

The constitutive proteasome is a multiprotease complex expressed in all eukaryotic cells and plays an important role in regulating intracellular protein homeostasis via ubiquitin-mediated degradation of proteins.[1–3] Mammalian cells can also express an alternative form of the proteasome called as the immunoproteasome.[4] The immunoproteasome is expressed in cells of hematopoietic origin but can also be induced in other types of cells upon exposure to cytokines such as interferon-γ (IFN-γ) or tumor necrosis factor-α (TNF-α). Exposure to these stimuli induces the synthesis of catalytic subunits LMP2 (β1i), MECL1 (β2i) and LMP7 (β5i), which replace their constitutive counterparts Y (β1), Z (β2) and X (β5), respectively, to form the immunoproteasome.[5] The primary function of the immunoproteasome is thought to be the promotion of adaptive immune responses, but recent studies also show that the immunoproteasome plays an important role in other cellular processes[6–8] as well as certain pathological states such as cancer, neurodegenerative and autoimmune diseases.[4,9–13] Despite recent studies indicating the distinct role of the immunoproteasome in normal and disease states, its functions remain poorly understood, in large part due to the lack of appropriate molecular probes.
Over the past decade, chemical probes targeting functionally active proteins have provided great tools for protein functional study.[14–17] For proteasome biology, this type of probe can be particularly useful, since there are two forms of β subunits in cells: catalytically inactive immature (propeptide containing) and active mature forms.[18] The immature β subunits undergo maturation only in the fully assembled proteasome complex.[18] Therefore, proteasome inhibitors which contain active-site-targeting pharmacophores, such as the vinyl sulfone[19] or epoxyketone functional groups,[20] can function as activity-based probes, targeting only the catalytically active β subunits in the fully assembled proteasome. The use of these pharmacophores led to the design of the first generation activity-based fluorescent probes targeting proteasomes, which proved to be useful in functional studies or for inhibitor screening.[21–23] However, due to a high degree of primary sequence and structural homology between the immunoproteasome and the constitutive proteasome,[24] these probes typically target both proteasome subtypes, adding to the difficulty in deciphering the distinct functions of the immunoproteasome. Therefore, in order to properly study functional intricacies of the immunoproteasome, it will be necessary to develop a new generation of molecular probes specific for the immunoproteasome.
Here we report the synthesis and application of LKS01-B650, an imaging probe that selectively binds and visualize the catalytically active LMP7 subunit of immunoproteasome in living cells. Since LKS01-B650 is designed for near-infrared (NIR) ranges, it can be potentially implemented as an imaging agent to monitor the activity and subcellular localization of the immunoproteasome in vivo.
In our current study, we used IPSI as our lead compound to develop an LMP7-specific imaging probe (Fig. 1). IPSI has been recently reported to selectively inhibit the LMP7 subunit in a dose-dependent manner.[25] To design an LMP7-specific fluorescent molecular probe, we first examined how IPSI binds with the LMP7 subunit by performing in silico molecular modeling studies utilizing a 3D model of the immunoproteasome previously described by us.[26] These studies of the interaction between LMP7 and IPSI predicted that the tryptophan residue at P2 segment of IPSI is not involved in any specific interaction with LMP7, thus providing a potential anchor site to introduce a fluorophore without disrupting the LMP7 binding specificity of IPSI (Fig. 1).
Figure 1.

Chemical structure of IPSI (A) and its predicted LMP7 binding mode (B). The LMP7:IPSI binding structure depicted here was the last snapshot of the molecular dynamics (MD) simulation of the LMP7:IPSI complex in water. In this model, the tryptophan side chain is exposed towards the surface of LMP7 through the open channel extending from the active site. The blue or white circle highlights the tryptophan group at P2, which is modified to yield LKS01.
Based on these predictions, we set out to synthesize an IPSI analog which was derivatized with a fluorophore at the P2 position. Specifically, the synthesis of IPSI derivative was started with Boc-Phe-OH (1), which was converted to an α,β-unsaturated ketone 2 by treatment with N,O-dimethylhydroxylamine hydrochloride followed by a reaction with isopropenyl magnesium bromide (Scheme 1). Epoxidation of 2 with dimethyldioxirane (DMDO) afforded 3a and 3b with a ratio of 2.5 to 1 and subsequent deprotection of tert-butyloxycarbonyl (Boc) group by trifluoroacetic acid (TFA) yielded 4. Coupling of H-Lys(Z)-OtBu (5) with Fmoc-D-Ala-OH using HBTU, followed by Fmoc removal and coupling to 3-methylindene-2-carboxylic acid, and then acid deprotection yielded 6. Finally, a standard peptide coupling between compounds 4 and 6 yielded LKS01 (7).
Scheme 1. Synthetic Route to LKS01 (7) and LKS01-B650 (9).

aReagents and conditions: (a) N,O-dimethylhydroxylamine hydrochloride (1.2 equiv), HBTU (1.05 equiv), HOBt (1.05 equiv), DIPEA (5 equiv), DCM, rt, 15 hr, 98%; (b) Isopropenyl magnesium bromide (2.5 equiv), THF, 0°C, 3 hr, 84%; (c) DMDO (1.5 equiv), rt, 15 hr, 98%, ratio 3a:3b (2.c5:1); (d) TFA-DCM (1:1), 30 min., rt (e) Fmoc-D-Ala-OH (1 equiv), HBTU (1.5 equiv), HOBt (1.5 equiv), DIPEA (5 equiv), DCM, rt, 3 hr, 96%; (f) 20% piperidine, 2 hr; (g) 3-Methylindene-2-carboxylic acid (1 equiv), HBTU (1.5 equiv), HOBt (1.5 equiv), DIPEA (5 equiv), DCM, rt, 3 hr, 95% for 2 steps; (h) TFA (90% in DCM), 1 hr; (i) 4 (1 equiv), HBTU (1.5 equiv), HOBt (1.5 equiv), DIPEA (5 equiv), DCM, rt, 3 hr, 70% for 3 steps (d,h,i). (j) H2-Pd/C, MeOH, rt, 2 h; (k) BODIPY®650/665-X-succinimidyl ester, DMF, rt, 2 hr, 90% for 2 steps.
Before attaching a fluorescent residue to LKS01 (7), we wanted to test whether LKS01 still maintained the LMP7 binding selectivity of IPSI. To test this, we performed a mobility shift assay using Panc-1 cells (derived from human pancreatic ductal adenocarcinoma), since these cells abundantly express both the immunoproteasome and the constitutive proteasome (Suppl. Fig. S2). Following LKS01 treatment at varying concentrations, the Panc-1 cells were lysed and subjected to immunoblotting analyses using antibodies specific for different proteasome catalytic subunits. Our results showed that LKS01 maintains the LMP7 binding specificity of IPSI at low concentrations, indicated by a shift of the LMP7 band mobility on the immunoblot (Fig. 2A). At high concentrations, LKS01 also modifies the X and LMP2 subunits, which is the same result as observed with IPSI (Suppl. Fig. S2). Overall, these results indicated that LKS01 maintains the proteasome subunit binding pattern of IPSI.
Figure 2.
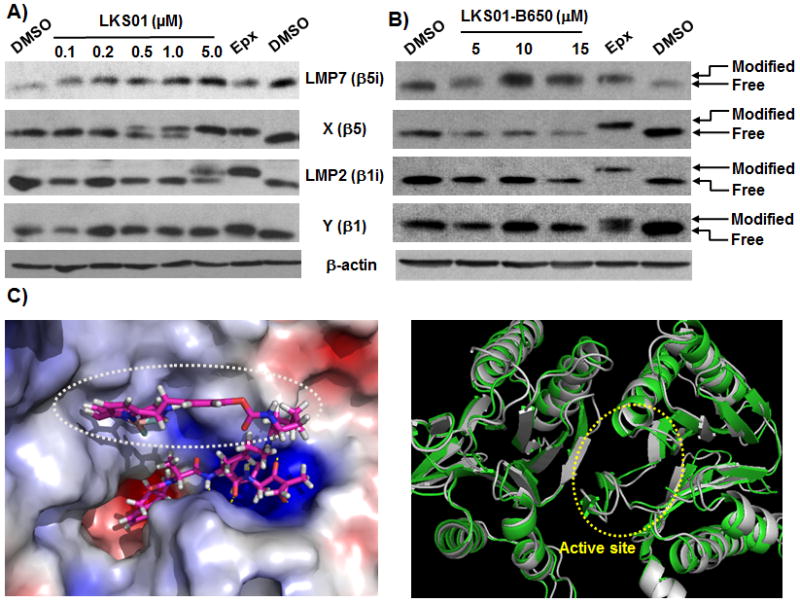
Selective modification of LMP7 by LKS01 (A) and LKS01-B650 (B) in Panc-1 cells. Panc-1 cells were incubated with LKS01 (0.1, 0.2, 0.5, 1 or 5 μM) or LKS01-B650 (5, 10 or 15 μM) for 1.5 hr and lysed. The resulting cell extracts were electrophoresed and subjected to immunoblotting. LKS01-B650 selectively modified LMP7, but not other proteasome catalytic subunits at the concentrations tested. Epx (epoxomicin, used as positive control). (C) Binding mode of LKS01-B650 with LMP7 active site from X-ray crystal structure.24 An overlay of the LMP7 homology model (gray) used for initial design of LKS01 derivatives with X-ray structure (green) showed a good alignment (right panel). The root-mean-square deviation (RMSD) of the atomic positions of the homology model from the X-ray crystal structure is 1.41 Å which is very small compared to the resolution of the X-ray crystal structure (2.9 Å).
After confirming the selectivity of LKS01 towards LMP7, we then coupled BODIPY® 650/665 to dihydro-LKS01 (8) using BODIPY® 650/665-X succinimidyl ester to yield LKS01-B650 (9) (Scheme 1). We then examined whether LKS01-B650 selectively modifies LMP7 by performing a mobility shift assay using Panc-1 cells. As shown in Fig. 2B, LKS01-B650 (9) modified LMP7 at 5 μM, but not other proteasome subunits including X and LMP2 in concentrations of up to 15μM. These results indicate that the introduction of the BODIPY® 650/665 fluorescent group at the P2 position does not alter the binding specificity of LKS01 to LMP7. This result is consistent with the LKS01-B650 binding study with the LMP7 X-ray structure (Fig. 2C), which indicated that the S2 pocket of LMP7 can accomodate the BODIPY fluorescent residue of LKS01-B650 without disrupting interactions between LMP7 and LKS01-B650.
To further examine whether LKS01-B650 labels any other proteasomal catalytic subunits or non-proteasomal proteins, we performed in-gel fluorescence assay in immunoproteasome-expressing parental T1 cells and T1-derived T2 lymphoblast cells lacking the immunoproteasome. Of note, inhibitor concentrations used in these studies were determined by mobility shift assays (Suppl. Figs. S3 & S4). When T1 cell lysate was treated with LKS01-B650, a strong fluorescence signal was observed around 23 kD (Fig. 3, left panel). This signal was completely abolished by the pretreatment of cells with LKS01 or epoxomicin, a broadly acting proteasome inhibitor (Fig. 3, left panel). However, the fluorescent signal was not affected by the pretreatment of cells with YU-102, an LMP2 selective inhibitor (Suppl. Fig. S5).[27] Importantly, no significant fluorescent bands except the 23kD one were observed, suggesting the specificity of LKS01-B650 towards LMP7. Similarly, no fluorescent signals were detected in the in-gel fluorescence assay with T2 cell lysate in approximately 20 to 30 kD ranges (Fig. 3, right panel), suggesting that LKS01-B650 does not target constitutive proteasomal catalytic subunits such as β1 (Y), β2 (Z) and β5 (X). Of note, fluorescence signals were observed at above 100 kD ranges, but these appeared to be non-specific and were removed by extensive washing with buffers. In addition, we performed western blotting analyses and our results confirmed that LKS01-B650 leads to selective labeling of LMP7 in the T1 cells, but not in the LMP7-deficient T2 cells. (Suppl. Fig. S6). Taken together, these results strongly support that LMP7 is the only major target of LKS01-B650 in the concentration range tested.
Figure 3.

Visualization of LMP7 bound with LKS01-B650 in T1 & T2 lymphoblast cell lysate. Cell lysate was pretreated with LKS01, YU102, epoxomicin (epx) or DMSO for 1 hr, followed by incubation of LKS01-B650 for 1 hr at room temperature. LKS01-B650 binding was then visualized using Typhoon FLA 9000 imaging system. Pretreatment of LKS01 (a parent compound of LKS01-B650), or epoxomicin (a general proteasome inhibitor) effectively competed away fluorescent signals.
To test the ability of LKS01-B650 to visualize the catalytically active LMP7 in living cells in a subunit-specific manner, we captured fluorescent images of Panc-1 cells treated with LKS01, followed by LKS01-B650. As shown in Fig. 4, pretreatment of Panc-1 cells with LKS01 effectively abolished fluorescent signals. These are consistent with the results obtained from the in-gel fluorescence assay (Fig. 3), demonstrating the selective labeling and visualization of catalytically active LMP7 using LKS01-B650.
Figure 4.

Visualization of LMP7 using LKS01-B650 in Panc-1 cells. Cells were pretreated with DMSO or LKS01 (5 μM) for 1.5 hr, followed by incubation of LKS01-B650 (10 μM) for 1.5 hr. LKS01-B650 binding was then visualized via fluorescence microscopy. Pretreatment of LKS01, a parent compound of LKS01-B650, effectively competed away fluorescent signals. LKS01-B650 (Red, λex = 635 nm, λem = 665 nm), DAPI (Blue) = Nucleus.
Subcellular localization of proteins may provide important functional clues and such information is obtained by using fluorescently labeled antibody or by green fluorescent protein (GFP)-tagged proteins of interest.[28] However, it is difficult to investigate the localization of the catalytically active immunoproteasome using immunofluorescence labeling since the antibodies often detect both catalytically active and propeptide-containing inactive forms of proteasome subunits. With this in mind, we wanted to test the utility of LKS01-B650 as an imaging probe by investigating the localization of catalytically active immunoproteasome. Specifically, Panc-1 cells were incubated with LKS01-B650 for 2 hours and after extensive washing were stained with fluorescein-labeled LMP7 antibodies. As shown in Fig. 5A, fluorescent signals from LKS01-B650 were mainly localized in the perinuclear area, where the assembled immunoproteasome complex is known to be located. Fluorescence signals derived from the LMP7 antibody displayed a similar localization pattern in the perinuclear area, but did not completely overlap with the fluorscence signals from LKS01-B650. These observed differences between the fluorscence signals from LKS01-B650 and LMP7 antibody may be attributed to the presence of LMP7 precursors and/or differing sensitivity/specificity of the two visualization methods. As a complementary approach to verify whether the fluorescent signals detected by LKS01-B650 are derived from the catalytically active LMP7, we examined whether the fluorescent signals from LKS01-B650 would overlap with those from UK101-Fluor, a fluorescent probe targeting the catalytic active LMP2.[29] When Panc-1 cells were cotreated with LKS01-B650 and UK101-Fluor, the red fluorescent signals from LKS01-B650 were extensively colocalized with the green fluorescent signals derived from UK101-Fluor (Fig. 5B). These results indicate that LKS01-B650 is likely to visualize the catalytically active, LMP7 as a component of the fully assembled immunoproteasome.
Figure 5.

Localization of LKS01-B650-bound LMP7. (A) Panc-1 cells were incubated with LKS01-B650 (10 μM) for 1.5 hr, and subsequently stained with antibodies recognizing LMP7. Fluorescent signals from LKS01-B650 were mainly localized in the perinuclear area, where the assembled immunoproteasome complex is known to be located. Fluorescence signals derived from the LMP7 antibody displayed a similar localization pattern in the perinuclear area, but did not completely overlap with the fluorscence signals from LKS01-B650. (B) Panc-1 cells treated with LKS01-B650 and UK101-Fluor, a LMP2-specific fluorescent imaging probe. LMP7 signals are colocalized with signals from LMP2, indicating LKS01-B650-bound LMP7 is located in fully assembled immunoproteasomes.
In summary, we have developed a NIR fluorescent probe that selectively targets the catalytically active LMP7 subunit of the immunoproteasome. Our results show that this probe can be used to visualize the catalytically active LMP7 in living cells, providing a tool to monitor the catalytically active LMP7/immunoproteasome in real-time. Given the implications of the immunoproteasome in cancer and inflammatory diseases, this imaging agent will provide a useful tool to understand the function of the immunoproteasome in these disease states.
Experimental Section
All details about the Experimental Procedures and materials used in this study are given in accompanying Supplemental Information.
Supplementary Material
Acknowledgments
We gratefully acknowledge the National Institutes of Health for their financial support of this work through Grants R01 CA128903 (KBK), R15 CA156601 (WL), and RC1 MH088480 (CGZ). We thank the members of the Kim and Lee laboratories for their helpful comments on the manuscript.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
Contributor Information
Prof. Wooin Lee, Email: wooin.lee@uky.edu.
Prof. Kyung-Bo Kim, Email: kyung.kim@uky.edu.
References
- 1.Navon A, Ciechanover A. J Biol Chem. 2009;284(49):33713–33718. doi: 10.1074/jbc.R109.018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jung T, Catalgol B, Grune T. Mol Aspects Med. 2009;30(4):191–296. doi: 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Marques AJ, Palanimurugan R, Matias AC, Ramos PC, Dohmen RJ. Chem Rev. 2009;109(4):1509–1536. doi: 10.1021/cr8004857. [DOI] [PubMed] [Google Scholar]
- 4.Groettrup M, Kirk CJ, Basler M. Nat Rev Immunol. 2010;10(1):73–78. doi: 10.1038/nri2687. [DOI] [PubMed] [Google Scholar]
- 5.Strehl B, Seifert U, Kruger E, Heink S, Kuckelkorn U, Kloetzel PM. Immunol Rev. 2005;207:19–30. doi: 10.1111/j.0105-2896.2005.00308.x. [DOI] [PubMed] [Google Scholar]
- 6.Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schroter F, Prozorovski T, Lange N, Steffen J, Rieger M, Kuckelkorn U, Aktas O, Kloetzel PM, Kruger E. Cell. 2010;142(4):613–624. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 7.Rockwell CE, Monaco JJ, Qureshi N. Pharmacology. 2012;89(3–4):117–126. doi: 10.1159/000336335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angeles A, Fung G, Luo H. Front Biosci. 2012;17:1904–1916. doi: 10.2741/4027. [DOI] [PubMed] [Google Scholar]
- 9.Basler M, Groettrup M. Methods Mol Biol. 2012;832:391–401. doi: 10.1007/978-1-61779-474-2_27. [DOI] [PubMed] [Google Scholar]
- 10.Lee W, Kim KB. Current Topics in Medicinal Chemistry. 2011;11(23):2923–2930. doi: 10.2174/156802611798281348. [DOI] [PubMed] [Google Scholar]
- 11.Kuhn DJ, Orlowski RZ, Bjorklund CC. Curr Cancer Drug Targets. 2011;11(3):285–295. doi: 10.2174/156800911794519725. [DOI] [PubMed] [Google Scholar]
- 12.Wehenkel M, Hong JT, Kim KB. Mol Biosyst. 2008;4(4):280–286. doi: 10.1039/b716221a. [DOI] [PubMed] [Google Scholar]
- 13.Yewdell JW. Proc Natl Acad Sci U S A. 2005;102(26):9089–9090. doi: 10.1073/pnas.0504018102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cravatt BF, Wright AT, Kozarich JW. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 15.Nomura DK, Dix MM, Cravatt BF. Nat Rev Cancer. 2010;10(9):630–638. doi: 10.1038/nrc2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadaghiani AM, Verhelst SH, Bogyo M. Curr Opin Chem Biol. 2007;11(1):20–28. doi: 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 17.Berger AB, Vitorino PM, Bogyo M. Am J Pharmacogenomics. 2004;4(6):371–381. doi: 10.2165/00129785-200404060-00004. [DOI] [PubMed] [Google Scholar]
- 18.Murata S, Yashiroda H, Tanaka K. Nature reviews Molecular cell biology. 2009;10(2):104–115. doi: 10.1038/nrm2630. [DOI] [PubMed] [Google Scholar]
- 19.Bogyo M, McMaster JS, Gaczynska M, Tortorella D, Goldberg AL, Ploegh H. Proc Natl Acad Sci U S A. 1997;94(13):6629–6634. doi: 10.1073/pnas.94.13.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Groll M, Kim KB, Kairies N, Huber R, Crews CM. J Am Chem Soc. 2000;122(6):1237–1238. [Google Scholar]
- 21.Berkers CR, van Leeuwen FW, Groothuis TA, Peperzak V, van Tilburg EW, Borst J, Neefjes JJ, Ovaa H. Mol Pharm. 2007 doi: 10.1021/mp0700256. [DOI] [PubMed] [Google Scholar]
- 22.Ovaa H. Nat Rev Cancer. 2007;7(8):613–620. doi: 10.1038/nrc2128. [DOI] [PubMed] [Google Scholar]
- 23.Luker GD, Pica CM, Song J, Luker KE, Piwnica-Worms D. Nat Med. 2003;9(7):969–973. doi: 10.1038/nm894. [DOI] [PubMed] [Google Scholar]
- 24.Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, Groll M. Cell. 2012;148(4):727–738. doi: 10.1016/j.cell.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 25.Parlati F, Lee SJ, Aujay M, Suzuki E, Levitsky K, Lorens JB, Micklem DR, Ruurs P, Sylvain C, Lu Y, Shenk KD, Bennett MK. Blood. 2009;114(16):3439–3447. doi: 10.1182/blood-2009-05-223677. [DOI] [PubMed] [Google Scholar]
- 26.Lei B, Abdul Hameed MD, Hamza A, Wehenkel M, Muzyka JL, Yao XJ, Kim KB, Zhan CG. The journal of physical chemistry B. 2010;114(38):12333–12339. doi: 10.1021/jp1058098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myung J, Kim KB, Lindsten K, Dantuma NP, Crews CM. Molecular Cell. 2001;7:411–420. doi: 10.1016/s1097-2765(01)00188-5. [DOI] [PubMed] [Google Scholar]
- 28.Crivat G, Taraska JW. Trends Biotechnol. 2012;30(1):8–16. doi: 10.1016/j.tibtech.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carmony KC, Lee DM, Wu Y, Lee NR, Wehenkel M, Lee J, Lei B, Zhan CG, Kim KB. Bioorg Med Chem. 2012;20(2):607–613. doi: 10.1016/j.bmc.2011.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.