaxo-smith kline, Astra-Zeneca, Nycomed, Boehringer and Chiesi; has received funds for research from Novartis, Glaxo-smith kline and Nycomed; and travel to the ERS and ATS congress was funded by Novartis, Glaxo-smith kline, Astra-Zeneca.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Busse WW, Lemanske RF., Jr Asthma. N Engl J Med. 2001;344:350–62. doi: 10.1056/NEJM200102013440507. [DOI] [PubMed] [Google Scholar]
- 2.Huber HL, Koessler KK. The pathology of bronchial asthma. Arch Int Med. 1922;30:689–760. [Google Scholar]
- 3.Bousquet J, Chanez P, Lacoste JY, et al. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323:1033–9. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 4.Brightling CE, Bradding P, Symon FA, et al. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med. 2002;346:1699–705. doi: 10.1056/NEJMoa012705. [DOI] [PubMed] [Google Scholar]
- 5.Ramos-Barbon D, Fraga-Iriso R, Brienza NS, et al. T Cells localize with proliferating smooth muscle alpha-actin+ cell compartments in asthma. Am J Respir Crit Care Med. 2010;182:317–24. doi: 10.1164/rccm.200905-0745OC. [DOI] [PubMed] [Google Scholar]
- 6.Bara I, Ozier A, Tunon de Lara JM, et al. Pathophysiology of bronchial smooth muscle remodelling in asthma. Eur Respir J. 2010;36:1174–84. doi: 10.1183/09031936.00019810. [DOI] [PubMed] [Google Scholar]
- 7.Pepe C, Foley S, Shannon J, et al. Differences in airway remodeling between subjects with severe and moderate asthma. J Allergy Clin Immunol. 2005;116:544–9. doi: 10.1016/j.jaci.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 8.Kaminska M, Foley S, Maghni K, et al. Airway remodeling in subjects with severe asthma with or without chronic persistent airflow obstruction. J Allergy Clin Immunol. 2009;124:45–51. doi: 10.1016/j.jaci.2009.03.049. [DOI] [PubMed] [Google Scholar]
- 9.Seow CY, Schellenberg RR, Pare PD. Structural and functional changes in the airway smooth muscle of asthmatic subjects. Am J Respir Crit Care Med. 1998;158:S179–86. doi: 10.1164/ajrccm.158.supplement_2.13tac160. [DOI] [PubMed] [Google Scholar]
- 10.James A, Carroll N. Airway smooth muscle in health and disease; methods of measurement and relation to function. Eur Respir J. 2000;15:782–9. doi: 10.1034/j.1399-3003.2000.15d25.x. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt D, Ruehlmann E, Branscheid D, et al. Passive sensitization of human airways increases responsiveness to leukotriene C4. Eur Respir J. 1999;14:315–9. doi: 10.1034/j.1399-3003.1999.14b13.x. [DOI] [PubMed] [Google Scholar]
- 12.Black J, Marthan R, Armour CL, et al. Sensitization alters contractile responses and calcium influx in human airway smooth muscle. J Allergy Clin Immunol. 1989;84:440–7. doi: 10.1016/0091-6749(89)90356-4. [DOI] [PubMed] [Google Scholar]
- 13.Marthan R, Crevel H, Guénard H, et al. Responsiveness to histamine in human sensitized airway smooth muscle. Respir Physiol. 1992;90:239–50. doi: 10.1016/0034-5687(92)90084-a. [DOI] [PubMed] [Google Scholar]
- 14.Roux E, Hyvelin JM, Savineau JP, et al. Calcium signaling in airway smooth muscle cells is altered by in vitro exposure to the aldehyde acrolein. Am J Respir Cell Mol Biol. 1998;19:437–44. doi: 10.1165/ajrcmb.19.3.3048. [DOI] [PubMed] [Google Scholar]
- 15.Amrani Y, Panettieri RA., Jr Modulation of calcium homeostasis as a mechanism for altering smooth muscle responsiveness in asthma. Curr Opin Allergy Clin Immunol. 2002;2:39–45. doi: 10.1097/00130832-200202000-00007. [DOI] [PubMed] [Google Scholar]
- 16.Amrani Y, Panettieri RA., Jr Cytokines induce airway smooth muscle cell hyperresponsiveness to contractile agonists. Thorax. 1998;53:713–6. doi: 10.1136/thx.53.8.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao FC, Shah S, Pradhan AA, et al. Enhanced calcium signaling to bradykinin in airway smooth muscle from hyperresponsive inbred rats. Am J Physiol Lung Cell Mol Physiol. 2003;284:L90–9. doi: 10.1152/ajplung.00023.2002. [DOI] [PubMed] [Google Scholar]
- 18.Deshpande DA, Walseth TF, Panettieri RA, et al. CD38/cyclic ADP-ribose-mediated Ca2+ signaling contributes to airway smooth muscle hyper-responsiveness. Faseb J. 2003;17:452–4. doi: 10.1096/fj.02-0450fje. [DOI] [PubMed] [Google Scholar]
- 19.Wills-Karp M. IL-12/IL-13 axis in allergic asthma. J Allergy Clin Immunol. 2001;107:9–18. doi: 10.1067/mai.2001.112265. [DOI] [PubMed] [Google Scholar]
- 20.Deshpande DA, Dogan S, Walseth TF, et al. Modulation of calcium signaling by interleukin-13 in human airway smooth muscle: role of CD38/cyclic adenosine diphosphate ribose pathway. Am J Respir Cell Mol Biol. 2004;31:36–42. doi: 10.1165/rcmb.2003-0313OC. [DOI] [PubMed] [Google Scholar]
- 21.Deshpande DA, White TA, Dogan S, et al. CD38/cyclic ADP-ribose signaling: role in the regulation of calcium homeostasis in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2005;288:L773–88. doi: 10.1152/ajplung.00217.2004. [DOI] [PubMed] [Google Scholar]
- 22.Kellner J, Gamarra F, Welsch U, et al. IL-13Ralpha2 reverses the effects of IL-13 and IL-4 on bronchial reactivity and acetylcholine-induced Ca+ signaling. Int Arch Allergy Immunol. 2007;142:199–210. doi: 10.1159/000097022. [DOI] [PubMed] [Google Scholar]
- 23.Tliba O, Deshpande D, Chen H, et al. IL-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. Br J Pharmacol. 2003;140:1159–62. doi: 10.1038/sj.bjp.0705558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Syed F, Panettieri RA, Jr, Tliba O, et al. The effect of IL-13 and IL-13R130Q, a naturally occurring IL-13 polymorphism, on the gene expression of human airway smooth muscle cells. Respir Res. 2005;6:9. doi: 10.1186/1465-9921-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergner A, Sanderson MJ. Airway contractility and smooth muscle Ca(2+) signaling in lung slices from different mouse strains. J Appl Physiol. 2003;95:1325–32. doi: 10.1152/japplphysiol.00272.2003. [DOI] [PubMed] [Google Scholar]
- 26.Amrani Y, Chen H, Panettieri RA., Jr Activation of tumor necrosis factor receptor 1 in airway smooth muscle: a potential pathway that modulates bronchial hyper-responsiveness in asthma? Respir Res. 2000;1:49–53. doi: 10.1186/rr12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito S, Kume H, Honjo H, et al. Possible involvement of Rho kinase in Ca2+ sensitization and mobilization by MCh in tracheal smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1218–24. doi: 10.1152/ajplung.2001.280.6.L1218. [DOI] [PubMed] [Google Scholar]
- 28.Hunter I, Cobban HJ, Vandenabeele P, et al. Tumor necrosis factor-alpha-induced activation of RhoA in airway smooth muscle cells: role in the Ca2+ sensitization of myosin light chain20 phosphorylation. Mol Pharmacol. 2003;63:714–21. doi: 10.1124/mol.63.3.714. [DOI] [PubMed] [Google Scholar]
- 29.Parris JR, Cobban HJ, Littlejohn AF, et al. Tumour necrosis factor-alpha activates a calcium sensitization pathway in guinea-pig bronchial smooth muscle. J Physiol. 1999;518 ( Pt 2):561–9. doi: 10.1111/j.1469-7793.1999.0561p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakai H, Otogoto S, Chiba Y, et al. Involvement of p42/44 MAPK and RhoA protein in augmentation of ACh-induced bronchial smooth muscle contraction by TNF-alpha in rats. J Appl Physiol. 2004;97:2154–9. doi: 10.1152/japplphysiol.00752.2003. [DOI] [PubMed] [Google Scholar]
- 31.Sakai H, Otogoto S, Chiba Y, et al. TNF-alpha augments the expression of RhoA in the rat bronchus. J Smooth Muscle Res. 2004;40:25–34. doi: 10.1540/jsmr.40.25. [DOI] [PubMed] [Google Scholar]
- 32.Kume H, Takeda N, Oguma T, et al. Sphingosine 1-phosphate causes airway hyper-reactivity by rho-mediated myosin phosphatase inactivation. J Pharmacol Exp Ther. 2007;320:766–73. doi: 10.1124/jpet.106.110718. [DOI] [PubMed] [Google Scholar]
- 33.Rosenfeldt HM, Amrani Y, Watterson KR, et al. Sphingosine-1-phosphate stimulates contraction of human airway smooth muscle cells. Faseb J. 2003;17:1789–99. doi: 10.1096/fj.02-0836com. [DOI] [PubMed] [Google Scholar]
- 34.Sakai J, Oike M, Hirakawa M, et al. Theophylline and cAMP inhibit lysophosphatidic acid-induced hyperresponsiveness of bovine tracheal smooth muscle cells. J Physiol. 2003;549:171–80. doi: 10.1113/jphysiol.2003.039024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith PG, Roy C, Zhang YN, et al. Mechanical stress increases RhoA activation in airway smooth muscle cells. Am J Respir Cell Mol Biol. 2003;28:436–42. doi: 10.1165/rcmb.4754. [DOI] [PubMed] [Google Scholar]
- 36.Liu C, Tazzeo T, Janssen LJ. Isoprostane-induced airway hyperresponsiveness is dependent on internal Ca2+ handling and Rho/ROCK signaling. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1177–84. doi: 10.1152/ajplung.00142.2006. [DOI] [PubMed] [Google Scholar]
- 37.Liu HW, Halayko AJ, Fernandes DJ, et al. The RhoA/Rho kinase pathway regulates nuclear localization of serum response factor. Am J Respir Cell Mol Biol. 2003;29:39–47. doi: 10.1165/rcmb.2002-0206OC. [DOI] [PubMed] [Google Scholar]
- 38.Gosens R, Schaafsma D, Meurs H, et al. Role of Rho-kinase in maintaining airway smooth muscle contractile phenotype. Eur J Pharmacol. 2004;483:71–8. doi: 10.1016/j.ejphar.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 39.Ito S, Kume H, Oguma T, et al. Roles of stretch-activated cation channel and Rho-kinase in the spontaneous contraction of airway smooth muscle. Eur J Pharmacol. 2006;552:135–42. doi: 10.1016/j.ejphar.2006.08.067. [DOI] [PubMed] [Google Scholar]
- 40.Takeda N, Kondo M, Ito S, et al. Role of RhoA inactivation in reduced cell proliferation of human airway smooth muscle by simvastatin. Am J Respir Cell Mol Biol. 2006;35:722–9. doi: 10.1165/rcmb.2006-0034OC. [DOI] [PubMed] [Google Scholar]
- 41.Schaafsma D, Zuidhof AB, Nelemans SA, et al. Inhibition of Rho-kinase normalizes nonspecific hyperresponsiveness in passively sensitized airway smooth muscle preparations. Eur J Pharmacol. 2006;531:145–50. doi: 10.1016/j.ejphar.2005.12.043. [DOI] [PubMed] [Google Scholar]
- 42.Reithmann C, Gierschik P, Werdan K, et al. Tumor necrosis factor alpha up-regulates Gi alpha and G beta proteins and adenylyl cyclase responsiveness in rat cardiomyocytes. Eur J Pharmacol. 1991;206:53–60. doi: 10.1016/0922-4106(91)90146-9. [DOI] [PubMed] [Google Scholar]
- 43.Hakonarson H, Herrick DJ, Grunstein MM. Mechanism of impaired beta-adrenoceptor responsiveness in atopic sensitized airway smooth muscle. Am J Physiol. 1995;269:L645–52. doi: 10.1152/ajplung.1995.269.5.L645. [DOI] [PubMed] [Google Scholar]
- 44.Amrani Y, Krymskaya V, Maki C, et al. Mechanisms underlying TNF-alpha effects on agonist-mediated calcium homeostasis in human airway smooth muscle cells. Am J Physiol. 1997;273:L1020–8. doi: 10.1152/ajplung.1997.273.5.L1020. [DOI] [PubMed] [Google Scholar]
- 45.Hall IP, Donaldson J, Hill SJ. Modulation of fluoroaluminate-induced inositol phosphate formation by increases in tissue cyclic AMP content in bovine tracheal smooth muscle. Br J Pharmacol. 1990;100:646–50. doi: 10.1111/j.1476-5381.1990.tb15861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hardy E, Farahani M, Hall IP. Regulation of histamine H1 receptor coupling by dexamethasone in human cultured airway smooth muscle. Br J Pharmacol. 1996;118:1079–84. doi: 10.1111/j.1476-5381.1996.tb15509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsu YM, Chiu CT, Wang CC, et al. Tumour necrosis factor-alpha enhances bradykinin-induced signal transduction via activation of Ras/Raf/MEK/MAPK in canine tracheal smooth muscle cells. Cell Signal. 2001;13:633–43. doi: 10.1016/s0898-6568(01)00182-6. [DOI] [PubMed] [Google Scholar]
- 48.Yang CM, Chien CS, Wang CC, et al. Interleukin-1beta enhances bradykinin-induced phosphoinositide hydrolysis and Ca2+ mobilization in canine tracheal smooth-muscle cells: involvement of the Ras/Raf/mitogen-activated protein kinase (MAPK) kinase (MEK)/MAPK pathway. Biochem J. 2001;354:439–46. doi: 10.1042/0264-6021:3540439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidlin F, Scherrer D, Daeffler L, et al. Interleukin-1beta induces bradykinin B2 receptor gene expression through a prostanoid cyclic AMP-dependent pathway in human bronchial smooth muscle cells. Mol Pharmacol. 1998;53:1009–15. [PubMed] [Google Scholar]
- 50.Pype JL, Xu H, Schuermans M, et al. Mechanisms of interleukin 1beta-induced human airway smooth muscle hyporesponsiveness to histamine. Involvement of p38 MAPK NF-kappaB. Am J Respir Crit Care Med. 2001;163:1010–7. doi: 10.1164/ajrccm.163.4.9911091. [DOI] [PubMed] [Google Scholar]
- 51.Emala CW, Kuhl J, Hungerford CL, et al. TNF-alpha inhibits isoproterenol-stimulated adenylyl cyclase activity in cultured airway smooth muscle cells. Am J Physiol. 1997;272:L644–50. doi: 10.1152/ajplung.1997.272.4.L644. [DOI] [PubMed] [Google Scholar]
- 52.Shore SA, Laporte J, Hall IP, et al. Effect of IL-1 beta on responses of cultured human airway smooth muscle cells to bronchodilator agonists. Am J Respir Cell Mol Biol. 1997;16:702–12. doi: 10.1165/ajrcmb.16.6.9191472. [DOI] [PubMed] [Google Scholar]
- 53.Moore PE, Lahiri T, Laporte JD, et al. Selected contribution: synergism between TNF-alpha and IL-1 beta in airway smooth muscle cells: implications for beta-adrenergic responsiveness. J Appl Physiol. 2001;91:1467–74. doi: 10.1152/jappl.2001.91.3.1467. [DOI] [PubMed] [Google Scholar]
- 54.Laporte JC, Moore PE, Baraldo S, et al. Direct effects of interleukin-13 on signaling pathways for physiological responses in cultured human airway smooth muscle cells. Am J Respir Crit Care Med. 2001;164:141–8. doi: 10.1164/ajrccm.164.1.2008060. [DOI] [PubMed] [Google Scholar]
- 55.Amrani Y, Tliba O, Deshpande DA, et al. Bronchial hyperresponsiveness: insights into new signaling molecules. Curr Opin Pharmacol. 2004;4:230–4. doi: 10.1016/j.coph.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 56.Hall IP, Widdop S, Townsend P, et al. Control of cyclic AMP levels in primary cultures of human tracheal smooth muscle cells. Br J Pharmacol. 1992;107:422–8. doi: 10.1111/j.1476-5381.1992.tb12762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ammit AJ, Hoffman RK, Amrani Y, et al. Tumor necrosis factor-alpha-induced secretion of RANTES and interleukin-6 from human airway smooth-muscle cells. Modulation by cyclic adenosine monophosphate. Am J Respir Cell Mol Biol. 2000;23:794–802. doi: 10.1165/ajrcmb.23.6.4184. [DOI] [PubMed] [Google Scholar]
- 58.Pang L, Knox AJ. Regulation of TNF-alpha-induced eotaxin release from cultured human airway smooth muscle cells by beta2-agonists and corticosteroids. Faseb J. 2001;15:261–9. doi: 10.1096/fj.00-0103com. [DOI] [PubMed] [Google Scholar]
- 59.Hallsworth MP, Twort CH, Lee TH, et al. beta(2)-adrenoceptor agonists inhibit release of eosinophil-activating cytokines from human airway smooth muscle cells. Br J Pharmacol. 2001;132:729–41. doi: 10.1038/sj.bjp.0703866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pang L, Knox AJ. Synergistic inhibition by beta(2)-agonists and corticosteroids on tumor necrosis factor-alpha-induced interleukin-8 release from cultured human airway smooth-muscle cells. Am J Respir Cell Mol Biol. 2000;23:79–85. doi: 10.1165/ajrcmb.23.1.3985. [DOI] [PubMed] [Google Scholar]
- 61.Ammit AJ, Hastie AT, Edsall LC, et al. Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. Faseb J. 2001;15:1212–4. doi: 10.1096/fj.00-0742fje. [DOI] [PubMed] [Google Scholar]
- 62.Ammit AJ, Lazaar AL, Irani C, et al. Tumor necrosis factor-alpha-induced secretion of RANTES and interleukin-6 from human airway smooth muscle cells: modulation by glucocorticoids and beta-agonists. Am J Respir Cell Mol Biol. 2002;26:465–74. doi: 10.1165/ajrcmb.26.4.4681. [DOI] [PubMed] [Google Scholar]
- 63.DiCosmo BF, Geba GP, Picarella D, et al. Airway epithelial cell expression of interleukin-6 in transgenic mice. Uncoupling of airway inflammation and bronchial hyperreactivity. J Clin Invest. 1994;94:2028–35. doi: 10.1172/JCI117556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lazzeri N, Belvisi MG, Patel HJ, et al. Effects of prostaglandin E2 and cAMP elevating drugs on GM-CSF release by cultured human airway smooth muscle cells. Relevance to asthma therapy. Am J Respir Cell Mol Biol. 2001;24:44–8. doi: 10.1165/ajrcmb.24.1.4027. [DOI] [PubMed] [Google Scholar]
- 65.Bonazzi A, Bolla M, Buccellati C, et al. Effect of endogenous and exogenous prostaglandin E(2) on interleukin-1 beta-induced cyclooxygenase-2 expression in human airway smooth-muscle cells. Am J Respir Crit Care Med. 2000;162:2272–7. doi: 10.1164/ajrccm.162.6.2003127. [DOI] [PubMed] [Google Scholar]
- 66.Kanehiro A, Ikemura T, Makela MJ, et al. Inhibition of phosphodiesterase 4 attenuates airway hyperresponsiveness and airway inflammation in a model of secondary allergen challenge. Am J Respir Crit Care Med. 2001;163:173–84. doi: 10.1164/ajrccm.163.1.2001118. [DOI] [PubMed] [Google Scholar]
- 67.Oppenheimer-Marks N, Kavanaugh AF, Lipsky PE. Inhibition of the transendothelial migration of human T lymphocytes by prostaglandin E2. J Immunol. 1994;152:5703–13. [PubMed] [Google Scholar]
- 68.To SS, Schrieber L. Effect of leukotriene B4 and prostaglandin E2 on the adhesion of lymphocytes to endothelial cells. Clin Exp Immunol. 1990;81:160–5. doi: 10.1111/j.1365-2249.1990.tb05308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Panettieri RA, Jr, Lazaar AL, Pure E, et al. Activation of cAMP-dependent pathways in human airway smooth muscle cells inhibits TNF-alpha-induced ICAM-1 and VCAM-1 expression and T lymphocyte adhesion. J Immunol. 1995;154:2358–65. [PubMed] [Google Scholar]
- 70.Berger P, Girodet PO, Tunon De Lara JM. Mast cell myositis: a new feature of allergic asthma? Allergy. 2005;60:1238–40. doi: 10.1111/j.1398-9995.2005.00898.x. [DOI] [PubMed] [Google Scholar]
- 71.Siddiqui S, Mistry V, Doe C, et al. Airway hyperresponsiveness is dissociated from airway wall structural remodeling. J Allergy Clin Immunol. 2008;122:335–41. doi: 10.1016/j.jaci.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berry M, Morgan A, Shaw DE, et al. Pathological features and inhaled corticosteroid response of eosinophilic and non-eosinophilic asthma. Thorax. 2007;62:1043–9. doi: 10.1136/thx.2006.073429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saha SK, Berry MA, Parker D, et al. Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J Allergy Clin Immunol. 2008;121:685–91. doi: 10.1016/j.jaci.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carroll NG, Mutavdzic S, James AL. Distribution and degranulation of airway mast cells in normal and asthmatic subjects. Eur Respir J. 2002;19:879–85. doi: 10.1183/09031936.02.00275802. [DOI] [PubMed] [Google Scholar]
- 75.Kaur D, Saunders R, Hollins F, et al. Mast cell fibroblastoid differentiation mediated by airway smooth muscle in asthma. J Immunol. 2010;185:6105–14. doi: 10.4049/jimmunol.1000638. [DOI] [PubMed] [Google Scholar]
- 76.Amin K, Janson C, Boman G, et al. The extracellular deposition of mast cell products is increased in hypertrophic airways smooth muscles in allergic asthma but not in non-allergic asthma. Allergy. 2005 doi: 10.1111/j.1398-9995.2005.00823.x. in press. [DOI] [PubMed] [Google Scholar]
- 77.Berger P, Girodet PO, Begueret H, et al. Tryptase-stimulated human airway smooth muscle cells induce cytokine synthesis and mast cell chemotaxis. Faseb J. 2003;17:2139–41. doi: 10.1096/fj.03-0041fje. [DOI] [PubMed] [Google Scholar]
- 78.Brightling CE, Ammit AJ, Kaur D, et al. The CXCL10/CXCR3 axis mediates human lung mast cell migration to asthmatic airway smooth muscle. Am J Respir Crit Care Med. 2005;171:1103–8. doi: 10.1164/rccm.200409-1220OC. [DOI] [PubMed] [Google Scholar]
- 79.Sutcliffe A, Kaur D, Page S, et al. Mast cell migration to Th2 stimulated airway smooth muscle from asthmatics. Thorax. 2006;61:657–62. doi: 10.1136/thx.2005.056770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.El-Shazly A, Berger P, Girodet PO, et al. Fraktalkine produced by airway smooth muscle cells contributes to mast cell recruitment in asthma. J Immunol. 2006;176:1860–8. doi: 10.4049/jimmunol.176.3.1860. [DOI] [PubMed] [Google Scholar]
- 81.Yang W, Kaur D, Okayama Y, et al. Human lung mast cells adhere to human airway smooth muscle, in part, via Tumor Suppressor in Lung Cancer-1. J Immunol. 2006;176:1238–43. doi: 10.4049/jimmunol.176.2.1238. [DOI] [PubMed] [Google Scholar]
- 82.Hollins F, Kaur D, Yang W, et al. Human airway smooth muscle promotes human lung mast cell survival, proliferation, and constitutive activation: cooperative roles for CADM1, stem cell factor, and IL-6. J Immunol. 2008;181:2772–80. doi: 10.4049/jimmunol.181.4.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Girodet PO, Ozier A, Trian T, et al. Mast cell adhesion to bronchial smooth muscle in asthma specifically depends on CD51 and CD44 variant 6. Allergy. 2010;65:1004–12. doi: 10.1111/j.1398-9995.2009.02308.x. [DOI] [PubMed] [Google Scholar]
- 84.Begueret H, Berger P, Vernejoux JM, et al. Inflammation of bronchial smooth muscle in allergic asthma. Thorax. 2007;62:8–15. doi: 10.1136/thx.2006.062141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carter RJ, Bradding P. The role of mast cells in the structural alterations of the airways as a potential mechanism in the pathogenesis of severe asthma. Curr Pharm Des. 2011;17:685–98. doi: 10.2174/138161211795428975. [DOI] [PubMed] [Google Scholar]
- 86.Brightling CE, Symon FA, Holgate ST, et al. Interleukin-4 and -13 expression is co-localized to mast cells within the airway smooth muscle in asthma. Clin Exp Allergy. 2003;33:1711–6. doi: 10.1111/j.1365-2222.2003.01827.x. [DOI] [PubMed] [Google Scholar]
- 87.Bradding P, Walls AF, Holgate ST. The role of the mast cell in the pathophysiology of asthma. J Allergy Clin Immunol. 2006;117:1277–84. doi: 10.1016/j.jaci.2006.02.039. [DOI] [PubMed] [Google Scholar]
- 88.Woodman L, Siddiqui S, Cruse G, et al. Mast cells promote airway smooth muscle cell differentiation via autocrine up-regulation of TGF-beta 1. J Immunol. 2008;181:5001–7. doi: 10.4049/jimmunol.181.7.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kaur D, Hollins F, Saunders R, et al. Airway smooth muscle proliferation and survival is not modulated by mast cells. Clin Exp Allergy. 2010;40:279–88. doi: 10.1111/j.1365-2222.2009.03423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thangam EB, Venkatesha RT, Zaidi AK, et al. Airway smooth muscle cells enhance C3a-induced mast cell degranulation following cell-cell contact. Faseb J. 2005;19:798–800. doi: 10.1096/fj.04-2797fje. [DOI] [PubMed] [Google Scholar]
- 91.Berger P, Tunon-de-Lara JM, Savineau JP, et al. Tryptase-induced PAR-2-mediated Ca(2+) signaling in human airway smooth muscle cells. J Appl Physiol. 2001;91:995–1003. doi: 10.1152/jappl.2001.91.2.995. [DOI] [PubMed] [Google Scholar]
- 92.Berger P, Compton SJ, Molimard M, et al. Mast cell tryptase as a mediator of hyperresponsiveness in human isolated bronchi. Clin Exp Allergy. 1999;29:804–12. doi: 10.1046/j.1365-2222.1999.00580.x. [DOI] [PubMed] [Google Scholar]
- 93.Molinari JF, Scuri M, Moore WR, et al. Inhaled tryptase causes bronchoconstriction in sheep via histamine release. Am J Respir Crit Care Med. 1996;154:649–53. doi: 10.1164/ajrccm.154.3.8810600. [DOI] [PubMed] [Google Scholar]
- 94.Berger P, Perng DW, Thabrew H, et al. Tryptase and agonists of PAR-2 induce the proliferation of human airway smooth muscle cells. J Appl Physiol. 2001;91:1372–9. doi: 10.1152/jappl.2001.91.3.1372. [DOI] [PubMed] [Google Scholar]
- 95.Kaur D, Saunders R, Berger P, et al. Airway smooth muscle and mast cell-derived CC chemokine ligand 19 mediate airway smooth muscle migration in asthma. Am J Respir Crit Care Med. 2006;174:1179–88. doi: 10.1164/rccm.200603-394OC. [DOI] [PubMed] [Google Scholar]
- 96.Alkhouri H, Hollins F, Moir LM, et al. Human lung mast cells modulate the functions of airway smooth muscle cells in asthma. Allergy. 2011;66:1231–41. doi: 10.1111/j.1398-9995.2011.02616.x. [DOI] [PubMed] [Google Scholar]
- 97.Saunders R, Sutcliffe A, Woodman L, et al. The airway smooth muscle CCR3/CCL11 axis is inhibited by mast cells. Allergy. 2008;63:1148–55. doi: 10.1111/j.1398-9995.2008.01684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ramos-Barbon D, Presley JF, Hamid QA, et al. Antigen-specific CD4+ T cells drive airway smooth muscle remodeling in experimental asthma. J Clin Invest. 2005;115:1580–9. doi: 10.1172/JCI19711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lazaar AL, Albelda SM, Pilewski JM, et al. T lymphocytes adhere to airway smooth muscle cells via integrins and CD44 and induce smooth muscle cell DNA synthesis. J Exp Med. 1994;180:807–16. doi: 10.1084/jem.180.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hakonarson H, Kim C, Whelan R, et al. Bi-directional activation between human airway smooth muscle cells and T lymphocytes: role in induction of altered airway responsiveness. J Immunol. 2001;166:293–303. doi: 10.4049/jimmunol.166.1.293. [DOI] [PubMed] [Google Scholar]
- 101.Veler H, Hu A, Fatma S, et al. Superantigen presentation by airway smooth muscle to CD4+ T lymphocytes elicits reciprocal proasthmatic changes in airway function. J Immunol. 2007;178:3627–36. doi: 10.4049/jimmunol.178.6.3627. [DOI] [PubMed] [Google Scholar]
- 102.Lazaar AL, Reitz HE, Panettieri RA, Jr, et al. Antigen receptor-stimulated peripheral blood and bronchoalveolar lavage- derived T cells induce MHC class II and ICAM-1 expression on human airway smooth muscle. Am J Respir Cell Mol Biol. 1997;16:38–45. doi: 10.1165/ajrcmb.16.1.8998077. [DOI] [PubMed] [Google Scholar]
- 103.Lazaar AL, Amrani Y, Hsu J, et al. CD40-mediated signal transduction in human airway smooth muscle. J Immunol. 1998;161:3120–7. [PubMed] [Google Scholar]
- 104.Burgess JK, Blake AE, Boustany S, et al. CD40 and OX40 ligand are increased on stimulated asthmatic airway smooth muscle. J Allergy Clin Immunol. 2005;115:302–8. doi: 10.1016/j.jaci.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 105.Krimmer DI, Loseli M, Hughes JM, et al. CD40 and OX40 ligand are differentially regulated on asthmatic airway smooth muscle. Allergy. 2009;64:1074–82. doi: 10.1111/j.1398-9995.2009.01959.x. [DOI] [PubMed] [Google Scholar]
- 106.Burgess JK, Carlin S, Pack RA, et al. Detection and characterization of OX40 ligand expression in human airway smooth muscle cells: a possible role in asthma? J Allergy Clin Immunol. 2004;113:683–9. doi: 10.1016/j.jaci.2003.12.311. [DOI] [PubMed] [Google Scholar]
- 107.Siddiqui S, Mistry V, Doe C, et al. Airway wall expression of OX40/OX40L and interleukin-4 in asthma. Chest. 2010;137:797–804. doi: 10.1378/chest.09-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu Y, Song G. The role of CD40-CD154 interaction in cell immunoregulation. J Biomed Sci. 2004;11:426–38. doi: 10.1007/BF02256091. [DOI] [PubMed] [Google Scholar]
- 109.Fujita T, Ukyo N, Hori T, et al. Functional characterization of OX40 expressed on human CD8+ T cells. Immunol Lett. 2006;106:27–33. doi: 10.1016/j.imlet.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 110.Lazaar AL, Plotnick MI, Kucich U, et al. Mast cell chymase modifies cell-matrix interactions and inhibits mitogen-induced proliferation of human airway smooth muscle cells. J Immunol. 2002;169:1014–20. doi: 10.4049/jimmunol.169.2.1014. [DOI] [PubMed] [Google Scholar]
- 111.Durrani SR, Viswanathan RK, Busse WW. What effect does asthma treatment have on airway remodeling? Current perspectives. J Allergy Clin Immunol. 2011;128:439–48. doi: 10.1016/j.jaci.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 112.Lambert RK, Wiggs BR, Kuwano K, et al. Functional significance of increased airway smooth muscle in asthma and COPD. J Appl Physiol. 1993;74:2771–81. doi: 10.1152/jappl.1993.74.6.2771. [DOI] [PubMed] [Google Scholar]
- 113.Ebina M, Takahashi T, Chiba T, et al. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma. A 3-D morphometric study. Am Rev Respir Dis. 1993;148:720–6. doi: 10.1164/ajrccm/148.3.720. [DOI] [PubMed] [Google Scholar]
- 114.Bai TR. Evidence for airway remodeling in chronic asthma. Curr Opin Allergy Clin Immunol. 2010;10:82–6. doi: 10.1097/ACI.0b013e32833363b2. [DOI] [PubMed] [Google Scholar]
- 115.Tschumperlin DJ, Drazen JM. Mechanical stimuli to airway remodeling. Am J Respir Crit Care Med. 2001;164:S90–4. doi: 10.1164/ajrccm.164.supplement_2.2106060. [DOI] [PubMed] [Google Scholar]
- 116.Mohamed JS, Lopez MA, Boriek AM. Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3beta. J Biol Chem. 2010;285:29336–47. doi: 10.1074/jbc.M110.101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Woodruff PG, Dolganov GM, Ferrando RE, et al. Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am J Respir Crit Care Med. 2004;169:1001–6. doi: 10.1164/rccm.200311-1529OC. [DOI] [PubMed] [Google Scholar]
- 118.Johnson PR, Roth M, Tamm M, et al. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med. 2001;164:474–7. doi: 10.1164/ajrccm.164.3.2010109. [DOI] [PubMed] [Google Scholar]
- 119.Trian T, Benard G, Begueret H, et al. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J Exp Med. 2007;204:3173–81. doi: 10.1084/jem.20070956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ward JE, Harris T, Bamford T, et al. Proliferation is not increased in airway myofibroblasts isolated from asthmatics. Eur Respir J. 2008;32:362–71. doi: 10.1183/09031936.00119307. [DOI] [PubMed] [Google Scholar]
- 121.Mahn K, Hirst SJ, Ying S, et al. Diminished sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) expression contributes to airway remodelling in bronchial asthma. Proc Natl Acad Sci U S A. 2009;106:10775–80. doi: 10.1073/pnas.0902295106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mahn K, Ojo OO, Chadwick G, et al. Ca(2+) homeostasis and structural and functional remodelling of airway smooth muscle in asthma. Thorax. 2010;65:547–52. doi: 10.1136/thx.2009.129296. [DOI] [PubMed] [Google Scholar]
- 123.Lee JH, Johnson PR, Roth M, et al. ERK activation and mitogenesis in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1019–29. doi: 10.1152/ajplung.2001.280.5.L1019. [DOI] [PubMed] [Google Scholar]
- 124.Chung KF. p38 mitogen-activated protein kinase pathways in asthma and COPD. Chest. 2011;139:1470–9. doi: 10.1378/chest.10-1914. [DOI] [PubMed] [Google Scholar]
- 125.Burgess JK, Lee JH, Ge Q, et al. Dual ERK and phosphatidylinositol 3-kinase pathways control airway smooth muscle proliferation: differences in asthma. J Cell Physiol. 2008;216:673–9. doi: 10.1002/jcp.21450. [DOI] [PubMed] [Google Scholar]
- 126.Hassan M, Jo T, Risse PA, et al. Airway smooth muscle remodeling is a dynamic process in severe long-standing asthma. J Allergy Clin Immunol. 2010;125:1037–45. doi: 10.1016/j.jaci.2010.02.031. [DOI] [PubMed] [Google Scholar]
- 127.Yan H, Deshpande DA, Misior AM, et al. Anti-mitogenic effects of beta-agonists and PGE2 on airway smooth muscle are PKA dependent. Faseb J. 2011;25:389–97. doi: 10.1096/fj.10-164798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roth M, Johnson PR, Borger P, et al. Dysfunctional interaction of C/EBPalpha and the glucocorticoid receptor in asthmatic bronchial smooth-muscle cells. N Engl J Med. 2004;351:560–74. doi: 10.1056/NEJMoa021660. [DOI] [PubMed] [Google Scholar]
- 129.Damera G, Fogle HW, Lim P, et al. Vitamin D inhibits growth of human airway smooth muscle cells through growth factor-induced phosphorylation of retinoblastoma protein and checkpoint kinase 1. Br J Pharmacol. 2009;158:1429–41. doi: 10.1111/j.1476-5381.2009.00428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Grainge CL, Lau LC, Ward JA, et al. Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med. 2011;364:2006–15. doi: 10.1056/NEJMoa1014350. [DOI] [PubMed] [Google Scholar]
- 131.Kelly MM, O'Connor TM, Leigh R, et al. Effects of budesonide and formoterol on allergen-induced airway responses, inflammation, and airway remodeling in asthma. J Allergy Clin Immunol. 2010;125:349–56. e13. doi: 10.1016/j.jaci.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 132.Martinez FD. Asthma treatment and asthma prevention: a tale of 2 parallel pathways. J Allergy Clin Immunol. 2007;119:30–3. doi: 10.1016/j.jaci.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 133.An SS, Bai TR, Bates JH, et al. Airway smooth muscle dynamics: a common pathway of airway obstruction in asthma. Eur Respir J. 2007;29:834–60. doi: 10.1183/09031936.00112606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bai TR, Cooper J, Koelmeyer T, et al. The effect of age and duration of disease on airway structure in fatal asthma. Am J Respir Crit Care Med. 2000;162:663–9. doi: 10.1164/ajrccm.162.2.9907151. [DOI] [PubMed] [Google Scholar]
- 135.Johnson PR, Black JL, Carlin S, et al. The production of extracellular matrix proteins by human passively sensitized airway smooth-muscle cells in culture: the effect of beclomethasone. Am J Respir Crit Care Med. 2000;162:2145–51. doi: 10.1164/ajrccm.162.6.9909111. [DOI] [PubMed] [Google Scholar]
- 136.Laitinen LA, Laitinen A, Altraja A, et al. Bronchial biopsy findings in intermittent or “early” asthma. J Allergy Clin Immunol. 1996;98:S3–6. discussion S33–40. [PubMed] [Google Scholar]
- 137.Laitinen A, Altraja A, Kampe M, et al. Tenascin is increased in airway basement membrane of asthmatics and decreased by an inhaled steroid. Am J Respir Crit Care Med. 1997;156:951–8. doi: 10.1164/ajrccm.156.3.9610084. [DOI] [PubMed] [Google Scholar]
- 138.Roberts CR, Burke AK. Remodelling of the extracellular matrix in asthma: proteoglycan synthesis and degradation. Can Respir J. 1998;5:48–50. [PubMed] [Google Scholar]
- 139.Roche WR, Beasley R, Williams JH, et al. Subepithelial fibrosis in the bronchi of asthmatics. Lancet. 1989;1:520–4. doi: 10.1016/s0140-6736(89)90067-6. [DOI] [PubMed] [Google Scholar]
- 140.Bousquet J, Chanez P, Lacoste JY, et al. Asthma: a disease remodeling the airways. Allergy. 1992;47:3–11. doi: 10.1111/j.1398-9995.1992.tb02242.x. [DOI] [PubMed] [Google Scholar]
- 141.Johnson PR, Burgess JK, Underwood PA, et al. Extracellular matrix proteins modulate asthmatic airway smooth muscle cell proliferation via an autocrine mechanism. J Allergy Clin Immunol. 2004;113:690–6. doi: 10.1016/j.jaci.2003.12.312. [DOI] [PubMed] [Google Scholar]
- 142.Lau JY, Oliver BG, Baraket M, et al. Fibulin-1 is increased in asthma--a novel mediator of airway remodeling? PLoS One. 2010;5:e13360. doi: 10.1371/journal.pone.0013360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schuliga M, Ong SC, Soon L, et al. Airway smooth muscle remodels pericellular collagen fibrils: implications for proliferation. Am J Physiol Lung Cell Mol Physiol. 2010;298:L584–92. doi: 10.1152/ajplung.00312.2009. [DOI] [PubMed] [Google Scholar]
- 144.Bourke JE, Li X, Foster SR, et al. Collagen remodelling by airway smooth muscle is resistant to steroids and beta-agonists. Eur Respir J. 2011;37:173–82. doi: 10.1183/09031936.00008109. [DOI] [PubMed] [Google Scholar]
- 145.Araujo BB, Dolhnikoff M, Silva LF, et al. Extracellular matrix components and regulators in the airway smooth muscle in asthma. Eur Respir J. 2008;32:61–9. doi: 10.1183/09031936.00147807. [DOI] [PubMed] [Google Scholar]
- 146.Watson ML, Grix SP, Jordan NJ, et al. Interleukin 8 and monocyte chemoattractant protein 1 production by cultured human airway smooth muscle cells. Cytokine. 1998;10:346–52. doi: 10.1006/cyto.1997.0350. [DOI] [PubMed] [Google Scholar]
- 147.Pype JL, Dupont LJ, Menten P, et al. Expression of monocyte chemotactic protein (MCP)-1, MCP-2, and MCP-3 by human airway smooth-muscle cells. Modulation by corticosteroids and T-helper 2 cytokines. Am J Respir Cell Mol Biol. 1999;21:528–36. doi: 10.1165/ajrcmb.21.4.3660. [DOI] [PubMed] [Google Scholar]
- 148.John M, Hirst SJ, Jose PJ, et al. Human airway smooth muscle cells express and release RANTES in response to T helper 1 cytokines: regulation by T helper 2 cytokines and corticosteroids. J Immunol. 1997;158:1841–7. [PubMed] [Google Scholar]
- 149.Amrani Y, Ammit AJ, Panettieri RA., Jr Tumor necrosis factor receptor (TNFR) 1, but not TNFR2, mediates tumor necrosis factor-alpha-induced interleukin-6 and RANTES in human airway smooth muscle cells: role of p38 and p42/44 mitogen-activated protein kinases. Mol Pharmacol. 2001;60:646–55. [PubMed] [Google Scholar]
- 150.Chung KF, Patel HJ, Fadlon EJ, et al. Induction of eotaxin expression and release from human airway smooth muscle cells by IL-1beta and TNFalpha: effects of IL-10 and corticosteroids. Br J Pharmacol. 1999;127:1145–50. doi: 10.1038/sj.bjp.0702660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ghaffar O, Hamid Q, Renzi PM, et al. Constitutive and Cytokine-Stimulated Expression of Eotaxin by Human Airway Smooth Muscle Cells. Am J Respir Crit Care Med. 1999;159:1933–42. doi: 10.1164/ajrccm.159.6.9805039. [DOI] [PubMed] [Google Scholar]
- 152.John M, Au BT, Jose PJ, et al. Expression and release of interleukin-8 by human airway smooth muscle cells: inhibition by Th-2 cytokines and corticosteroids. Am J Respir Cell Mol Biol. 1998;18:84–90. doi: 10.1165/ajrcmb.18.1.2813. [DOI] [PubMed] [Google Scholar]
- 153.Kassel O, Schmidlin F, Duvernelle C, et al. Human bronchial smooth muscle cells in culture produce stem cell factor. Eur Respir J. 1999;13:951–4. doi: 10.1034/j.1399-3003.1999.13e04.x. [DOI] [PubMed] [Google Scholar]
- 154.McKay S, de Jongste JC, Saxena PR, et al. Angiotensin II induces hypertrophy of human airway smooth muscle cells: expression of transcription factors and transforming growth factor- beta1. Am J Respir Cell Mol Biol. 1998;18:823–33. doi: 10.1165/ajrcmb.18.6.2924. [DOI] [PubMed] [Google Scholar]
- 155.Hakonarson H, Maskeri N, Carter C, et al. Regulation of TH1- and TH2-type cytokine expression and action in atopic asthmatic sensitized airway smooth muscle. J Clin Invest. 1999;103:1077–87. doi: 10.1172/JCI5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hallsworth MP, Soh CP, Twort CH, et al. Cultured human airway smooth muscle cells stimulated by interleukin- 1beta enhance eosinophil survival. Am J Respir Cell Mol Biol. 1998;19:910–9. doi: 10.1165/ajrcmb.19.6.3275. [DOI] [PubMed] [Google Scholar]
- 157.Saunders MA, Mitchell JA, Seldon PM, et al. Release of granulocyte-macrophage colony stimulating factor by human cultured airway smooth muscle cells: suppression by dexamethasone. Br J Pharmacol. 1997;120:545–6. doi: 10.1038/sj.bjp.0700998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Elias JA, Wu Y, Zheng T, et al. Cytokine- and virus-stimulated airway smooth muscle cells produce IL-11 and other IL-6-type cytokines. Am J Physiol. 1997;273:L648–55. doi: 10.1152/ajplung.1997.273.3.L648. [DOI] [PubMed] [Google Scholar]
- 159.Prefontaine D, Lajoie-Kadoch S, Foley S, et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. 2009;183:5094–103. doi: 10.4049/jimmunol.0802387. [DOI] [PubMed] [Google Scholar]
- 160.Amrani Y, Lazaar AL, Panettieri RA., Jr Up-regulation of ICAM-1 by cytokines in human tracheal smooth muscle cells involves an NF-kappa B-dependent signaling pathway that is only partially sensitive to dexamethasone. J Immunol. 1999;163:2128–34. [PubMed] [Google Scholar]
- 161.Grunstein MM, Hakonarson H, Maskeri N, et al. Intrinsic ICAM-1/LFA-1 activation mediates altered responsiveness of atopic asthmatic airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1154–63. doi: 10.1152/ajplung.2000.278.6.L1154. [DOI] [PubMed] [Google Scholar]
- 162.Panettieri RA, Jr, Goldie RG, Rigby PJ, et al. Endothelin-1-induced potentiation of human airway smooth muscle proliferation: an ETA receptor-mediated phenomenon. Br J Pharmacol. 1996;118:191–7. doi: 10.1111/j.1476-5381.1996.tb15385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Johnson PR, Armour CL, Carey D, et al. Heparin and PGE2 inhibit DNA synthesis in human airway smooth muscle cells in culture. Am J Physiol. 1995;269:L514–9. doi: 10.1152/ajplung.1995.269.4.L514. [DOI] [PubMed] [Google Scholar]
- 164.Kelleher MD, Abe MK, Chao TS, et al. Role of MAP kinase activation in bovine tracheal smooth muscle mitogenesis. Am J Physiol. 1995;268:L894–901. doi: 10.1152/ajplung.1995.268.6.L894. [DOI] [PubMed] [Google Scholar]
- 165.Hirst SJ, Barnes PJ, Twort CH. PDGF isoform-induced proliferation and receptor expression in human cultured airway smooth muscle cells. Am J Physiol. 1996;270:L415–28. doi: 10.1152/ajplung.1996.270.3.L415. [DOI] [PubMed] [Google Scholar]
- 166.Risse PA, Jo T, Suarez F, et al. Interleukin-13 inhibits proliferation and enhances contractility of human airway smooth muscle cells without change in contractile phenotype. Am J Physiol Lung Cell Mol Physiol. 2011;300:L958–66. doi: 10.1152/ajplung.00247.2010. [DOI] [PubMed] [Google Scholar]
- 167.Hawker KM, Johnson PRA, Hughes JM, et al. Interleukin-4 inhibits mitogen-induced proliferation of human airway smooth muscle cells in culture. Am J Physiol. 1998;275:L469–77. doi: 10.1152/ajplung.1998.275.3.L469. [DOI] [PubMed] [Google Scholar]
- 168.Maruno K, Absood A, Said SI. VIP inhibits basal and histamine-stimulated proliferation of human airway smooth muscle cells. Am J Physiol. 1995;268:L1047–51. doi: 10.1152/ajplung.1995.268.6.L1047. [DOI] [PubMed] [Google Scholar]
- 169.Lew DB, Dempsey BK, Zhao Y, et al. beta-hexosaminidase-induced activation of p44/42 mitogen-activated protein kinase is dependent on p21Ras and protein kinase C and mediates bovine airway smooth-muscle proliferation. Am J Respir Cell Mol Biol. 1999;21:111–8. doi: 10.1165/ajrcmb.21.1.3542. [DOI] [PubMed] [Google Scholar]
- 170.Huang CD, Chen HH, Wang CH, et al. Human neutrophil-derived elastase induces airway smooth muscle cell proliferation. Life Sci. 2004;74:2479–92. doi: 10.1016/j.lfs.2003.07.059. [DOI] [PubMed] [Google Scholar]
- 171.Panettieri RA, Jr, Hall IP, Maki CS, et al. alpha-Thrombin increases cytosolic calcium and induces human airway smooth muscle cell proliferation. Am J Respir Cell Mol Biol. 1995;13:205–16. doi: 10.1165/ajrcmb.13.2.7626288. [DOI] [PubMed] [Google Scholar]
- 172.Panettieri RA, Tan EM, Ciocca V, et al. Effects of LTD4 on human airway smooth muscle cell proliferation, matrix expression, and contraction In vitro: differential sensitivity to cysteinyl leukotriene receptor antagonists. Am J Respir Cell Mol Biol. 1998;19:453–61. doi: 10.1165/ajrcmb.19.3.2999. [DOI] [PubMed] [Google Scholar]
- 173.Ravasi S, Citro S, Viviani B, et al. CysLT1 receptor-induced human airway smooth muscle cells proliferation requires ROS generation, EGF receptor transactivation and ERK1/2 phosphorylation. Respir Res. 2006;7:42. doi: 10.1186/1465-9921-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Nguyen TT, Ward JP, Hirst SJ. beta1-Integrins mediate enhancement of airway smooth muscle proliferation by collagen and fibronectin. Am J Respir Crit Care Med. 2005;171:217–23. doi: 10.1164/rccm.200408-1046OC. [DOI] [PubMed] [Google Scholar]
- 175.Hirst SJ, Martin JG, Bonacci JV, et al. Proliferative aspects of airway smooth muscle. J Allergy Clin Immunol. 2004;114:S2–17. doi: 10.1016/j.jaci.2004.04.039. [DOI] [PubMed] [Google Scholar]