

Background: proMMP-9 binds to B-CLL cells and contributes to malignant cell migration/arrest and survival.
Results: The VPLDTHDVFQ sequence from the proMMP-9 PEX9 domain inhibits α4β1 integrin-mediated proMMP-9-B-CLL interaction, transendothelial migration, and proMMP-9-induced survival signaling.
Conclusion: The VPLDTHDVFQ sequence is an α4β1 integrin binding site in proMMP-9 and blocks proMMP-9 effects on B-CLL cells.
Significance: The identified sequence may be a novel therapeutic target in B-CLL.
Keywords: Apoptosis, Cell Death, Cell Invasion, Cell Signaling, Chemotaxis, Integrin, Leukemia, Hemopexin Domain, Matrix Metalloproteinase 9
Abstract
We previously showed that pro-matrix metalloproteinase-9 (proMMP-9) binds to B chronic lymphocytic leukemia (B-CLL) cells and contributes to B-CLL progression by regulating cell migration and survival. Induction of cell survival involves a non-proteolytic mechanism and the proMMP-9 hemopexin domain (PEX9). To help design specific inhibitors of proMMP-9-cell binding, we have now characterized B-CLL cell interaction with the isolated PEX9. B-CLL cells bound soluble and immobilized GST-PEX9, but not GST, and binding was mediated by α4β1 integrin. The ability to recognize PEX9 was observed in all 20 primary samples studied irrespective of their clinical stage or prognostic marker phenotype. By preparing truncated forms of GST-PEX9 containing structural blades B1B2 or B3B4, we have identified B3B4 as the primary α4β1 integrin-interacting region within PEX9. Overlapping synthetic peptides spanning B3B4 were then tested in functional assays. Peptide P3 (FPGVPLDTHDVFQYREKAYFC), a sequence present in B4 or smaller versions of this sequence (peptides P3a/P3b), inhibited B-CLL cell adhesion to GST-PEX9 or proMMP-9, with IC50 values of 138 and 279 μm, respectively. Mutating the two aspartate residues to alanine rendered the peptides inactive. An anti-P3 antibody also inhibited adhesion to GST-PEX9 and proMMP-9. GST-PEX9, GST-B3B4, and P3/P3a/P3b peptides inhibited B-CLL cell transendothelial migration, whereas the mutated peptide did not. B-CLL cell incubation with GST-PEX9 induced intracellular survival signals, namely Lyn phosphorylation and Mcl-1 up-regulation, and this was also prevented by the P3 peptides. The P3 sequence may, therefore, constitute an excellent target to prevent proMMP-9 contribution to B-CLL pathogenesis.
Introduction
B-cell chronic lymphocytic leukemia (B-CLL)5 is characterized by the accumulation in the peripheral blood of CD5+ B lymphocytes, which progressively infiltrate the bone marrow and secondary lymphoid tissues (1, 2). As per most cell types, these migration processes include chemokine receptors, integrins, and matrix metalloproteinases (MMPs) (3–7).
The major MMP found in B-CLL cells is proMMP-9 (92 kDa) (8), and we and others have shown that (pro)MMP-9 (the proform and/or the active enzyme) plays an important role in B-CLL transendothelial migration and basement membrane invasion (9–11). Additionally, the constitutive levels of (pro)MMP-9 in B-CLL cells are significantly higher than in normal B-cells, and elevated intracellular levels of (pro)MMP-9 correlate with advanced stage and poor patient survival (9). Another difference with normal B cells is the consistent presence of (pro)MMP-9 at the B-CLL cell surface (9, 12). We previously demonstrated that α4β1 integrin and a 190-kDa CD44 variant (CD44v) constitute a docking complex for (pro)MMP-9 in B-CLL cells and that binding of (pro)MMP-9 to this complex results in inhibition of B-CLL cell migration (12). We also recently showed that surface-bound proMMP-9 induces B-CLL cell survival by a non-proteolytic mechanism that involves Lyn and STAT3 activation and Mcl-1 up-regulation (13). The proMMP-9 hemopexin domain (PEX9) was required for (pro)MMP-9 cell binding and subsequent effects, as truncated proMMP-9 forms lacking PEX9 did not bind to B-CLL cells. Murine PEX9 also bound to these cells and induced survival, although the receptor involved has not been characterized (13).
These previous studies indicate that (pro)MMP-9 contributes to B-CLL pathogenesis by proteolytic and non-proteolytic mechanisms and may constitute a therapeutic target. Given the lower homology among MMP hemopexin domains compared with catalytic domains (14), targeting PEX9, in particular its interaction with cell surface receptors, may be a useful and more specific approach to prevent B-CLL cell dissemination and survival. These strategies are already in progress in other cell systems. For example, phage display analyses have identified a synthetic peptide (CRVYGPYLLC) that binds to the PEX9 domain, disrupts its binding to α5β1/αVβ5 integrins in fibrosarcoma cells, and inhibits cell migration (15). Two synthetic peptides (SRPQGPFL and NQVDQVGY) mimicking sequences in blade 1 and blade 4 of PEX9, respectively, were shown to affect MMP-9-CD44 interaction, MMP-9 dimerization, and migration of fibrosarcoma and carcinoma cells (16). A recent in silico approach has identified two small-molecule compounds that bind to PEX9 and inhibit tumor growth and metastasis (17). The isolated murine PEX9 on the other hand was shown to inhibit MMP-9 activity and invasion of melanoma cells (18), adhesion and migration of colorectal cancer cells (19), and angiogenesis and tumor growth in a glioblastoma model (20).
The interaction between human PEX9 and B-CLL cells has not been characterized. In this study we show that human PEX9 binds to B-CLL cells via α4β1 integrin and inhibits transendothelial migration. Moreover, we have identified an amino acid sequence within PEX9 that is involved in PEX9/proMMP-9-B-CLL cell interaction and functional consequences and may thus constitute a therapeutic target in B-CLL.
EXPERIMENTAL PROCEDURES
Patients and Cells
Approval was obtained from the Consejo Superior de Investigaciones Científicas Bioethics Review Board for these studies. Peripheral blood samples from 20 B-CLL patients (Table 1) were obtained after informed consent. CD5+ B-lymphocytes were purified by Ficoll-Hypaque (Nycomed, Oslo, Norway) centrifugation and (if needed) negative selection with anti-CD3-conjugated Dynabeads (Invitrogen). The resulting B cell population was >92% CD19+ and >72% CD5+, determined on a Coulter Epics XL flow cytometer (Beckman Coulter, Fullerton, CA). The MEC-1 cell line, established from a B-CLL patient (23), was obtained from Dr. Enrique Ocio (Cancer Research Center, Salamanca, Spain) and maintained in IMDM medium (Lonza, Basel, Switzerland), 10% fetal bovine serum. K562 and K562-α4 cells were obtained from Dr. Joaquín Teixidó (Centro de Investigaciones Biológicas, Madrid) and cultured in RPMI 1640, 10% fetal bovine serum. Human umbilical vein endothelial cells (HUVEC) were purchased from Lonza and cultured as reported (10–12).
TABLE 1.
Clinical characteristics of B-CLL patients
α4 integrin subunit expression was determined for each sample and is indicated as the percentage of positive cells. ND, not determined.
| Patient | Sex/Age (y) | Stagea | α4 (%) | CD38/ZAP70b | Ig status |
|---|---|---|---|---|---|
| 1 | F/67 | B/II | 71 | −/ND | ND |
| 2 | F/54 | B/II | 55 | −/ND | Unmutated |
| 3 | M/43 | A/0 | 29 | −/− | Mutated |
| 4 | M/81 | A/0 | 57 | −/ND | Mutated |
| 5 | M/56 | C/IV | 88 | −/− | Unmutated |
| 6 | M/58 | B/II | 48 | +/+ | Unmutated |
| 7 | M/67 | B/II | 20 | −/+ | Mutated |
| 8 | M/69 | C/IV | 37 | +/+ | ND |
| 9 | F/83 | C/IV | 74 | −/+ | Mutated |
| 10 | M/70 | B/II | 75 | +/+ | Unmutated |
| 11 | M/64 | A/I | 78 | −/− | Mutated |
| 12 | M/80 | A/I | 81 | −/+ | Unmutated |
| 13 | M/77 | B/I | 82 | +/+ | ND |
| 14 | M/83 | B/I | 43 | +/+ | Unmutated |
| 15 | M/68 | B/II | 12 | −/+ | Mutated |
| 16 | M/59 | C/IV | 30 | +/+ | Unmutated |
| 17 | M/85 | C/IV | 25 | + / ND | Mutated |
| 18 | F/73 | A/II | 76 | −/− | Mutated |
| 19 | M/60 | A/0 | 37 | +/− | Mutated |
| 20 | M/48 | B/I | 20 | +/− | ND |
Antibodies, Reagents, Proteins, and Peptides
Monoclonal antibodies (mAbs) HP2/1 (anti-α4 integrin subunit, function-blocking), HP1/7 (anti-α4 integrin subunit, non-blocking), HP2/9 (anti-CD44 function blocking), and TS2/16 (anti-β1 integrin subunit) were obtained from Dr. Francisco Sánchez-Madrid (Hospital de la Princesa, Madrid, Spain); mAb P1D6 (anti-α5 integrin subunit, function-blocking) has been previously described (10). Rabbit polyclonal antibodies (RpAbs) to Mcl-1 (sc-819), glutathione S-transferase (GST, sc-459), and MMP-9 (sc-6841-R) were from Santa Cruz Biotechnology (Santa Cruz, CA). RpAb to Lyn (#2732) was from Cell Signaling Technology, Inc. (Beverly, MA); RmAb to phospho-Lyn (Tyr-396) was from Abcam (Cambridge, UK). mAb to actin (#3853) was from Sigma). HRP-labeled Abs to rabbit or mouse IgG were from Dako (Glostrup, Denmark). Alexa488- and Alexa568-labeled Abs were from Molecular Probes (Eugene, OR). Hyaluronan was from Sigma. CXCL12 and TNF-α were from R&D Systems (Minneapolis, MN). proMMP-9 was isolated from THP-1 cell cultures as described (24) or purchased from Calbiochem. The fibronectin fragment FN-H89, containing the CS1 site (see below) and vascular cell adhesion molecule-1 (VCAM-1) were prepared as described (10). Fibronectin-derived synthetic peptides CS1 (α4β1 integrin ligand) and CS3 (inactive control) and PEX9-derived peptides (Table 2) were synthesized on an automated multiple peptide synthesizer (AMS 422, ABIMED Analysen-Technik GmbH, Langenfeld, Germany). Fluorescein-labeled peptides (Table 2) were synthesized using 5(6)-carboxyfluorescein (CF, Merck). Spatial localization of peptides within PEX9 was determined using the Chimera 1.5.3 Program (RBVI, UCSF, San Francisco, CA).
TABLE 2.
Synthetic peptides prepared in this study
P3arv, reversed P3a peptide; P3am, mutated P3a peptide.
| Blades B3B4-derived peptides | |
| P1 (621–640)a | RGKMLLFSGRRLWRFDVKAQ |
| P2 (638–657) | KAQMVDPRSASEVDRMFPGV |
| P3 (654–674) | PGVPLDTHDVFQYREKAYFC |
| P4 (672–691) | YFCQDRFYWRVSSRSELNQV |
| P5 (689–707) | NQVDQVGYVTYDILQCPED |
| P3-derived peptides | |
| P3a | FPGVPLDTHDVFQYREKb |
| P3b | VPLDTHDVFQb |
| P3arv | KERYQFVDHTDLPVGPF |
| P3am | FPGVPLATHAVFQYREKb |
| CS1, CS3 peptides | |
| CS1 | LVTLPHPNLHGPEILDVPST |
| CS3 | TSGQQPSVGQQMIFEEHGFR |
| 5(6)-Carboxyfluorescein-labeled peptides | |
| CF-P3a | CF-FPGVPLDTHDVFQYREK |
| CF-P3am | CF-FPGVPLATHAVFQYREK |
| CF-CS1 | CF-LVTLPHPNLHGPEILDVPST |
| CF-CS3 | CF-TSGQQPSVGQQMIFEEHGFR |
a Numbers in parenthesis indicate the position within proMMP-9 of the amino acid residues contained in each peptide.
b The two aspartate residues (D) mutated to alanine (A) are highlighted in bold.
Construction of Plasmids
A 624-bp sequence corresponding to the hemopexin domain in the full-length human MMP-9 DNA (cloned in pEGFP-N1) was obtained from Dr. Santos Mañes (Centro Nacional de Biotecnología, Madrid, Spain) (25) and amplified by PCR. The forward primer 5′-GAATTCCCCTTTGAGTCCGGTGGACG-3′ was engineered with an internal EcoRI site, and the reverse primer 5′-CTCGAGCTAGTCCTCAGGGCACTGCA-3′ was engineered with an internal XhoI site. All primers were custom-made by Sigma. Amplification of DNAs was performed using AmpliTaqR DNA polymerase (Applied Biosystems, Foster City, CA), and the integrity of the final construct was confirmed by sequence analysis (Secugen, Madrid). The resultant PCR fragment was then inserted into the pGEX4T3 vector (GE Healthcare) to generate the GST-PEX9 DNA. PEX9 mutants were generated using the following primers: GST-B1B2 forward (5′-GAATTCCCCTTTGAGTCCGGTGGACG-3′) and reverse (5′-CTCGAGTCACCTGGGCCACGTC-3′); GST-B3B4 forward (5′-GAATTCAGCCGACGTGGCCCAG-3′) and reverse (5′ CTCGAGCTAGTCCTCAGGGCACTGCA-3′). All sequences were confirmed by DNA sequencing.
Expression and Purification of GST and GST Fusion Proteins
GST and GST fusion proteins were expressed in DH5α Escherichia coli competent cells by induction with isopropyl-1-thio-β-d-galactopyranoside. Bacteria cultures were lysed by sonication in 1.5 m NaCl, 0.5 m Tris, 50 mm Na2EDTA, 10% Triton, and centrifuged. GST was soluble in this buffer and was purified using a glutathione-agarose matrix (Sigma). The GST fusion proteins appeared in inclusion bodies and were solubilized in PBS, 1% sarkosyl. These fusion proteins did not bind to glutathione-agarose under several experimental conditions and were purified by SDS-PAGE and electroelution. Purity and identity of electroeluted proteins was confirmed by SDS-PAGE and Western blotting. Purified fusion proteins were renatured by extensive dialysis against PBS and proved to be functionally active.
Preparation of the Anti-P3 Peptide Polyclonal Antibody
To prepare the immunogen, keyhole limpet hemocyanin (Calbiochem) dissolved in PBS was first coupled to sulfo-succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (1:6000 molar ratio, 1 h, 30 °C). After removing excess reagents by gel filtration on a PD10 column, keyhole limpet hemocyanin-succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate was mixed with the P3 synthetic peptide at a 1:3000 ratio in 0.1 m phosphate buffer, pH 7.6. The mixture was rotated overnight at 4 °C in the dark and dialyzed against PBS. A New Zealand rabbit was immunized with 250 μg of P3-keyhole limpet hemocyanin Y in complete Freund's adjuvant by subcutaneous injections at several regions. Subsequent boosters were with 125 μg of antigen in incomplete Freund's adjuvant at 3-week intervals. After testing for the presence of antibodies, the whole blood was obtained by intra-cardiac canaling after two boosters, serum was collected, and the anti-P3 Ab was purified using a protein A-Sepharose column (GE Healthcare). These protocols were approved by the Consejo Superior de Investigaciones Científicas Ethics Board. The specificity of the antibody was confirmed by ELISA assays with immobilized peptides (8 μm) or proteins (2 μm) using the pre-immune IgG fraction as control.
RNA Interference Experiments
The following siRNA sequences were custom-made by Ambion (Austin, TX): targeting the human β1 integrin subunit, predesigned siRNA #109879, sense (5′-GGAAUGUUCCUAUUUUAACdTT-3′); targeting human CD44, sense (5′-GAACGAAUCCUGAAGACAUCUdTT-3′) (target bases 1087–1108) (26); control siRNA sequence, sense (5′-AUUGUAUGCGAUCGCAGACdTT-3′). For transfection of B-CLL cells, 2 μg of siRNA in 100 μl of RPMI were incubated at room temperature for 10 min with 4.5 μl of HiPerfect Transfection Reagent (Qiagen, Hilden, Germany). The mixture was added dropwise to 2 × 106 cells in RPMI, 10% FBS, and transfected cells were used after 16 h (β1 siRNAs) or 24 h (CD44 siRNAs). Viability of transfected cells was determined by annexin V and propidium iodide as described (13).
Cell Adhesion and Soluble Binding Assays
Adhesion assays were performed on 96-well plates coated with 0.5% BSA or various concentrations of appropriate proteins. 2 × 105 primary B-CLL cells or 1 × 105 MEC-1 cells were incubated with 1.4 ng/ml 2′,7′-bis(carboxyethyl)-5(6′)-carboxyfluorescein-acetoxymethyl ester (BCECF-AM, Molecular Probes) for 20 min, suspended in RPMI 1640, 0.5% BSA (adhesion medium), and added to the coated wells. After 60 min at 37 °C, attached cells were lysed with PBS, 0.1% SDS and quantified using a fluorescence analyzer (BMG Labtech, Offenburg, Germany). For inhibition experiments, cells were incubated (30 min, 37 °C) with appropriate Abs (10 μg/ml) or peptides (500 μg/ml, 183–427 μm) before adding to the wells. For binding assays in solution, 1 × 105 cells were incubated in 100 μl of adhesion medium (for constitutive proMMP-9 expression) or medium containing the various proteins, peptides, or Abs for 30 min at 4 °C. After washing with ice-cold medium, cells were incubated with anti-MMP-9 pAbs (30 min, 4 °C), washed with cold PBS, and incubated (30 min, 4 °C) with Alexa488-labeled secondary Abs. Surface-bound proteins were analyzed by flow cytometry. To calculate IC50 values, cells were incubated with increasing concentrations of peptides (P3a, P3am, CS1, average range 1.8–510 μm) before the adhesion or soluble binding assays. Values were determined using the SigmaPlot program (Systat software Inc., San José, CA).
Measurement of Peptide Cell Binding Affinity
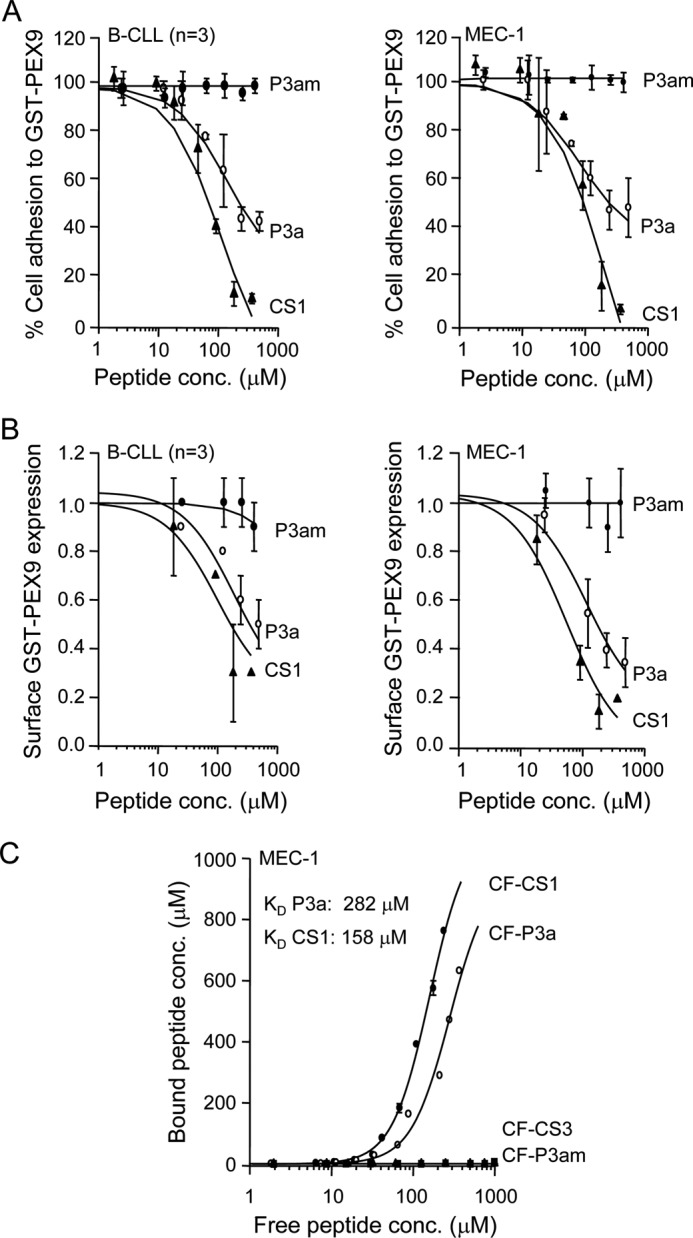
1 × 106 MEC-1 cells in PBS, 1% BSA were incubated at room temperature with mixtures of 1 μm fluorescent peptide (P3a, P3am, CS1, CS3) and increasing concentrations (1–1000 μm) of the respective unlabeled peptides in a final volume of 100 μl. After 60 min, cells were washed and separated by centrifugation, and the fluorescence remaining in the supernatant (free peptide) was determined in a fluorescence analyzer. The amount of bound peptide was determined by subtracting the free peptide values from the input fluorescence. Binding analyses were performed using the empirical Hill function (27),
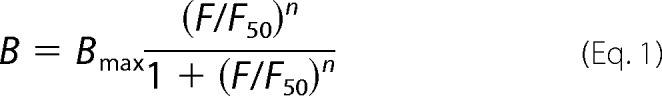 |
where B is the concentration of bound peptide/cell at a given free peptide concentration, Bmax is the maximal binding capacity, F is the concentration of free peptide, F50 is Bmax/2, and n is a Hill coefficient. This analysis allows the estimation of an apparent KD value (by means of the F50 values) and the comparison of the binding properties of the various peptides used in the study. A MATLAB model script was written for fitting this model to the binding data.
Transendothelial Migration Assays
0.75 × 105 HUVEC were plated on fibronectin-coated (10 μg/ml) Transwell filters (Costar, New York, NY), and confluent monolayers were stimulated with 15 ng/ml TNF-α for 16 h before the assay. B-CLL cells with or without previous incubation with the appropriate proteins or peptides were added to the HUVEC monolayer and allowed to migrate toward medium alone or containing 150 ng/ml CXCL12 in the lower chamber. After 24 h at 37 °C, transmigrated cells were counted by flow cytometry. Cells that migrated through HUVEC were expressed as the percentage of the total number of cells added, also counted by flow cytometry.
Immunofluorescence Analyses
Glass coverslips were coated with 10 μg/ml poly-lysine Lys for 2 h at 37 °C, washed, and blocked with 1% BSA for 30 min. 5 × 105 MEC-1 cells with or without previous incubation with GST-PEX9 were added to the coverslips and incubated at 37 °C for 1 h. Cells were fixed with paraformaldehyde, permeabilized with PBS, 0.5% Triton X-100, and incubated with appropriate primary Abs (5 μg/ml) for 45 min. Cells were washed with ice-cold PBS, 1% BSA and incubated at room temperature with Alexa488- or Alexa568-labeled secondary Abs for 45 min. Images were acquired using a Leica TCS-SP2-AOBS-UV microscope with ×63 oil immersion objective. The “Dye-Separation Leica software” was used for colocalization studies. Image pixels were depicted as dot-plot representations, where X and Y correspond to the fluorescence intensity value of each fluorescence channel per pixel. A colocalization region was assigned to pixels displaying high levels of fluorescence for the two analyzed colors.
Western Blotting
B-CLL cells were incubated on 24-well plates (2.5 × 106/well) coated with 0.2 μm GST, 0.2 μm GST-PEX9, or 60 nm proMMP-9 for either 30 min (p-Lyn) or 24 h (Mcl-1). For inhibition experiments, cells were incubated (30 min, 37 °C) with 500 μg/ml peptides before adding to the wells. Cells were lysed in cold 50 mm Tris, pH 7.4, 100 mm NaCl, 1% Nonidet P-40, 2 mm MgCl2, 10% glycerol, and protease inhibitors. Cell lysates were resolved by 10% SDS-PAGE and analyzed by Western blotting as described (13).
Statistical Analyses
Statistical significance of the data was determined using the two-tailed Student's t test. A p value of ≤0.05 was considered significant. Analyses were performed using the GraphPad InStat v3.05 software (GraphPad Software, San Diego, CA). All values are expressed as the means ± S.D.
RESULTS
The Human proMMP-9 Hemopexin Domain Binds to α4β1 Integrin and Supports Adhesion of B-CLL Cells
We previously showed that proMMP-9 supports B-CLL cell adhesion via α4β1 integrin and CD44v and that removal of the hemopexin domain (PEX9) in proMMP-9 significantly impairs this interaction (12). To establish if PEX9 was indeed responsible for the adhesion-mediating property of proMMP-9, we prepared a GST-PEX9 fusion protein comprising residues 508–707 of proMMP-9 (HPRD NP004985.2) by E. coli expression (Fig. 1A). After purification, GST-PEX9 ran as a 50-kDa band in SDS gels, and its identity was confirmed by Western blotting with an anti-MMP-9 Ab (Fig. 1B). Recombinant GST (25 kDa) was also prepared by bacteria expression (Fig. 1B) and used as control in subsequent experiments. Cell adhesion to various concentrations of these proteins was then analyzed. Primary B-CLL cells and MEC-1 cells attached to GST-PEX9 in a dose-dependent manner with maximum average values of 84 and 59% adhesion, respectively. No adhesion to GST was observed at any concentration tested (Fig. 1C).
FIGURE 1.

B-CLL cells bind to immobilized GST-PEX9 via α4β1 integrin. A, a schematic drawing shows the various domains of proMMP-9 and the four structural blades (B1-B4) of the hemopexin domain (PEX9). The GST fusion protein prepared in this study (GST-PEX9) contained amino acid residues 508–707 of proMMP-9. B, shown is analysis of GST-PEX9 by 10% SDS-PAGE followed by Coomassie Blue staining (CB) or Western blotting (WB) with anti-GST or anti-MMP-9 polyclonal antibodies. C, BCECF-AM-labeled primary B-CLL cells (patients 5, 7, 10) or MEC-1 cells (3 different experiments) were added to wells coated with the indicated concentrations of GST or GST-PEX9. After 60 min at 37 °C, attached cells were quantitated using a fluorescence analyzer. D, BCECF-AM-labeled cells with or without previous incubation with the indicated Abs or peptides were added to wells coated with 0.2 μm (B-CLL, patients 3, 10, 15) or 0.4 μm (MEC-1, 3 different experiments) GST-PEX9 or 4 μm hyaluronic acid (HA), and adhesion was quantitated as explained. E, B-CLL cells (patients 11, 17, 18) were transfected with control siRNA or siRNA for the β1 integrin subunit or for CD44, labeled with BCECF-AM, and tested for adhesion to 0.2 μm GST-PEX9 as explained. F, 1 × 105 BCECF-AM-labeled K562 cells, transfected or not with α4 integrin subunit and preincubated or not with the indicated Abs or peptides, were added to wells coated with either 0.2 μm GST, 0.036 μm fibronectin (FN), or 0.4 μm GST-PEX9. After 60 min, adhesion was quantitated as explained. All values represent the percentage of the total number of cells added. **, p ≤ 0.01; ***, p ≤ 0.001.
To identify the receptor involved in B-CLL cell interaction with GST-PEX9, we first tested the effect of blocking α4β1 integrin or CD44 function with antibodies or peptides on cell adhesion to this substrate. Fig. 1D shows that cell preincubation with the HP2/1 mAb or the CS1 peptide, two well known inhibitors of α4 integrin function (28–30), significantly (p ≤ 0.001) inhibited cell adhesion to GST-PEX9, whereas the control HP1/7 mAb or the CS3 peptide had no effect. In contrast, the anti-CD44 mAb HP2/9 did not affect cell adhesion to GST-PEX9 but efficiently blocked (>90% inhibition) B-CLL cell adhesion to hyaluronic acid (Fig. 1D), confirming that the Ab was functional. To confirm these results, we transfected B-CLL cells (samples 11, 17, and 18) with siRNA specific for the β1 integrin subunit or for CD44 or with a control siRNA. Flow cytometry analyses confirmed the reduction of β1 and CD44 surface expression by 73 and 75%, respectively (shown for two representative patients in supplemental Fig. S1A). Transfection efficiency was also validated by Western blot analyses of cell lysates (shown for patient 18 and quantitated for patients 11 and 18 in supplemental Fig. S1B). Transfected cells were then tested for adhesion to 0.2 μm GST-PEX9. Gene silencing β1 reduced cell adhesion by 84%, whereas CD44 gene silencing had a minor effect, diminishing cell adhesion by 19% (Fig. 1E). In contrast, and in agreement with our previous results (12), both β1 and CD44 were required for B-CLL cell adhesion to proMMP-9 (results not shown).
To further establish that α4β1 integrin was the receptor for PEX9, we used K562 cells before and after transfection with the α4 integrin subunit. Parental K562 cells, which lack α4β1, were unable to attach to GST-PEX9 but efficiently adhered to fibronectin via α5β1 integrin (Fig. 1F). Transfecting K562 cells with the α4 integrin subunit resulted in efficient adhesion to GST-PEX9, which was blocked by the specific α4β1 inhibitors HP2/1 and CS1 but not by their corresponding controls (Fig. 1F). Altogether these results indicated that α4β1 is the primary adhesion receptor for PEX9 in B-CLL cells.
Soluble proMMP-9 Hemopexin Domain Binds B-CLL Cells via α4β1 Integrin
To determine whether B-CLL cells bound soluble PEX9, as previously observed for soluble proMMP-9 (12), we incubated primary B-CLL cells (three different patients) or MEC-1 cells with GST-PEX9 in suspension (0.4 or 0.6 μm, respectively) and measured the binding by flow cytometry using an anti-MMP-9 antibody. As shown in Fig. 2A for a representative sample and quantitated for all three patients studied, primary B-CLL cells bound soluble GST-PEX9, increasing 2.5-fold the constitutive expression of surface-bound MMP-9 (12). MEC-1 cells lacked basal MMP-9 surface expression but also efficiently bound soluble GST-PEX9 (Fig. 2A). In both cases and as observed in the adhesion experiments, binding was completely inhibited by the HP2/1 mAb and the CS1 peptide but not by their respective controls (Fig. 2A). Moreover, gene silencing β1 prevented GST-PEX9 cell binding, reducing the surface expression of (pro)MMP-9 to below the constitutive levels; CD44 gene silencing, however, had no effect (supplemental Fig. S1C). GST-PEX9 binding was due to PEX9, as GST alone was unable to bind B-CLL cells, as analyzed with an anti-GST antibody, which in turn efficiently detected surface-bound GST-PEX9 (Fig. 2A).
FIGURE 2.

α4β1 integrin mediates binding of soluble GST-PEX9 to B-CLL cells. A, primary B-CLL cells (patients 1, 2, 11) and MEC-1 cells (3 different experiments) with or without previous incubation with the indicated Abs or peptides were incubated for 30 min with or without GST-PEX9 (0.4 or 0.8 μm for B-CLL or MEC-1 cells, respectively) and analyzed by flow cytometry using anti-MMP-9 or anti-GST Abs. A representative B-CLL sample (patient 1) out of the three studied is shown. White areas represent constitutive (pro)MMP-9 expression; gray areas, (pro)MMP-9 expression upon GST-PEX9 binding. Constitutive MMP-9 expression was normalized to 1, and average quantitative values are shown. *, p ≤ 0.05; **, p ≤ 0.01. B, MEC-1 cells with or without previous incubation with 0.6 μm GST-PEX9 were added to glass coverslips coated with 10 μg/ml poly-lysine Lys. After 1 h at 37 °C, cells were fixed and analyzed by confocal microscopy using specific Abs for α4, β1, CD44, or MMP-9 followed by Alexa488- or Alexa568-labeled secondary Abs. Colocalization of GST-PEX9 with α4β1 integrin was further demonstrated using dot-plot analyses. The bar represents 5 μm.
To further confirm these results, we performed immunofluorescence analyses using confocal microscopy. MEC-1 cells were used for this purpose because, as shown in Fig. 2A, they lack constitutive expression of surface-bound MMP-9, thus facilitating detection of exogenously added PEX9. Consequently, in the absence of GST-PEX9, only the α4 or β1 integrin subunits were detected at the cell surface (Fig. 2B). The addition of GST-PEX9 resulted in a clear colocalization with both subunits at the cell periphery, as documented by dot-plot analyses (Fig. 2B). In contrast, GST-PEX9 did not colocalize with CD44 (Fig. 2B), in agreement with the cell adhesion results shown in Fig. 1D.
Characterization of the PEX9 Region Responsible for the Interaction with α4β1 Integrin in B-CLL Cells
The PEX9 domain contains a four-bladed β-propeller structure (31) as schematically shown in Fig. 3A. To define the PEX9 region involved in α4β1 integrin binding, we prepared two GST fusion proteins containing deletions of blades 1–2 (GST-B3B4 protein, proMMP-9 residues 609–707) or blades 3–4 (GST-B1B2 protein, proMMP-9 residues 508–613) (Fig. 3B). After purification and SDS-PAGE analyses (Fig. 3C), purified proteins were tested in adhesion and soluble binding assays. Fig. 3D shows for MEC-1 cells that the GST-B1B2 construct was a poor adhesion substrate, whereas the GST-B3B4 protein supported cell adhesion as effectively as GST-PEX9. As observed for parental GST-PEX9, adhesion to GST-B3B4 was completely inhibited by the HP2/1 but not the HP1/7 mAbs (Fig. 3D), confirming that α4β1 integrin was the receptor for GST-B3B4. In agreement with these results, the soluble GST-B1B2 protein did not bind to MEC-1 cells, whereas the soluble GST-B3B4 protein bound 2.9-fold over the MMP-9 basal expression (Fig. 3E). The anti-GST Ab was used in these experiments, as ELISA assays demonstrated that the anti-MMP-9 Ab did not recognize the GST-B1B2 protein (not shown). GST-B3B4 soluble binding was also inhibited by blocking α4β1 function with specific antibodies or peptides (not shown).
FIGURE 3.

Characterization of the PEX9 region that interacts with α4β1 integrin. A, a ribbon diagram of the monomeric PEX9 domain (PDB entry 1ITV) shows the position of the four structural blades B1-B4. B, shown is a schematic drawing of the recombinant GST-fusion proteins prepared in this study. The GST-PEX9 fusion protein contains all four blades (B1-B4), whereas truncated proteins contain either B3B4 (B3B4 protein) or B1B2 (B1B2 protein). C, SDS-gel analysis of the purified GST-fusion proteins shown in B is visualized by Coomassie Blue staining. D, 1 × 105 BCECF-AM-labeled MEC-1 cells were added to wells coated with the indicated proteins. After 60 min at 37 °C, attached cells were quantitated using a fluorescence analyzer. Cells with or without previous incubation with the indicated Abs were also added to wells coated with GST-B3B4, and adhesion was analyzed as explained. 1 × 105 MEC-1 cells (E) or primary B-CLL cells (patients 9, 10, 17) (F) were incubated for 30 min with soluble GST-PEX9, GST-B1B2, or GST-B3B4 and analyzed by flow cytometry using an anti-GST antibody. A representative sample for B-CLL (patient 17) of the three studied with identical results is shown. White areas represent constitutive (pro)MMP-9 expression; gray areas, (pro)MMP-9 expression upon GST-PEX9 binding. Quantitative values represent the average of three different experiments with triplicate determinations (MEC-1) or three different samples after normalizing constitutive values (Const.) to 1. *, p ≤ 0.05.
Although the differential adhesion to the GST-B1B2 and GST-B3B4 constructs was not so obvious for primary B-CLL cells (average 54% versus 57% adhesion), there were clear and significant differences in the ability to bind these proteins in soluble form. Fig. 3F shows, for a representative sample out of the four samples studied, that soluble GST-B1B2 was unable to bind to B-CLL cells. In contrast, soluble GST-B3B4 bound, increasing 2.1-fold the constitutive MMP-9 surface expression. Altogether these results established that the PEX9 region represented by blades 3–4 contained binding sites for α4β1 integrin.
Identification of an Amino Acid Sequence within PEX9 Blades 3–4 Recognized by α4β1 Integrin
To further identify the specific sequence within PEX9 blades 3–4 that interacted with α4β1 integrin, we prepared five overlapping synthetic peptides (P1-P5) spanning residues 621–707 of proMMP-9 (Table 2 and Fig. 4A) and tested their ability to block cell adhesion to GST-PEX9. For this initial characterization, peptides were used at 500 μg/ml, equivalent to 202.8 μm (P1), 225.2 μm (P2), 197.5 μm (P3), 193.5 μm (P4), and 227.3 μm (P5) concentrations; thus all in a similar molar range. Average control adhesion values in these experiments were 78 and 49% for primary B-CLL and MEC-1 cells, respectively, and were normalized to 100. Cell preincubation with the P3 peptide significantly abolished cell adhesion to GST-PEX9 (55 and 70% average inhibition for B-CLL and MEC-1, respectively), whereas preincubation with any of the other peptides did not (Fig. 4B). Narrowing the P3 sequence to FPGVPLDTHDVFQYREK (peptide P3a, 244 μm) or to VPLDTHDVFQ (peptide P3b, 427.4 μm) also significantly inhibited cell adhesion to GST-PEX9 (Fig. 4C) or to proMMP-9 (Fig. 4D). This effect was specific as the reverse P3a amino acid sequence (peptide P3arv, 244 μm) was inactive (Fig. 4, C and D). To determine if the two aspartic acid residues of P3a were important for its function, as observed for many integrin ligands (32), we mutated the Asp-660 and Asp-663 residues to alanine (peptide P3am) and tested its effect of cell adhesion. Fig. 4, C and D, show that P3am at 255 μm was unable to inhibit B-CLL cell adhesion to either GST-PEX9 or proMMP-9, thus establishing that Asp-660 and Asp-663 are involved in α4β1 binding. The spatial localization of these Asp residues within PEX9 is depicted in Fig. 4E.
FIGURE 4.

Effect of synthetic peptides derived from the PEX9 domain on B-CLL cell adhesion. A, a ribbon diagram of the monomeric PEX9 domain shows the location in blades 3–4 of the synthetic peptides P1-P5 prepared in this study (see Table 2). B, BCECF-AM-labeled primary B-CLL cells (patients 8, 14, 19) and MEC-1 cells (3 different experiments) with or without previous incubations with the indicated peptides (500 μg/ml, equivalent to 202.8 μm P1, 225.2 μm P2, 197.5 μm P3, 193.5 μm P4, and 227.3 μm P5 concentrations) were added to wells coated with 0.2 μm (B-CLL) or 0.4 μm (MEC-1) GST-PEX9. After 60 min, cell adhesion was quantitated as explained. C and D, BCECF-AM-labeled primary B-CLL cells (patients 4, 8, 14) and MEC-1 cells with or without previous incubation with the indicated peptides (500 μg/ml, equivalent to 197.5 μm P3, 244 μm P3a, 427.4 μm P3b, 244 μm P3arv, 255 μm P3am concentrations) were added to wells coated with GST-PEX9 (C) or proMMP-9 (D), and cell adhesion was measured as explained. E, spatial localization of the Asp-660 and Asp-663 residues of the P3 peptide (shown in red) was determined by the Chimera 1.5.3 Program (RBVI, UCSF). F, BCECF-AM-labeled MEC-1 cells with or without previous incubation with the indicated peptides (all at 500 μg/ml; CS1 at 183.0 μm), were added to wells coated with GST-PEX9 (0.4 μm), FN-H89 fragment (0.2 μm), or VCAM-1 (0.1 μm), and cell adhesion was analyzed as explained. Values are the average of two different experiments with triplicate determinations and were obtained after normalizing control values to 100. **, p ≤ 0.01; ***, p ≤ 0.001.
We also studied the functional effect of P3a on α4β1 binding to other ligands, such as VCAM-1 or FN-H89. Fig. 4F shows that P3a (244 μm) did not block MEC-1 cell adhesion to either VCAM-1 or FN-H89, whereas the CS1 peptide (183 μm) effectively inhibited these interactions. In the same experiment, P3a significantly blocked adhesion to proMMP-9 (Fig. 4F) or to GST-PEX9 (not shown), thus confirming its specificity for these ligands.
We next analyzed if the P3 peptides also blocked cell binding of soluble GST-PEX9 or proMMP-9. B-CLL cells or MEC-1 cells were treated with the mentioned μm concentration of P3, P3a, P3b, or P3am peptides followed by incubation with GST-PEX9 or proMMP-9 and analysis by flow cytometry. Fig. 5, A and B, show for a representative sample that P3, P3a, and P3b significantly prevented binding of soluble GST-PEX9 to B-CLL or MEC-1 cells. These peptides also inhibited soluble proMMP-9 binding to B-CLL cells (Fig. 5, A and B) or MEC-1 cells (95% inhibition, not shown). As observed for cell adhesion, the P3am peptide had no effect on either GST-PEX9 or proMMP-9 binding.
FIGURE 5.

Further effects of P3, P3-derived peptides, and anti-P3 antibodies on B-CLL cells. A, primary B-CLL or MEC-1 cells treated or not for 30 min with the indicated peptides (at the same μm concentrations used in Fig. 4), were incubated with soluble GST-PEX9 (0.4 and 0.6 μm, respectively) or proMMP-9 (60 nm). After 30 min, binding was analyzed by flow cytometry using an anti-MMP-9 antibody. A representative experiment (patient 18 for B-CLL) is shown. White areas represent constitutive (pro)MMP-9 expression; gray areas, (pro)MMP-9 expression upon GST-PEX9 binding. Binding of soluble proMMP-9 to MEC-1 cells was also inhibited by the P3/P3a/P3b peptides and is not shown. B, quantitative values of the soluble binding assays represent the average of four (patients 8, 14, 17, 18; GST-PEX9) or three (patients 14, 17, 18; proMMP-9) B-CLL samples or the average of three different experiments with triplicate determinations (MEC-1). C, the specificity of the anti-P3 antibody was determined by ELISA assays with the indicated immobilized peptides (8 μm) or proteins (2 μm). Negative control (Neg. ctrl), average absorbance of peptides or proteins with only secondary antibody. D, BCECF-AM-labeled B-CLL cells (patients 15, 18, 20) with or without previous incubation with preimmune Ig or anti-P3 antibody were added to wells coated with GST-PEX-9 or proMMP9 and adhesion measured as explained. *, p ≤ 0.05.
A Polyclonal Antibody Raised against the FPGVPLDTHDVFQYREKAYFC proMMP-9 Sequence Inhibits B-CLL Cell Adhesion to PEX9 and proMMP-9
To further confirm the function of the sequence represented by the P3 peptide, we prepared a polyclonal antibody by immunizing a rabbit with P3 covalently coupled to keyhole limpet hemocyanin. ELISA analyses demonstrated that this antibody specifically recognized P3 but not P1, P2, P4, or P5 peptides (Fig. 5C). It also recognized P3a, GST-PEX9 (but not GST), and to a lesser yet specific extent, proMMP-9. The mutated peptide P3am showed 67% reduced reactivity against this antibody compared with P3a. The anti-P3 antibody was next tested in cell adhesion assays. Average control adhesion values in these experiments were 76 and 39% for PEX9 and proMMP-9, respectively, and were normalized to 100 (Fig. 5D). Treatment of immobilized GST-PEX9 or proMMP-9 with anti-P3 antibody (15 μg/ml) significantly inhibited B-CLL cell adhesion to both substrata by 45 and 55%, respectively. The preimmune Ig fraction (15 μg/ml) had no effect (Fig. 5D). These results confirmed the involvement of the P3 sequence in proMMP-9 interaction with B-CLL cells and the accessibility of this sequence in PEX9 and proMMP-9.
Further Characterization of the P3a Sequence Interaction with B-CLL Cells
Having identified and established the specificity of the sequence represented by the P3/P3a/P3b peptides, we carried out dose dependence inhibition analyses. The P3a and P3am peptides were chosen for these studies and, for comparison, the previously described α4β1 ligand CS1. Fig. 6A shows that P3a, but not the control P3am, inhibited primary B-CLL and MEC-1 cell adhesion to PEX9 in a dose-dependent manner, with an average IC50 value of 138 and 178 μm for primary B-CLL and MEC-1 cells, respectively. The average IC50 for CS1 in these experiments was 80 μm (B-CLL) and 98 μm (MEC-1). P3a inhibition of soluble PEX9 cell binding was also dose-dependent, resulting in IC50 values of 199 and 114 μm for primary B-CLL and MEC-1 cells, respectively (Fig. 6B). CS1 inhibition in this case rendered IC50 values of 100 and 55 μm, respectively. In both cases, cell adhesion and soluble binding, the mutated peptide P3am had no effect at any concentration tested (Fig. 6, A and B).
FIGURE 6.
Dose-response inhibitory effect and cell binding affinity of the P3a peptide. Comparison with the CS1 peptide. A, BCECF-AM-labeled primary B-CLL cells (patients 10, 11, 20) or MEC-1 cells (3 different experiments) were incubated (30 min, 37 °C) with the indicated concentrations of P3a, P3am, or CS1 peptides and added to wells coated with 0.2 μm (B-CLL) or 0.4 μm (MEC-1) GST-PEX9. After 60 min at 37 °C, attached cells were quantitated using a fluorescence analyzer. Adhesion values in the absence of peptides were normalized to 100. B, the same B-CLL samples listed in A or MEC-1 cells were treated for 30 min with the indicated peptide concentrations and incubated with 0.4 μm (B-CLL) or 0.6 μm (MEC-1) soluble GST-PEX9. After 30 min, binding was analyzed by flow cytometry using an anti-MMP-9 antibody. Binding in the absence of peptides was normalized to 1. IC50 values for A and B were calculated using the SigmaPlot program. C, increasing concentrations of the indicated fluoresceinated peptides (CF) were incubated with MEC-1 cells (106/100 μl) for 60 min at room temperature. After centrifugation, the fluorescence of free and bound peptide was determined as explained. Experimental data were estimated according to the Hill equation using the MATLAB software. This analysis rendered a Bmax of 960 μm (P3a) and 1100 μm (CS1), Hill n values of 1.8 (P3a) and 1.9 (CS1), and F50 values of 2.45 (P3a) and 2.2 (CS1).
Similar results were obtained for inhibition of cell binding to proMMP-9 either in immobilized or soluble form. The IC50 values for P3a inhibition of cell adhesion to proMMP-9 were 279 and 109 μm for B-CLL and MEC-1 cells, respectively, whereas these values were 102 and 74 μm, respectively, for CS1 (supplemental Fig. S2). Binding of soluble proMMP-9 to cells was also inhibited by P3a in a dose-dependent manner and with IC50 values of 219 μm (B-CLL) and 282 μm (MEC-1), whereas the P3am peptide had no effect (supplemental Fig. S2). The IC50 values for CS1 in this case were 158 μm (B-CLL) and 84 μm (MEC-1). Altogether these results indicated that the inhibitory activity of the P3a peptide was slightly lower but comparable with that of the well known α4β1 ligand CS1.
We next performed direct cell binding analyses using CF-labeled P3a and P3am peptides as well as CF-CS1 and CF-CS3 (inactive control) for comparison. MEC-1 cells were chosen for these studies because of their more homogeneous α4β1 integrin expression compared with primary B-CLL cells. The CF-P3a and CF-CS1 peptides, but not their corresponding controls CF-P3am or CF-CS3, inhibited cell adhesion to GST-PEX9 or proMMP-9 as efficiently as their unlabeled counterparts (not shown) and thus were considered suitable for these studies. Fig. 6C shows that both CF-P3a and CF-CS1 peptides bound to MEC-1 cells in a dose-dependent manner, whereas the control CF-P3am and CF-CS3 peptides did not. The calculated apparent KD values obtained from these assays was 282 and 158 μm for CF-P3a and CF-CS1, respectively. Saturation was not reached under the conditions used, probably reflecting low affinity interactions. Importantly, these results indicated that the apparent KD for the interaction of P3a with MEC-1 cells was of the same order as that of the well known α4β1 integrin ligand CS1.
Peptides Containing the VPLDTHDVFQ Sequence Block B-CLL Cell Transendothelial Migration and PEX9/proMMP-9-induced Intracellular Signaling
We and others previously showed that endogenous (pro)MMP-9 plays a crucial role in B-CLL cell migration and invasion (9, 10). In this study we tested whether PEX9 or the P3 peptide modulated the MMP-9 migratory function. B-CLL cells migrated through endothelial cells in response to CXCL12 with average values of 22–25% after 24 h, which were normalized to 100. Cell preincubation with GST-PEX9 or GST-B3B4 significantly inhibited this migration, whereas GST alone had no effect (Fig. 7A). Importantly, the synthetic peptides P3 (197.5 μm), P3a (244 μm), and P3b (427.4 μm) also significantly hampered B-CLL cell migration (80–85% inhibition, Fig. 7A). As observed for cell adhesion, reversing the amino acid sequence or mutating the Asp-660 and Asp-663 residues in P3a abolished the inhibitory effect (Fig. 7A). To confirm that the inhibitory effect of PEX9 or the P3 peptides was not due to induction of apoptosis after the 24 h exposure, we examined cell viability in parallel samples using annexin V and propidium iodide (13). This analysis showed that average viability values (n = 4) were 73, 69, and 77% for control, GST-PEX9, or P3/P3a peptide-treated cells, respectively, thus indicating that inhibition of migration was not due to apoptosis.
FIGURE 7.
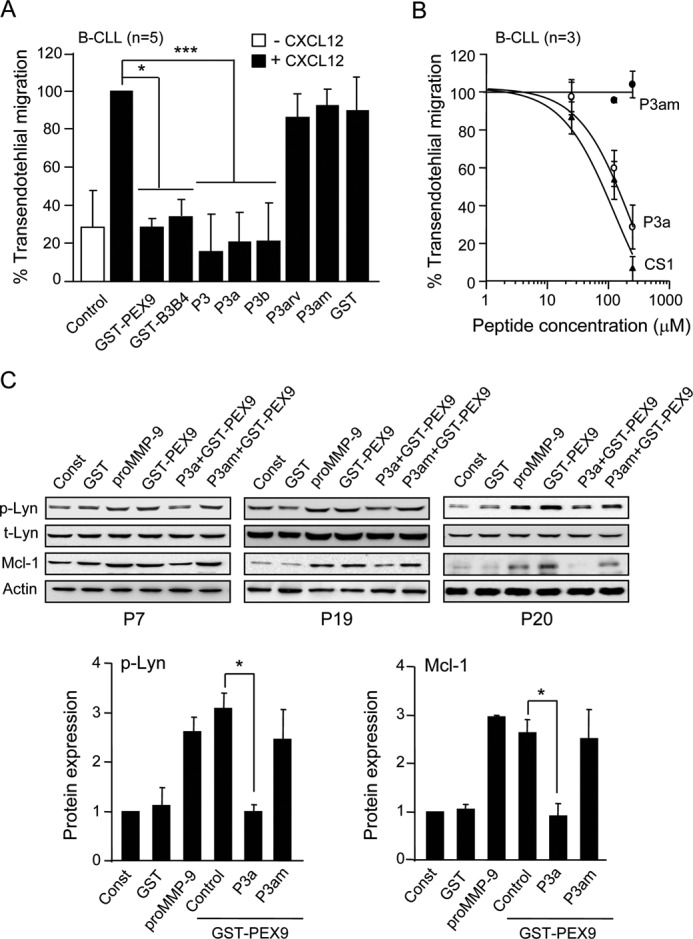
Functional effects of P3 peptides on B-CLL cell transendothelial migration and proMMP-9/GST-PEX9-induced intracellular signaling. A, primary B-CLL cells (patients 8, 11, 13, 14, 16) treated or not with the indicated proteins for 30 min were added to the upper chamber of transwell filters coated with TNF-α-activated HUVEC. 150 ng/ml CXCL12 was added to the medium in the bottom chamber, and migrated cells were counted after 24 h by flow cytometry. Average values represent the percentage of the total number of cells added. B, the same five samples used in A were preincubated or not with the indicated peptides at 197.5 μm P3, 244 μm P3a, 427.4 μm P3b, 255 μm P3am, and transendothelial migration was measured as explained. Average values (% of total cells added) are shown. C, 2.5 × 106 B-CLL cells from the three patients indicated were added to wells coated with GST, proMMP-9, or GST-PEX9. Cells were also treated with either 244 μm P3a or 255 μm P3am peptides before adding to GST-PEX9-coated wells. After 30 min (p-Lyn/Lyn) or 24 h (Mcl-1), cells were lysed, and lysates were analyzed by Western blotting. Constitutive protein levels (Const) are also shown. Normalized average values of p-Lyn and Mcl-1 are shown. *, p ≤ 0.05; ***, p ≤ 0.001.
We next determined whether the observed peptide inhibitory effect on migration was dose-dependent, as was the case for inhibition of cell adhesion and soluble binding. As above, the P3a and P3am peptides and, for comparison, the CS1 peptide were chosen for these studies. Fig. 7B shows that both P3a and CS1 inhibited B-CLL cell migration in a dose-dependent manner, with IC50 values of 190 and 97 μm, respectively. The control P3am peptide had no effect. These results, therefore, indicated that the IC50 values for P3a and CS1 in these assays were of the same order of magnitude.
We recently showed that B-CLL cell adhesion to proMMP-9 via α4β1 integrin initiates a survival pathway that includes activation of the Src-family kinase Lyn and up-regulation of Mcl-1 (13). To determine if the P3a sequence interfered with this signaling, we first studied if cell culturing on GST-PEX9 also activated Lyn. B-CLL cells from three different patients were incubated on GST, GST-PEX9, or proMMP-9 (positive control) for 30 min, lysed, and analyzed by Western blotting. Fig. 7C shows that proMMP-9 and GST-PEX9 increased Lyn phosphorylation 2.6- and 3.1-fold, respectively, compared with the constitutive values, whereas GST was inactive. Cell preincubation with the P3a peptide (244 μm) completely inhibited this activation, whereas the mutated peptide P3am (255 μm) had no effect (Fig. 7C), in agreement with the above results on cell adhesion and soluble binding.
We also studied the effect of GST-PEX9 on Mcl-1 regulation. B-CLL cells from the same three patients used for the Lyn analyses were cultured on GST, GST-PEX9, or proMMP-9. After 24 h, cells were lysed, and lysates were analyzed by Western blotting. Fig. 7C shows that incubation on GST-PEX9 increased the levels of Mcl-1 2.6-fold over the constitutive values, similar to the effect of proMMP-9 (2.9-fold increase). Cell preincubation with the P3a peptide completely abolished Mcl-1 up-regulation (Fig. 7C). As observed for Lyn phosphorylation, the P3am peptide was inactive. Altogether these results established that the sequence represented by the P3/P3a/P3b peptides inhibited B-CLL cell binding to proMMP-9/GST-PEX9 and subsequent intracellular signaling.
DISCUSSION
Our recent studies have shown that (pro)MMP-9 binds to B-CLL cells via its hemopexin domain (PEX9) and contributes to B-CLL pathogenesis by modulating cell migration and survival (10–13). These studies demonstrated that the survival-inducing effect does not require MMP-9 catalytic activity but simply binding to its cell surface receptors (13). In an effort to help design specific inhibitors that prevent (pro)MMP-9-B-CLL cell binding, in this report we have biochemically characterized B-CLL cell interaction with the isolated PEX9 domain. Our major findings are 1) PEX9 supports cell adhesion and soluble binding to B-CLL cells via α4β1 integrin and inhibits B-CLL cell transendothelial migration, 2) synthetic peptides (P3/P3a/P3b) containing the VPLDTHDVFQ sequence of PEX9 blade 4 specifically inhibit cell interaction with PEX9 and proMMP-9, and the two Asp residues in VPLDTHDVFQ are critical for its function, 3) a polyclonal antibody raised against P3 blocks B-CLL cell adhesion to PEX9 and proMMP-9, 4) the P3 peptides inhibit B-CLL cell transendothelial migration and PEX9/proMMP-9-induced intracellular signaling.
The GST-PEX9 fusion protein prepared in this study, either immobilized or in soluble form, specifically and efficiently bound to B-CLL cells, and α4β1 integrin was the receptor involved. This was clearly demonstrated by blocking α4β1 function or gene expression and by using K562 cells with or without α4β1 expression. B-CLL cell interaction with isolated PEX9 thus appears to differ from binding to (pro)MMP-9, which we previously showed (and confirmed here) to require α4β1 and CD44v engagement (12). Indeed, CD44H is a well known receptor for MMP-9 in many cell types (14, 33, 34), and a recent study using MMP-9-transfected COS-1 cells has located a CD44 binding region in blade I of the MMP-9 hemopexin domain (16). Our present results show that CD44 was not involved in B-CLL cell adhesion to GST-PEX9 and that both proteins did not colocalize at the cell surface. Interaction with one or both receptors may, therefore, depend on the conformation (and/or affinity) adopted by PEX9 when present in (pro)MMP-9 or as an isolated protein. It may also depend on the cell type studied as, in the above mentioned report (12), we showed that B-CLL cells differ from other cell types in that CD44v, but not CD44H, interacts with proMMP-9. Additionally, we also recently reported that the survival pathway induced by proMMP-9 in B-CLL cells apparently involves only α4β1 integrin (13).
We further show that binding of GST-PEX9 to B-CLL cells results in inhibition of B-CLL cell transendothelial migration, as previously observed for murine PEX9 (18–20) and human PEX2 (the MMP-2 hemopexin domain) (35) in other cell types. This function may have special relevance as naturally occurring PEX2 has been detected in vivo and produced in vitro upon proMMP-2 activation with p-aminophenyl-mercuric acetate (35). It is not known if PEX9 is naturally formed in B-CLL, but truncated forms of (pro)MMP-9 consisting of a catalytically active fragment and the hemopexin domain were recently found in the conditioned medium of breast cancer cells and produced in vitro by treatment with kallikrein-related peptidase-7 (36).
(pro)MMP-9 has been shown to interact with various integrins in different cell systems, and targeting these interactions has been the aim of several previous reports. Using phage display libraries, peptides containing the sequence DELW, present in the catalytic domain of MMP-9 (and MMP-2), were shown to disrupt the interaction of these MMPs with the I domain of αLβ2/αMβ2 integrins in THP-1 monocytic cells (37). The related peptide HFDDDE, also from the catalytic domain, inhibited in vivo cell extravasation in an acute myeloid leukemia xenograft model (38). The same authors also reported that peptides containing the CRV motif, a mimic of an integrin activation epitope, inhibited binding of MMP-9 to the β subunit of αVβ5 integrin in fibrosarcoma cells (15). In this study we have used a different approach to define the specific sequences within the proMMP-9 hemopexin domain involved in α4β1 binding in B-CLL cells. Using recombinant constructs containing truncated forms of PEX9 and a series of overlapping synthetic peptides, we have identified the FPGVPLDTHDVFQYREKAYFC sequence, located in PEX9 blade 4, as a novel binding site for α4β1 integrin. The synthetic peptide P3, containing this sequence, or the related peptides P3a and P3b, representing smaller versions of this sequence, completely abrogated B-CLL cell transendothelial migration and soluble binding of GST-PEX9 and proMMP-9 to B-CLL cells. Their effect on cell adhesion to GST-PEX9 or proMMP-9 was also dose-dependent and very significant yet partial, suggesting that other cell binding sites may exist within PEX9. Importantly, this partial effect was sufficient to completely inhibit the survival signaling pathway induced by cell adhesion to proMMP-9 or GST-PEX9, as the P3a peptide reduced the up-regulated levels of p-Lyn and Mcl-1 to constitutive values.
Further evidence for the specificity and integrin ligand function of the FPGVPLDTHDVFQYREKAYFC sequence came from two findings; 1) the functional effect of an antibody raised against the P3 sequence and 2) the fact that mutating the two Asp residues (Asp-660 and Asp-663) in this sequence abolished the inhibitory effect of the P3a peptide. It is well established that Asp residues are key recognition sites for many integrins, including α4β1 (29, 30). Moreover, the LDT motif present in the P3 sequence resembles motifs found in well known α4β1 ligands, namely LDV and LDA, found in the CS1 region and the III5 repeat of fibronectin, respectively (29, 39, 40), or the IDS sequence of VCAM-1 (41). Despite this homology, the P3a peptide, at concentrations proven effective for inhibiting adhesion to PEX9 or proMMP-9, did not affect cell adhesion to the α4β1 ligands VCAM-1 or FN-H89. Moreover, our KD and IC50 analyses indicated that the binding of P3a to B-CLL cells is of low affinity, and this may in fact represent an advantage for specifically targeting (pro)MMP-9-cell interactions (presumably also of low affinity). In agreement with this, the CS1 peptide, which in this study showed similar inhibitory and binding activity as P3a, efficiently inhibited B-CLL cell binding to VCAM-1 or FN-H89, perhaps reflecting different binding characteristics that affect more α4β1/ligand interactions.
Targeting the PEX9 domain is becoming an important focus for drug development, and a recent study (17) has identified a small-molecule compound that binds to PEX9 and inhibits carcinoma cell migration, proliferation, and metastasis. Using an in silico docking approach, these authors mapped the binding site of this compound to the central cavity of PEX9, in fact in close proximity to the VPLDTHDVFQ sequence identified in our study. Although it is not known if the compound affects B-CLL cells, this study strongly supports our present findings. Moreover, VPLDTHDVFQ, in contrast to previously identified sites located in the MMP-9 catalytic region, is not present in other MMPs as a BLAST analysis revealed. Additionally, we show in our study that this sequence was effective in all B-CLL cases studied, irrespectively of the clinical stage or prognostic marker expression. All these findings make the VPLDTHDVFQ sequence identified here an excellent target to specifically block (pro)MMP-9-B-CLL cell interactions and subsequent intracellular events that contribute to B-CLL progression.
Supplementary Material
Acknowledgments
We thank the B-CLL patients who donated samples for this research, Dr. Silvia Zorrilla (Instituto de Química-Física Rocasolano, Consejo Superior de Investigaciones Científicas) for expert advice and analyses of the peptide binding data, Dr. Pedro Lastres and Teresa Seisdedos (Centro de Investigaciones Biológicas) for valuable help with flow cytometry and confocal microscopy, respectively, and Jorge Huertas (Centro de Investigaciones Biológicas Animal Facility) for help with the immunization procedures.
This work was supported by Grants SAF2009–07035 and RTICC RD06/0020/0011 (to A. G.-P.) and RTICC RD06/0020/0080 (to M. J. T.) from the Ministerio de Ciencia e Innovación, Spain, and by a grant from the Fundación Puerta de Hierro (to J. A. G. M.).

This article contains supplemental Figs. S1 and S2.
- B-CLL
- B-cell chronic lymphocytic leukemia
- MMP
- matrix metalloproteinase
- HUVEC
- human umbilical vein endothelial cells
- VCAM-1
- vascular cell adhesion molecule-1
- CF
- 5(6)-carboxyfluorescein
- BCECF-AM
- 2′,7′-bis(carboxyethyl)-5(6′)-carboxyfluorescein-acetoxymethyl ester
- Ab
- antibody.
REFERENCES
- 1. Chiorazzi N., Rai K. R., Ferrarini M. (2005) Chronic lymphocytic leukemia. N. Engl. J. Med. 352, 804–815 [DOI] [PubMed] [Google Scholar]
- 2. Zenz T., Mertens D., Küppers R., Döhner H., Stilgenbauer S. (2010) From pathogenesis to treatment of chronic lymphocytic leukemia. Nat. Rev. Cancer 10, 37–50 [DOI] [PubMed] [Google Scholar]
- 3. Till K. J., Lin K., Zuzel M., Cawley J. C. (2002) The chemokine receptor CCR7 and α4 integrin are important for migration of chronic lymphocytic leukemia cells into lymph nodes. Blood 99, 2977–2984 [DOI] [PubMed] [Google Scholar]
- 4. López-Giral S., Quintana N. E., Cabrerizo M., Alfonso-Pérez M., Sala-Valdés M., De Soria V. G., Fernández-Rañada J. M., Fernández-Ruiz E., Muñoz C. (2004) Chemokine receptors that mediate B cell homing to secondary lymphoid tissues are highly expressed in B cell chronic lymphocytic leukemia and non-Hodgkin lymphomas with widespread nodular dissemination. J. Leukoc. Biol. 76, 462–471 [DOI] [PubMed] [Google Scholar]
- 5. Till K. J., Spiller D. G., Harris R. J., Chen H., Zuzel M., Cawley J. C. (2005) CLL, but not normal, B cells are dependent on autocrine VEGF and α4β1 integrin for chemokine-induced motility on and through endothelium. Blood 105, 4813–4819 [DOI] [PubMed] [Google Scholar]
- 6. Kessenbrock K., Plaks V., Werb Z. (2010) Matrix metalloproteinases. Regulators of the tumor microenvironment. Cell 141, 52–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deryugina E. I., Quigley J. P. (2010) Pleiotropic roles of matrix metalloproteinases in tumor angiogenesis. Contrasting, overlapping, and compensatory functions. Biochim. Biophys. Acta 1803, 103–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bauvois B., Dumont J., Mathiot C., Kolb J. P. (2002) Production of matrix metalloproteinase-9 in early stage B-CLL. Suppression by interferons. Leukemia 16, 791–798 [DOI] [PubMed] [Google Scholar]
- 9. Kamiguti A. S., Lee E. S., Till K. J., Harris R. J., Glenn M. A., Lin K., Chen H. J., Zuzel M., Cawley J. C. (2004) The role of matrix metalloproteinase-9 in the pathogenesis of chronic lymphocytic leukemia. Br. J. Haematol. 125, 128–140 [DOI] [PubMed] [Google Scholar]
- 10. Redondo-Muñoz J., Escobar-Díaz E., Samaniego R., Terol M. J., García-Marco J. A., García-Pardo A. (2006) MMP-9 in B-cell chronic lymphocytic leukemia is up-regulated by α4β1 integrin or CXCR4 engagement via distinct signaling pathways, localizes to podosomes, and is involved in cell invasion and migration. Blood 108, 3143–3151 [DOI] [PubMed] [Google Scholar]
- 11. Redondo-Muñoz J., José Terol M., García-Marco J. A., García-Pardo A. (2008) Matrix metalloproteinase-9 is up-regulated by CCL21/CCR7 interaction via extracellular signal-regulated kinase-1/2 signaling and is involved in CCL21-driven B-cell chronic lymphocytic leukemia cell invasion and migration. Blood 111, 383–386 [DOI] [PubMed] [Google Scholar]
- 12. Redondo-Muñoz J., Ugarte-Berzal E., García-Marco J. A., del Cerro M. H., Van den Steen P. E., Opdenakker G., Terol M. J., García-Pardo A. (2008) α4β1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood 112, 169–178 [DOI] [PubMed] [Google Scholar]
- 13. Redondo-Muñoz J., Ugarte-Berzal E., Terol M. J., Van den Steen P. E., Hernández del Cerro M., Roderfeld M., Roeb E., Opdenakker G., García-Marco J. A., García-Pardo A. (2010) Matrix metalloproteinase-9 promotes chronic lymphocytic leukemia B cell survival through its hemopexin domain. Cancer Cell 17, 160–172 [DOI] [PubMed] [Google Scholar]
- 14. Piccard H., Van den Steen P. E., Opdenakker G. (2007) Hemopexin domains as multifunctional liganding modules in matrix metalloproteinases and other proteins. J. Leukoc. Biol. 81, 870–892 [DOI] [PubMed] [Google Scholar]
- 15. Björklund M., Heikkilä P., Koivunen E. (2004) Peptide inhibition of catalytic and noncatalytic activities of matrix metalloproteinase-9 blocks tumor cell migration and invasion. J. Biol. Chem. 279, 29589–29597 [DOI] [PubMed] [Google Scholar]
- 16. Dufour A., Zucker S., Sampson N. S., Kuscu C., Cao J. (2010) Role of matrix metalloproteinase-9 dimers in cell migration. Design of inhibitory peptides. J. Biol. Chem. 285, 35944–35956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dufour A., Sampson N. S., Li J., Kuscu C., Rizzo R. C., Deleon J. L., Zhi J., Jaber N., Liu E., Zucker S., Cao J. (2011) Small-molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase-9. Cancer Res. 71, 4977–4988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roeb E., Schleinkofer K., Kernebeck T., Pötsch S., Jansen B., Behrmann I., Matern S., Grötzinger J. (2002) The matrix metalloproteinase 9 (MMP-9) hemopexin domain is a novel gelatin binding domain and acts as an antagonist. J. Biol. Chem. 277, 50326–50332 [DOI] [PubMed] [Google Scholar]
- 19. Burg-Roderfeld M., Roderfeld M., Wagner S., Henkel C., Grötzinger J., Roeb E. (2007) MMP-9-hemopexin domain hampers adhesion and migration of colorectal cancer cells. Int. J. Oncol. 30, 985–992 [DOI] [PubMed] [Google Scholar]
- 20. Ezhilarasan R., Jadhav U., Mohanam I., Rao J. S., Gujrati M., Mohanam S. (2009) The hemopexin domain of MMP-9 inhibits angiogenesis and retards the growth of intracranial glioblastoma xenograft in nude mice. Int. J. Cancer 124, 306–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Binet J. L., Auquier A., Dighiero G., Chastang C., Piguet H., Goasguen J., Vaugier G., Potron G., Colona P., Oberling F., Thomas M., Tchernia G., Jacquillat C., Boivin P., Lesty C., Duault M. T., Monconduit M., Belabbes S., Gremy F. (1981) A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate analysis. Cancer 48, 198–206 [DOI] [PubMed] [Google Scholar]
- 22. Rai K. R., Sawitsky A., Cronkite E. P., Chanana A. D., Levy R. N., Pasternack B. S. (1975) Clinical staging of chronic lymphocytic leukemia. Blood 46, 219–234 [PubMed] [Google Scholar]
- 23. Stacchini A., Aragno M., Vallario A., Alfarano A., Circosta P., Gottardi D., Faldella A., Rege-Cambrin G., Thunberg U., Nilsson K., Caligaris-Cappio F. (1999) MEC1 and MEC2. Two new cell lines derived from B-chronic lymphocytic leukemia in prolymphocytoid transformation. Leuk. Res. 23, 127–136 [DOI] [PubMed] [Google Scholar]
- 24. Morodomi T., Ogata Y., Sasaguri Y., Morimatsu M., Nagase H. (1992) Purification and characterization of matrix metalloproteinase-9 from U937 monocytic leukemia and HT1080 fibrosarcoma cells. Biochem. J. 285, 603–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mira E., Lacalle R. A., Buesa J. M., de Buitrago G. G., Jiménez-Baranda S., Gómez-Moutón C., Martínez-A C., Mañes S. (2004) Secreted MMP9 promotes angiogenesis more efficiently than constitutively active MMP9 bound to the tumor cell surface. J. Cell Sci. 117, 1847–1857 [DOI] [PubMed] [Google Scholar]
- 26. Ghatak S., Misra S., Toole B. P. (2005) Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J. Biol. Chem. 280, 8875–8883 [DOI] [PubMed] [Google Scholar]
- 27. Hulme E. C. (ed) (1992) Receptor-Ligand Interactions: A Practical Approach. pp. 63–176, Oxford University Press, Oxford [Google Scholar]
- 28. Pulido R., Elices M. J., Campanero M. R., Osborn L., Schiffer S., García-Pardo A., Lobb R., Hemler M. E., Sánchez-Madrid F. (1991) Functional evidence for three distinct and independently inhibitable adhesion activities mediated by the human integrin VLA-4. Correlation with distinct α4 epitopes. J. Biol. Chem. 266, 10241–10245 [PubMed] [Google Scholar]
- 29. Humphries M. J., Komoriya A., Akiyama S. K., Olden K., Yamada K. M. (1987) Identification of two distinct regions of the type III connecting segment of human plasma fibronectin that promote cell type-specific adhesion. J. Biol. Chem. 262, 6886–6892 [PubMed] [Google Scholar]
- 30. Garcia-Pardo A., Wayner E. A., Carter W. G., Ferreira O. C., Jr. (1990) Human B lymphocytes define an alternative mechanism of adhesion to fibronectin. The interaction of the α4β1 integrin with the LHGPEILDVPST sequence of the type III-connecting segment is sufficient to promote cell attachment. J. Immunol. 144, 3361–3366 [PubMed] [Google Scholar]
- 31. Cha H., Kopetzki E., Huber R., Lanzendörfer M., Brandstetter H. (2002) Structural basis of the adaptive molecular recognition by MMP9. J. Mol. Biol. 320, 1065–1079 [DOI] [PubMed] [Google Scholar]
- 32. Arnaout M. A., Goodman S. L., Xiong J. P. (2002) Coming to grips with integrin binding to ligands. Curr. Opin. Cell Biol. 14, 641–651 [DOI] [PubMed] [Google Scholar]
- 33. Bauvois B. (2012) New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers. Outside-in signaling and relationship to tumor progression. Biochim. Biophys. Acta. 1825, 29–36 [DOI] [PubMed] [Google Scholar]
- 34. Murphy G., Nagase H. (2011) Localizing matrix metalloproteinase activities in the pericellular environment. FEBS J. 278, 2–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brooks P. C., Silletti S., von Schalscha T. L., Friedlander M., Cheresh D. A. (1998) Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell 92, 391–400 [DOI] [PubMed] [Google Scholar]
- 36. Ramani V. C., Kaushal G. P., Haun R. S. (2011) Proteolytic action of kallikrein-related peptidase 7 produces unique active matrix metalloproteinase-9 lacking the C-terminal hemopexin domains. Biochem. Biophys. Acta 1813, 1525–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stefanidakis M., Bjorklund M., Ihanus E., Gahmberg C. G., Koivunen E. (2003) Identification of a negatively charged peptide motif within the catalytic domain of progelatinases that mediates binding to leukocyte b2 integrins. J. Biol. Chem. 278, 34674–34684 [DOI] [PubMed] [Google Scholar]
- 38. Stefanidakis M., Karjalainen K., Jaalouk D. E., Gahmberg C. G., O'Brien S., Pasqualini R., Arap W., Koivunen E. (2009) Role of leukemia invadosome in extramedullary infiltration. Blood 114, 3008–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wayner E. A., Kovach N. L. (1992) Activation-dependent recognition by hematopoietic cells of the LDV sequence in the V region of fibronectin. J. Cell Biol. 116, 489–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moyano J. V., Carnemolla B., Domínguez-Jiménez C., García-Gila M., Albar J. P., Sánchez-Aparicio P., Leprini A., Querzé G., Zardi L., Garcia-Pardo A. (1997) Fibronectin type III5 repeat contains a novel cell adhesion sequence, KLDAPT, which binds activated α4β1 and α4β7 integrins. J. Biol. Chem. 272, 24832–24836 [DOI] [PubMed] [Google Scholar]
- 41. Clements J. M., Newham P., Shepherd M., Gilbert R., Dudgeon T. J., Needham L. A., Edwards R. M., Berry L., Brass A., Humphries M. J. (1994) Identification of a key integrin-binding sequence in VCAM-1 homologous to the LDV active site in fibronectin. J. Cell Science 107, 2127–2135 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.