

Background: GlfT2 is a bifunctional galactofuranosyltransferase essential for mycobacterial cell wall biosynthesis.
Results: Crystal structures of free and UDP-bound GlfT2, along with mutagenesis and kinetic studies, reveal novel details underlying substrate binding and catalysis.
Conclusion: The homotetrameric architecture, distinctive nucleotide-binding site, and unprecedented structural features underlying the bifunctional polymerase activity of GlfT2 are revealed.
Significance: Novel insights into polymerizing glycosyltransferases and the design of anti-mycobacterial therapeutics are obtained.
Keywords: Cell Wall, Enzyme Structure, Glycosyltransferases, Membrane Proteins, Mycobacterium tuberculosis, X-ray Crystallography, Carbohydrate polymerase, Glycan Biosynthesis
Abstract
Biosynthesis of the mycobacterial cell wall relies on the activities of many enzymes, including several glycosyltransferases (GTs). The polymerizing galactofuranosyltransferase GlfT2 (Rv3808c) synthesizes the bulk of the galactan portion of the mycolyl-arabinogalactan complex, which is the largest component of the mycobacterial cell wall. We used x-ray crystallography to determine the 2.45-Å resolution crystal structure of GlfT2, revealing an unprecedented multidomain structure in which an N-terminal β-barrel domain and two primarily α-helical C-terminal domains flank a central GT-A domain. The kidney-shaped protomers assemble into a C4-symmetric homotetramer with an open central core and a surface containing exposed hydrophobic and positively charged residues likely involved with membrane binding. The structure of a 3.1-Å resolution complex of GlfT2 with UDP reveals a distinctive mode of nucleotide recognition. In addition, models for the binding of UDP-galactofuranose and acceptor substrates in combination with site-directed mutagenesis and kinetic studies suggest a mechanism that explains the unique ability of GlfT2 to generate alternating β-(1→5) and β-(1→6) glycosidic linkages using a single active site. The topology imposed by docking a tetrameric assembly onto a membrane bilayer also provides novel insights into aspects of processivity and chain length regulation in this and possibly other polymerizing GTs.
Introduction
Tuberculosis (TB),3 a disease that remains a significant worldwide health threat, is caused by infection with Mycobacterium tuberculosis. In common with all mycobacteria, M. tuberculosis possesses a unique glycolipid-rich cell wall structure that provides substantial protection from the environment, including preventing the passage of antibiotics into the organism (1). The standard TB drug regimen (2) therefore involves multiple antibiotics, including some that target cell wall biosynthesis and others that have intracellular targets (3). Concerns about drug resistance (4) have led to heightened interest in identifying novel therapeutic agents and developing a more detailed understanding of mycobacterial biochemical pathways to facilitate this task. In this regard, cell wall assembly has received particular attention (1).
The largest component of the mycobacterial cell wall is the mycolyl-arabinogalactan complex. This glycolipid has at its core a galactan domain composed of 30–35 galactofuranose (Galf) residues attached in alternating β-(1→5) and β-(1→6) linkages, which, in turn, is covalently bound to cell wall peptidoglycan through a linker consisting of rhamnose and N-acetylglucosamine phosphate (1). The galactan serves as the attachment site for three arabinan domains, each containing ∼30 α-(1→5)-, α-(1→3)-, and β-(1→2)-linked arabinofuranose residues. Esterified to the nonreducing termini of the arabinan are mycolic acids, branched long-chain (C60–C90) lipids that are characteristic to mycobacteria and related organisms such as nocardia and corynebacteria. Two clinically used anti-TB drugs target enzymes involved in the assembly of the mycolyl-arabinogalactan complex (1). Ethambutol inhibits at least one of the arabinosyltransferases responsible for arabinan formation (5), whereas isoniazid is a prodrug that targets an enoyl-acyl carrier protein reductase required for mycolic acid biosynthesis (6). Although none of the drugs in use or in development for the treatment of TB are known to target enzymes responsible for galactan biosynthesis, these enzymes are likely suitable targets, as the galactan forms the foundation upon which both the arabinan and mycolate moieties are incorporated. This proposal is further supported by gene knock-out studies demonstrating that galactan formation is essential for mycobacterial viability (7). Because a clearer, more detailed picture of galactan biosynthesis is a necessary prerequisite for identifying novel therapeutics, this area is now receiving increasing attention (8).
Previous work has revealed that mycobacterial galactan biosynthesis requires two bifunctional GTs, GlfT1 and GlfT2, which use UDP-α-d-galactofuranose (UDP-Galf) as the donor and a growing polyprenol-bound oligosaccharide as the acceptor (9, 10). The α-stereochemistry in UDP-Galf and the β-stereochemistry of the newly synthesized glycosidic linkages in the products generated by GlfT1 and GlfT2 indicate that catalysis follows an inverting mechanism (9, 11). GlfT1 adds the first two Galf moieties, whereas GlfT2, a polymerizing glycosyltransferase that produces both β-(1→5) and β-(1→6) linkages between Galf residues (10–12), introduces the remaining ∼30 monosaccharides (Fig. 1). GlfT2 has been more extensively studied than GlfT1. Of particular note are saturation-transfer difference-NMR studies suggesting that GlfT2 synthesizes both the β-(1→5) and β-(1→6) linkages using a single active site (13), and mass spectrometry studies indicate that the enzyme is processive (14). A very recent study examining GlfT2 mutants provides additional support for the presence of a single active site that carries out both glycosylations (15).
FIGURE 1.

A, alternating glycosylation reactions catalyzed by GlfT2. The β-(1→5)-transferase activity of the enzyme first glycosylates the galactofuranosyl residue at the nonreducing terminus of the polyprenol-bound tetrasaccharide acceptor. The resulting product is then a substrate for the β-(1→6)-transferase activity of GlfT2. Repetition of these two alternating glycosylations generates the elongated product. B, chemical structures of synthetic acceptor 1 and 2 used in enzyme kinetics and modeling studies.
In addition to the importance of understanding how GlfT2 works as a key enzyme in mycobacterial cell wall synthesis, GlfT2 is a member of the Carbohydrate-Active enZyme (CAZy) GT-2 family (16, 17), which also contains other polymerizing GTs, including cellulose synthase (18) and chondroitin polymerase (19). GlfT2 has recently become of interest as a model for studying processivity and chain length control in a wide range of polymerizing GTs (14, 20).
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
For crystallographic studies, GlfT2 was produced from Escherichia coli C41(DE3) transformed with pET-15b/Rv3808c (9). Cells stored in glycerol stocks were grown overnight on MDG-agar (21). Single colonies were added to MDG (100 ml) and grown for 36 h at 25 °C. These starter cultures were added to Overnight Express Instant TB medium (Novagen, 1 liter) and grown for 24 h at 25 °C. Cells were harvested by centrifugation (5000 × g, 20 min) and stored at −70 °C. All purification steps were performed at 4 °C. Thawed cells (∼16 g) were suspended in 50 ml of disruption buffer (50 mm NaH2PO4, pH 7.6, 400 mm NaCl, 15% (v/v) glycerol, 20 mm imidazole, 5 mm β-ME, and 0.2% (v/v) Triton X-100), disrupted by two passes through a Model 110 Microfluidizer (Microfluidics), and clarified by centrifugation (60 min, 100,000 × g). The supernatant was loaded onto nickel-Sepharose HP (7 ml, GE Healthcare) equilibrated with buffer A (50 mm NaH2PO4, pH 7.6, 400 mm NaCl, 15% (v/v) glycerol, 5 mm β-ME) plus 20 mm imidazole. The column was washed with 30 ml of buffer A plus 50 mm imidazole and eluted with a 90-ml gradient of buffer A plus 50–400 mm imidazole. Fractions containing GlfT2 were identified by SDS-PAGE and dialyzed overnight against 2 liters of buffer B (50 mm MOPS, pH 7.6, 15% (v/v) glycerol, 5 mm β-ME). The dialysate was centrifuged (12,000 × g, 15 min), and the supernatant was concentrated to ∼3.5 mg/ml (Bradford assay, Bio-Rad) using Ultrafree-15 centrifugal filters (10,000 molecular weight cutoff, Millipore). Aliquots were flash-frozen in liquid nitrogen and stored at −80 °C.
Gel filtration chromatography was performed using a Sephacryl S-200 HR 16/60 column (GE Healthcare) equilibrated with buffer C (50 mm MOPS, pH 7.6, 100 mm NaCl, 10 mm MgCl2, 5 mm β-ME). GlfT2 (1 ml, 8 mg/ml) or a mixture of standard proteins, including blue dextran 2000, was eluted at 0.5 ml/min and monitored by absorbance at 280 nm.
Crystallization
Crystals were grown by hanging-drop vapor diffusion at room temperature by mixing GlfT2 (2 μl, 2.2 mg/ml, buffer B plus 0.2% (v/v) Triton X-100) with precipitant A (2 μl, 5–7% (w/v) PEG 4000, 100 mm sodium cacodylate, pH 6.5, 100 mm sodium acetate). Crystals (∼350 × 70 × 70 μm) grew to full size in 7–14 days. Crystals were cryoprotected by transferring to precipitant A plus 1% (w/v) PEG 4000 and successively 5, 10, 15, and 20% (v/v) glycerol. Crystals were equilibrated at least 15 min between transfers and flash-cooled under nitrogen gas at 100 K.
A lutetium derivative was grown by mixing GlfT2 (1.8 μl, 2.2 mg/ml, buffer C plus 1.5 mm LuCl3, 0.2% (v/v) Triton X-100, 5 mm TCEP, and no β-ME) with precipitant B (1.8 μl, 3–5% (w/v) PEG 4000, 100 mm sodium acetate, 5 mm LuCl3, and 5 mm TCEP). Crystals (∼200 × 100 × 25 μm) grew to full size in 7–10 days and were adapted to precipitant B plus 1% (w/v) PEG 4000 and 20% (v/v) glycerol, as described for native crystals.
A complex of GlfT2 and UDP was prepared by mixing GlfT2 (2 μl, 2.2 mg/ml, buffer B plus 5 mm TCEP, 0.2% (v/v) Triton X-100) with precipitant C (2 μl, 6–7% (w/v) PEG 4000, 100 mm sodium acetate, 10% (v/v) glycerol, 5 mm TCEP). Crystals (∼500 × 50 × 50 μm) grew to full size in 3–7 days. Crystals were adapted to precipitant C plus 10 mm MgCl2, 1% (w/v) PEG 4000, and successively 12, 16, and 20% (v/v) glycerol. Crystals were allowed to equilibrate at least 15 min between transfers. Immediately before flash cooling, crystals were soaked for 10 min in the final cryoprotectant solution plus 5 mm UDP.
Structure Determination and Refinement
Diffraction intensities were measured at 100 K at Stanford Synchrotron Research Laboratory (SSRL), beamline 9-2, using a MAR 325 CCD detector. Data were processed and scaled using HKL-2000 (22). All crystals belonged to space group P4212 with two protein molecules per asymmetric unit. Data collection and refinement statistics are presented in Table 1. The data were somewhat anisotropic, which is reflected by the higher value of Rsym in the high resolution shell, and the high resolution cutoff was chosen as a compromise that indicates the average diffraction limit in all directions without sacrificing significant data in the direction of stronger high resolution diffraction. Three lutetium sites were located, and multiple wavelength anomalous dispersion phases were determined to 3.1 Å using SOLVE (23). The initial figure of merit was only 0.37, but nearly the entire model could be built using PHENIX (24) after density modification, including 2-fold noncrystallographic averaging and phase extension to 2.45 Å using amplitudes from a higher resolution native data set. The final model of the native structure was refined by iterative cycles of model building with COOT (25) and reciprocal space refinement with REFMAC (26). Noncrystallographic restraints were not applied during refinement. Structural comparisons were performed using DALI (27) and structural alignments with LSQMAN (28). PyMOL (Schrödinger, LLC) was used for graphical structural analysis and to generate figures.
TABLE 1.
Crystallographic statistics
| Data collection | Lu3+ | Apoenzyme | UDP complex | ||
|---|---|---|---|---|---|
| Unit cell dimensions | |||||
| a = b, c | 150.2, 145.2 Å | 150.8, 148.0 Å | 150.4, 147.4 Å | ||
| Wavelength | 1.34046 Å | 1.34085 Å | 0.91837 Å | 1.0442 Å | 0.97911 Å |
| Resolutiona | 40 to 3.0 Å (3.11 to 3.0 Å) | 40.0 to 2.45 Å (2.54 to 2.45 Å) | 40.0 to 3.1 Å (3.21 to 3.1 Å) | ||
| Rsymb | 0.147 (0.837) | 0.144 (0.897) | 0.153 (0.888) | 0.104 (0.942) | 0.125 (1.00) |
| I/σI | 18.2 (1.2) | 16.7 (1.0) | 13.6 (1.8) | 27.2 (2.0) | 22.0 (1.9) |
| Completeness | 98.0% (82.3%) | 96.9% (76.1%) | 99.9% (99.7%) | 99.9% (99.4%) | 99.8% (99.2%) |
| Redundancy | 6.7 (3.1) | 6.7 (2.8) | 6.2 (4.9) | 11.9 (9.4) | 8.8 (7.0) |
| Refinement | |||||
| Resolution | 40.0 to 2.45 Å | 40.0 to 3.1 Å | |||
| Unique reflections | 59,863 | 30,138 | |||
| Rworkc/Rfreed | 0.186/0.222 | 0.204/0.279 | |||
| No. of atoms | |||||
| Total | 10,410 | 10,032 | |||
| Protein | 9966 | 9956 | |||
| Glycerol | 30 | 6 | |||
| Mn2+ | 4 | 2 | |||
| UDP | 0 | 50 | |||
| Water atoms | 414 | 14 | |||
| Average temperature factors | |||||
| Protein | 41.3 | 49.9 | |||
| Glycerol | 72.6 | 66.1 | |||
| Mn2+ | 74.1 | 108.4 | |||
| UDP | 113.9 | ||||
| Water | 38.7 | 31.5 | |||
| Root mean square deviation from ideal geometry | |||||
| Bond lengths | 0.008 | 0.008 | |||
| Bond angles | 1.06° | 1.08° | |||
a Values from the outermost resolution shell are given in parentheses.
b Rsym = Σi|Ii − 〈I〉|/ΣiIi, where Ii is the ith integrated intensity of a given reflection, and 〈I〉 is the weighted mean of all measurements of I.
c Rwork = Σ‖Fo| − |Fc‖/Σ|Fo| for 95% of reflection data used in refinement.
d Rfree = Σ‖Fo| − |Fc‖/Σ|Fo| for 5% of reflection data excluded from refinement.
Mutagenesis and Kinetics
GlfT2 mutants were expressed from a chemically synthesized gene with codon usage optimized for E. coli (Genscript). Mutagenesis was performed using the QuikChange II XL kit (Stratagene) with the oligonucleotide primer sequences given in supplemental Table 1. GlfT2 mutants were expressed and purified as described previously (29). Enzyme activity was measured using a coupled spectrophotometric assay (29) and a radiochemical assay (9) as described previously.
The spectrophotometric assay was performed in a 384-well microtiter plate with an incubation mixture containing 50 mm MOPS, pH 7.6, 50 mm KCl, 20 mm MgCl2, 1.1 mm NADH, 3.5 mm phosphoenolpyruvate, 7.5 units of pyruvate kinase (PK, EC 2.7.1.40), 16.8 units of lactate dehydrogenase (EC 1.1.1.27), acceptor 1 (Fig. 1B), and UDP-Galf. Each reaction was initiated by the addition of 15 μg of GlfT2 protein or the related mutant to the assay mixture. Assays were performed at 37 °C in a total volume of 20 μl and continuously monitored at 37 °C using a Spectra Max 340PC microplate reader in the kinetics read mode. Each reaction was monitored at 12–15-s intervals for up to 15 min at 340 nm. The rate of NADH oxidation was converted to picomoles by using a path length of 0.37 cm and an extinction coefficient of 6300 (m·cm)−1.
The radiochemical assay mixtures consisted of 50 mm MOPS, pH 7.6, 5 mm β-mercaptoethanol, 10 mm MgCl2, 62.5 μm ATP, 10 mm NADH, 5 μm UDP-Galp, 4 μl of purified UDP-Galp mutase (4 mg/ml), 6 μl of purified E371S or E372S mutant (0.5 mg/ml), and uridine-diphosphate-galactose[6-3H] (American Radiolabeled Chemicals, Inc., 20 Ci/mmol, 0.1 μl) in a total reaction volume of 20 μl. The reaction mixtures were incubated at 37 °C for 1 and 16 h, respectively. Radiolabeled products were isolated by reverse-phase chromatography on SepPak C18 cartridges (Waters) and eluted with methanol (3.5 ml). The reaction products in the eluants were quantified by liquid scintillation counting on a Beckman LS6500 Scintillation Counter using 10 ml of Ecolite mixture.
Molecular Modeling of Donor and Acceptor Substrate Conformations
The starting point for the modeling of donor and acceptor molecules was to generate conformations consistent with the preferences expected in solution. Low energy conformations of methyl β-d-galactofuranoside from DFT calculations (B3LYP/6–31+G** (30–33)) in the gas phase4 indicated a preference for the 4E ring pucker. As a result, two conformers of 1 and 2 were generated by restraining the galactofuranose rings to 4E in minimizations with the MM2 forcefield (34).
Comparisons between the structure of GlfT2 and related GT enzymes containing bound acceptor molecules suggested a way to place 1 and 2 into the putative acceptor-binding site of GlfT2. LSQMAN (28) was used to superimpose the GT domains of related enzymes and to superimpose parts of 1 and 2 onto acceptor molecules bound to these related enzymes. The torsion angles for the glycosidic linkages of 1 and 2 were adjusted to avoid steric clashes within the glycan and between the glycan and the protein. The hydroxyl group expected to react with the anomeric carbon of UDP-Galf was positioned near the predicted general base catalyst (Asp-372) and the anomeric carbon of UDP-Galf.
The conformation adopted by UDP-Galf was modeled by adding a galactofuranose ring onto the end of the UDP molecule seen in the product complex crystal structure. The conformation of the pyrophosphate moiety and general position of the Galf ring were modeled to be consistent with crystal structures determined at high resolution, as well as NMR and molecular mechanics calculations (35). The conformation adopted by the galactofuranose ring was modeled in the 2T1 conformation at the energy minimum from molecular mechanics calculations and near the broad minimum consistent with NMR and molecular dynamics simulations of α-d-galactofuranosyl phosphate and methyl α-d-galactofuranoside.5 The torsion angle of the glycosidic bond was manually adjusted to minimize steric clashes with the protein, and the position and torsion angles of the exocyclic moiety at C-4 were rotated to minimize steric clashes and to form favorable hydrogen bonds with the protein.
RESULTS
Structure of the GlfT2 Protomer
Crystal structures were determined for the unliganded form of GlfT2 as well as for the enzyme in complex with the product UDP. Crystals of GlfT2 contain two protein chains per asymmetric unit. Apart from the N-terminal histidine tag and the final eight (native) or nine (UDP complex) residues, the entire polypeptide backbone of both chains was clearly defined by electron density. The structures of the two chains in both the unliganded and product-bound enzyme are nearly identical (root mean square deviation of 0.18 and 0.29 Å, respectively), even though noncrystallographic symmetry restraints were not imposed during refinement. Each protein chain forms a kidney-shaped structure with four domains (Fig. 2A). A central GT family A (GT-A) (36) domain is preceded by an N-terminal β-sandwich domain and followed by an α-helical domain and a C-terminal mixed α + β domain (Fig. 2A and supplemental Fig. 1).
FIGURE 2.

GlfT2 bound to UDP. A ribbon diagram of a single protomer, with domains 1–4 drawn in different colors, with Mn2+ drawn as a gray sphere and UDP drawn in stick representation.
The N-terminal domain contains two short helices preceding a 10-stranded β-sandwich with jelly roll topology. Structural alignments using DALI (37) indicate similarities with carbohydrate-binding modules (CBM) from a variety of glycosidases, with the CBM from ManA (38) scoring the highest at 9.9, despite sharing only ∼11% sequence identity. Although the arrangement of a CBM domain N-terminal to a GT-A domain has not yet been described, enzymes from the GT family 27 (GT-27) have trefoil-fold CBMs appended C-terminal to GT-A domains (39). By binding to peptide-linked glycans, the CBMs in some GT-27 enzymes direct the GT domain to specific sites of glycosylation (40). In contrast, the N-terminal β-sandwich domain in GlfT2 has not yet been shown to act as a CBM. Indeed, the most commonly conserved binding site for carbohydrates in CBMs, formed by one of the faces of the β-sandwich (41), is part of the interface between protomers in the GlfT2 tetramer. A second, less commonly observed binding site in CBMs is formed by long loops at one of the edges of the β-sandwich (42). In GlfT2, the analogous loops are short, lack homology with known carbohydrate-binding sites, and form the N-terminal face of the protein, which is distant from the active site.
The GT-A domain in GlfT2 is most similar to enzymes in CAZy families 2, 27, and 78 and is less similar to those from families 13 and 64. Although sequence identity is less than 15% to other GTs with known three-dimensional structures, the GT-A fold is conserved. Common features of the GT-A domain that are found in GlfT2 include the 3214657 topology of the parallel β-sheet core and a “DXD” (here DDD) motif following β4 (43, 44). GT-A domain proteins commonly contain a less highly conserved “variable region” following β5. In GlfT2, this region consists of a β-hairpin followed by a long loop and a short α-helix. Although this region differs from other GT-2 proteins, a similar β-hairpin structure is also found in GT-27 (39) and GT-64 proteins. The β-hairpin extends away from the GT-A core to contact a helix-loop-helix motif at the beginning of domain 3. In the GT-27 UDP-N-acetyl-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase, this structural motif forms one of the walls of the substrate-binding channel for the extended peptide acceptor, and it may play a similar function for an extended carbohydrate acceptor in GlfT2, as described below. The loop and helix following the β-hairpin also forms a key interface with domain 1. As a result, this entire variable region forms key interactions between domain 2 and both domains 1 and 3.
Immediately following the GT-A domain is a long loop (loop 1, residues 397–407), which, in various GT-A enzymes, can adopt alternate conformations to cover and uncover the active site (45). Although loop 1 is often poorly ordered in GT-A structures, the complete chain can be traced in GlfT2, and it adopts a similar conformation in both the presence and absence of UDP. Loop 1 is proposed to have an important role in the binding of acceptor substrates (below).
The third domain of GlfT2 consists of eight α-helices, five of which immediately follow the GT domain and form the central portion of domain 3. Three helices at the C terminus of the polypeptide chain wrap around these core helices to complete the domain. The first helix in domain 3 anchors loop 1 and is conserved in many GT-A proteins (43). However, for the rest of the domain, structural comparisons using DALI indicate only minor similarities (Z <5.3) to proteins unrelated in overall structure and function. The fourth and last domain consists of a three-stranded β-sheet surrounded by three α-helices. The DALI algorithm fails to find any structures with significant similarity to this fold.
The accessory domains flanking the central GT domain in GlfT2 appear to be conserved in a small family of closely related proteins, but they differ significantly from all other GT-A proteins of known structure. Sequence comparisons indicate that the flanking domains and many hydrophobic residues found at the interface between domains 2 and 3 are conserved in >100 proteins from mycobacteria, related Actinobacteria, and even more distantly related species, thus suggesting conserved structure and function relationships (supplemental Figs. 2 and 3). Outside this family, however, there appear to be less closely related enzymes, such as the nonpolymerizing mycobacterial GT GlfT1, which contain a homologous GT-A domain but no flanking regions (10).
Structure of the GlfT2 Homotetramer
Each GlfT2 protomer is 90 × 50 × 50 Å in size, and each of the two protomers in the asymmetric unit forms a separate C4-symmetric homotetramer in the crystal (Fig. 3). This 295-kDa oligomer is consistent with the elution profile of the protein in gel filtration chromatography. Approximately 10.4% of the accessible surface area (2740 Å2 out of 26,400 Å2) for each protomer is buried in the tetramer. The amount and percentage of total buried surface area are both lower than the average (3900 Å2) seen in homodimeric proteins (46) and the average percentage of buried surface area seen in multimeric proteins (18%) (47). However, each of the two independent protomers in the asymmetric unit forms a separate but essentially identical tetramer, thus arguing that the tetramer is likely the form seen in solution and not a crystallographic artifact.
FIGURE 3.

A, N-face view of the GlfT2 tetramer, with each protomer colored as in Fig. 1, and a single protomer drawn with semi-transparent surface representation superimposed. B, C-face view of the tetramer. C, view rotated 90° from the views in A and B, showing a side view of the tetramer. D and E, stereoscopic views of the C-face view of the unliganded GlfT2 tetramer, showing the extent of the hollow central core of the enzyme. D, hydrophobic surface potential representation (red, hydrophobic; white, polar) (53). E, electrostatic surface potential representation (red, negatively charged; white, neutral; blue, positively charged).
The dimensions of the tetramer are 100 × 100 × 75 Å. A hollow funnel-shaped pore with a diameter of less than 10 Å at the face formed primarily by the N-terminal portion of each protomer (N-face, Fig. 3A) expands to over 40 Å at the opposing face (C-face, Fig. 3B). The C-face contains large hydrophobic patches (Fig. 3D), and the charged residues exposed are predominantly positive (Fig. 3E). Notably, the hydrophobic and positively charged residues on the C-face of GlfT2 are quite highly conserved in GlfT2 homologues predicted to have flanking C-terminal domains. For example, Trp-555 is perfectly conserved in the 110 most closely related sequences in GenBankTM, whereas Arg-554 and Lys-445 are positively charged in nearly all of the ∼80 most closely related sequences. Many surface-exposed leucine, alanine, isoleucine, and tryptophan residues are also generally conserved in the ∼80 most closely related sequences. These features suggest that the C-face associates with the hydrophobic acyl chains and negatively charged head groups of membrane phospholipids. This inference is consistent with the observation that GlfT2 is localized in the membrane fractions of mycobacteria and heterologous hosts (11, 12). Assuming this orientation of the tetramer relative to the cell membrane, the N-face contains a small pore at its center, as well as grooves between adjacent subunits (Fig. 3C). Each of these grooves provides an opening toward the active site, which allows for the entry of UDP-Galf and the release of UDP after glycosyl transfer.
Substrate Binding and Catalysis
Similar to other GT-A enzymes, the active site of GlfT2 contains a divalent metal ion (Fig. 4A). Anomalous difference Fourier electron density maps indicate that Mn2+ is bound to the active site, even though Mg2+ and not Mn2+ was present in purification and crystallization solutions. The Mn2+-binding site is formed by Asp-256 and Asp-258 from the “DXD” motif found in the loop following β4, as well as His-396 at the C-terminal end of the GT domain. This arrangement of coordinating residues resembles that seen in many GT-A enzymes, especially those from families 2, 27, and 78, although the details of metal ion coordination vary substantially among these enzymes.
FIGURE 4.

Binding site for UDP-Galf donor. A, structure of UDP bound to GlfT2 and showing interactions with highly conserved residues and Mn2+ (gray sphere), as well as the coordinating side chains. The difference electron density map (contoured at 3σ) was calculated with phases after the model of UDP was removed, and the structure of GlfT2 was subjected to 20 cycles of positional and temperature factor refinement. B, model of the structure of UDP-Galf (magenta) and a Galf acceptor residue (gray) showing possible hydrogen bonding interactions (dashed red lines) with highly conserved residues. In this model, the 6-OH of the acceptor sugar is poised for interaction with the side chain of Asp-372 (general base catalysis) and the anomeric carbon of the donor sugar-nucleotide.
The structure of the GlfT2-UDP complex also reveals a mode of UDP recognition with similarities to other GT-A enzymes, as well as some unique features (Fig. 4A). The side chain –NH2 and =O groups of Asn-229 donate and accept hydrogen bonds from the O4 and N3 atoms of the uracil base, and the side chain –NH2 group of Gln-200 donates a hydrogen bond to the O2 atom. This hydrogen-bonding arrangement resembles the interaction between the side chain of Asn-130 and the uracil base in the human GT-64 enzyme EXTL2 (48), and these two amino acids occupy a similar position in the loop preceding α3 in the GT-A fold. However, this hydrogen-bonding arrangement differs from the one most commonly seen in enzymes from the GT-2, -13, and -27 families, where Asp takes the place of Gln-200 and accepts a hydrogen bond from the uracil ring nitrogen. The aromatic rings of Phe-173 and Phe-367 flank the two faces of the uracil ring, forming face-to-face and edge-to-face stacking interactions, respectively. The former aromatic residue is highly conserved in other GTs, whereas the latter is more variable. In addition to these contacts, the uracil base as well as the ribose C1 and O4 atoms also contact the main chain of Gly-231 and Gly-232. The ribose moiety only forms a single hydrogen bond with the protein; the ring oxygen accepts an H-bond from the backbone –NH group of Gly-232. These Gly residues lie at the N-terminal end of α3 in the GT domain and are part of a highly conserved motif found only in the family of actinobacterial GTs to which GlfT2 and GlfT1 belong. The lack of side chains at these key positions provides a larger space for accommodating the ribose moiety than seen in most other GTs of known three-dimensional structure. In most other GTs, such as SpsA and EXTL2, for example, the other side of the ribose interacts with Asp residues equivalent to Asp-257, the middle X residue in the DXD motif. Although Asp-257 in GlfT2 is positioned similarly to the corresponding residue in these other GTs, the ribose moiety is too distant to interact (over 5.5 Å). Saturation-transfer difference-NMR studies confirm the distinctive binding mode seen in the GlfT2-UDP complex, as the UDP protons making the most intimate contacts with the protein are attached to the C1 atom of the ribose and the uracil base (49). Each phosphate group in the diphosphate moiety coordinates to Mn2+ through a single oxygen atom, as seen for UDP in solution and when bound to enzymes (35).
The conformation of the diphosphate group allows for the construction of a model for UDP-Galf by placing a furanose ring adopting a low energy 2T1 conformer in a “folded-back” conformation relative to the diphosphate group (Fig. 4B). This conformation allows hydroxyl groups on the Galf residue to form hydrogen bonds with the side chains of His-396, Asp-371, and Asp-372. In this model, the plane of the Galf ring is roughly perpendicular to the axis formed by the diphosphate moiety. A similar conformation is seen in nearly all structures of GT-donor complexes (36, 45). In other inverting GTs, a carboxylate group in the active site is proposed to act as a general base to activate an oxygen atom of the acceptor sugar for reaction with the anomeric carbon of the sugar nucleotide (36). In GlfT2, Asp-372 is positioned appropriately for this function (Fig. 4B). To evaluate the role of the carboxylate side chain of Asp-372, we replaced this residue by Ser using site-directed mutagenesis. Using both a coupled spectrophotometric assay (29) and a more sensitive radiochemical assay (9), the D372S mutant showed no detectable activity, even following an overnight incubation (Table 2). In comparison, replacing the nearby Asp-371 residue with Ser also leads to a dramatic loss of activity. However, a very small amount of residual activity can be detected by overnight incubation of this enzyme in the radiochemical assay. Recently reported mutagenesis results using a mass spectrometric method that is less sensitive than the radiochemical assay are also consistent with these observations (15). The enzyme-substrate interactions inferred from the structure and mutagenesis experiments are also consistent with the findings from a recent study in which a panel of methyl- and deoxy-UDP-Galf analogs were evaluated against the enzyme. In particular, UDP-Galf derivatives methylated at O2, O5, or O6 were all inactive as substrates, although those deoxygenated at C5 or C6 were very weak substrates (50).
TABLE 2.
Kinetic analysis
Kinetic parameters were determined using the coupled spectrophotometric assay (29) with acceptor 1 (Fig. 1B). To determine Km, app values for UDP-Galf, [acceptor 1] = 3.0 mm and [UDP-Galf] = 0.0625–4.0 mm. To determine Km, app values for acceptor 1, [UDP-Galf] = 4 mm and [acceptor 1] = 0.094–6.0 mm (0.125–8.0 mm for H413S).
|
Km, app |
kcat | ||
|---|---|---|---|
| UDP-Galf | Acceptor 1 | ||
| mm | min−1 | ||
| Wild type | 0.38 ± 0.06 | 0.60 ± 0.20 | 430 ± 35 |
| E371S | a | a | a |
| E372S | a | a | a |
| E300S | 0.42 ± 0.06 | 1.78 ± 0.12 | 0.30 ± 0.01 |
| W399S | 0.41 ± 0.04 | 1.78 ± 0.10 | 0.35 ± 0.04 |
| H413S | 0.43 ± 0.05 | 1.54 ± 0.14 | 0.25 ± 0.01 |
a For E371S and E372S, activity was not detectable using the spectrophotometric assay. The radiochemical assay (9) detected only a slight amount of activity (565 versus 236 dpm background, 1 h of incubation; 2329 dpm versus 212 dpm background, 16 h of incubation) for E371S, but only background counts were detected during incubations with E372S. Activity measurements using acceptor 2 were also attempted, but the lower level of activity of this substrate compared with acceptor 1 prevented the determination of Km and kcat parameters for any of the mutant enzymes. The lower level of activity with acceptor 2, and other compounds that first act as substrates for the α-(1→5)-transferase activity of GlfT2, has previously been reported (9, 14, 15, 20, 29) and is a topic of current study.
Based on homology with other GT-A structures, the structure of GlfT2 suggests that the acceptor binding site is located in a region containing an intriguing “ring” of mostly aromatic side chains from Trp-309, Lys-369, Trp-370, Trp-399, and His-413 (Fig. 5, A and B). Trp-399 and adjacent residues are part of loop 1, which is likely flexible and may close more tightly over the active site during catalysis as seen in related GTs (45). Supporting the importance of Trp-399 in acceptor binding, the apparent Km value for a trisaccharide acceptor 1 (Fig. 1B) increases severalfold, and kcat value decreases by more than 1000-fold when Trp-399 is replaced by Ser. Similar effects on Km and kcat values are also seen when His-413 is replaced by Ser. This putative binding site allows the hydroxyl group that reacts with the sugar nucleotide to be positioned next to the carboxylate side chain of Asp-372, the catalytic base. Nearby, His-296, Glu-300, and Tyr-344 compose a highly conserved block of residues that form a binding pocket for either the 5-OH or the 6-OH group of the terminal Galf residue in the nascent polysaccharide chain; this binding pocket is proposed to bind the other terminal hydroxyl group not involved in glycosyl transfer (Fig. 4, A and B). Consistent with this model, replacing Glu-300 with Ser again increases the apparent Km value for the acceptor by severalfold, while decreasing kcat by over 1000-fold. The importance of Glu-300 is further supported by the observation that acceptor substrates deoxygenated at these positions are inactive as substrates (50). This mode of binding also positions the growing polysaccharide into the central hollow core of the homotetrameric complex. These initial mutagenesis results and models for acceptor substrates support the involvement of this region of the protein in positioning the acceptor substrate for glycosyl transfer. However, it is clear that additional experimentally determined structural information will be needed to establish the details of acceptor recognition and the positioning of reactive groups to promote catalysis.
FIGURE 5.

Model of acceptor 1 (β-d-Galf-(1→5)-β-d-Galf-(1→6)-β-d-Galf-octyl) (gray) (A) and acceptor 2 (β-d-Galf-(1→6)-β-d-Galf-(1→5)-β-d-Galf-octyl) (white) bound to GlfT2, Mn2+ (gray sphere) and UDP-Galf (magenta) (B). Loop 1 is colored yellow.
DISCUSSION
One of the key puzzles central to the biological function of GlfT2 is its ability to generate alternating β-(1→5) and β-(1→6) glycosidic linkages through a single active site. Although the structures of GlfT2 bound to acceptor substrate molecules have not yet been determined, the structure of the GlfT2-UDP complex reveals a narrow channel near loop 1, which can accommodate low energy conformers of trisaccharide substrates (Fig. 1), as well as the nonreducing ends of longer galactan substrates. The structure of this channel, which may adopt a different conformation in the presence of acceptor, suggests a simple explanation for the formation of alternating β-(1→5) and β-(1→6) linkages by GlfT2. The hydrophobic ring of residues can accommodate both β-(1→5) and β-(1→6) linkages, but the difference in length of the two types of linkages leads to differing positions of the terminal residue at the nonreducing end of the growing chain. The more extended β-(1→6) linkage positions the terminal residue deeper into the active site, which promotes reaction with UDP-Galf with the 5-OH. In contrast, the less extended β-(1→5) linkage positions the terminal residue less deep in the active site, thus promoting reaction with the 6-OH (Fig. 5, A and B). This mechanism is consistent with the mutagenesis results described above, the presence of a single GT-A domain, and a single active site per GT protomer, in addition to saturation-transfer difference-NMR studies (13).
Studies with a series of deoxygenated acceptor oligosaccharide analogs are also consistent with these binding interactions.4 Up to 8-fold higher activity (kcat/Km) for some of these analogs when compared with the parent acceptors may reflect the improved binding of specific deoxy analogs with the primarily hydrophobic and aromatic residues in the acceptor ring binding site.
The structure of GlfT2 also suggests how interactions with the lipid bilayer membrane may affect how the lipid-linked acceptor is presented to the enzyme active site. As mentioned above, the C-face of the GlfT2 tetramer contains an abundance of exposed hydrophobic and positively charged residues (Figs. 3, D and E, and 6; supplemental Fig. 3). The patch of exposed hydrophobic residues contributed by the α2- and α6-helices from domain 3 and the α1-helix from domain 4 likely generate a binding site for the hydrophobic acyl chains of membrane phospholipids or possibly the hydrophobic decaprenol group of the acceptor substrate. This structural feature provides support for an earlier proposal for the direct binding of GlfT2 to hydrophobic aglycones of synthetic acceptor substrates in vitro, in the absence of a phospholipid bilayer (20). In vivo, because the decaprenol group of the acceptor substrate is likely buried in the hydrophobic membrane bilayer, it is more plausible that the exposed hydrophobic residues of GlfT2 would interact with the acyl groups of the membrane phospholipids. However, it is also possible that some interactions with the decaprenol group could help guide the acceptor into the active site. If the hydrophobic residues in the C-face of GlfT2 interact with the membrane bilayer, the positively charged lysine and arginine side chains located between this hydrophobic patch and loop 1 are positioned appropriately for interacting with the negatively charged phosphate groups of membrane phospholipids.
FIGURE 6.
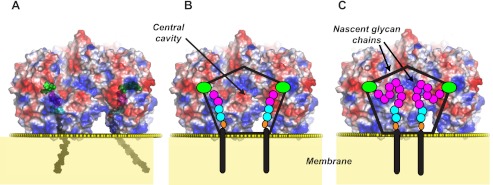
A, model showing the location of UDP-Galf (green) and β-d-Galf-(1→5)-β-d-Galf-(1→4)-α-l-Rhap-(1→3)-α-d-GlcpNAc-decaprenyl-pyrophosphate located inside the central cavity of the GlfT2 tetramer during glycan synthesis. Different parts of the lipid-linked acceptor are colored as follows: Galf residues magenta; Rhap cyan; GlcpNAc cyan; pyrophosphate orange, and decaprenol black. The approximate location of the junction between the hydrophilic and hydrophobic portions of the membrane is denoted by yellow spheres, as calculated by the PPM server (54). Semi-transparent electrostatic surface potential representation of GlfT2 (red, negatively charged; white, neutral; blue, positively charged). B, pentagon overlaid onto GlfT2 shows the approximate extent of the hollow core inside the tetramer. The locations of different parts of the lipid-linked acceptor and UDP-Galf are shown schematically and colored as in A. C, schematic diagram illustrates how two nascent chains (each eight Galf residues long in this example) are expected to form a loop extending into the central cavity formed by the tetramer and the lipid bilayer.
The role of GlfT2 in galactan biosynthesis is to add ∼30 Galf residues to a β-d-Galf-(1→5)-β-d-Galf-(1→4)-α-l-Rhap-(1→3)-α-d-GlcpNAc-decaprenyl-pyrophosphate acceptor substrate (10). Models of the lipid-linked tetrasaccharide substrate and longer nascent glycan chains indicate that the growing polymer would be located in the hollow core of the GlfT2 tetramer, anchored to the membrane by the decaprenol group (Figs. 3 and 6; supplemental Fig. 3). The structure of GlfT2 indicates a distance of ∼30 Å from the location of the hydrophobic residues in the C-face to the active site. The pyrophosphate group and carbohydrate residues of the nascent glycan chain span most of this distance. Some distortions in the plasma membrane from protein binding or transient interactions between the acceptor decaprenyl-pyrophosphate moiety and the protein may also be present. Specific interactions between the carbohydrate residues of the acceptor and the protein were not detected in an earlier study (20), which is consistent with the apparent lack of a well formed carbohydrate-binding site with highly conserved residues in the hollow core.
The molecular mechanisms underlying the polymerase activity of GlfT2 have been the subject of recent investigations (15, 20). Considered in aggregate, the model outlined above is generally consistent with some features of the tethering model proposed by Kiessling and co-workers (20). However, the structure of GlfT2 suggests that instead of a direct interaction between the decaprenol group of the natural acceptor substrate and the enzyme, the lipid-linked acceptor may diffuse freely in the portion of the membrane bilayer underlying the hollow central core of GlfT2 while still being confined by the surrounding protein. This topology suggests that GlfT2 acts to promote polymerization processivity through a mechanism akin to metabolic channeling.
In addition, the geometric restrictions of the central core on the growing polysaccharide may assist in limiting the extent of polymerization to the lengths of glycans observed in vivo (∼30 Galf residues). The volume of the central cavity depends on assumptions about the location of the tetramer relative to the membrane bilayer, something that is not well defined at present. However, calculations using VOIDOO (51) estimate a volume of ∼60,000 Å3, which is sufficient to accommodate at least 100–150 residues of Galf. Thus, the dimensions of the internal cavity appear to provide only sufficient space to accommodate four nascent chains of ∼30 Galf residues each. Further studies are clearly needed to define the specific mechanisms underlying the polymerization activity of GlfT2 and possibly other GTs using membrane-embedded, lipid-linked acceptor substrates. Notably, the structure of GlfT2 indicates for the first time that the topology imposed by docking a tetrameric assembly onto a membrane bilayer is likely important for the function of the enzyme as a polymerase in vivo.
The efficacy of ethambutol, an arabinosyltransferase inhibitor, for the treatment of TB has prompted the search for other inhibitors of polysaccharide biosynthetic GTs as therapeutics for mycobacterial diseases (16). Knock-out studies also indicate that GlfT2 is essential for growth (7), and the structure of the enzyme reported here suggests at least three novel approaches for designing inhibitors that may be suitable lead compounds for therapeutic development. First, the recognition of uracil by Asn-229 in GlfT2 is unusual, as a negatively charged Asp residue occupies this position in most other GTs using UDP donors, with the exception of GT64 enzyme EXTL2, which has Gln (48). This feature could be combined with modifications of the uridine base to take advantage of a potential binding site provided by surface-exposed hydrophobic residues (e.g. Trp-408 from loop 1, which packs against Leu-480 in domain 3) adjacent to C5 in the uridine base to generate specificity and affinity (Fig. 3B). Related to this idea, modifications of the uridine base at C5 were recently shown to produce novel GT inhibitors with allosteric effects (52). Second, the larger binding pocket surrounding the ribose moiety of the UDP donor may be a distinctive feature of GlfT2 that could be exploited for inhibitor design. Increased specificity for GlfT2 could possibly be generated by incorporating a bulky functional group that would occupy this distinctive binding pocket in GlfT2 but lead to steric clashes with the smaller pocket found in many other GTs (45). Finally, the structure suggests that patches of surface-exposed hydrophobic and positively charged residues may also provide a target for inhibitor design. If these regions on the C-face of the GlfT2 tetramer are important for membrane attachment and confining the acceptor substrate within the central cavity, an inhibitor that bound to these regions with high affinity could prevent GlfT2 from forming some of the critical binding interactions required for galactan synthesis.
Supplementary Material
Acknowledgments
We acknowledge the help of the staff at the Stanford Synchrotron Radiation Laboratory and the Canadian Light Source for providing access to the synchrotron radiation beamlines used for crystal screening and data collection at various stages of this project. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the United States Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research, and by National Institutes of Health Grant P41GM103393 from NIGMS and Grant P41RR001209 from NCRR. Portions of the research described in this paper were performed using beamline 08ID-1 at the Canadian Light Source, which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan.
This work was supported by funding from the Alberta Glycomics Centre, the Natural Sciences and Engineering Research Council Discovery grants (to T. L. L. and K. K. S. N.), and the Alberta Heritage Foundation for Medical Research Senior Scholar Award (to K. K. S. N.).

This article contains supplemental Movies 1 and 2, Figs. 1–3, Table 1, and additional references.
The atomic coordinates and structure factors (codes 4FIX and 4FIY) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
T. L. Lowary, unpublished data.
M. R. Richards and T. L. Lowary, unpublished data.
- TB
- tuberculosis
- CAZy
- carbohydrate-active enzyme
- Galf
- galactofuranose
- GlcpNAc
- 2-acetamido-2-deoxy-glucopyranose
- GT
- glycosyltransferase
- Rhap
- rhamnopyranose
- β-ME
- β-mercaptoethanol
- TCEP
- tris(2-carboxyethyl)phosphine
- CBM
- carbohydrate-binding module.
REFERENCES
- 1. Kaur D., Guerin M. E., Skovierová H., Brennan P. J., Jackson M. (2009) Chapter 2. Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv. Appl. Microbiol. 69, 23–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bass J. B., Jr., Farer L. S., Hopewell P. C., O'Brien R., Jacobs R. F., Ruben F., Snider D. E., Jr., Thornton G. (1994) Treatment of tuberculosis and tuberculosis infection in adults and children. American Thoracic Society and The Centers for Disease Control and Prevention. Am. J. Respir. Crit. Care Med. 149, 1359–1374 [DOI] [PubMed] [Google Scholar]
- 3. Dover L. G., Coxon G. D. (2011) Current status and research strategies in tuberculosis drug development. J. Med. Chem. 54, 6157–6165 [DOI] [PubMed] [Google Scholar]
- 4. Nachega J. B., Chaisson R. E. (2003) Tuberculosis drug resistance: a global threat. Clin. Infect. Dis. 36, S24–S30 [DOI] [PubMed] [Google Scholar]
- 5. Belanger A. E., Besra G. S., Ford M. E., Mikusová K., Belisle J. T., Brennan P. J., Inamine J. M. (1996) The embAB genes of Mycobacterium avium encode an arabinosyltransferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc. Natl. Acad. Sci. U.S.A. 93, 11919–11924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Banerjee A., Dubnau E., Quemard A., Balasubramanian V., Um K. S., Wilson T., Collins D., de Lisle G., Jacobs W. R., Jr. (1994) inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263, 227–230 [DOI] [PubMed] [Google Scholar]
- 7. Pan F., Jackson M., Ma Y., McNeil M. (2001) Cell wall core galactofuran synthesis is essential for growth of mycobacteria. J. Bacteriol. 183, 3991–3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Richards M. R., Lowary T. L. (2009) Chemistry and biology of galactofuranose-containing polysaccharides. ChemBioChem 10, 1920–1938 [DOI] [PubMed] [Google Scholar]
- 9. Rose N. L., Completo G. C., Lin S. J., McNeil M., Palcic M. M., Lowary T. L. (2006) Expression, purification, and characterization of a galactofuranosyltransferase involved in Mycobacterium tuberculosis arabinogalactan biosynthesis. J. Am. Chem. Soc. 128, 6721–6729 [DOI] [PubMed] [Google Scholar]
- 10. Belánová M., Dianisková P., Brennan P. J., Completo G. C., Rose N. L., Lowary T. L., Mikusová K. (2008) Galactosyltransferases in mycobacterial cell wall synthesis. J. Bacteriol. 190, 1141–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kremer L., Dover L. G., Morehouse C., Hitchin P., Everett M., Morris H. R., Dell A., Brennan P. J., McNeil M. R., Flaherty C., Duncan K., Besra G. S. (2001) Galactan biosynthesis in Mycobacterium tuberculosis. Identification of a bifunctional UDP-galactofuranosyltransferase. J. Biol. Chem. 276, 26430–26440 [DOI] [PubMed] [Google Scholar]
- 12. Mikusová K., Yagi T., Stern R., McNeil M. R., Besra G. S., Crick D. C., Brennan P. J. (2000) Biosynthesis of the galactan component of the mycobacterial cell wall. J. Biol. Chem. 275, 33890–33897 [DOI] [PubMed] [Google Scholar]
- 13. Szczepina M. G., Zheng R. B., Completo G. C., Lowary T. L., Pinto B. M. (2009) STD-NMR studies suggest that two acceptor substrates for GlfT2, a bifunctional galactofuranosyltransferase required for the biosynthesis of Mycobacterium tuberculosis arabinogalactan, compete for the same binding site. ChemBioChem 10, 2052–2059 [DOI] [PubMed] [Google Scholar]
- 14. Levengood M. R., Splain R. A., Kiessling L. L. (2011) Monitoring processivity and length control of a carbohydrate polymerase. J. Am. Chem. Soc. 133, 12758–12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. May J. F., Levengood M. R., Splain R. A., Brown C. D., Kiessling L. L. (2012) A processive carbohydrate polymerase that mediates bifunctional catalysis using a single active site. Biochemistry 51, 1148–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berg S., Kaur D., Jackson M., Brennan P. J. (2007) The glycosyltransferases of Mycobacterium tuberculosis. Roles in the synthesis of arabinogalactan, lipoarabinomannan, and other glycoconjugates. Glycobiology 17, 35R–56R [DOI] [PubMed] [Google Scholar]
- 17. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) The Carbohydrate-Active EnZymes database (CAZy). An expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Somerville C. (2006) Cellulose synthesis in higher plants. Annu. Rev. Cell Dev. Biol. 22, 53–78 [DOI] [PubMed] [Google Scholar]
- 19. Osawa T., Sugiura N., Shimada H., Hirooka R., Tsuji A., Shirakawa T., Fukuyama K., Kimura M., Kimata K., Kakuta Y. (2009) Crystal structure of chondroitin polymerase from Escherichia coli K4. Biochem. Biophys. Res. Commun. 378, 10–14 [DOI] [PubMed] [Google Scholar]
- 20. May J. F., Splain R. A., Brotschi C., Kiessling L. L. (2009) A tethering mechanism for length control in a processive carbohydrate polymerization. Proc. Natl. Acad. Sci. U.S.A. 106, 11851–11856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 22. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 23. Terwilliger T. C., Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 27. Holm L., Park J. (2000) DaliLite workbench for protein structure comparison. Bioinformatics 16, 566–567 [DOI] [PubMed] [Google Scholar]
- 28. Kleywegt G. J., Jones T. A. (1997) Detecting folding motifs and similarities in protein structures. Methods Enzymol. 277, 525–545 [DOI] [PubMed] [Google Scholar]
- 29. Rose N. L., Zheng R. B., Pearcey J., Zhou R., Completo G. C., Lowary T. L. (2008) Development of a coupled spectrophotometric assay for GlfT2, a bifunctional mycobacterial galactofuranosyltransferase. Carbohydr. Res. 343, 2130–2139 [DOI] [PubMed] [Google Scholar]
- 30. Becke A. D. (1993) Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 [Google Scholar]
- 31. Rassolov V. A., Ratner M. A., Pople J. A., Redfern P. C., Curtiss L. A. (2001) 6–31G* basis set for third-row atoms. J. Comput. Chem. 22, 976–984 [Google Scholar]
- 32. Clark T., Chandrasekhar J., Spitznagel G. W., Schleyer P. V. (1983) Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first row elements, Li-F. J. Comput. Chem. 4, 294–301 [Google Scholar]
- 33. Frisch M. J., Pople J. A., Binkley J. S. (1984) Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 80, 3265–3269 [Google Scholar]
- 34. Burkert U., Allinger N. L. (1982) Molecular Mechanics, American Chemical Society, Washington, D. C [Google Scholar]
- 35. Petrová P., Monteiro C., Hervé du Penhoat C., Koca J., Imberty A. (2001) Conformational behavior of nucleotide-sugar in solution. Molecular dynamics and NMR study of solvated uridine diphosphate-glucose in the presence of monovalent cations. Biopolymers 58, 617–635 [DOI] [PubMed] [Google Scholar]
- 36. Lairson L. L., Henrissat B., Davies G. J., Withers S. G. (2008) Glycosyltransferases. Structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555 [DOI] [PubMed] [Google Scholar]
- 37. Holm L., Kääriäinen S., Rosenström P., Schenkel A. (2008) Searching protein structure databases with DaliLite version 3. Bioinformatics 24, 2780–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bae B., Ohene-Adjei S., Kocherginskaya S., Mackie R. I., Spies M. A., Cann I. K., Nair S. K. (2008) Molecular basis for the selectivity and specificity of ligand recognition by the family 16 carbohydrate-binding modules from Thermoanaerobacterium polysaccharolyticum ManA. J. Biol. Chem. 283, 12415–12425 [DOI] [PubMed] [Google Scholar]
- 39. Fritz T. A., Hurley J. H., Trinh L. B., Shiloach J., Tabak L. A. (2004) The beginnings of mucin biosynthesis. The crystal structure of UDP-GalNAc:polypeptide α-N-acetylgalactosaminyltransferase-T1. Proc. Natl. Acad. Sci. U.S.A. 101, 15307–15312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Raman J., Fritz T. A., Gerken T. A., Jamison O., Live D., Liu M., Tabak L. A. (2008) The catalytic and lectin domains of UDP-GalNAc:polypeptide α-N-acetylgalactosaminyltransferase function in concert to direct glycosylation site selection. J. Biol. Chem. 283, 22942–22951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sunna A., Gibbs M. D., Bergquist P. L. (2001) Identification of novel β-mannan- and β-glucan-binding modules. Evidence for a superfamily of carbohydrate-binding modules. Biochem. J. 356, 791–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Czjzek M., Bolam D. N., Mosbah A., Allouch J., Fontes C. M., Ferreira L. M., Bornet O., Zamboni V., Darbon H., Smith N. L., Black G. W., Henrissat B., Gilbert H. J. (2001) The location of the ligand-binding site of carbohydrate-binding modules that have evolved from a common sequence is not conserved. J. Biol. Chem. 276, 48580–48587 [DOI] [PubMed] [Google Scholar]
- 43. Liu J., Mushegian A. (2003) Three monophyletic superfamilies account for the majority of the known glycosyltransferases. Protein Sci. 12, 1418–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Breton C., Snajdrová L., Jeanneau C., Koca J., Imberty A. (2006) Structures and mechanisms of glycosyltransferases. Glycobiology 16, 29R–37R [DOI] [PubMed] [Google Scholar]
- 45. Qasba P. K., Ramakrishnan B., Boeggeman E. (2005) Substrate-induced conformational changes in glycosyltransferases. Trends Biochem. Sci. 30, 53–62 [DOI] [PubMed] [Google Scholar]
- 46. Janin J., Bahadur R. P., Chakrabarti P. (2008) Protein-protein interaction and quaternary structure. Q. Rev. Biophys. 41, 133–180 [DOI] [PubMed] [Google Scholar]
- 47. Ponstingl H., Kabir T., Gorse D., Thornton J. M. (2005) Morphological aspects of oligomeric protein structures. Prog. Biophys. Mol. Biol. 89, 9–35 [DOI] [PubMed] [Google Scholar]
- 48. Pedersen L. C., Dong J., Taniguchi F., Kitagawa H., Krahn J. M., Pedersen L. G., Sugahara K., Negishi M. (2003) Crystal structure of an α1,4-N-acetylhexosaminyltransferase (EXTL2), a member of the exostosin gene family involved in heparan sulfate biosynthesis. J. Biol. Chem. 278, 14420–14428 [DOI] [PubMed] [Google Scholar]
- 49. Szczepina M. G., Zheng R. B., Completo G. C., Lowary T. L., Pinto B. M. (2010) STD-NMR studies of two acceptor substrates of GlfT2, a galactofuranosyltransferase from Mycobacterium tuberculosis. Epitope mapping studies. Bioorg. Med. Chem. 18, 5123–5128 [DOI] [PubMed] [Google Scholar]
- 50. Poulin M. B., Zhou R., Lowary T. L. (2012) Synthetic UDP-galactofuranose analogs reveal critical enzyme-substrate interactions in GlfT2-catalyzed mycobacterial galactan assembly. Org. Biomol. Chem. 10, 4074–4087 [DOI] [PubMed] [Google Scholar]
- 51. Kleywegt G. J., Jones T. A. (1994) Detection, delineation, measurement, and display of cavities in macromolecular structures. Acta Crystallogr. D Biol. Crystallogr. 50, 178–185 [DOI] [PubMed] [Google Scholar]
- 52. Pesnot T., Jørgensen R., Palcic M. M., Wagner G. K. (2010) Structural and mechanistic basis for a new mode of glycosyltransferase inhibition. Nat. Chem. Biol. 6, 321–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Eisenberg D., Schwarz E., Komaromy M., Wall R. (1984) Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 179, 125–142 [DOI] [PubMed] [Google Scholar]
- 54. Lomize M. A., Pogozheva I. D., Joo H., Mosberg H. I., Lomize A. L. (2012) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.