

Abstract
Transcription is a sophisticated multi-step process in which RNA polymerase II (Pol II) transcribes a DNA template into RNA in concert with a broad array of transcription initiation, elongation, capping, termination, and histone modifying factors. Recent global analyses of Pol II distribution have indicated that many genes are regulated during the elongation phase, shedding light on a previously underappreciated mechanism for controlling gene expression. Understanding how various factors regulate transcription elongation in living cells has been greatly aided by chromatin immunoprecipitation (ChIP) studies, which can provide spatial and temporal resolution of protein-DNA binding events. The coupling of ChIP with DNA microarray and high-throughput sequencing technologies (ChIP-chip and ChIP-seq) has significantly increased the scope of ChIP studies and genome-wide maps of Pol II or elongation factor binding sites can now be readily produced. However, while ChIP-chip/ChIP-seq data allow for high-resolution localization of protein-DNA binding sites, they are not sufficient to dissect protein function. Here we describe techniques for coupling ChIP-chip/ChIP-seq with genetic, chemical, and experimental manipulation to obtain mechanistic insight from genome-wide protein-DNA binding studies. We have employed these techniques to discern immature promoter-proximal Pol II from productively elongating Pol II, and infer a critical role for the transition between initiation and full elongation competence in regulating development and gene induction in response to environmental signals.
Keywords: transcription elongation, gene expression, ChIP-chip, ChIP-seq
1. Introduction
1.1. Background
Synthesis of messenger RNA by RNA polymerase II (Pol II) is a carefully orchestrated process. Although the regulated recruitment of the transcription machinery to a gene promoter has been studied for decades (1), recent evidence suggests that regulation can occur at many steps in the transcription cycle, and may be particularly prevalent during transcription elongation (2–5). Transcription begins with promoter recognition and binding by the pre-initiation complex (PIC) consisting of Pol II and general transcription factors including TFIID and TFIIH. The C-terminal domain (CTD) of the Rpb1 subunit of Pol II, consisting of multiple copies of the consensus sequence YSPTSPS, is largely unphosphorylated during initial promoter binding, which favors interactions between the CTD and activators such as the Mediator complex (6, 7). Unwinding of DNA by TFIIH allows Pol II to access the template DNA strand and begin incorporating nucleotides into a nascent RNA chain. As the nascent RNA is extended, TFIIH phosphorylates Serine-5 of the CTD, which is thought to positively influence the association of the mRNA capping machinery (8, 9). Factors such as the Negative Elongation Factor, or NELF, complex in collaboration with the heterodimeric DSIF complex (comprised of Spt4/Spt5) can impede elongation through the promoter-proximal region (10–14).
Recruitment of the P-TEFb kinase signals the transition to productive elongation, by phosphorylating the CTD at Serine-2 and helping to overcome NELF-dependent stalling of early elongation (15–17). The Serine-2 phosphorylated, fully elongation-competent form of Pol II is then bound by RNA processing and termination factors as it transcribes in a highly processive manner towards the poly-adenylation site, which signals for termination. In addition to this coordinated regulation of the phosphorylation status of the Pol II CTD and the association of transcription elongation and processing factors, histone remodeling and modifying factors are specifically recruited to facilitate efficient polymerase elongation (18).
Each aspect of transcription is governed by interactions between the largely proteinaceous transcription machinery and the DNA template. Chromatin immunoprecipitation (ChIP) is capable of providing high resolution spatial and temporal information about the interactions between proteins and DNA in living cells. It is therefore well suited for dissecting phases of the transcription cycle by placing individual protein complexes at specific genomic locations at biologically significant times. For example, biochemical studies had identified the NELF complex as capable of inhibiting Pol II elongation, in a manner that can be reversed by the kinase activity of P-TEFb (10–13), leading to the hypothesis that NELF played a role in regulating the efficiency of transcript elongation by Pol II prior to the transition to full elongation competence. Subsequent ChIP studies have shown that the NELF complex is associated broadly with unphosphorylated or Serine-5 phosphorylated Pol II near transcription start sites. This association is not maintained in downstream regions where the polymerase is Serine-2-phosphorylated and productive elongation occurs, thereby confirming that the biochemically determined activity of the NELF complex is relevant and placing it in a global in vivo context (19, 20).
The power of ChIP has been tremendously increased by its coupling with DNA microarray technology. In traditional ChIP assays, protein complexes are localized to genomic loci by querying immunoprecipitated DNA with quantitative or semi-quantitative PCR reactions using primer pairs designed to amplify specific regions of interest. In ChIP-chip, immunoprecipitated material is labeled with fluorescent dyes (with or without prior amplification) and hybridized to DNA microarrays containing several hundred thousand, to several million probes (Figure 1). Performing ChIP coupled with DNA microarrays has several significant advantages over traditional ChIP. First, instead of querying a limited number of loci selected by researchers with inherent biases, large contiguous genomic regions are probed in a single experiment, eliminating bias and permitting discovery of unanticipated sites of protein-DNA binding, as well as regions where binding is unexpectedly absent. Second, localization of protein binding can be accomplished with optimized commercially available platforms, eliminating time spent designing and testing primer pairs and running expensive large-scale quantitative PCR assays. In addition, the use of the same platforms by different research groups facilitates direct comparison of binding data obtained for many individual proteins; groups such as the ENCODE consortium have used this to a great advantage (e.g. 21). Third, the parallel analysis of thousands of genes allows one to parse the data into distinct classes of genes based on different binding distributions or behaviors, and permits statistical comparisons to be made between classes.
Figure 1. Workflow for ChIP-chip and ChIP-seq Experiments.

Following experimental manipulation (yellow boxes), cells are crosslinked with formaldehyde, sonicated to fragment chromatin, and protein-DNA complexes immunoprecipitated with antibodies targeting the protein or modification of interest (here, Pol II). Following quality control qPCR to confirm expected ChIP signal at control regions, immunoprecipitated DNA is processed specifically for either ChIP-chip or ChIP-seq. ChIP-chip can provide information about all immunoprecipitated DNA sequences complementary to tiling array probes in a strand-insensitive manner. ChIP-seq provides information about all mappable sequences located at the 5′-ends of immunoprecipitated DNA (red and blue boxes).
Genomic distribution of Pol II and other transcription elongation factors has also been determined through a process referred to as ChIP-seq (22), which offers an appealing complementary or alternative method for mapping protein-DNA interactions. The strategy is similar to ChIP-chip but instead of labeling immunoprecipitated material and hybridizing it to a microarray, immunoprecipitated material is used to construct a library of millions of individual DNA fragments which are amplified and then sequenced in parallel (Figure 1). Massively parallel sequencing technology, also referred to as deep, or high-throughput sequencing, is now widely available on a variety of platforms, each with distinct characteristics (see section 2.6.2 below).
1.2. Genome-wide analyses illuminate novel aspects of transcription elongation
Global analyses of Pol II distribution have provided insight into mechanisms of regulation of transcription elongation that are unattainable with either traditional ChIP or biochemical techniques. In particular, ChIP-chip studies have detected a widespread decoupling of Pol II recruitment to a promoter and mature transcript formation in vivo. Preinitiation complexes (PICs) were mapped across the human genome by Ren and coworkers using ChIP against Pol II and TFIID. Surprisingly, 13% of genes with PIC-bound promoters did not produce detectable transcripts, though PIC occupancy was confirmed through a variety of other comparisons (3). A subsequent study detected Pol II at the majority of promoters of protein-coding genes in human embryonic stem cells, though only a subset of these genes produced full-length transcripts detectable by expression microarray (2). The limited correlation between transcription initiation and mature transcript production suggests that promoter-proximal pausing or stalling, as described at the Drosophila heat-shock loci (23–25), may occur at many more genes than previously appreciated. Promoter-proximal stalling is a phenomenon wherein Pol II is recruited to a gene promoter and initiates transcription, but slows or stops during elongation through the promoter-proximal region (26). Escape of stalled Pol II into the gene is rate-limiting for expression of genes like Drosophila Hsp70, but this was thought to be a relatively unique regulatory strategy.
To investigate how widespread promoter-proximal stalling is in Drosophila, our laboratory employed ChIP-chip to map the total Pol II distribution in S2 cells (using an antibody against the small Pol II subunit Rbp3) across the Drosophila genome. Analysis of signal intensities for Pol II (Rpb3)-binding at promoters versus downstream regions revealed that more than one thousand genes exhibit significant promoter-proximal enrichment of Pol II, a key hallmark of stalled polymerase (4). Very similar results were obtained by the Young and Levine laboratories, who performed ChIP-chip on Pol II using different Pol II-specific antibodies in Drosophila embryos, indicating that these results were not specific to one experimental or biological system (5). Importantly, in both S2 cells and embryos, validation by subsequent ChIP-chip studies in genetically manipulated backgrounds as well as permanganate footprinting (a technique that allows one to localize open transcription bubbles associated with engaged, stalled Pol II, described in ref. 27) confirmed that Pol II stalling is a widespread phenomenon (4, 5). Strikingly, Gene Ontology analysis of promoters with stalled Pol II revealed a significant enrichment in Drosophila genes that are induced in response to developmental or environmental stimuli, indicating that the transition to productive elongation may be a critical developmental regulatory step (4, 5).
2. Experimental design for performing ChIP-chip/ChIP-seq analyses
2.1 Antibody selection
Investigating transcription elongation with ChIP-chip/ChIP-seq demands an antibody that recognizes a biologically-relevant epitope with high affinity and selectivity. Moreover, it is important for a rigorous analysis of Pol II distribution that one employs an antibody that recognizes total Pol II regardless of phosphorylation state; for example, the commonly used 8WG16 antibody (Abcam, Cambridge, MA) that specifically recognizes unphosphorylated Pol II CTD has a higher affinity for the initiating polymerase than for the hyperphosphorylated, elongation-competent polymerase. ChIP material derived from immunoprecipitation with 8WG16 will thus be inherently and substantially biased towards enrichment in promoter-proximal Pol II signal, making such material not well suited for analyses of transcription elongation or Pol II stalling (see section 3.4 below). We detect total Pol II signal using a rabbit polyclonal antibody raised by our laboratory against the Rpb3 subunit of Pol II, which recognizes Pol II regardless of the phosphorylation state of the CTD of the Rpb1 subunit (4, 28). Antibodies that recognize total Pol II in mammalian systems are commercially available (for example, H-224, raised against the N-terminus of Rpb1, Santa Cruz Biotechnology, Santa Cruz, CA).
To contrast the distribution of total Pol II with that of productively elongating polymerase, we also use a commercially available rabbit polyclonal antibody raised against the Serine-2 phosphorylated CTD (Abcam, ab5095; we note that, given the high level of conservation of the CTD among species, this antibody works well in our hands in Drosophila and mammalian systems). Since Serine-2 phosphorylation of the CTD has been shown to occur concomitantly with the release of Pol II from the promoter-proximal region and the transition to full elongation competence, ChIP-signal from this antibody does not show significant enrichment near promoters, but instead reveals polymerase within the bodies of active genes (4).
Both antibodies share features critical for obtaining meaningful ChIP-chip/ChIP-seq data; high specificity (as demonstrated by western blotting); high affinity (quantitative PCR assays of enriched regions frequently produce signals of 2–10% of input DNA); and low background (signals in ‘background’ regions frequently produce qPCR signals in the range of 0.01–0.2% of input DNA). Employing antibodies that produce low signal and/or high backgrounds reduces the ability to discern areas of biologically meaningful protein enrichment from noise inherent in ChIP-chip/ChIP-seq methods.
The ultimate test for antibody specificity is to immunoprecipitate DNA from wild-type cells and cells that lack the factor of interest, obtained either through depletion using RNA interference or in an isogenic knock-out cell population. In these cases, if the ChIP signal arises from the desired target, the signal at enriched sites should disappear entirely. This may not be feasible when immunoprecipitating essential proteins such as Pol II subunits, but can be employed when determining binding sites for factors that are readily depleted such as the NELF complex. A complementary method for testing antibody specificity is to induce recruitment of the antigenic protein to specific sites and detect increased enrichment at these sites but not in background regions.
By using several different antibodies to perform immunoprecipitation on the same samples, one can compare the distributions of different proteins, or protein modifications. This strategy has been employed extensively in the study of histone modifications (29–31), as well as modifications of the Pol II CTD (6). By comparing the distribution of total Pol II with Serine-2 phosphorylated Pol II, we were able to infer that the bulk of promoter-proximally enriched Pol II was not hyperphosphorylated on Serine-2 and thus not engaged in productive elongation (4). Moreover, we established that while the amount of Serine-2 phosphorylated Pol II-signal at a gene correlates well with transcript levels, the amount of total Pol II does not, again suggesting that the level of Pol II recruitment to the promoter is not a good indicator of transcription levels and suggesting that gene expression can be regulated post-recruitment of polymerase (4, and D.G. and K.A, unpublished observations).
2.2 Coupling ChIP-chip/ChIP-seq with genetic and experimental manipulation
The comparison of gene expression in wild-type versus mutant or factor-depleted backgrounds has been possible using expression microarray platforms for many years; however the underlying mechanisms governing observed changes in transcription output have remained elusive. Now, ChIP-chip/ChIP-seq techniques allow one to probe the global distribution of the polymerase, transcription factors and histone modifications, shedding light on transcription regulation. Each of the ChIP-chip studies of transcriptional elongation cited above benefited from one of the most powerful methods for confirming specificity of ChIP-chip data and placing the data in a meaningful biological context: the use of complementary genome-wide analyses. By coupling ChIP-chip/ChIP-seq with genetic or chemical manipulation or by probing the same samples with antibodies recognizing distinct protein complexes, one may advance from merely mapping protein binding sites across the genome to gaining mechanistic insight.
For example, our ChIP-chip localization of polymerase identified genes with promoter-proximal enrichment of Pol II and suggested that Pol II stalling might occur at many more promoters than previously appreciated (4). To evaluate this possibility, a complementary experiment was performed in which microarrays were probed with total Pol II immunoprecipitated DNA from S2 cells that had been either mock-depleted or depleted of the NELF complex using RNA interference (RNAi). Since NELF had been implicated in regulating promoter-proximal stalling at several Drosophila genes (20, 32), we reasoned that depletion of NELF would identify promoters that harbored stalled polymerase by significantly decreasing the promoter-proximal enrichment of Pol II ChIP signal observed at those genes. In agreement with our predictions, NELF-depletion led to greatly reduced Pol II signal at genes identified as candidates for Pol II stalling (Figure 2 and ref. 4), but not at genes with more uniform Pol II distribution. These data indicated that NELF is an important regulator of early elongation at many genes, and that promoter-proximal enrichment of Pol II is an excellent predictor of Pol II stalling. The reduction in Pol II signal was significantly greater in the promoter region (where NELF is thought to be active) than in downstream regions, providing confidence that biologically meaningful changes were detected. Also, the loss of promoter-proximal Pol II signal at promoters with stalled Pol II was reproducible across replicate ChIP-chip experiments and confirmed using traditional ChIP assays from independently prepared ChIP material (ref. 4).
Figure 2. Global Changes in Pol II Promoter Occupancy Upon NELF-depletion Detected with ChIP-chip.

ChIP-chip was performed on Agilent Drosophila Partial Genome arrays using Pol II (Rpb3) ChIP material from Drosophila S2 cells that were mock-treated or depleted of NELF with RNAi (1099 Pol II-bound promoters; (4). A) Average Pol II signal at probes in the promoter-proximal regions (−250 to +500 bp with respect to the transcription start site) of Pol II-bound genes (log2 ratio of IP to input). B) Change in average promoter-proximal Pol II signal upon depletion of NELF, calculated for each bound promoter as ((Average Promoter SignalNELF-depleted) − (Average Promoter SignalMock-treated)). Pol II signal decreased at 1065 of 1099 promoters in NELF-depleted cells relative to mock-treated cells. C) Pol II signal detected near the Hsp70Ab and Timp promoters. Note the large decrease in Pol II signal at Timp and the smaller but appreciable decrease at Hsp70Ab in response to NELF-depletion; Pol II stalling was detected at both genes.
The comparison of two or more genetic backgrounds can be accomplished by the use of deletion strains of yeast, through RNA interference in higher eukaryotes, or by using cells derived from mutant or knock-out organisms. RNAi knockdown of proteins in Drosophila S2 cells can provide a straightforward, rapid and robust depletion of protein complexes thought to influence transcription elongation. We have depleted cells of NELF using double-stranded RNA (dsRNA) targeting two of the four subunits of the NELF complex and compared ChIP samples from these cells to ChIP material from cells that were mock-depleted using dsRNA targeting β-galactosidase (LacZ). Because determining changes in ChIP-chip signals that occur following genetic manipulation requires a firmly established baseline, we also frequently include untreated cells as a second control population in addition to mock-depletion, and include biological replicates for all treatment conditions.
To achieve highly efficient RNAi, we typically design dsRNAs complementary to a 1 kb region corresponding to the last exon of the target gene. Large quantities of dsRNA required for ChIP-chip experiments can be produced using commercially available kits, such as the MEGAscript T7 kit (Applied Biosystems/Ambion, Austin, TX). Serum-starvation of S2 cells followed by serum re-introduction induces rapid uptake of dsRNA, which is then processed by the Dicer/R2D2 complex into small interfering RNAs (siRNAs) to initiate RNA interference. Prior to performing ChIP-chip experiments with cells depleted of the NELF-complex, we extensively characterized the RNAi depletion and found it well-suited for these studies for two reasons. First, depletion of one or two subunits of the 4-subunit NELF complex effectively destabilized the entire complex, and thus diminished concerns about effects of partial NELF complexes. Second, time-courses studies established that the complex was efficiently depleted as early as 40 hours after the addition of dsRNA targeting two NELF-subunits. Depletion time courses allow one to conduct experiments at a time selected to maximize efficiency of depletion while minimizing the impact of secondary/indirect/downstream effects. We typically make ChIP material 70–90 hours following treatment with dsRNA.
Similar strategies can be employed to elicit RNAi in mammalian cells, using transfections of short interfering RNA (siRNA), plasmids that express short hairpin RNA (shRNA) or through infection with lentiviral vectors bearing an shRNA expression cassette. In systems that require transfection to achieve RNAi it is often preferable to create stable cell lines for analysis, rather than performing transient transfection of cell populations, where low or variable transfection efficiency can introduce significant heterogeneity that is problematic when dealing with techniques as sensitive as ChIP-chip/ChIP-seq.
Another powerful approach is to couple ChIP-chip/ChIP-seq with experimental manipulation. For example, Zhao and colleagues used ChIP-seq and micrococcal nuclease digestion/sequencing (MNase-seq) to compare nucleosome locations and Pol II binding in resting and activated CD4+ T-cells (33). This dynamic view of polymerase and nucleosome redistribution in response to signaling events provided insight about the relationship between Pol II occupancy, transcription activation, and promoter nucleosome occupancy. In a separate study, ChIP-chip was employed to determine estrogen-receptor and Pol II binding events that occur following reintroduction of estrogen to MCF-7 cells (34). Similar experiments combining induction of biological pathways or drug treatments with time-resolved ChIP-chip/ChIP-seq mapping of Pol II and transcription factor binding may offer great insight into the mechanisms that regulate transcription elongation.
2.3 Immunoprecipitation of protein-DNA complexes
Whereas traditional ChIP assays usually require 2.5–5×106 cells per immunoprecipitation, ChIP-chip experiments require a larger number of cells (typically 1–5×107 S2 cells per biological replicate; including samples for negative/no-antibody controls, quality control, etc.). Thus, obtaining a sufficient cell population often renders ChIP-chip experiments more time consuming, expensive or technically challenging than traditional ChIP. Once suitable cell populations are obtained, formaldehyde is added to form protein-DNA and protein-protein crosslinks. Crosslinking conditions must be determined empirically to maximize signal-to-noise ratio for the protein of interest. For proteins that are in close association with DNA such as Pol II or histones, short crosslinking times of 2–10 minutes with 1% formaldehyde provide good results. For factors more distant from DNA such as the NELF complex, increasing the duration of crosslinking to 30 minutes can substantially improve signal with a minimal increase in noise. When immunoprecipitating Pol II (Rpb3) we find that cells can be crosslinked for 10 to 30 minutes with negligible impact on the signal to noise ratio as determined by comparing peak enrichment to enrichment in background regions assumed to be devoid of Pol II (e.g. intergenic regions and heterochromatin).
Crosslinked protein-DNA complexes are subsequently fragmented by sonication. We obtain optimal results using the Bioruptor (Diagenode, Belgium) to sonicate DNA to a range spanning from 250–600 bp. Though fragment size may theoretically be a critical determinant of the width of the detected peak of enrichment at a protein-DNA binding site, we find similar results are obtained with average fragment size distributions centered anywhere from 300 to 600 bp. When comparing ChIP-chip data across different cell populations it is perhaps most important that the DNA fragment size range correspond closely between all samples investigated in one study. Size distributions of fragmented chromatin may be quantitatively compared across experiments using an instrument such as the Bioanalyzer (Agilent, Santa Clara, CA). Overly sonicated DNA produces poor results, in part because DNA is denatured or damage is done to the epitopes targeted for immunoprecipitation.
Antibodies must be titrated to ensure that peak signal intensities are biologically meaningful and do not plateau at an arbitrary level due to saturation of the available antibody. Our laboratory titrates antibodies over concentrations spanning an order of magnitude and compares the signal obtained at each antibody concentration by qPCR at several well-characterized loci. Optimum antibody concentration is selected as slightly above that where the signal plateaus at multiple loci. After overnight binding of antibodies to protein-DNA complexes, complexes are isolated with protein-A or -G agarose beads and washed using a protocol such as that suggested by Upstate/Millipore (http://www.millipore.com/techpublications/tech1/mcproto407; our laboratory has obtained reproducible results employing the wash buffers recommended in this protocol, but we include three, rather than one, high salt washes. This minimizes noise with little detriment to signal).
2.4 Quality control
Because a single ChIP experiment involves so many steps that may influence the final signal, it is critical that one validate immunoprecipitated material at control genomic regions with known distribution and occupancy of the protein in question. Prior to proceeding with ChIP-chip/ChIP-seq, we perform qPCR analyses using several primer pairs spanning the Tl and other similarly characterized loci (Figure 3; ChIP, blue line, is compared to data from ChIP-chip in red and ChIP-seq in green). In these regions, we have determined that Pol II signal shows high enrichment at the promoter-proximal primer pair but at least 10-fold lower enrichment in regions 400 bp upstream and downstream, as well as distant regions where Pol II binding is negligible. This quality control assay validates that crosslinking, fragmentation, immunoprecipitation, and washing were performed as in previous experiments. For example, when DNA has been inadequately fragmented, the ratio of enrichment in the promoter-proximal region to the upstream or downstream region is diminished. Similarly, detection of apparent Pol II enrichment in a distant background region may indicate sub-optimal washing or immunoprecipitation conditions.
Figure 3. Pol II Distribution Detected by ChIP, ChIP-chip, and ChIP-seq.
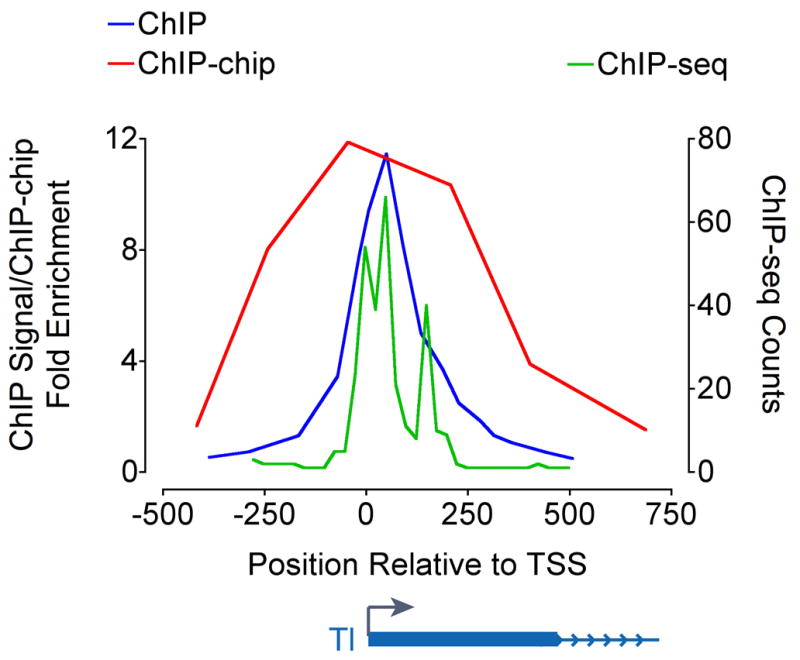
Drosophila S2 cells were crosslinked, sonicated, and total Pol II (Rpb3) was immunoprecipitated. Pol II ChIP signal at the Tl promoter region was quantified with qPCR using primer pairs spaced on average every 100 bp (blue line), ChIP-chip using Agilent Drosophila Whole Genome 2-ChIP sets with average probe spacing of 250 bp (red line), or ChIP-seq reads sequenced with the Illumina Genome Analyzer (green line), binned in 25 nucleotide windows. Genomic positions are reported as bp × 10−2, and represent the center point of primer pairs used, probe sequence, or window.
As an initial validation of the specificity of our ChIP-chip method, we probed Drosophila genomic microarrays with material precipitated with protein-A agarose in the absence of antibody. While only 13 ‘bound’ promoters were detected in the absence of antibody, Pol II (Rpb3) immunoprecipitation performed in parallel detected 7,037 ‘bound’ promoters, giving us high confidence that bona fide Pol II binding events were detected in our Pol II (Rpb3)-ChIP samples.
2.5 Preparation of DNA for genome-wide analyses
While ChIP with antibodies detecting very abundant, tightly associated proteins such as histones may precipitate sufficient quantities of DNA to directly label for probing of arrays (~2 micrograms of material), ChIP-chip detection of less abundant proteins may require amplification. Amplification can generate adequate quantities for labeling when starting with as little as 10–100 ng of immunoprecipitated material, which is comparable to the amounts needed for ChIP-seq. Rigorous comparisons of amplified and unamplified material have shown that standard whole genome amplification (WGA) or ligation-mediated PCR (LM-PCR) protocols introduce minimal bias, although they may somewhat decrease sensitivity (35). In agreement with this, we have identified very similar Pol II binding sites from amplified and unamplified material (see section 3.3 below); however, when amplifying, we employ the minimal number of cycles of amplification required to generate the required quantities of material.
DNA immunoprecipitated using a standard ChIP protocol is of adequate purity for dilution and quantitative PCR analysis but often contains contaminants that interfere with subsequent steps required for ChIP-chip or ChIP-seq analysis. Purification of immunoprecipitated DNA through commercially available nucleic acid clean-up kits typically eliminates these contaminants though substantially decreases yield. To ensure uniformity, control (input) DNA that will function as a reference should be amplified and purified in parallel to immunoprecipitated DNA. Following purification and amplification, we perform labeling, hybridization, washing, and scanning according to the protocols suggested by the manufacturer of the array platform used.
2.6 Choice of genome-wide mapping technique
Whereas the resolution of ChIP-chip is limited by the constraints of microarray probe spacing, ChIP-seq can theoretically offer single base-pair resolution. ChIP-seq also bypasses the labeling, hybridization, and washing steps required for ChIP-chip, which all may introduce experimental bias. However, while massively parallel sequencing remains expensive, microarray-based platforms are by comparison inexpensive, have proven robust, and the use of two-color arrays allows for an important internal control; any enrichment seen with immunoprecipitated DNA is reported relative to reference input genomic DNA. The inclusion of input DNA with each experiment provides a convenient measure of ‘baseline’ signal and allows one to measure changes in enrichment between experimental conditions. Whereas several methods exist for normalization of ChIP-chip data (36, 37), straightforward methods for performing similar normalization of ChIP-seq data are not yet widely available. Additionally, because microarray technology is now approaching its third decade, a wide variety of commercial and publicly available tools for data processing and analysis exist, and analogous tools for ChIP-seq are still in their infancy. However, expanding interest in ChIP-seq studies will undoubtedly lead to a variety of data analysis solutions in the near future (38).
2.6.1 ChIP-chip and choice of array platform
When performing ChIP-chip, the experimental question and model system will largely determine the choice of arrays, but there are important trade-offs that should be considered when selecting a tiling array platform. Arrays are available at a wide range of tiling densities, and offer anything from whole-genome to promoter-specific coverage. Arrays with more closely spaced probes are capable of delivering higher resolution binding data, but additional probe density comes at the cost of reduced breadth of coverage with the same number of probes. In addition, the more probes an array comprises, the more computing power is required for analysis; high-density array formats generate files that, even after compression, are often too large to upload to publicly available genome browsers in their entirety. Splitting the ChIP-chip data into smaller, more manageable files, for example by individual chromosome, is often helpful for overcoming this difficulty but requires some modifications to a standard work-flow (see section 3.1).
In addition to probe density, there are several differences between array platforms that make them better or worse suited for particular applications. For example, arrays are available in either one-color or two-color formats: whereas both involve labeling and hybridization of immunoprecipitated material, the two color format permits simultaneous hybridization of differentially labeled input DNA on the same slide, providing an internal control that is often useful for normalization. Probe length is also a consideration, as it has been shown that longer probes (50–75 nt) are more sensitive at detecting lower levels of enrichment (35). While we have obtained high-quality data using platforms offered by Affymetrix (Santa Clara, CA), Agilent (Santa Clara, CA) and NimbleGen (Madison, WI) in the past, we are using the NimbleGen HD2 Drosophila whole genome tiling arrays (2.1 million probes per array, covering the Drosophila genome at 55 bp resolution) for our current studies, as they represent a good balance of sensitivity, coverage and cost-effectiveness.
2.6.2 Methods for performing ChIP-seq
ChIP-seq offers extremely high spatial resolution by identifying the sequences present at the 5′-ends of either DNA strand of immunoprecipitated material, but with this resolution comes unique computing challenges. Convenient visualization and statistical analysis of 107 reads across the Drosophila genome requires binning of data into 10–50 nucleotide windows, resulting in an effective loss of resolution. Such difficulties are magnified when working with mammalian genomes. ChIP-seq is extremely sensitive, and has a superior signal: noise ratio to ChIP-chip, allowing for quantitative detection of protein-binding at both high- and low-affinity sites.
Several choices of massively parallel sequencing platforms are currently available, including 454 Life Sciences (39), Illumina (Solexa), or the Applied Biosystems SOLiD System. The 454 sequencing technology allows for long sequence reads (up to 400 nt) from ~400,000 individual molecules. Aligning these reads to the reference genome is relatively straightforward, but sequencing depth is often insufficient for optimal ChIP-seq analysis. In contrast, Illumina and SOLiD platforms can provide tens of million reads per sample lane; however, these reads tend to be 30–40 nt in length, making them potentially difficult to map uniquely to the genome assembly (see section 3.1 below).
3. Data analysis: extracting mechanistic insight from genome-wide binding studies
Moving from raw ChIP-chip or ChIP-seq data (scanned array images or unfiltered sequence reads) to biological insight requires sequential data analysis steps: 1) mapping signals (ChIP-chip) or sequence reads (ChIP-seq) across the genome; 2) defining “bound” regions where signal is significantly greater than background; 3) determining how peaks are distributed relative to interesting genomic elements (e.g. transcription start sites); and 4) parsing data structure to gain additional insight (Figure 5). Finally, data must be considered in light of experimental manipulation such as RNAi, drug treatment, or gene induction.
Figure 5. ChIP-chip and ChIP-seq Data Analysis Workflow.

ChIP-chip data (fold enrichment of immunoprecipitated material over genomic DNA) and/or ChIP-seq data are mapped to a reference genome. Control bound and unbound regions are visually inspected and validated by comparison to standard ChIP and qPCR. Genomic regions where signal is significantly greater than expected by chance (user-defined threshold) are identified as ‘bound.’ Bound regions are then compared to a database of genomic elements of interest (e.g. promoters) to identify bound elements. Note that absence of detected binding from a genomic region may result from absence of complementary probes upon the array (ChIP-chip), masking of repetitive regions (ChIP-chip and ChIP-seq), or unmappable regions (ChIP-seq).
3.1 Mapping signals to the reference genome and visualization
ChIP-chip data are readily normalized and mapped to specific genomic loci using commercially available software tailored to each array platform. Two-color ChIP-chip experiments produce Log2-scale ratios of enrichment of immunoprecipitated DNA relative to genomic DNA (input) at each probe queried, which can be converted to a fold-enrichment score for each probe of known location, and output as a .bed, .wig or other tab-delimited text format. These data may be uploaded and viewed with genome browsers such as the UCSC Genome Browser (http://genome.ucsc.edu/; (40), SignalMap (NimbleGen, Madison, WI) or the Integrated Genome Browser (IGB, Affymetrix; http://www.affymetrix.com/partners_programs/programs/developer/tools/download_igb.affx).
As noted above, uniquely mapping ChIP-seq data to the reference genome assembly presents unique computing challenges. The number of reads generated in massively parallel sequencing experiments is of the order, frequently extending to 107 or 108 reads, such that ‘traditional’ alignment tools Blast or Blat are inefficient and impractical. Alignment of short reads generated in massively parallel sequencing experiments is greatly facilitated by computationally efficient tools which allow researchers to take advantage of multiple processors on high RAM machines. As with traditional alignment algorithms, the ability to place sequences with imperfect but still useful alignments on a reference genome is a key feature. The current Illumina (Solexa) technology produces sequence reads with considerably position dependent error rates. It is known that the error rate near the 3′ end is highest and the rate near the 5′ end is also relatively elevated (41). Many of the commonly employed efficient short read alignment algorithms (ELAND, Illumina, San Diego, CA; and MAQ, publically available at http://maq.sourceforge.net/index.shtml, (42) facilitate mismatched alignment by allowing mismatch errors (often two per read) randomly throughout the query sequences. Other efficient tools such as SOAP accommodate for the increased error rate at the 3′ end by trimming the most error-prone base calls in that direction, but fail to correct for the elevated 5′ error rate (43). These algorithms may fail to align ‘useful’ sequences by not accounting for the known position specific error. MOM (for Maximal Oligonucleotide Mapping) is a seed based search tool that accounts for elevated errors at both the 3′ and 5′ ends, and thus substantially improves alignment (44). MOM is more sensitive, aligning a greater percentage of reads, while still aligning sequences at rates as high or higher than many other efficient alignment tools. MOM has been created in JAVA and is easily installed and used on JavaSE JRE 1.6 or later. We run MOM to map sequence reads derived from the Illumina Genome Analyzer on an 8 CPU 32 GB RAM machine running a 64bit JRE for increased performance and the ability to utilize larger memory.
Both ChIP-chip and ChIP-seq have limitations associated with characterizing protein distribution across repetitive sequences. With ChIP-chip technologies, repetitive sequence probes are typically excluded from the arrays. Arrays typically afford the ability to query repeat-masked genomes and often do not include probes that target multiple locations. An advantage to this design is simplification of downstream analyses. However, excluding probes that target repetitive sequences eliminates the opportunity to estimate protein binding at these locations.
In contrast, ChIP-seq technologies query unmasked total reference genomes. With ChIP-seq, variations in experimental design including read length, number of mismatches accepted, the nature of the reference genome(s) investigated, and the genome elements being queried can impact the number of hits that map to unique or multiple locations. ChIP-seq alignment algorithms typically identify each read as mapping uniquely; mapping to multiple locations (repetitive), not mapping to the reference genome, or quality control failure (functionally reads with stretches of poly N). A typical ChIP-seq experiment will uniquely map 30 to 60% of the raw reads to the reference genome. In addition, most alignment algorithms return the number but not the set of locations for multiple hitting reads. It is often desirable to determine the hit locations for reads that map to between 2–10 sites on the genome, as this allows one to gain information about protein distribution at multi-copy genes (such as the Hsp70 genes in Drosophila). The location set for such multi-hitting reads can be determined using alternative alignment algorithms such as Blat. Thus, the potential distribution of multi-hitting reads may be examined, although there are complications involved in assigning these reads in a quantitative fashion across several potential binding sites. Paired-end techniques and variation of the ChIP-seq read length can be used to increase the unique search space in the genome of interest. Repetitive elements of interest such as paralogous genes or conserved regulatory or structural elements remain difficult to query and are typically outside of the standard ChIP-seq work flow but estimation of coverage is possible and the absence of reads across a given repetitive search domain may afford useful data.
Regardless of platform, both ChIP-chip and ChIP-seq experiments can generate similar Pol II binding distributions and provide a wealth of information about regulation of transcription. Simple visual inspection of ChIP-chip/ChIP-seq data at several loci allows for comparison of genomic ChIP data with expected results defined by traditional ChIP, underscores the diversity in Pol II-binding profiles at different genes, and highlights some of the challenges of analyzing genome-wide data sets. ChIP-chip and ChIP-seq each reveal Pol II occupancy at the lace promoter and throughout the gene with significant enrichment detectable near the 3′ end (Figure 4A), consistent with the idea that lace is a highly expressed gene that does not undergo Pol II stalling (4). At the Drosophila kay locus, ChIP-chip and ChIP-seq data sets demonstrate Pol II recruitment to multiple alternative transcription start sites (Figure 4B). Both data sets also show substantial Pol II occupancy downstream of only one kay promoter, suggesting it is from this promoter that most transcription occurs under these experimental conditions. However, differentiating between elongating and promoter-bound Pol II at genes with nested transcription start sties is difficult; thus genes like kay may need to be excluded from downstream analyses. Likewise, the smi35A promoter displays significant Pol II enrichment, but with background levels of Pol II-binding detected within the gene (Figure 4C). In addition, unanticipated binding events, such as the apparently unannotated transcription start site upstream of the smi35A gene, are also readily detected by both platforms (Figure 4B). Both ChIP-chip and ChIP-seq data are capable of resolving Pol II binding at the closely spaced CLIP-190 and CG6860 promoters (Figure 4D). The presence of independent promoters located less than 750 bp apart is a common feature of the Drosophila genome, and requires high-resolution data sets to ensure that Pol II-binding data can be unambiguously assigned to a specific transcription start site.
Figure 4. Comparison of Pol II Distribution as Determined by ChIP-chip and ChIP- seq.

The distribution of Pol II (Rpb3)-binding at the: A) lace; B) kay; C) smi35A and; D) CG6860/CLIP-190 genes was determined by ChIP-chip (NimbleGen HD2 Drosophila whole genome arrays) or ChIP-seq (Illumina Genome Analyzer reads binned in 25 nucleotide windows).
3.2 Identifying peaks of Pol II binding across the genome
Biological meaning may be derived in identifying genomic regions where ChIP-chip probes are bound by the protein of interest at levels significantly above background or where ChIP-seq read density is significantly enriched above background. A variety of commercial and publicly available software packages exist to determine regions of significant ChIP enrichment (peak-detection), and best peak finding algorithm is often platform dependent (36). Peak detection algorithms typically employ normalized signals and P values for each genomic probe on the array to identify genomic regions where multiple adjacent probes show significant enrichment that is unlikely to have occurred by chance. Most algorithms allow for user-defined false discovery rates, and output text files containing a list of regions bound at a given level of confidence.
Defining bound regions with ChIP-seq data may be substantially more challenging because optimized commercial software is not widely available. In principle, after binning sequence reads in appropriately sized windows, regions of ChIP-seq enrichment may be identified in a manner analogous to that employed for ChIP-chip. Enriched regions are identified as areas where sequence reads are concentrated in a particular window at a frequency that is highly unlikely to have occurred by chance. This cut-off for ‘read density’ may be determined for a chosen P value; alternatively a false-discovery-rate approach may be employed. There are several publically available ChIP-seq peak finding tools including F-Seq, a Java package that efficiently generates a continuous tag sequence density estimation (45), and ERANGE, a multi-utility python suite that supports read directionality models and multi-reads may be employed for rapid determination and mapping of local bound regions (46).
When identifying enriched regions using ChIP-seq, it is important to consider two issues: a determination of background, and how “mappable” individual sequences are. Background, or noise, in ChIP-seq experiments is very low: even with very deep sequencing, most windows of 100–1000 bp will contain between zero and one read. Nonetheless, a careful estimate of background signal permits a statistically rigorous evaluation of which windows have a significantly enriched read density (e.g. 47). The second consideration arises because each ~35 nt block of sequence across the genome is not unique (especially when allowing for mismatches), leading to sites where reads could not be expected to align. Thus, any calculation of read density should also correct for the possibility of a given position to be represented in the data set.
The ability to uniquely map each position in the genome or in regions of interest can be determined in silico by extracting the appropriate sized subsequence at single base intervals on each strand. For example, for a 35mer read length one would extract the sequence from each chromosome corresponding to positions 1–35, 2–36, 3–37, etc. Mapping these extracted sequences back to the reference genome using the same parameters as the experimental data defines those genomic locations that are uniquely mappable. This is obviously a computationally intensive task for the global examination of large reference genomes such as mammals (5–6 × 109 total subsequences corresponding to 4–5 × 109 unique subsequences from both strands). However, adjusting local read hit density by the ratio of mappable/unmappable locations may improve the understanding of relative signal. Additionally, the set of unmappable locations affords the ability to differentiate between locations where signal is absent, and locations to which sequences could not be uniquely mapped (e.g. 47).
3.3 Mapping bound regions to genomic elements
We employ a custom program to globally identify where regions of Pol II-binding are located with respect to Drosophila genomic elements of interest. Two input files are constructed; the first contains genomic locations for all detected ChIP-chip or ChIP-seq peaks (chromosome, strand, plus start and stop coordinates). The second file contains genomic positions for the transcription start and poly-adenylation sites for ~21,000 “elements”, each comprising one unique Pol II transcript (including mRNA, snoRNA, ncRNA, etc.). Pairwise comparisons are made for all bound regions and all elements, and a list of all elements that overlap with a bound region is output. This list also contains specific information about where bound regions are located with respect to the transcription start site, allowing for determination of “bound” promoters, as well as evaluating binding levels within downstream gene regions. This method has the advantage of easy adaptation to enrichment data obtained with any ChIP-chip or ChIP-seq platform, facilitating between-method comparisons and combination of data sets. This type of ‘all-by-all’ comparison generates a computationally large search space that can be mitigated by parallelizing the data (for example by chromosome) or in our case by employing an efficient C++ program operating on a high RAM server.
ChIP-chip coupled with this type of analysis is a robust method for identifying genomic elements bound by Pol II. We identified similar sets of Pol II-bound promoters with two very different protocols (Figure 6). In the first, Pol II (Rpb3)-immunoprecipitated DNA was directly labeled (without amplification) and used to probe low-density Agilent Drosophila Whole Genome 2-ChIP sets (4). Pol II-bound genomic regions were defined using a custom algorithm as previously described (48) and mapped to the Drosophila Release 3 Genomic sequence (Berkeley Drosophila Genome Project) (4). In the second, immunoprecipitated DNA was amplified by WGA before labeling and used to probe high-density NimbleGen HD2 Drosophila whole genome arrays. Pol II-bound genomic regions were determined with NimbleScan software (FDR < 0.05) and mapped against all annotated Drosophila promoter regions from the Drosophila Release 5 Genomic sequence. Both methods identified approximately 5400 Pol II-bound promoters; 4363 (>80%) of these were identified as bound by both methods, despite differences in amplification and labeling, array platform, analysis method, and Drosophila genomic assembly.
Figure 6. Pol II Binding Detected with Differing ChIP-chip Methods and Platforms.

Pol II (Rpb3) ChIP was performed with material generated from Drosophila S2 cells and binding was detected with NimbleGen HD2 Drosophila whole genome arrays or Agilent Drosophila Whole Genome 2-ChIP sets. Pol II-bound genomic regions were determined for the NimbleGen array with NimbleScan software (FDR < 0.05) and mapped against promoter regions from the Drosophila Release 5 Genomic sequence. Pol II-bound genomic regions were determined for the Agilent arrays with the Drosophila Release 3 Genomic sequence as previously described (4, 5, 48).
3.4 Distinguishing Pol II stalling from productive elongation
While it is useful to distinguish bound promoters from those devoid of binding, visual inspection of Pol II (Rpb3) ChIP data reveals that the distribution of Pol II across bound genes varied greatly (examples shown in Figure 4). At some bound genes Pol II was rather uniformly distributed throughout the gene body, while at others Pol II was enriched near the transcription start site. As discussed above, we wished to quantify the levels of promoter-proximal polymerase enrichment in order to identify genes with stalled Pol II. This was accomplished by calculating a “stalling index”, which was the ratio of the average probe signal near each bound promoter (between positions −250 and +500 with respect to the transcription start site) to the average signal in the downstream transcribed region (between +501 and the site of termination). A frequency distribution of this ratio at all bound genes was approximated by a Gaussian; however, a substantial group of outliers displayed significant Pol II-enrichment at the promoter versus downstream regions (4). Those genes whose stalling indices fell more than two standard deviations above the mean were considered to be good candidates for Pol II stalling, and further analysis of these genes confirmed that the majority of them did indeed harbor stalled Pol II.
Recently, a similar method has been used to identify genes with stalled Pol II using massively parallel sequencing data (47). After identifying genes with promoter-proximal peaks of signal, the Fisher’s exact test was used to compare the density of reads within this promoter-proximal window to the density of the reads within the body of the gene. Genes with a statistically significant enrichment of read density near the promoter were defined as being significantly paused or stalled.
4. Discussion and future directions
Because the process of transcription is fundamentally a series of highly-coordinated interactions between the DNA template and protein factors, ChIP studies of protein-DNA interactions have greatly aided the in-vivo dissection of transcription regulation. Global ChIP-chip and ChIP-seq analyses have accelerated the pace of discovery, allowed insights unattainable with other methods, and have played an important role in identifying the elongation phase of transcription as a critical point of biological regulation. These assays are powerful, robust, and likely to become more economically and computationally feasible. When coupled with genetic and experimental variation, they are capable of providing a great breadth of mechanistic insight; thus they should be considered an important part of the tool-kit for investigating regulation of transcription elongation.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences to K.A. (Z01 ES101987).
References
- 1.Roeder RG. FEBS Lett. 2005;579:909–15. doi: 10.1016/j.febslet.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. Nature. 2005;436:876–80. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, Adelman K. Nat Genet. 2007;39:1507–11. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, Young RA. Nat Genet. 2007;39:1512–6. doi: 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komarnitsky P, Cho EJ, Buratowski S. Genes Dev. 2000;14:2452–60. doi: 10.1101/gad.824700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun X, Zhang Y, Cho H, Rickert P, Lees E, Lane W, Reinberg D. Mol Cell. 1998;2:213–22. doi: 10.1016/s1097-2765(00)80131-8. [DOI] [PubMed] [Google Scholar]
- 8.Cho EJ, Takagi T, Moore CR, Buratowski S. Genes Dev. 1997;11:3319–26. doi: 10.1101/gad.11.24.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCracken S, Fong N, Rosonina E, Yankulov K, Brothers G, Siderovski D, Hessel A, Foster S, Shuman S, Bentley DL. Genes Dev. 1997;11:3306–18. doi: 10.1101/gad.11.24.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng B, Price DH. J Biol Chem. 2007;282:21901–12. doi: 10.1074/jbc.M702936200. [DOI] [PubMed] [Google Scholar]
- 11.Renner DB, Yamaguchi Y, Wada T, Handa H, Price DH. J Biol Chem. 2001;276:42601–9. doi: 10.1074/jbc.M104967200. [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi Y, Inukai N, Narita T, Wada T, Handa H. Mol Cell Biol. 2002;22:2918–27. doi: 10.1128/MCB.22.9.2918-2927.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamaguchi Y, Wada T, Watanabe D, Takagi T, Hasegawa J, Handa H. J Biol Chem. 1999;274:8085–92. doi: 10.1074/jbc.274.12.8085. [DOI] [PubMed] [Google Scholar]
- 14.Narita T, Yamaguchi Y, Yano K, Sugimoto S, Chanarat S, Wada T, Kim DK, Hasegawa J, Omori M, Inukai N, Endoh M, Yamada T, Handa H. Mol Cell Biol. 2003;23:1863–73. doi: 10.1128/MCB.23.6.1863-1873.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marshall NF, Peng J, Xie Z, Price DH. J Biol Chem. 1996;271:27176–83. doi: 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- 16.Marshall NF, Price DH. J Biol Chem. 1995;270:12335–8. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- 17.Peterlin BM, Price DH. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 18.Saunders A, Core LJ, Lis JT. Nat Rev Mol Cell Biol. 2006;7:557–67. doi: 10.1038/nrm1981. [DOI] [PubMed] [Google Scholar]
- 19.Lee C, Li X, Hechmer A, Eisen M, Biggin MD, Venters BJ, Jiang C, Li J, Pugh BF, Gilmour DS. Mol Cell Biol. 2008;28:3290–300. doi: 10.1128/MCB.02224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li B, Weber JA, Chen Y, Greenleaf AL, Gilmour DS. Mol Cell Biol. 1996;16:5433–43. doi: 10.1128/mcb.16.10.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H, Giresi PG, Goldy J, Hawrylycz M, Haydock A, Humbert R, James KD, Johnson BE, Johnson EM, Frum TT, Rosenzweig ER, Karnani N, Lee K, Lefebvre GC, Navas PA, Neri F, Parker SC, Sabo PJ, Sandstrom R, Shafer A, Vetrie D, Weaver M, Wilcox S, Yu M, Collins FS, Dekker J, Lieb JD, Tullius TD, Crawford GE, Sunyaev S, Noble WS, Dunham I, Denoeud F, Reymond A, Kapranov P, Rozowsky J, Zheng D, Castelo R, Frankish A, Harrow J, Ghosh S, Sandelin A, Hofacker IL, Baertsch R, Keefe D, Dike S, Cheng J, Hirsch HA, Sekinger EA, Lagarde J, Abril JF, Shahab A, Flamm C, Fried C, Hackermuller J, Hertel J, Lindemeyer M, Missal K, Tanzer A, Washietl S, Korbel J, Emanuelsson O, Pedersen JS, Holroyd N, Taylor R, Swarbreck D, Matthews N, Dickson MC, Thomas DJ, Weirauch MT, Gilbert J, Drenkow J, Bell I, Zhao X, Srinivasan KG, Sung WK, Ooi HS, Chiu KP, Foissac S, Alioto T, Brent M, Pachter L, Tress ML, Valencia A, Choo SW, Choo CY, Ucla C, Manzano C, Wyss C, Cheung E, Clark TG, Brown JB, Ganesh M, Patel S, Tammana H, Chrast J, Henrichsen CN, Kai C, Kawai J, Nagalakshmi U, Wu J, Lian Z, Lian J, Newburger P, Zhang X, Bickel P, Mattick JS, Carninci P, Hayashizaki Y, Weissman S, Hubbard T, Myers RM, Rogers J, Stadler PF, Lowe TM, Wei CL, Ruan Y, Struhl K, Gerstein M, Antonarakis SE, Fu Y, Green ED, Karaoz U, Siepel A, Taylor J, Liefer LA, Wetterstrand KA, Good PJ, Feingold EA, Guyer MS, Cooper GM, Asimenos G, Dewey CN, Hou M, Nikolaev S, Montoya-Burgos JI, Loytynoja A, Whelan S, Pardi F, Massingham T, Huang H, Zhang NR, Holmes I, Mullikin JC, Ureta-Vidal A, Paten B, Seringhaus M, Church D, Rosenbloom K, Kent WJ, Stone EA, Batzoglou S, Goldman N, Hardison RC, Haussler D, Miller W, Sidow A, Trinklein ND, Zhang ZD, Barrera L, Stuart R, King DC, Ameur A, Enroth S, Bieda MC, Kim J, Bhinge AA, Jiang N, Liu J, Yao F, Vega VB, Lee CW, Ng P, Shahab A, Yang A, Moqtaderi Z, Zhu Z, Xu X, Squazzo S, Oberley MJ, Inman D, Singer MA, Richmond TA, Munn KJ, Rada-Iglesias A, Wallerman O, Komorowski J, Fowler JC, Couttet P, Bruce AW, Dovey OM, Ellis PD, Langford CF, Nix DA, Euskirchen G, Hartman S, Urban AE, Kraus P, Van Calcar S, Heintzman N, Kim TH, Wang K, Qu C, Hon G, Luna R, Glass CK, Rosenfeld MG, Aldred SF, Cooper SJ, Halees A, Lin JM, Shulha HP, Zhang X, Xu M, Haidar JN, Yu Y, Ruan Y, Iyer VR, Green RD, Wadelius C, Farnham PJ, Ren B, Harte RA, Hinrichs AS, Trumbower H, Clawson H, Hillman-Jackson J, Zweig AS, Smith K, Thakkapallayil A, Barber G, Kuhn RM, Karolchik D, Armengol L, Bird CP, de Bakker PI, Kern AD, Lopez-Bigas N, Martin JD, Stranger BE, Woodroffe A, Davydov E, Dimas A, Eyras E, Hallgrimsdottir IB, Huppert J, Zody MC, Abecasis GR, Estivill X, Bouffard GG, Guan X, Hansen NF, Idol JR, Maduro VV, Maskeri B, McDowell JC, Park M, Thomas PJ, Young AC, Blakesley RW, Muzny DM, Sodergren E, Wheeler DA, Worley KC, Jiang H, Weinstock GM, Gibbs RA, Graves T, Fulton R, Mardis ER, Wilson RK, Clamp M, Cuff J, Gnerre S, Jaffe DB, Chang JL, Lindblad-Toh K, Lander ES, Koriabine M, Nefedov M, Osoegawa K, Yoshinaga Y, Zhu B, de Jong PJ. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson DS, Mortazavi A, Myers RM, Wold B. Science. 2007;316:1497–502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 23.Gilmour DS, Lis JT. Mol Cell Biol. 1986;6:3984–9. doi: 10.1128/mcb.6.11.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rougvie AE, Lis JT. Cell. 1988;54:795–804. doi: 10.1016/s0092-8674(88)91087-2. [DOI] [PubMed] [Google Scholar]
- 25.Rougvie AE, Lis JT. Mol Cell Biol. 1990;10:6041–5. doi: 10.1128/mcb.10.11.6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Core LJ, Lis JT. Science. 2008;319:1791–2. doi: 10.1126/science.1150843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cartwright IL, Cryderman DE, Gilmour DS, Pile LA, Wallrath LL, Weber JA, Elgin SC. Methods Enzymol. 1999;304:462–96. doi: 10.1016/s0076-6879(99)04028-8. [DOI] [PubMed] [Google Scholar]
- 28.Adelman K, Marr MT, Werner J, Saunders A, Ni Z, Andrulis ED, Lis JT. Mol Cell. 2005;17:103–12. doi: 10.1016/j.molcel.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 29.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. Nature. 2007;448:553–60. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, Young RA. Cell. 2005;122:517–27. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 31.Rando OJ. Curr Opin Genet Dev. 2007;17:94–9. doi: 10.1016/j.gde.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Lee C, Gilmour DS, Gergen JP. Genes Dev. 2007;21:1031–6. doi: 10.1101/gad.1521207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Cell. 2008;132:887–98. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. Nat Genet. 2006;38:1289–97. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 35.Johnson DS, Li W, Gordon DB, Bhattacharjee A, Curry B, Ghosh J, Brizuela L, Carroll JS, Brown M, Flicek P, Koch CM, Dunham I, Bieda M, Xu X, Farnham PJ, Kapranov P, Nix DA, Gingeras TR, Zhang X, Holster H, Jiang N, Green RD, Song JS, McCuine SA, Anton E, Nguyen L, Trinklein ND, Ye Z, Ching K, Hawkins D, Ren B, Scacheri PC, Rozowsky J, Karpikov A, Euskirchen G, Weissman S, Gerstein M, Snyder M, Yang A, Moqtaderi Z, Hirsch H, Shulha HP, Fu Y, Weng Z, Struhl K, Myers RM, Lieb JD, Liu XS. Genome Res. 2008;18:393–403. doi: 10.1101/gr.7080508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson WE, Li W, Meyer CA, Gottardo R, Carroll JS, Brown M, Liu XS. Proc Natl Acad Sci U S A. 2006;103:12457–62. doi: 10.1073/pnas.0601180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smyth GK, Speed T. Methods. 2003;31:265–73. doi: 10.1016/s1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- 38.Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH. Nat Biotechnol. 2008;26:1293–300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, Weber RJ, Haussler D, Kent WJ. Nucleic Acids Res. 2003;31:51–4. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Nucleic Acids Res. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Ruan J, Durbin R. Genome Res. 2008;18:1851–8. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li R, Li Y, Kristiansen K, Wang J. Bioinformatics. 2008;24:713–4. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 44.Eaves HL, Gao Y. Bioinformatics. 2009 doi: 10.1093/bioinformatics/btp092. [DOI] [PubMed] [Google Scholar]
- 45.Boyle AP, Guinney J, Crawford GE, Furey TS. Bioinformatics. 2008;24:2537–8. doi: 10.1093/bioinformatics/btn480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 47.Core LJ, Waterfall JJ, Lis JT. Science. 2008 doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi Y, Rolfe A, MacIsaac KD, Gerber GK, Pokholok D, Zeitlinger J, Danford T, Dowell RD, Fraenkel E, Jaakkola TS, Young RA, Gifford DK. Nat Biotechnol. 2006;24:963–70. doi: 10.1038/nbt1233. [DOI] [PubMed] [Google Scholar]