

Abstract
Rationale: The L-type calcium channels (LTCC) are critical for maintaining Ca2+-homeostasis. In heterologous expression studies, the RGK-class of Ras-related G-proteins regulates LTCC function; however, the physiological relevance of RGK–LTCC interactions is untested.
Objective: In this report we test the hypothesis that the RGK protein, Rem, modulates native Ca2+ current (ICa,L) via LTCC in murine cardiomyocytes.
Methods and Results: Rem knockout mice (Rem−/−) were engineered, and ICa,L and Ca2+-handling properties were assessed. Rem−/− ventricular cardiomyocytes displayed increased ICa,L density. ICa,L activation was shifted positive on the voltage axis, and β-adrenergic stimulation normalized this shift compared with wild-type ICa,L. Current kinetics, steady-state inactivation, and facilitation was unaffected by Rem−/−. Cell shortening was not significantly different. Increased ICa,L density in the absence of frank phenotypic differences motivated us to explore putative compensatory mechanisms. Despite the larger ICa,L density, Rem−/− cardiomyocyte Ca2+ twitch transient amplitude was significantly less than that compared with wild type. Computer simulations and immunoblot analysis suggests that relative dephosphorylation of Rem−/− LTCC can account for the paradoxical decrease of Ca2+ transients.
Conclusions: This is the first demonstration that loss of an RGK protein influences ICa,L in vivo in cardiac myocytes.
Keywords: L-type calcium channel, Ras-GTPase, calcium current, patch-clamp
Introduction
L-type Ca channels (LTCC) are critical for maintaining Ca-homeostasis, providing trigger Ca for Ca-induced Ca-release, and regulating electrical function in cardiac myocytes. Therefore, precise control of LTCC function has profound implications for cardiac myocyte physiology. LTCC exist as a multi-protein complex in cardiac myocytes.1 Thus, understanding of LTCC function requires evaluation of LTCC Ca current (ICa,L) in native complexes. Cardiac myocyte LTCC consists of the main pore-forming subunit in complex with accessory proteins including CaVβ2 family members, α2δ, and calmodulin (CaM; reviewed by 2). In addition, we, and others have recently extensively characterized the ability of the RGK-family of monomeric G-proteins to modulate ICa,L.3-6
The RGK family of Ras-related monomeric G-proteins includes Rem, Rem2, Rad and Gem/Kir.7 Rem and Rad are expressed in myocardium.8,9 Rad co-expressed with recombinant LTCC show block of current,10 and therefore, reduction of Rad in vivo is expected to increase ICa,L. Rad is downregulated in experimental induced heart failure (thoracic aorta constriction, TAC) in mice.11 Rad knockout mice show no functional phenotype at baseline, but following TAC CaMKII increases.11 Rad knock-down via RNAi results in an increase of ICa,L, Ca transients, and contractility.12
Heterologous expression studies document Rem blockade of ICa,L5,10,13-15; however, there are no reports that assess in vivo Rem function. Overexpression of Rem, and other RGK proteins, results in prominent blockade of cardiac myocyte ICa,L.4,16 Therefore, we tested the hypothesis that depletion of Rem results in an increase of ICa,L. In this report, we examined cardiac myocyte ICa,L in Rem-null mice (Rem−/−). Cardiac myocyte ICa,L density increases in Rem−/− compared with that from wild-type mice. Nevertheless, Rem-deletion results in no frank cardiac phenotype at baseline. The increased ICa,L density may be offset by a positive shift of the steady-state activation curve. Thus, our data support the notion that compensatory changes in Ca homeostasis preserve cardiac function.
Results
ICa,L in Rem−/− cardiac myocytes
Rem overexpression blocks ICa,L in heterologous expression systems,14 and in cardiac myocytes.16 These results suggested that Rem governs ICa,L in vivo. To test this idea, we generated a Rem1 knockout mouse (Fig. 1). Rem null mutant mice (Rem−/−) were born at the expected Mendelian ratio, and grew to adulthood without showing any discernible abnormalities.

Figure 1. Rem knockout mouse. (A) Schematic representation of the strategy used to generate the Rem knockout mouse. The coding region of Exon 2 was targeted and replaced by a LacZ expressing cassette, followed by the neomycin gene flanked by FRT sites. Arrows indicate the location of genotyping primers (a, b, c, and d). (B) Genomic DNA was isolated from mouse tail biopsies, and subjected to PCR analysis using the oligonucleotide primer pairs indicated in panel A. A 348 base pair band represents the mutant allele (lower panel; amplification using primer pair c/d), while a 500 base pair band represents the wild-type allele (upper panel; amplification using primer pair a/b). (C) Total protein was isolated from hearts obtained from Rem mutant or WT-littermates, and subjected to anti-Rem immunoblot analysis. Wild-type Rem expressed in HEK293 cells was included as a positive control. Note that Rem protein levels are completely eliminated in Rem−/− cardiac muscle, while reduced in Rem+/− animals.
To examine the contribution of Rem to LTCC function, we measured ICa,L in Rem−/− cardiac myocytes. The expectation is that ICa,L should increase in the absence of Rem blockade of current. As the analog traces of Figure 2A shows the ICa,L density was significantly greater in Rem−/− compared with age-matched wild-type controls (peak ICa,L density amplitudes at + 10 mV: -19.0 ± 0.5 pA/pF (n = 55) for Rem−/−, and -16.0 ± 1.0 pA/pF (n = 40) for the age-matched wild-type controls; p < 0.05). The current-voltage relationships (I(V)) show that the differences in calcium current densities were statistically significant in the membrane potential range between 0 mV and +45 mV (Fig. 2B). These data are consistent with Rem governing ICa,L amplitude in cardiomyocytes.

Figure 2. ICa,L density for wild-type and Rem−/− cardiac ventricular myocytes. (A) Representative original traces from wild-type and Rem−/− mice. (B) Average ICa,L-V relationships for wild-type and Rem−/− cardiac myocytes. (C) The voltage dependence of steady-state activation and steady-state inactivation of ICa,L for wild-type and Rem−/− cardiac myocytes. V½ of steady-state activation is significantly shifted +4mV for Rem−/− compared with wild type. V½ was obtained from fitting the current-voltage data as in panel B to a modified Boltzmann distribution of the form: I(V) = Gmax*(V-Erev)/(1+exp(V½ - V)/k), where Gmax is maximal conductance, Erev is reversal potential, V½ is activation midpoint potential, and k is the slope factor. The voltage dependence of steady-state inactivation of ICa,L was measured by a double-pulse protocol. The overlap of steady-state activation and steady-state inactivation curves (cross-hatched) illustrates decrease of window ICa,L in Rem−/− compared with wild-type cardiac myocytes.
The maximal ICa,L was greater in Rem−/− compared with wild-type control, and examination of the I(V) curves reveals a shift in the voltage dependence of ICa,L (Fig. 2B). Figure 2C shows that the midpoint of activation of Rem−/− ICa,L is significantly shifted relative to control myocytes (-7.2 ± 0.3 mV and -3.3 ± 0.2 mV at wild-type and Rem−/− myocytes respectively, p < 0.05). The voltage dependence of steady-state inactivation was measured by a double pulse protocol. As Figure 2C demonstrates Rem deletion does not affect the voltage dependence of steady-state inactivation. Thus the theoretical steady-state window Ca-current was reduced in Rem−/− cardiac myocytes (Fig. 2C, shaded area). Taken together, there is an increase of maximal-activated ICa,L that is potentially compensated by a shift of steady-state activation.
ICa,L kinetics are key determinants of total Ca2+-entry into the cytosol, and in heterologous expression systems Rem alteration of kinetics depends on exogenous CaM expression.5 To evaluate whether Rem alters native ICa,L we measured ICa,L kinetics in cardiomyocytes. ICa,L kinetics were unaffected by Rem−/−. The effect of Rem on the time dependent inactivation of ICa,L was determined by bi-exponential fitting to the decay of current. The tau1 values were 24.7 ± 2.2 ms, and 25.5 ± 1.8 ms for wild type, and Rem−/−, respectively. The tau2 values were 5.0 ± 0.8 ms, and 5.3 ± 0.6 ms (Fig. S1). As expected, the fast and slow component amplitudes of Rem−/− were significantly larger than that of the age-matched wild-type controls (p < 0.05) but the relative amplitudes of the fast/slow components were not different between Rem−/− and control. The effect of Rem was also tested on the facilitation of ICa,L . During these measurements ICa,L was elicited by 16 consecutive pulses at 0.5Hz to 0 mV from -80 mV holding potential. The current amplitudes were normalized to the ICa,L amplitude elicited by the first pulse. Facilitation was not significantly different for Rem−/− (8+4%) compared with that in wild-type cardiac myocytes (6+/−3%; Fig. S2).
β-adrenergic stimulation increases Rem blockade of ICa,L in heterologous expression systems.16 Also, it is well-established that β-adrenergic stimulation increases the ICa,L amplitude, and causes a negative shift of ICa,L activation. Thus, in the absence of Rem, we predict there is a greater dynamic range of ICa,L responsiveness to β-adrenergic stimulation. To test this hypothesis, we superfused isoproterenol onto cardiomyocytes during the whole-cell recording. The ICa,L response to isoproterenol was greater for Rem−/− compared with wild-type cardiac myocytes. 10−8 M isoproterenol caused a 28 ± 4% and 22 ± 3% increase in ICa,L amplitude of Rem−/− and wild-type myocytes, respectively. Isoproterenol treatment caused no significant shift in the midpoint (V1/2) of steady-state inactivation curve. In the presence of 10−8 M isoproterenol inactivation V1/2 was -29.8 ± 1.0 mV for Rem−/−, and -28.9 ± 0.5 mV for wild-type cells. Isoproterenol shifted the voltage dependence of steady-state activation by -6.2 ± 1.0 mV for Rem−/−, but only -3.9 ± 1.3mV for wild type (Fig. 3). As a consequence, isoproterenol-modulated channel voltage-dependence was no longer significantly different between Rem−/− and wild type. These data are consistent with the notion that Rem serves as a governor of β-adrenergic stimulated ICa,L.
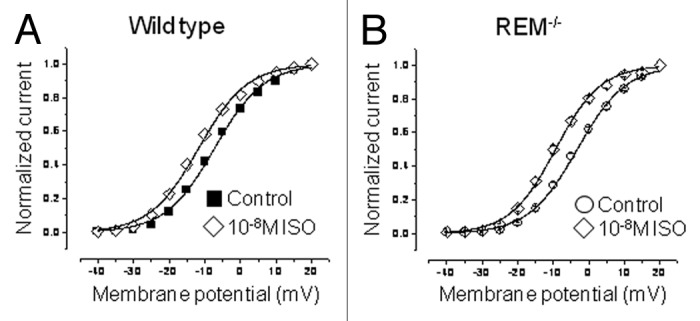
Figure 3. Isoproterenol normalizes the shift of activation V½ for Rem−/− compared with wild type. (A) Activation curves for wild-type ICa,L in control bath and 10nM isoproterenol (V½ for control and isoproterenol were -7.2 ± 0.3mV and -11.7 ± 0.3mV, respectively; p < 10−3; n = 12). (B) Activation curves for Rem−/− ICa,L in control bath, and following 10 nM isoproterenol (V½ for control and isoproterenol were -3.3 ± 0.2mV and -9.4 ± 0.3mV, respectively; p < 10−4; n = 24).
The Na+-Ca2+ exchange current (INCX) is the major efflux pathway that homeostatically balances cytosolic Ca-entry via ICa,L.17,18 The increased peak ICa,L can be offset by a positive shift of LTCC activation. Therefore, as a surrogate for assessing net trans-sarcolemmal ICa,L entry we measured INCX. INCX was recorded as a Ni2+-sensitive current using the descending limb of a ramp pulse changing from +60 to −100 mV. Outward and inward INCX were determined at +40 and −80 mV, respectively (Fig. 4). Rem−/− have a significantly smaller inward INCX (0.90 ± 0.16 pA/pF) than for the wild type (1.92 ± 0.4 pA/pF; p < 0.05, n = 7; Figure 4B). The mean outward current was larger in wild-type cells compared with that of Rem−/− cells, but the difference was not significant statistically (1.19 ± 0.24 pA/pF vs. 0.73 ± 0.11 pA/pF for wild-type and for Rem−/− myocytes, respectively, N.S., n = 7; Figure 4A). These results are consistent with the notion that Rem−/− net LTCC trans-sarcolemmal Ca flux may be reduced relative to wild-type cardiac myocytes.

Figure 4. INCX in wild-type and Rem−/− ventricular myocytes. Upper panel shows voltage protocol. Outward INCX measured at +40 mV, and inward INCX measured at -80mV as indicated on hyperpolarizing ramp. Mean INCX is greater in wild type compared with control for (A) outward and (B) inward current. *p < 0.05
Ca2+-transients and contractility in Rem−/− cardiac myocytes
Despite the increase of maximal peak ICa,L in Rem−/− cardiomyocytes, the shift of activation, and the decrease of INCX suggest that in intact cells Rem deletion leads to reduced LTCC function. To determine the impact of Rem deletion on Ca2+-handling, we simultaneously measured whole-cell Ca2+ transients and cell shortening (Fig. 5). Isolated ventricular myocytes were paced with 1 Hz field stimulation. The amplitude of twitch Ca2+ was significantly smaller for Rem−/− compared with wild type (0.35 ± 0.03 vs. 0.27 ± 0.02 for wild-type and for Rem−/− myocytes, respectively, p < 0.01, n = 25 and n = 19 for Rem−/− and wt, respectively; Figure 5A, B). Ten nM isoproterenol significantly increased twitch Ca amplitude in both Rem−/− and wild type, but in the presence of isoproterenol the twitch Ca amplitude remained significantly smaller for Rem−/− relative to wild type.

Figure 5. Ca-transients in intact ventricular myocytes. One Hz pacing reveals decreased twitch amplitude, without an effect on SR load in Rem−/− cardiomyocytes. (A) Representative single twitch Ca2+-transient from myocytes 1 paced at 1Hz. (B) Rem−/− twitch Ca-amplitude is significantly smaller than that in wild type. SR Ca-load was assessed by rapid application of 10mM caffeine. SR Ca load is not significantly different between Rem−/− and wild type. n = 25 and 19, Rem−/−, and wild type, respectively; *p < 0.01 control; *p < 0.05 in isoproterenol. (C) Decay kinetics of twitch Ca transients are not significantly different between Rem−/− and wild type. (D) Simultaneous measure of fractional cell shortening revealed no significant different between Rem−/− and wild type in either control or isoproterenol bath solution. (E and F) Computer simulations capture the decreased Ca transient amplitude in response to a +4mV shift of ICa,L steady-state activation. (E) Superimposed steady-state inactivation curve normalized to maximal conductance, and steady-state activation curve. For the activation curve, Gmax = 0.45 pS/pF for Rem−/− and 0.35pS/pF for wt. Inset shows activation curves expanded for voltage ranging from -40 to -10mV. (F) Simulated Ca2+-transients shows smaller peak Ca2+-transient amplitude in Rem−/− compared with wt.
The decaying phase of Ca transients was fitted to an exponential function to summarize the kinetics of Ca extrusion. The absence of Rem had no effect on the decay time constant (0.14 ± 0.01 sec for Rem−/− and 0.15 ± 0.01 sec, for wild-type cells, N.S., n = 25 and n = 19 for Rem−/− and wt, respectively, Figure 5C). Ten nM isoproterenol did not significantly alter the rate of Ca extrusion. The calcium load of SR was evaluated by a pulse of 5 mM caffeine, which was applied at the end of each experiment. SR Ca2+ load was unchanged by Rem knockout (Fig. 5B).
In spite of the smaller twitch Ca amplitude fractional cell shortening was not significantly different between Rem−/− and wild type (Fig. 5D). Time to peak and relaxation kinetics of cell shortening was faster in Rem−/− compared with wild-type myocytes (Fig. S3). The time to peak values of Rem−/− and wild type were 110 ± 4 ms and 132 ± 7ms (n = 21; p < 0.05), while the half relaxation time was 77 ± 5 ms and 124 ± 16 ms (n = 21; p < 0.05), respectively. 10nM isoproterenol treatment decreased the time-to-peak values compared with normal pre-drug treatment values in both animal groups (Fig. S3B). Isoproterenol treatment reduced the half relaxation time by 20 ± 8 ms in wild-type animals (n = 21; p < 0.05), but we did not observe significant change of this parameter in of Rem−/− (Fig. S3B). Cell shortening kinetics is a measure of a complex process. Shortening time reflects Ca-induced Ca release, Ca diffusion, and contractile apparatus response. Relaxation kinetics is dominated by cytosolic Ca clearance. Taken together these results suggest that the effect of the chronic absence of Rem extends beyond modulation of ICa,L.
Computer simulations
To explain our results we employed a newly developed computational model of mouse ventricular myocytes.19 Our approach was to re-adjust LTCC-related parameters based on data from ICa,L recordings in Rem−/− vs. wild-type myocytes, and then have the model generate Ca2+ transients. Figure 5E shows the activation and steady-state inactivation curves for ICa,L superimposed with the model output. The computer simulations quantitatively capture the resulting decrease of 1-Hz stimulated Ca transients (Fig. 5F). In addition, the tendency for the mean sarcoplasmic reticulum Ca reduction was also present in the model output. Cell-size adjustments had only modest effects on Ca transients (data not shown). These results are consistent with the contention that a +4mV shift of LTCC activation results in a decreased Ca-transient, despite a larger maximal conductance.
Compensatory responses
The positive shift of basal ICa,L activation suggests that CaV1.2 protein might be less phosphorylated in Rem−/− compared with wild type. The CaV1.2 distal carboxyl terminus (DCT) Ser1928 is a known substrate for the β-adrenergic-PKA signaling axis in cardiomyocytes.20,21 Cardiomyocyte lysates show that DCT migrates at about 37kDa.22 The specificity for the antibody used to probe for Ser1928P is demonstrated in Figure S4. Fractional DCT-S1928P/total-DCT was significantly less in Rem−/− (51+/−3%; n = 4) compared with that in wild-type ventricular lysates (78+/−4%; n = 4; p < 0.002; Figure 6A). We also tested phospholamban (PLB) phosphorylation to assess whether this was a general elevation of PKA signaling. PLB-Ser16 is a PKA substrate site,23 and there was no difference in PLB-Ser16P between wt and Rem−/− (Fig. 6B and C). PLB-Thr17 is an established CaMKII substrate. Although the mean PLB-Thr17P was greater in wt than Rem−/−, the differences were not statistically significant (Fig. 6B and C).

Figure 6. Analysis of CaV1.2 and phospholamban phosphorylation in wt and Rem−/−. (A) Protein extracts from wild-type and Rem−/− hearts were subjected to western blotting and probed with antibodies recognizing CaV1.2 distal carboxyl terminus (DCT) and DCT-Ser1928P. Size marker (horizontal line) is 37kDa. n = 4 Rem−/− and n = 4 wild type. (B) Protein extract was not boiled prior to gel loading to preserve multimeric PLB structure. Six bands for PLN are resolvable; Ser16P, and Thr17P are confined to upper two bands. (C) Bar graph summarizes fractional phosphorylation. *p < 0.002; n = 4 Rem−/− and n = 4 wild type. PLB phosphorylation was not significantly different. n = 6. (D) CaV1.2 expression is downregulated in Rem−/− vs. wt heart; 74+/− 4% decrease in Rem−/− vs. wild type; n = 3 Rem−/− and n = 3 wild type; p < 0.01.
We postulated that an increase of ICa,L density in the absence of Rem expression, could in theory, be offset by a compensatory decrease of CaV1.2 protein expression. To test this straightforward notion, we evaluated immunoblots from isolated ventricular membrane fractions. We observed a significant decrease of full-length CaV1.2 protein for Rem−/− compared with wild type (74+4%; p < 0.01; Figure 6D). The other major RGK protein in the myocardium, Rad, was not different in Rem−/− hearts (Fig. S5).
Discussion
Overexpressed Rem blocks ICa,L in myocytes suggesting that endogenous Rem might provide tonic blockade of ICa,L. Thus, eliminating Rem expression was expected to increase ICa,L. The major finding of this study is that Rem knockout results in an increased ICa,L density. This is the first time that RGK GTPase modification of ICa,L has been shown following in vivo knockout. Rem deletion also resulted in a depolarizing shift of channel activation, and this shift was normalized to control by β-adrenergic stimulation.
Rem is one of three RGK proteins (Rem, Rad, and Gem) expressed by cardiac myocytes. To date, all RGK proteins share the functional property of blockade of ICa,L when overexpressed in either cardiac myocytes,4,16 or other cells,6,14 including those expressing recombinant proteins.3 In contrast, loss-of-function studies reveal that Rad knockdown via shRNA,12 or Rad-dominant negative mutant overexpression24 leads to increased ICa,L density. A Rad-null mouse has been described,11 but unfortunately no information regarding ICa,L is available in these studies. At baseline, Rad-null mice have no phenotypic differences from control cardiac myocytes and hearts11; however, in view of our present findings it would be reasonable to re-evaluate Rad-null mice. Such a study might shed light on possible non-redundant functions of Rad vs. Rem in cardiac myocytes.
A partial decrease of Rad via shRNA increases ICa,L and leads to a similar increase of SR Ca-release.12 The parallels between depressing Rad function and the present report of Rem knockout are partial. Elimination of Rem also increases ICa,L density, but does not alter SR Ca-load. However, twitch Ca release was diminished. There are at least two non-competing mechanistic interpretations of how Ca-transients can be diminished despite elevated ICa,L density in Rem−/− cardiac myocytes. Rem may interfere with excitation-contraction coupling directly; alternatively, ICa,L driven by the action potential may be reduced in Rem−/− cardiac myocytes. Computer simulations support this notion. The +4mV activation shift is sufficient to decrease ICa,L, despite an elevated maximally-activated conductance, and this activation shift may responsible for the reduced twitch Ca amplitude. Interestingly the reduced twitch Ca amplitude did not cause a change of shortening amplitude. The activation shift in Rem-null cardiac myocytes is consistent with relatively fewer de-phosphorylated LTCC. Elevated ICa,L leads to cardiac hypertrophy.25 Thus, it is plausible to view the shift of ICa,L activation as a compensatory protective effect against increased LTCC Gmax in the absence of Rem expression.
Other compensatory responses that might occur in Rem−/− mice are more complex. For example, there is an elevated in vivo heart rate (data not shown) consistent with elevated basal sympathetic tone. In this light, the relative de-phosphorylation of CaV1.2 could protect against Ca-overload via hyper-active ICa,L. However, we could not detect a change in PLB phosphorylation status, despite assessing conditions that preserved pentameric PLB. Note that such conditions show more pronounced changes in the phosphorylation status of Ser16 and Thr17.26 The difference in relative phosphorylation status of CaV1.2 and PLB can reside in a number of factors, including but not limited to differential accessibilities to PKA and differential modulation by a variety of phosphatases.
We used CaV1.2-Ser1928 phosphorylation as a surrogate for the capacity of LTCC to serve as a substrate for PKA activity. The function of CaV1.2-Ser1928P is controversial, but the conservation of Ser1928 across species indirectly argues for physiological relevance. Two recent studies argue convincingly that Ser1700P and Ser1704P confer β-adrenergic receptor initiated modulation,21 whereas Ser1928P is not necessary for ICa,L modulation.27,28 Nonetheless, data are also convincing that Ser1928 is a substrate for PKA in cardiac myocytes.20
The mechanism for Rem alteration of ICa,L has been the focus of several recent studies. Early suggestions were that Rem blocks current via ‘sponging’ CaVβ; however, several lines of evidence argue in favor of a model whereby a Rem-LTCC interaction occurs at the cell surface. Despite RGK overexpression, CaV1.2 can be detected at the cell surface in heterologous expression systems,13 neurons,29 and other excitable cells.13 In addition, Ca2+-channel agonists causes rescue of ICa,L despite RGK overexpression in cardiac myocytes.4 Thus, it follows that in the absence of Rem, CaV1.2 channels at the surface are unmasked. This does not preclude alternative mechanisms of RGK modulation of LTCC function, particularly changes in stability of LTCC complexes on the plasma membrane whereby Rem promotes CaV1.2 internalization.15 We measured less total CaV1.2 protein concomitantly with more maximally activated ICa,L. This would be consistent with less CaV1.2 internalization in the absence of Rem expression. Unfortunately we were not able to directly assess endogenous CaV1.2 – this will be an important prediction to evaluate in future studies. To counterbalance higher maximal ICa,L the activation curve shift prevents excessive Ca2+ load as discussed above. It will be interesting to explore additional compensatory mechanisms possibly operating to maintain Ca-homeostasis. In summary our data are consistent with Rem null resulting in increased ICa,L; this is the opposite result to that in response to Rem overexpression in cardiac myocytes.16
In conclusion, the major finding of this study is that Rem-null mice display increased ICa,L. The increased ICa,L density is accompanied by a positive shift of the activation curve resulting in a predicted, yet paradoxical decrease of ICa,L entry at voltages corresponding to the plateau of the action potential. Thus, Rem modulates ICa,L in vivo, and cardiac myocytes compensate for modulated ICa,L to maintain Ca-homeostasis.
Methods
Generation of Rem−/− mice
Mice were housed in a pathogen-free facility and handled in accordance with standard use protocols, animal welfare regulations, and the NIH Guide for the Care and Use of Laboratory Animals. All protocols were approved by the University of Kentucky Institutional Animal Care and Use Committee. A targeting vector (Fig. 1) containing approximately 11.4 kb of the murine Rem1 gene was constructed from 129/Sv strain mouse genomic DNA. Additional details are provided in the Supp. Methods.
Adult ventricular myocyte cell isolation, electrophysiology, and calcium imaging
Single ventricular myocytes from female mice were enzymatically isolated following a modified AfCS protocol PP00000125 as previously described.30 External Ca was added incrementally back to the solution to 2.0 mM. Only rod-shaped, quiescent myocytes with clear edges were selected for current recording.
Ionic currents were recorded from Ca-tolerant female mouse ventricular cells at 37°C, 1 to 6 h post-isolation. Currents were recorded with the whole-cell configuration of the patch-clamp technique.
ICa,L was elicited by 500 ms long depolarizations to various test potentials arising from the holding potential of −40 mV. The stimulation frequency was 0.2 Hz. Peak current amplitude was defined as a difference between the peak value of ICa,L and its pedestal measured at the end of the pulse. ICa,L was normalized to cell capacitance.
Na+/Ca2+ exchanger (NCX) current was recorded in voltage-clamped ventricular myocytes as described previously.31 Briefly, NCX current was recorded using ramp pulses (shown in inset to Figure 3). Outward and inward NCX currents were determined during the descending limb of the ramp at +40 and –80 mV, respectively. After taking the control record in K+-free solution, the cell was superfused with 10 mM NiCl2 in order to fully block the current. Thus, total NCX current was determined at both membrane potentials as a Ni2+-sensitive current.
The measurements of intracellular calcium concentration were done on isolated cardiac myocytes. The cells were loaded with 2 μM fura-2-a.m. for 45 min. All recordings were performed at 20–22°C. Cells were paced for a minimum of 2 min at 1Hz before recordings to allow steady-state Ca loading. At the end of each measurement, the intracellular calcium store was emptied by a pulse of 5 mM caffeine. During the caffeine pulse the cells were not stimulated. Cell shortening was recorded simultaneously using IonOptix myocam and software.
Additional recording conditions are presented in the Supp. Methods. In all recordings the mouse of origin genotype was encoded to blind the investigator.
Computer simulations
Computer modeling was performed using the framework of the previously developed electrophysiology model for murine ventricular myocytes at 35°C. The difference in cell volume between the wild type and Rem−/− was calculated based on average measurements of the cell surface area and cell length and under the assumption that the myocytes are cylindrical in shape. The volumes of the junctional SR, network SR and dyadic space were adjusted according to their percentages (0.35, 1.05 and 0.1%, respectively) of the myoplasmic volume. The differences in the cell capacitance, myoplasmic volume and the volumes of the intracellular compartments between the wild type and Rem−/− were incorporated into the model during parameterization of the ICa,L.
Parameters for ICa,L were fitted to experimental measurements obtained from the wild type and Rem−/−, including the voltage-dependence of activation and inactivation kinetics, the current-voltage relationship, and the decay kinetics of the current, as described previously.31 All other parameters for the wild-type and the Rem−/− models were unchanged from the existing model.19
Western blot analysis
Hearts were excised from wild-type and Rem−/− mice and the left ventricles were isolated. Additional details are in Supp. Methods. Anti-pan phospholamban (PLB) was obtained from Thermo Scientific (USA) and S16P and T17P-phospholamban antibodies were purchased from Badrilla Ltd (UK). The CaV1.2 distal carboxyl terminus (DCT) antibodies (pan-DCT and S1928P-DCT) custom designed (ECM Biosciences, Versailles, KY, USA). Figure S4 shows a representative blot demonstrating phospho-selectivity of the S1928P-antibody; for these experiments a group of treated cells was exposed to 10μM forskolin and 30 μM 3-isobutyl-1-methylxanthine (IBMX) for 10 min immediately prior to cell harvest. The Rad antibody was the kind gift of Dr. CR Kahn (Joslin Diabetes Center, Harvard Medical School, Boston). The anti-Rem antibody was monoclonal anti-Rem (24E4) from gamma-one laboratories (Lexington, KY). All antibodies were diluted in 5% milk blocking buffer and incubated either over night at 4°C or for at least 1 h at room temperature with rocking. The membranes were washed and incubated with secondary antibody for use on the Li-Cor Odyssey Imaging System (Lincoln, NB, USA) and analyzed using their software.
Supplementary Material
Acknowledgments
We thank Ling Jin for assisting in the generation and maintenance of the Rem−/− mouse colony. Gabriela Hernandez and Douglas Yozwiak provided outstanding technical support.
Glossary
Abbreviations:
- LTCC
L-type calcium channel
- Ca
Ca2+ or calcium ion
- RGK
Rem, Rad, Rem2, Gem/Kir subfamily of Ras-related small GTPase
- ICa,L
L-type calcium channel current
- TAC
thoracic aorta constriction
- CaM
calmodulin
- CaMKII
calmodulin kinase II
- Rem−/−
Rem-null mouse
- ECG
electrocardiogram
- NCX
sodium-calcium exchange protein
- PLB
phospholamban
- DCT
distal carboxyl terminus of CaV1.2
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Sources of Funding
Supported by NIH HL072936 (DAA, JS), HL07491 (JS), 2P20 RR020171 (DAA), OTKA 73160 (JM), MRC G0800980 (NS), and Challenge Grant ES018636.
Supplemental Material
Supplemental material may be found here: http://www.landesbioscience.com/journals/channels/article/20192/
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/20192
References
- 1.Dai S, Hall DD, Hell JW. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 2009;89:411–52. doi: 10.1152/physrev.00029.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benitah JP, Alvarez JL, Gómez AM. L-type Ca(2+) current in ventricular cardiomyocytes. J Mol Cell Cardiol. 2010;48:26–36. doi: 10.1016/j.yjmcc.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 3.Finlin BS, Mosley AL, Crump SM, Correll RN, Ozcan S, Satin J, et al. Regulation of L-type Ca2+ channel activity and insulin secretion by the Rem2 GTPase. J Biol Chem. 2005;280:41864–71. doi: 10.1074/jbc.M414261200. [DOI] [PubMed] [Google Scholar]
- 4.Xu X, Marx SO, Colecraft HM. Molecular mechanisms, and selective pharmacological rescue, of Rem-inhibited CaV1.2 channels in heart. Circ Res. 2010;107:620–30. doi: 10.1161/CIRCRESAHA.110.224717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pang C, Crump SM, Jin L, Correll RN, Finlin BS, Satin J, et al. Rem GTPase interacts with the proximal CaV1.2 C-terminus and modulates calcium-dependent channel inactivation. Channels (Austin) 2010;4:192–202. doi: 10.4161/chan.4.3.11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Béguin P, Nagashima K, Gonoi T, Shibasaki T, Takahashi K, Kashima Y, et al. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature. 2001;411:701–6. doi: 10.1038/35079621. [DOI] [PubMed] [Google Scholar]
- 7.Correll RN, Pang C, Niedowicz DM, Finlin BS, Andres DA. The RGK family of GTP-binding proteins: regulators of voltage-dependent calcium channels and cytoskeleton remodeling. Cell Signal. 2008;20:292–300. doi: 10.1016/j.cellsig.2007.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finlin BS, Andres DA. Rem is a new member of the Rad- and Gem/Kir Ras-related GTP-binding protein family repressed by lipopolysaccharide stimulation. J Biol Chem. 1997;272:21982–8. doi: 10.1074/jbc.272.35.21982. [DOI] [PubMed] [Google Scholar]
- 9.Reynet C, Kahn CR. Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science. 1993;262:1441–4. doi: 10.1126/science.8248782. [DOI] [PubMed] [Google Scholar]
- 10.Béguin P, Mahalakshmi RN, Nagashima K, Cher DH, Ikeda H, Yamada Y, et al. Nuclear sequestration of beta-subunits by Rad and Rem is controlled by 14-3-3 and calmodulin and reveals a novel mechanism for Ca2+ channel regulation. J Mol Biol. 2006;355:34–46. doi: 10.1016/j.jmb.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Chang L, Zhang J, Tseng YH, Xie CQ, Ilany J, Brüning JC, et al. Rad GTPase deficiency leads to cardiac hypertrophy. Circulation. 2007;116:2976–83. doi: 10.1161/CIRCULATIONAHA.107.707257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang G, Zhu X, Xie W, Han P, Li K, Sun Z, et al. Rad as a novel regulator of excitation-contraction coupling and beta-adrenergic signaling in heart. Circ Res. 2010;106:317–27. doi: 10.1161/CIRCRESAHA.109.208272. [DOI] [PubMed] [Google Scholar]
- 13.Correll RN, Pang C, Finlin BS, Dailey AM, Satin J, Andres DA. Plasma membrane targeting is essential for Rem-mediated Ca2+ channel inhibition. J Biol Chem. 2007;282:28431–40. doi: 10.1074/jbc.M706176200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finlin BS, Crump SM, Satin J, Andres DA. Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases. Proc Natl Acad Sci U S A. 2003;100:14469–74. doi: 10.1073/pnas.2437756100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang T, Xu X, Kernan T, Wu V, Colecraft HM. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J Physiol. 2010;588:1665–81. doi: 10.1113/jphysiol.2010.187203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crump SM, Correll RN, Schroder EA, Lester WC, Finlin BS, Andres DA, et al. L-type calcium channel alpha-subunit and protein kinase inhibitors modulate Rem-mediated regulation of current. Am J Physiol Heart Circ Physiol. 2006;291:H1959–71. doi: 10.1152/ajpheart.00956.2005. [DOI] [PubMed] [Google Scholar]
- 17.Lester WC, Schroder EA, Burgess DE, Yozwiak D, Andres DA, Satin J. Steady-state coupling of plasma membrane calcium entry to extrusion revealed by novel L-type calcium channel block. Cell Calcium. 2008;44:353–62. doi: 10.1016/j.ceca.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pott C, Yip M, Goldhaber JI, Philipson KD. Regulation of cardiac L-type Ca2+ current in Na+-Ca2+ exchanger knockout mice: functional coupling of the Ca2+ channel and the Na+-Ca2+ exchanger. Biophys J. 2007;92:1431–7. doi: 10.1529/biophysj.106.091538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Niederer SA, Idigo W, Zhang YH, Swietach P, Casadei B, et al. A mathematical model of the murine ventricular myocyte: a data-driven biophysically based approach applied to mice overexpressing the canine NCX isoform. Am J Physiol Heart Circ Physiol. 2010;299:H1045–63. doi: 10.1152/ajpheart.00219.2010. [DOI] [PubMed] [Google Scholar]
- 20.Hulme JT, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during β1-adrenergic regulation. Proc Natl Acad Sci U S A. 2006;103:16574–9. doi: 10.1073/pnas.0607294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci Signal. 2010;3:ra70. doi: 10.1126/scisignal.2001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schroder E, Byse M, Satin J. L-type calcium channel C terminus autoregulates transcription. Circ Res. 2009;104:1373–81. doi: 10.1161/CIRCRESAHA.108.191387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colyer J. Phosphorylation states of phospholamban. Ann N Y Acad Sci. 1998;853:79–91. doi: 10.1111/j.1749-6632.1998.tb08258.x. [DOI] [PubMed] [Google Scholar]
- 24.Yada H, Murata M, Shimoda K, Yuasa S, Kawaguchi H, Ieda M, et al. Dominant negative suppression of Rad leads to QT prolongation and causes ventricular arrhythmias via modulation of L-type Ca2+ channels in the heart. Circ Res. 2007;101:69–77. doi: 10.1161/CIRCRESAHA.106.146399. [DOI] [PubMed] [Google Scholar]
- 25.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–44. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sachan N, Dey A, Rotter D, Grinsfelder DB, Battiprolu PK, Sikder D, et al. Sustained hemodynamic stress disrupts normal circadian rhythms in calcineurin-dependent signaling and protein phosphorylation in the heart. Circ Res. 2011;108:437–45. doi: 10.1161/CIRCRESAHA.110.235309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganesan AN, Maack C, Johns DC, Sidor A, O’Rourke B. Beta-adrenergic stimulation of L-type Ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of alpha1C but not serine 1928. Circ Res. 2006;98:e11–8. doi: 10.1161/01.RES.0000202692.23001.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemke T, Welling A, Christel CJ, Blaich A, Bernhard D, Lenhardt P, et al. Unchanged beta-adrenergic stimulation of cardiac L-type calcium channels in Ca v 1.2 phosphorylation site S1928A mutant mice. J Biol Chem. 2008;283:34738–44. doi: 10.1074/jbc.M804981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen H, Puhl HL, 3rd, Niu SL, Mitchell DC, Ikeda SR. Expression of Rem2, an RGK family small GTPase, reduces N-type calcium current without affecting channel surface density. J Neurosci. 2005;25:9762–72. doi: 10.1523/JNEUROSCI.3111-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schroder E, Magyar J, Burgess D, Andres D, Satin J. Chronic verapamil treatment remodels ICa,L in mouse ventricle. Am J Physiol Heart Circ Physiol. 2007;292:H1906–16. doi: 10.1152/ajpheart.00793.2006. [DOI] [PubMed] [Google Scholar]
- 31.Birinyi P, Acsai K, Bányász T, Tóth A, Horváth B, Virág L, et al. Effects of SEA0400 and KB-R7943 on Na+/Ca2+ exchange current and L-type Ca2+ current in canine ventricular cardiomyocytes. Naunyn Schmiedebergs Arch Pharmacol. 2005;372:63–70. doi: 10.1007/s00210-005-1079-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.