

Background: SAP97 and CaM play a role in the regulation of connexin 32 (Cx32) gap junctions.
Results: SAP97 and CaM affect the same Cx32CT residues, calmodulin induces Cx32CT α-helical structure, and Cx32CT mutations that cause X-linked Charcot-Marie-Tooth disease (CMTX) affect the binding of SAP97 and CaM.
Conclusion: Cx32-protein partner interactions are important for channel regulation and myelin homeostasis.
Significance: Cx32CT mutations may cause CMTX by disrupting the binding of SAP97 and CaM.
Keywords: Biophysics, Calmodulin, Circular Dichroism (CD), Gap Junctions, NMR, CMTX, Cx32, SAP97
Abstract
In Schwann cells, connexin 32 (Cx32) can oligomerize to form intracellular gap junction channels facilitating a shorter pathway for metabolite diffusion across the layers of the myelin sheath. The mechanisms of Cx32 intracellular channel regulation have not been clearly defined. However, Ca2+, pH, and the phosphorylation state can regulate Cx32 gap junction channels, in addition to the direct interaction of protein partners with the carboxyl-terminal (CT) domain. In this study, we used different biophysical methods to determine the structure and characterize the interaction of the Cx32CT domain with the protein partners synapse-associated protein 97 (SAP97) and calmodulin (CaM). Our results revealed that the Cx32CT is an intrinsically disordered protein that becomes α-helical upon binding CaM. We identified the GUK domain as the minimal SAP97 region necessary for the Cx32CT interaction. The Cx32CT residues affected by the binding of CaM and the SAP97 GUK domain were determined as well as the dissociation constants for these interactions. We characterized three Cx32CT Charcot-Marie-Tooth disease mutants (R219H, R230C, and F235C) and identified that whereas they all formed functional channels, they all showed reduced binding affinity for SAP97 and CaM. Additionally, we report that in RT4-D6P2T rat schwannoma cells, Cx32 is differentially phosphorylated and exists in a complex with SAP97 and CaM. Our studies support the importance of protein-protein interactions in the regulation of Cx32 gap junction channels and myelin homeostasis.
Introduction
In the peripheral nervous system Schwann cells wrap around the axon multiple times forming an insulating envelope (i.e. myelin sheath) that benefits neuron function by increasing the transmission of nerve impulses. Schwann cell-axon interactions are important because axons regulate the survival, proliferation, and differentiation of Schwann cells that in turn determine the distribution of ion channels and support the maintenance and survival of axons (1–4). Schwann cell wrapping is followed by compaction where the majority of the cytoplasm from the myelin sheath is excluded resulting in two distinct domains, compact and noncompact myelin, each characterized by their own unique set of proteins. For example, areas of noncompact myelin, including the Schmidt-Lanterman incisures and paranodes, contain myelin-associated glycoprotein, E-cadherin, calmodulin (CaM),2 synapse-associated protein 97 (SAP97), and connexin 32 (Cx32) (5–7).
Cx32 is the most abundant connexin isoform in Schwann cells, as well as being expressed in the liver, lung, and several other tissues. Connexins are tetraspan transmembrane domain proteins with intracellular amino and carboxyl termini. These integral membrane proteins oligomerize to form hexameric connexons, which usually are contributed by two adjacent cells and interact across the extracellular space to form gap junction channels allowing for the exchange of ions and small metabolites. However, gap junctions in Schwann cells can form intracellular channels through the layers of myelin. Aggregates of these Cx32 channels are found predominantly at the Schmidt-Lanterman incisures and paranodal regions providing a direct/shorter route for the diffusion of second messengers and other small molecules from the Schwann cell perinuclear cytoplasm to the adaxonal region (8, 9). This pathway across the myelin sheath is important because it allows the Schwann cell and axon to maintain communication, which is likely necessary for proper myelin formation. In fact, demyelination is the first alteration seen in Cx32-deficient mice, further supporting the importance of Cx32 in Schwann cells (10, 11).
Although much of the protein sequence is conserved throughout the connexin family, the major divergence occurs in the cytoplasmic loop (CL) and carboxyl-terminal (CT) domains, which leads to their classification into subdivisions (α, β, and γ). CT domains, in particular, contain multiple sites for protein-protein interactions that play essential roles in channel localization and regulation (12–14). The cytoplasmic domains of transmembrane proteins are often intrinsically disordered, including the gap junction proteins Cx43CT and Cx40CT from the α subdivision (12). Intrinsically disordered proteins play important roles in regulation, signaling, and control pathways, where binding to multiple partners via high-specificity/low-affinity interactions is facilitated via a disordered to ordered conformational transition (15–17). Unaddressed to date is whether the CT domain of the β subdivision connexins (e.g. Cx32) have similar structure and function as their α subdivision counterparts.
There are several known binding partners to the Cx32CT, and specifically two that play a role in the regulation of Schwann cell function, the Ca2+-sensing protein CaM and the scaffolding protein SAP97. CaM is a Ca2+-binding protein that acts by transducing the effect of a rise in intracellular Ca2+ to create physiological responses; specifically it has been implicated in mediating the Ca2+-dependent down-regulation of Cx32 gap junction channels (18, 19). Cx32 has been shown to interact with CaM directly via the Cx32CT domain in a Ca2+-dependent manner (13). Additionally, elevated Ca2+ levels in the Schwann cell paranodes has been implicated in demyelination due to increased proliferation and impaired Schwann cell function (1, 20, 21). The observation of reduced myelination and Cx32 channel closure in response to elevated Ca2+, the direct interaction between Cx32 and CaM, and their overlapping expression in Schwann cells, suggests that this complex is important for Ca2+ regulation or sensing across the myelin sheath (6).
SAP97 was also found to be enriched at Schwann cell paranodes (5). SAP97 is a member of the membrane-associated guanylate kinase family, whose members are involved in the clustering of ion channels, regulation of cell adhesion molecules, as well as downstream signaling (22–24). These multidomain proteins are composed of three PDZ domains, a SH3 domain, a HOOK region, and a guanylate kinase-like domain (GUK domain). The SH3-HOOK-GUK domains of SAP97 are the primary sites for a number of protein-protein interactions. Our laboratory has previously shown that Cx32 interacts directly somewhere within the SH3-HOOK-GUK region of SAP97 and in mouse liver the loss of Cx32 altered the levels, localization, and interactions of the tumor suppressor protein SAP97 (25). SAP97 plays an important role in maintaining myelin homeostasis in Schwann cells via interactions with myotubularin-related protein 2 (Mtmr2) at sites of membrane remodeling and the exocyst protein Sec8 to promote membrane addition (5). The observation that Cx32 is important for SAP97 scaffolding, the necessity for SAP97 to localize at the Schwann cell membrane for myelin homeostasis, and their overlapping expression in Schwann cells, suggests that this complex is important for the regulation of the myelin sheath.
Here, we have characterized the structure and biophysical properties of the Cx32CT domain and examined its interaction with SAP97 and CaM. Our studies determined that the Cx32CT is intrinsically disordered; however, in the presence of trifluoroethanol (TFE), some Cx32CT residues had a propensity to form α-helical structure. The Cx32CT residues showing α-helical propensity were the same residues involved in binding to the SAP97 GUK domain and CaM. Unlike SAP97, CaM binding induced the α-helical structure within the Cx32CT and involved both the N- and C-lobes. We also characterized the structural and functional consequences, compared with wild type Cx32CT, of three Cx32CT Charcot-Marie-Tooth disease (CMTX) mutations: R219H, R230C, and F235C. All three mutants formed functional channels; however, they all showed reduced binding affinity for SAP97 and CaM. Finally, we confirmed the direct Cx32 interaction with SAP97 and CaM in RT4-D6P2T rat schwannoma cells. Thus, we propose that one mechanism by which Cx32 channels are regulated in Schwann cells is via the interactions with SAP97 and CaM.
MATERIALS AND METHODS
Expression and Purification of Recombinant GST-tagged Proteins
The rat Cx32CT(217–283) (and Cx32CT-R219H, Cx32CT-R230C, and Cx32CT-F235C mutants), mouse SAP97 GUK(701–764), mouse SAP97 GUK(701–927), mouse SAP97 END(569–764), and mouse SAP97 END(569–927) polypeptides as well as bovine CaM (unlabeled, 15N-labeled, or 15N,13C-labeled) were expressed and purified as described previously (26, 27). One exception, after digestion of SAP97 END(569–927) off of the GST, the supernatant was added to a 50,000 Amicon centrifuge filter to concentrate the protein down to 500 μl, which was equilibrated in 50 mm Tris-HCl, pH 7.0, buffer containing 100 mm NaCl, 1 mm EDTA, 1 mm DTT, and 10% ammonium sulfate using a NAP-5 column (GE Healthcare). The protein was then applied to a phenyl-Sepharose column (GE Healthcare) and eluted using ammonium sulfate-free buffer (50 mm Tris-HCl, pH 7.0, 100 mm NaCl, 1 mm EDTA, 1 mm DTT). All polypeptides were confirmed for purity and analyzed for degradation by SDS-PAGE and equilibrated in 25 mm Tris-HCl, pH 7.0, 100 mm KCl, and 1 mm DTT, using NAP-5 columns (GE Healthcare). Protein concentrations were determined using the Bradford assay and UV spectroscopy.
Circular Dichroism (CD)
CD experiments were performed on a JASCO-815 spectrometer at 7 °C with a 1-mm path length quartz cell using a bandwidth of 1 nm, an integration time of 1 s, and a scan rate of 50 nm/s. Each spectrum is the average of 5 scans. All spectra were corrected by subtracting the solvent spectrum acquired under identical conditions. The protein concentration for each sample was 200 μm. All CD data were processed using CDtool (28). In the Cx32CT titrations with SAP97 GUK and CaM, up to 60 μm Cx32CT in 50 mm Tris-HCl, pH 7.0, buffer containing 100 mm NaCl and 1 mm DTT was added to 30 μm GUK or CaM with 5 mm CaCl2 or 5 mm EGTA. The signals from the Cx32CT itself were subtracted. All of the measurements were carried out in triplicate and were baseline corrected. Mean residue ellipticity and molar ellipticity are defined by the units degree cm2 dmol−1 and 1 × 106 degree cm2 dmol−1 residues−1, respectively.
Nuclear Magnetic Resonance (NMR)
All NMR data were acquired at 7 °C using a 600-MHz Varian INOVA NMR spectrometer outfitted with a cryoprobe. Experimental data to determine the Cx32CT backbone sequential assignments have been described (29). Gradient-enhanced two-dimensional 15N-HSQC experiments (30) were used to observe all backbone amide resonances from the 15N-labeled Cx32CT and 15N-labeled CaM samples in the absence and presence of different combinations of unlabeled CaM (Sigma), Cx32CT, SAP97 GUK(701–927), and SAP97 END(569–927) domains. Data were acquired with 1024 complex points in the direct dimension and 512 complex points in the indirect dimension. NMR spectra were processed using NMRPipe (31) and analyzed using NMRView (32). Binding isotherms from the 15N-HSQC titration experiments were calculated with GraphPad Prism 5 software (GraphPad Software, Inc.) and Origin 6 software (Microcal).
Yeast Two-hybrid Assay (Y2H)
The Saccharomyces cerevisiae strain AH109 (BD Biosciences Clontech) was maintained on yeast extract, peptone, and dextrose agar plates. Co-transformation was done by the lithium acetate procedure as described in the manufacturer's instructions for the MATCHMAKER two-hybrid kit (BD Biosciences Clontech) with the following GAL4ad fusion constructs: GAL4ad-SH3 (SAP97(569–644)), GAL4ad-HOOK (SAP97(645–700)), GAL4ad-GUK (SAP97(701–764)), GAL4ad-END (SAP97(569–764)), GAL4ad-SV40 Large T antigen (control), and with the following GAL4bd fusion constructs: GAL4bd-Cx32CT(217–283) and GAL4bd-pVA3 murine p53 (control). For colony growth assays, AH109 co-transformants were streaked on plates lacking leucine and tryptophan and allowed to grow at 30 °C usually for 3 days or until colonies were large enough for further assays. An average of three to four colonies was then chosen and suspended in water, equilibrated to the same optical density at 600 nm, and replated on plates lacking leucine and tryptophan (+His) as well as plates also lacking histidine (−His).
Isothermal Titration Calorimetry (ITC)
The heat for the reaction of Cx32CT with CaM was measured directly by isothermal titration calorimetry using the iTC200 titration calorimeter from Microcal (Northampton, MA). ITC binding isotherms were obtained as follows; 21 injections of 1–2 μl of Cx32CT (400 μm) were used to titrate a 200-μl CaM solution (30 μm) in the reaction cell at 37 °C. The reaction heat of each injection was measured by integration of the area of the injection peak, corrected for the dilution heat of the Cx32CT, and normalized by the moles of Cx32CT added. The resulting calorimetric binding isotherm, heat/(mole of injectant) versus [Cx32CT]/[CaM], was fitted with the Origin 7 software, supplied by Microcal Inc. We used a model of one single binding site to fit each binding isotherm. These fits are characterized by a set of three parameters: binding constant (Kb), binding enthalpy (ΔHb), and number of sites (n). The Gibbs free energy (ΔG0b) was determined from the equation: ΔG0b = RT lnKb, where R is the molar gas constant and T is the temperature at which the experiment is performed. The entropy of binding (ΔSb) was calculated from the Gibbs equation.
Coimmunoprecipitation (Co-IP)
RT4-D6P2T rat schwannoma cells were lysed in complete lysis buffer (50 mm Tris-HCl, pH 7.4, 0.25 mm sodium deoxycholate, 150 mm NaCl, 2 mm EGTA, 1 mm Na3VO4, 1 mm NaF, 1 mm phenylmethylsulfonyl fluoride, 1% Triton X-100, and one-half tablet of Complete Protease Inhibitor (Roche Applied Science)), maintained on ice for 30 min, and then spun at 12,000 × g for 15 min. Total protein was assessed using the BCA protein assay kit (Pierce). The lysate (50 μg of total protein) was incubated with 2 μg of rabbit anti-SAP97 (Abcam), rabbit anti-CaM (Sigma), or rabbit anti-IgG (Jackson ImmunoResearch) for control. After a 4-h incubation with the antibody at 4 °C, 100 μl of protein A-Sepharose (GE Healthcare) was added to the samples and incubated overnight at 4 °C. The Sepharose was washed four times with cold lysis buffer, and the immunoprecipitated complex was resolved by SDS-PAGE and Western blot. Additional conditions tested were the presence of 5 mm EGTA or 5 mm CaCl2 to the lysate and lysis buffer, or the general phosphatase λPP (New England Biolabs) in the lysate prior to IP (30 °C for 1 h), then washed four times after IP with cold buffer (50 mm Tris-HCl, 150 mm NaCl).
For transfection of wild type Cx32 and mutants, GeneExpresso (Excellgen) and plasmid DNA were incubated separately in nonsupplemented Dulbecco's modified Eagle's medium (DMEM) for 10 min at room temperature, then combined for another 15 min. HeLa cells (∼80% confluent) were washed with DMEM and then incubated with combined GeneExpresso/DNA solution for 24 h at 37 °C. HeLa cells were lysed 24 h post-transfection in complete lysis buffer (50 mm Tris-HCl, pH 7.4, 0.25 mm sodium deoxycholate, 150 mm NaCl, 2 mm EGTA, 1 mm Na3VO4, 1 mm NaF, 1 mm phenylmethylsulfonyl fluoride, 1% Triton X-100), maintained on ice for 30 min, and then spun at 12,000 × g for 15 min. Plasmid DNA amounts used for transfection were optimized to acquire equal expression of each protein, which was confirmed by densitometric analysis of Western blots. Total protein was assessed using the BCA protein assay kit (Pierce) to normalize the amount of protein in each co-IP. The lysate (50 μg of total protein) was incubated with 2 μg of mouse anti-Cx32 (Sigma), or mouse anti-IgG (Jackson ImmunoResearch) for control. After a 4-h incubation with the antibody at 4 °C, 100 μl of protein A-Sepharose (GE Healthcare) was added to the samples and incubated overnight at 4 °C. The Sepharose was washed four times with cold lysis buffer, and the immunoprecipitated complex was resolved by SDS-PAGE and Western blot. All co-IP experiments were performed in triplicate.
Scrape-loading Assay
Cells were scrape-loaded and immunostaining were performed as described in (33, 34). Briefly, HeLa cells were seeded in six-well clusters at a density of 1.5 × 106/well and grew for 24 h prior and post-transfection. Cell culture medium from freshly confluent cells was removed and replaced with 1 ml of medium containing rhodamine-conjugated fluorescent dextran (10 kDa, 1 mg/ml; fixable) and Lucifer Yellow (0.5%). Cells were scrape-loaded using a sterile scalpel by two longitudinal scratches and incubated for 1 min at room temperature. Cells were washed quickly two to three times with warm PBS containing calcium and magnesium and returned to the incubator for 5 min, after which the medium was removed, cells were washed 2 times with PBS and fixed with 3.7% buffered formalin at room temperature. The autofluorescence of cells was quenched with 0.1 m glycine and after washing once with PBS, the coverslips were mounted on glass slides in a droplet of SlowFade. After scrape-loading, cells were immunostained at room temperature with rabbit anti-Cx32CL (Sigma) after fixing. Secondary antibody conjugated to Alexa-594 (Invitrogen) was used. Images of the scrape-loaded cells were captured using a CCD camera (Retiga 2000R, FAST 1394) with the aid of Volocity software (Improvision, Lexington, MA). The experiment was repeated 3 times and in each trial, 4 side by side images were captured and estimation of junction permeability was based on methods described previously where the number of fluorescent cells (containing Lucifer Yellow) were counted, excluding the cells containing dextran, which indicates the initially loaded cells (33, 34). Data were analyzed statistically by analysis of variance with Tukey post hoc tests.
RESULTS
Secondary Structure and Resonance Assignments of the Cx32CT
To evaluate the global structure of the soluble Cx32CT(217–283) domain, the secondary structure was determined using CD. Random or flexible protein structures are characterized by a peak minimum at 198 nm and α-helical content by two peak minima around 208 and 222 nm. The soluble Cx32CT showed a peak minimum near 198 nm and little ellipticity at 222 nm (Fig. 1A), characteristic of a flexible, intrinsically disordered protein.
FIGURE 1.

Secondary structure and resonance assignments of the Cx32CT. A, CD spectrum of Cx32CT. Mean residue ellipticity (MRE; deg cm2 dmol−1) as a function of wavelength was recorded at 7 °C at a protein concentration of 280 μm. B, 15N-HSQC NMR spectrum of the Cx32CT. The assignments are shown by the residue name and number according to the rat Cx32 sequence. C, CD spectra of Cx32CT were obtained in the absence and presence of varying concentrations of TFE (% displayed in each panel).
Although unstructured, the resonance assignments of the Cx32CT domain were determined to characterize the interactions with the molecular partners SAP97 and CaM (described later). The Cx32CT contains 67 amino acids with a molecular mass of 7.8 kDa. The assignments comprise 100% of all 1H-N, 15N, 13CO, 13Cα, and 13Cβ resonances covering 62 of the 62 nonproline residues. Fig. 1B shows the 15N-HSQC spectrum labeled with the assigned amino acids. Analysis of the 15N-NOESY spectra did not produce NOEs consistent with any secondary structure (data not shown). The intrinsically disordered structure for the soluble Cx32CT domain as determined by CD is consistent with the narrow 1H chemical shift dispersions (<1 ppm) observed in the 15N-HSQC (Fig. 1B).
Although stable secondary structure was not observed for the soluble Cx32CT, the possibility exists that the inherent secondary structure could form when tethered to the membrane (i.e. attached to 4th transmembrane domain) or when bound to a protein partner. Therefore, to identify if the Cx32CT contains an inherent propensity to adopt secondary structure, we acquired CD spectra in the absence and presence of various concentrations of TFE (Fig. 1C). TFE is capable of stabilizing intramolecular hydrogen bonding and is known to stabilize the intrinsic secondary structure in peptides (35, 36). Here, TFE is being used as a tool to address the ability of the Cx32CT domain to form structure. When the TFE concentration was increased to 15%, the weak absorption at 222 nm increased and the minimum absorption near 198 nm shifted to 205 nm. Moreover, this change was further enhanced upon the addition of 25% TFE. These results strongly suggest that the Cx32CT domain has a propensity to form α-helical structure.
Interaction between the Cx32CT Domain and SAP97
Previously, we identified that the Cx32CT interacts with SAP97 residues Val569–Ser764 (25). These SAP97 residues contain the SH3, HOOK, and part of the GUK domains (missing the C-terminal residues Cys765-Leu927 of the GUK domain) and were referred to as the END(569–764) domain. To identify the minimal domain of SAP97 END(569–764) that is involved in the Cx32CT interaction, we performed an Y2H assay. The Cx32CT and the SH3, HOOK, GUK(701–764), and END(569–764) regions of SAP97 (constructs depicted in Fig. 2A) were coexpressed in AH109 yeast cells as GAL4 DNA-binding domain (bd) fusion proteins and as GAL4 transactivation domain (ad) fusion proteins, respectively (Fig. 2B). The direct interaction previously observed between the SAP97 END(569–764) domain and the Cx32CT domain was confirmed, in addition to identifying the minimal region involved, the SAP97 GUK(701–764) domain. Originally, when the SAP97 END construct (and subsequent GUK only construct) was created the carboxyl-terminal residues Cys765-Leu927 were deleted due to their known involvement in the intramolecular interaction with the SAP97 SH3 domain, which could potentially block the binding of Cx32CT, as has been observed with other GUK-binding proteins (e.g. GKAP) (37). However, deleting portions of a protein to obtain minimal regions of interaction can disrupt secondary/tertiary structures that are important for the function of the native protein. Therefore, CD was used to determine whether the secondary structures of these minimal regions retain their native conformations.
FIGURE 2.
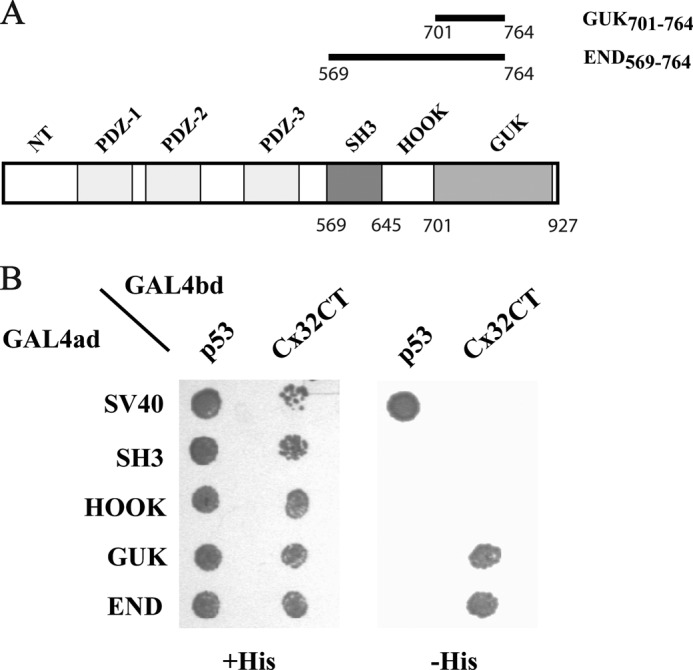
Characterization of the Cx32/SAP97 interaction by the Y2H. A, schematic representation of the SAP97 domains and constructs used in the Y2H assay. B, the Saccharomyces cerevisiae yeast strain AH109 was cotransformed with GAL4ad fusion constructs GAL4ad-SH3 (SAP97(569–644)), GAL4ad-HOOK (SAP97(645–700)), GAL4ad-GUK (SAP97(701–764)), GAL4ad-END (SAP97(569–764)), or GAL4ad-SV40 Large T antigen (control) and with GALbd fusion constructs GAL4bd-Cx32CT (Cx32(217–283)) or GAL4bd-pVA3 murine p53 (control). Cotransformants were assayed for growth on nonselective (+His) and selective (−His) media. Abbreviations are as follows; ad, activation domain; bd, DNA-binding domain.
The secondary structure of SAP97 GUK(701–764) shows a minimum near 198 nm indicating a random coiled structure (Fig. 3A). The crystal structure of the SH3-HOOK-GUK domains of SAP97 revealed that the GUK domain consists of an α-helical core and some β-strands, suggesting that deletion of the C-terminal residues is disrupting the secondary structure (38, 39). Additionally, even in the presence of TFE no secondary structure could be stabilized. Conversely, we observed that the presence of the C-terminal residues (Cys765-Leu927) was necessary for proper folding as evident by the minima around 208 and 220 nm, indicating the α-helical structure (Fig. 3B). The contribution of β-strands in far-UV is sometimes not visible and is overlapped by α-helical content, yet dicroweb was able to deconvolute the data to show that the contribution from β-strand was 13% and α-helix was 75% (40). A similar result was found when we investigated the SAP97 SH3-HOOK-GUK domains together (END). The C-terminal GUK residues in the END(569–927) construct (as compared with the END(569–764)) were necessary for a properly folded structure (Fig. 3, C versus D). In the presence of TFE, the α-helical structure was induced for the END(569–764), unlike GUK(701–764), suggesting the stabilization was in the SH3-HOOK domains (Fig. 3, C versus A).
FIGURE 3.

CD spectra of the SAP97 minimal regions in the presence of TFE. Mean residue ellipticity (MRE) as a function of wavelength was recorded at 7 °C at a protein concentration of 200 μm. CD spectra of SAP97 regions: A, GUK(701–764) B, GUK(701–927); C, END(569–764); and D, END(569–927) were collected in the absence and presence of varying concentrations of TFE (% displayed in each panel).
NMR titrations were used to confirm the Cx32CT interaction with the full-length SAP97 GUK(701–927) and END(569–927) domains and determine the KD. NMR experiments were performed with unlabeled SAP97 GUK(701–927) or END(569–927) titrated into a Tris-HCl buffer solution, pH 7.0, containing 15N-Cx32CT and 15N-HSQC spectra were acquired (Fig. 4, A and B). We calculated the KD for both the Cx32CT/GUK(701–927) and Cx32CT/END(569–927) interactions by holding the concentration of the 15N-labeled CT domain constant (100 μm) and titrating the unlabeled full-length GUK or END domain from 25 to 300 μm. The decreasing signal intensity for a subset of Cx32CT residues affected as a result of the increasing GUK or END concentrations (Fig. 4, A and B) were fit according to the nonlinear least squares method and the titration data provided a KD of 14.7 ± 5.9 μm (mean ± S.D.) for the Cx32CT/GUK(701–927) interaction and a KD of 25.3 ± 14.4 μm for the Cx32CT/END(569–927) interaction. The Cx32CT residues that are involved in the interaction with the SAP97 GUK(701–927) and END(569–927) domains were mapped onto the Cx32CT sequence (Fig. 4C). The affected residues for each interaction were similar and corresponded to the N-terminal portion of the Cx32CT (Cys217-Ser266). The strongly affected residues are suggested to be the direct site of SAP97 binding (Cys217-Gln245). The observation of similar dissociation kinetics between the Cx32CT and SAP97 domains with and without the SH3-HOOK suggest that the GUK domain alone is sufficient for the Cx32CT interaction consistent with the Y2H data. Additionally, the carboxyl-terminal residues Cys765-Leu927 of the GUK domain are necessary for a higher binding affinity with Cx32CT because the KD for the Cx32CT/GUK(701–764) interaction was in the low mm range (data not shown). Therefore, we used the SAP97 GUK(701–927) domain for differential CD studies to determine whether binding induces secondary structure for the Cx32CT domain. The addition of Cx32CT to SAP97 GUK(701–927) (1:1 molar ratio) results in no change in the CD spectrum, and the difference spectrum obtained by subtracting the GUK(701–927) spectrum from the GUK(701–927)/Cx32CT spectrum also showed no significant change because the minima remained at 200 nm (data not shown). These data suggest no change in the Cx32CT structure upon binding GUK(701–927).
FIGURE 4.

NMR studies characterizing the Cx32CT residue affected upon SAP97 binding. The Cx32CT was titrated with the SAP97 GUK(701–927) (A) or END(569–927) (B) domains. 15N-HSQC spectrum for Cx32CT alone (black) has been overlaid with spectra obtained in the presence of different concentrations of the SAP97 domains GUK(701–927) and END(569–927). The cross-peak color changes according to the concentration ratio (Cx32CT:SAP97 domain 1:0 (black), 1:0.25 (red), 1:0.75 (blue), 1:3 (green)). Titrations of 1:0.5 and 1:1 were also collected and used for calculating the dissociation constant (KD) (lower panels A and B), but were not included for easier visualization of the chemical shift changes. The KD for the Cx32CT/GUK(701–927) and Cx32CT/END(569–927) interaction was estimated by fitting the decrease in signal intensity for the Cx32CT residues Cys217, Ala218, Arg220-Asn226, Ser229, and Lys244 as a function of GUK(701–927) or END(569–927) concentration. C, schematic representation of the residues of Cx32CT affected by the binding of the SAP97 GUK(701–927) and END(569–927) domains. Highlighted in yellow are the residues that were most affected (peaks broaden beyond detection or shift with no overlap at 1:0.25 ratio) and those in green were less affected (shift at 1:0.75 ratio).
Interaction between the Cx32CT Domain and CaM
Gap junctions are regulated by intracellular Ca2+; such that high nanomolar to low micromolar concentrations inhibit gap junctional communication. CaM has been implicated in mediating the Ca2+-dependent regulation of gap junctions via interacting directly with the intracellular domains of connexins (13, 18, 19). A CaM-binding region has been identified in Cx32CT peptides (residues Ala216-Arg230 and Glu208-Asn226) and kinetic studies showed a KD of 2.1 and 3.5 μm, respectively (13, 41). Initially, ITC experiments were performed using our longer Cx32CT construct (Cys217-Cys283) to determine the stoichiometry of this interaction as well as confirm the KD. Our ITC results revealed that the Cx32CT/CaM interaction is 1:1 with a KD of 4.7 μm (Fig. 5A). In addition, the ITC results revealed that the binding of Cx32CT to CaM is exothermic and driven by an enthalpic component (Table 1).
FIGURE 5.

Thermodynamic and structural characterization of the interaction between the Cx32CT and CaM. A, ITC profile of the Cx32CT interaction with CaM. The ITC runs were performed at 37 °C. The upper halves show the original heat production upon injection and the lower show the integrated, dilution-corrected, and normalized peaks. B, 15N-HSQC titration of 15N-Cx32CT with unlabeled CaM in the presence of 5 mm Ca2+. Each titration contained the same concentration of 15N-Cx32CT (100 μm) with different concentrations of the CaM. The cross-peak color changes according to the concentration ratio (Cx32CT:CaM 1:0 (black), 1:0.25 (red), 1:0.5 (blue), 1:1.5 (green)). Titrations of 1:0.75, 1:1, and 1:3 were also collected, but were not included for easier visualization of the chemical shift changes. C, displayed is the amino acid sequence for the Cx32CT and the most affected residues due to titrated CaM have been highlighted in yellow (peaks broaden beyond detection or shift with no overlap at 1:0.25 ratio) and those less affected in green (peaks shift or broaden beyond detection at 1:0.5).
TABLE 1.
Thermodynamic parameters for the interaction of Cx32CT-WT and mutants with calmodulin
Experiments were done in buffer containing 25 mm Tris-HCl, 100 mm KCl, 1 mm β−ME, and 5 mm CaCl2 at pH 7.0. Experimental errors are as follows: ΔHb (±5%), ΔKb (±20%), ΔSb (±5%), and ΔGb (±7%).
| Protein | ΔHb | n | Kb × 104 | ΔGb0 | TΔS |
|---|---|---|---|---|---|
| cal/mol | m−1 | cal/mol | |||
| Cx32CT-WT | −8082 | 1.01 | 21.5 | −7567 | −514.6 |
| Cx32CT-R219H | −7376 | 1.00 | 9.6 | −7071 | −305.0 |
| Cx32CT-R230C | −6825 | 0.99 | 6.7 | −6851 | 26.1 |
| Cx32CT-F235C | −4558 | 1.00 | 2.6 | −6260 | 1701.9 |
The previously identified Cx32CT peptide (Glu208-Pro227) that interacts with CaM was shown to bind only one lobe of CaM at a time (41). Therefore, we performed NMR titration experiments using a longer, more native like Cx32CT construct (Cys217-Cys283) to test if both CaM lobes can bind simultaneously, consistent with the known binding mechanism for CaM. Unlabeled CaM was titrated into a Tris-HCl buffer solution, pH 7.0, containing 15N-Cx32CT, and 15N-HSQC spectra were acquired (Fig. 5B). We determined the Cx32CT residues affected by CaM binding and these are highlighted in Fig. 5C. Those residues most strongly affected (Cys217-Asn226) correspond to the previously identified CaM binding motif in the Cx32CT (13); however, we also observed residues affected downstream of this site within the Cx32CT sequence (Gly236-Ser266). These data, combined with the ITC data showing a 1:1 stoichiometry, suggest both the C- and N-lobes of CaM can interact at the same time with the Cx32CT domain. To confirm this observation, we investigated this interaction from the CaM point of view.
The NMR resonances of Ca2+-CaM have been assigned by several groups (42–44). By following their movements during titration of 15N-CaM with unlabeled Cx32CT with and without Ca2+, we were able to identify the affected CaM residues. In the absence of Ca2+, only small changes in a few CaM residues were observed (Fig. 6A); confirming the Ca2+ dependence for this interaction. In contrast, in the presence of Ca2+, a number of CaM resonances corresponding to residues in both the N- and C-lobes as well as the linker region showed significant intensity changes >10% following the addition of Cx32CT (Fig. 6, B and C). These NMR results strongly suggest that CaM folds around the Cx32CT, binding via both the N- and C-lobes.
FIGURE 6.

NMR studies characterizing the CaM residues affected upon Cx32CT binding. A, overlaid 15N-HSQC spectrum of apo-CaM (black) with apo-CaM-Cx32CT (red). B, change in chemical shift position on CaM binding to Cx32CT in the presence of 5 mm Ca2+. 15N-HSQC titration of 15N-CaM with unlabeled Cx32CT. Each titration contained the same concentration of 15N-CaM (70 μm) with different concentrations of the Cx32CT. The cross-peak color changes according to the concentration ratio (CaM:Cx32CT 1:0 (black), 1:0.5 (red), 1:0.75 (blue), 1:3 (green)). C, three-dimensional representation of perturbed residues in Ca2+-CaM (Protein Data Bank code 3CLN) upon binding to Cx32CT. Residues with amide proton resonance intensity changes over 10% are shown in blue (weaker interaction) and when peaks broaden beyond detection shown in red (stronger interaction). Calcium ions are shown in green.
A common mechanism upon CaM binding with both lobes is induction of α-helical structure for the target peptide/protein, as has been observed for the Cx43CL and Cx44CL (45, 46). To determine whether the α-helical structure can be induced for the Cx32CT upon CaM binding, the Cx32CT(217–283)/CaM interaction was examined structurally by differential CD. As seen in Fig. 7A, the addition of Cx32CT to Ca2+-CaM (1:1 molar ratio) results in a more negative signal at 222 nm in the CD spectrum. Studies have revealed no change in CaM secondary structure when bound to different peptides (47, 48), therefore the observed net change in the CD signal can be attributed to the CaM-bound Cx32CT. The difference spectrum obtained by subtracting the Ca2+-CaM spectrum from the Ca2+-CaM/Cx32CT spectrum showed the 198 nm minimum shift to 208 nm and the absorption at 222 nm increased, indicating a change to α-helical structure from the intrinsically disordered Cx32CT alone (Fig. 1A versus 7A). Comparison of the CaM/Cx32CT difference spectrum (Fig. 7A) with the spectrum of the Cx32CT in 25% TFE (Fig. 1C) reveals that both conditions stabilize the formation of similar α-helical structures because the minima are at 208 and 222 nm.
FIGURE 7.

CD studies of the interaction between Cx32CT and CaM. CD spectra of the CaM/wild-type Cx32CT (A), CaM/Cx32CT-R219H (B), CaM/Cx32CT-R230C (C), and CaM/Cx32CT-F235C (D) interactions. Top panels, CD spectra of CaM in the presence of 5 mm EGTA or 5 mm CaCl2 and a 1:1 CaM/Cx32CT mixture after subtracting the contribution from buffer and added Cx32CT. Spectra were recorded at 7 °C in 25 mm Tris, 100 mm KCl, pH 7.0. Lower panels, CD spectra of the Cx32CT (solid line) and the calculated difference spectrum (dashed line) by subtracting the spectrum of Ca2+-CaM from that of the Ca2+-CaM/Cx32CT mixture.
SAP97 and CaM Interaction with Cx32 in RT4-D6P2T Rat Schwannoma Cells
The importance of Cx32 within Schwann cells has been demonstrated by its involvement in a sensory and motor neuropathy called Charcot-Marie-Tooth disease (CMTX), which is caused by mutations throughout Cx32 (49, 50). Interestingly, the Cx32CT residues strongly affected in the presence of the GUK domain and CaM contain several residues that are mutated in CMTX (51). However, these interactions have not been identified in Schwann cells, which demyelinate as a result of the Cx32CT mutations.
Co-IP studies in the rat schwannoma cell line RT4-D6P2T were performed to identify if full-length Cx32 complexes with SAP97 and CaM (Fig. 8). The co-IP showed that SAP97 and CaM could pull-down Cx32 because the SAP97 and CaM antibody immunoprecipitates contained a 26-kDa protein that was immunoreactive to the Cx32 antibodies (Fig. 8, A and B). We also observed additional bands above the 26 kDa for the Cx32 protein, which suggests different phosphorylation states (52). Therefore, we added a general phosphatase (λ) to the lysate and Cx32 immunoprecipitates. The immunoblot revealed the loss of the upper band and retention of the lower 26-kDa band suggesting Cx32 is differentially phosphorylated in Schwann cells (Fig. 8C). To confirm the presence of phosphorylated Cx32, Cx32 antibody immunoprecipitates were immunoblotted with phospho-Tyr and phospho-Ser antibodies. In the presence of the phosphatase, no bands were detected. In the absence of phosphatase, the 26-kDa band was reactive to both the phospho-Tyr and phospho-Ser antibodies; however, the additional band above 26 kDa was only reactive to the phospho-Ser antibody (Fig. 8C). Because the Cx32/CaM interaction is known to be Ca2+-dependent we also added Ca2+ or EGTA to see if we could enhance/disrupt the interaction, respectively. This Cx32/CaM interaction was indeed sensitive to changes in Ca2+ levels in the precipitates suggesting that intracellular Ca2+ levels could increase this complex (Fig. 8B). The presence of Ca2+ in both pull-downs revealed a band lower than 26 kDa, which could be the result of Cx32 proteolysis caused by Ca2+-dependent calpains (53). Fig. 8D provides controls demonstrating that these proteins do not bind the protein A-Sepharose beads or the antibodies nonspecifically. The identification of these complexes using full-length proteins combined with the in vitro studies strongly suggests that Cx32 interacts directly with SAP97 and CaM in RT4-D6P2T rat schwannoma cells.
FIGURE 8.

Cx32, SAP97, and CaM coimmunoprecipitation studies using rat Schwannoma cell lysates. A, lysates of RT4-D6P2T cells were incubated with antibodies directed against SAP97 (SAP97 IP) and probed for Cx32 and SAP97. This co-IP was repeated in the presence of EGTA and Ca2+. B, lysates of RT4-D6P2T cells were incubated with antibodies directed against CaM (CaM IP) and probed for Cx32 and CaM. This co-IP was repeated in the presence of EGTA and Ca2+. C, lysates of RT4-D6P2T cells were incubated with antibodies directed against Cx32 (Cx32 IP) in the presence and absence of λ-phosphatase and probed for Cx32, phospho-Ser, and phospho-Tyr. D, lysates of RT4-D6P2T cells were incubated with antibodies directed against IgG (control IP), SAP97 (SAP97 IP), and CaM (CaM IP) and probed for Cx32, SAP97, and CaM. Input is the RT4-D6P2T lysate. Coimmunoprecipitation experiments were performed in triplicate.
Functional Studies of Cx32 Channels with CMTX Mutations in the CT Domain
The Cx32 mutations that cause CMTX affect every portion of the protein. The means by which many of these mutants lead to disease is fairly well understood (i.e. altered trafficking resulting in no channels, formation of nonfunctional channels or channels with altered biophysical properties at the plasma membrane) (51, 54). However, some CT mutations (F235C, R238H, C280G, and S281x) still form functional gap junction channels, suggesting other mechanisms cause CMTX (51, 55–57). One possibly, as in the case of F235C, is through altered interactions with protein partners that regulate channel function, such as CaM and SAP97 (57). Other Cx32CT CMTX mutations within the CaM and SAP97 binding domain form gap junctions, however, to the best of our knowledge, only the mutants mentioned above have been tested for functionality. Therefore, before testing if the CMTX mutations affect binding of CaM and SAP97 (next section), we performed functional studies on two additional Cx32CT CMTX mutations, R219H and R230C. These mutations were chosen to represent different areas of the SAP97 and CaM binding domain and different types of amino acid substitutions (e.g. change in charge, change in hydrophobicity, and a more conserved change). The junctional transfer of the fluorescent tracer, Lucifer Yellow (Mr 443) was measured by scrape-loading (Fig. 9), in transfected HeLa cells grown on glass coverslips. We found that the wild type Cx32 (Cx32-WT) transfected cells communicated extensively. The cells transfected with all three CT mutants (R219H, R230C, and F235C) were also able to form channels that were functional, however, the mean (± S.E.) number of fluorescent cells after scrape-loading was significantly reduced in the R219H- and R230C-transfected cells as compared with Cx32-WT (Fig. 9B). In HeLa cells, the Cx32-WT and mutants assembled into gap junctions at cell-cell contact regions, although some of the protein was also intracellular as observed by discrete puncta (Fig. 10). Interestingly, despite the formation of smaller gap junction plaques the F235C mutant transferred dye similar to WT.
FIGURE 9.

Gap junctional communication of Cx32-WT and CMTX mutants in HeLa cells. A, cells were grown on glass coverslips and scrape-loaded with Lucifer Yellow and Alexa 594-conjugated dextran. Note that control cells were not transfected and no transfer is observed, whereas Lucifer Yellow has transferred from scrape-loaded (red) cells to neighboring cells when transfected with Cx32-WT and all three mutants studied (R219H, R230C, and F235C). B, effect of Cx32 mutation on the gap junctional permeability expressed as the number of fluorescent cells. Cx32-WT, 45 ± 5.7%; Cx32-R219H, 34 ± 6.1%; Cx32-R230C, 30 ± 5.6%; Cx32-F235C, 53 ± 6.1%. Results are the mean ± S.E. values (n = 4). *, significantly (p < 0.05) different as compared with Cx32-WT and Cx32-F235C. Magnification at ×10.
FIGURE 10.

Localization of Cx32-WT and CMTX mutants in HeLa cells. HeLa cells were seeded on glass coverslips and immunostained for Cx32. Note that Cx32-WT and mutants (R219H, R230C, and F235C) assembled into gap junctions (white arrows). White dashed box within merge image was enlarged on the right to show the gap junctions at sites of cell-cell contact. The scrape line was located to the right of the imaged cells (white dashed line). Note also that some of the protein remains intracellular as vesicular puncta in HeLa cells. Magnification at ×63 oil using extended focus.
Interaction between Cx32CT CMTX Mutants and the SAP97 GUK Domain
SAP97 has been shown to maintain myelin homeostasis in Schwann cells and down-regulation of SAP97 leads to demyelination (5). We have identified that the SAP97 GUK domain interacts directly with the Cx32CT domain (Fig. 4) and the loss of Cx32 alters the levels, localization, and interactions of SAP97 (25). These findings suggest that any disruption of the Cx32/SAP97 interaction at the membrane, for example, by Cx32CT mutations, could alter myelin homeostasis.
To determine whether the Cx32CT mutations disrupt the binding of SAP97, we performed NMR titration experiments. The unlabeled SAP97 GUK domain was titrated into a Tris-HCl buffer solution, pH 7.0, containing 15N-labeled Cx32CT mutants and 15N-HSQC spectra were acquired (Fig. 11). We calculated the KD for these interactions by holding the concentration of the mutants constant (100 μm) and titrating the SAP97 GUK domain from 100 to 500 μm. Although the KD for the interaction between Cx32CT-WT and the SAP97 GUK domain was 14.7 μm (Fig. 4), all three of the mutations had a similar 8-fold decrease in the binding affinity for the GUK domain (Fig. 11).
FIGURE 11.

Characterizing the interaction between the Cx32CT CMTX mutants and the SAP97 GUK domain. Each titration contained the same concentration of 15N-Cx32CT mutant (100 μm) with different concentrations of the SAP97 GUK domain. 15N-HSQC spectrum for each Cx32CT mutant alone (black): R219H (A), R230C (B), and F235C (C) has been overlaid with spectra obtained in the presence of different concentrations of the SAP97 GUK (Cx32CT:SAP97 GUK domain 1:0 (black), 1:0.25 (red), 1:0.75 (blue), 1:3 (green), 1:5 (magenta)). Titrations of 1:0.5, 1:1, and 1:2.5 were also collected and used for calculating the dissociation constant (KD) (lower panels), but were not included for easier visualization of the resonance intensity changes. The KD for each interaction was estimated by fitting the normalized intensity change for the affected residues as a function of GUK concentration.
The crystal structure of the SAP97 SH3-HOOK-GUK domains in complex with a peptide revealed that the GUK domain can induce structure in its partner (38). Although no change in the Cx32CT-WT structure was observed upon binding to the SAP97 GUK domain, we used differential CD to test if the Cx32CT CMTX mutants behaved in a similar manner (data not shown). The data showed that the mutants interacted with the SAP97 GUK domain in a similar mode of binding as the Cx32CT-WT although with decreased affinity because no change in structure was observed.
Interaction between Cx32CT CMTX Mutants and CaM
Cx32 intracellular channels mediate the exchange of small molecules (e.g. Ca2+) across the Schwann cell myelin sheath. In response to increased Ca2+, CaM binds to the Cx32CT and Cx32NT domains closing Cx32 channels (13, 58). Interestingly, elevated Ca2+ in Schwann cell paranodes and incisures, where Cx32 gap junctions are located, is implicated in demyelination (20, 59). Therefore, if the Cx32/CaM interaction is disrupted, Ca2+ would be allowed to permeate throughout the layers of the myelin sheath ultimately leading to demyelination. NMR titrations were performed to determine whether the Cx32CT CMTX mutants affect the binding of CaM, as was observed for the SAP97 GUK domain.
NMR experiments were performed with unlabeled CaM titrated from 100 to 300 μm into a Tris-HCl buffer, pH 7.0, solution containing each 15N-Cx32CT CMTX mutant (100 μm) and 15N-HSQC spectra were acquired (Fig. 12, top). CaM interacted in the same general region of the CT for each of the mutants as compared with Cx32CT-WT; however, the peak intensities were different between the mutants and the Cx32CT-WT. Because the peaks broaden beyond detection even at the early titration points, the KD for each interaction could not be accurately determined; therefore, we used ITC experiments.
FIGURE 12.

Thermodynamic and structural characterization of the interaction between the Cx32CT CMTX mutants and CaM. NMR spectra and the ITC profile of the CaM/Cx32CT-R219H (A), CaM/Cx32CT-R230C (B), and CaM/Cx32CT-F235C (C) interactions. Top panels, each titration contained the same concentration of 15N-Cx32CT mutant (100 μm) with different concentrations of CaM. 15N-HSQC spectrum for each Cx32CT mutant alone (black) has been overlaid with spectra obtained in the presence of different concentrations of CaM (Cx32CT:CaM 1:0 (black), 1:0.25 (red), 1:0.5 (blue), 1:1.5 (green)). Lower panels, the ITC runs were performed at 37 °C. The upper halves show the original heat production upon injection and the lower panels show the integrated, dilution-corrected, and normalized peaks. IB, immunoblot.
Although the KD for the Cx32CT-WT interaction with CaM was 4.7 μm (Fig. 5), all three of the Cx32CT CMTX mutations caused a significant decrease in the binding affinity for CaM (Fig. 12, bottom, and Table 1). Among the CMTX mutants, Cx32CT-F235C caused the most significant effect, decreasing the KD to 388 μm, whereas the KD for the R230C and R219H mutants were 104 and 149 μm, respectively. Additionally, the ITC experiments revealed that the interaction between the Cx32CT and CaM is driven by exothermic enthalpy, which suggests that van der Waals, ionic bonds, and/or hydrogen bonds predominate over nonspecific hydrophobic forces (Table 1). This observation is consistent with the type of amino acids that were substituted (e.g. R219H and R230C, disrupting electrostatic interactions; F235C, disrupting hydrophobic interactions).
CaM was identified to induce the Cx32CT to form the α-helical structure (Fig. 7A). Because the mutations had a similar propensity as the Cx32CT-WT to form α-helical structure in the presence of TFE (data not shown), we used differential CD to test if the Cx32CT mutations alter the ability for the CT to adopt α-helical structure upon binding CaM. As seen in Fig. 7, B–D, the presence of CaM affected each of the mutants differently when compared with the Cx32CT-WT. The Cx32CT-R219H formed α-helical structure upon binding CaM although to a lesser extent than Cx32CT-WT. The decrease in α-helical constant is consistent with a lower KD (i.e. less Cx32CT binding CaM, less induced α-helical structure) and involves the binding of both the N- and C-lobes of CaM. On the other hand, the binding of CaM did not induce the formation of α-helical structure for the R230C and F235C mutants. The lack of structure and similar KD for the R230C mutant as compared with the R219H mutant suggests the binding of only one lobe of CaM. The lack of α-helical structure and even lower affinity for the F235C mutant also suggests that only one lobe of CaM can bind at a time. Additionally, the reduced binding affinity for the F235C mutant may reflect the importance of hydrophobic over electrostatic contacts for this direct interaction.
Effect of Cx32CT CMTX Mutants on Pull-downs of SAP97 and CaM
As a compliment to the in vitro studies using minimal domains, we assessed the ability of the full-length SAP97, CaM, and Cx32 proteins to interact when Cx32 contains the CT CMTX mutations. We performed co-IP in the connexin-deficient HeLa cell line transfected with the Cx32-WT and mutants (Fig. 13). The co-IP showed that the Cx32 mutants could all pull-down SAP97 as evident from the immunoprecipitates that contained a 130-kDa protein that was immunoreactive to the SAP97 antibodies (Fig. 13A). We also observed that CaM could pulldown the Cx32 mutants because the CaM antibody immunoprecipitates contained a 26-kDa protein that was immunoreactive to the Cx32 antibodies (Fig. 13B). However, all of the Cx32 mutations significantly reduced the amount of SAP97 that was pulled down and Cx32 that could be pulled down by CaM compared with Cx32-WT (Fig. 13, C and D). These results are consistent with the NMR and ITC data and suggest that the Cx32CT CMTX mutations tested in this study disrupt the binding of SAP97 and CaM in vivo providing a mechanism for demyelination.
FIGURE 13.

Cx32-WT and mutants, SAP97, and CaM co-IP studies using HeLa cell lysates. A, lysates of nontransfected and transfected (Cx32-WT and mutants) HeLa cells were incubated with antibodies directed against Cx32 (Cx32 IP) and probed for Cx32 and SAP97. B, lysates of nontransfected and transfected (Cx32-WT and mutants) HeLa cells were incubated with antibodies directed against CaM (CaM IP) and probed for Cx32 and CaM. Input is the HeLa lysate. Co-IP experiments were performed in triplicate. C, quantification of the data shown in A. SAP97 co-IP intensity was normalized to Cx32-WT IP (Cx32-WT, 100%; Cx32-R219H, 83.2 ± 2.5%; Cx32-R230C, 61.5 ± 3.3%; Cx32-F235C, 61.3 ± 2.1%). Results are the mean ± S.E. values (n = 3). a and b are significantly (p < 0.05) different as compared with Cx32-WT and each other. D, quantification of the data shown in B. Cx32 co-IP intensity was normalized to CaM IP (Cx32-WT, 100%; Cx32-R219H, 65.3 ± 3.1%; Cx32-R230C, 59.3 ± 3.3%; Cx32-F235C, 53.1 ± 3.6%). Results are the mean ± S.E. values (n = 3). c and d are significantly (p < 0.05) different as compared with Cx32-WT and each other.
DISCUSSION
In this study we have biophysically characterized the Cx32CT domain and its interaction with two molecular partners, SAP97 and CaM. The Cx32CT domain alone in solution exists as an intrinsically disordered protein; however, unlike the SA97 GUK domain, CaM induced α-helical structure. Both the SAP97 GUK domain and CaM interacted with similar Cx32CT residues, yet their mode of binding was different. These interactions were confirmed with full-length proteins in rat Schwannoma cells and Cx32 was found to be differentially serine and tyrosine phosphorylated. We also determined that three Cx32CT CMTX mutations significantly decreased the Cx32 binding affinity for SAP97 and CaM. These studies represent the first structural characterization of the CT domain of Cx32 alone and when integrated in a complex with molecular partners, providing insight into the molecular mechanisms involved in the regulation of Schwann cell myelination.
Functional Significance of the Cx32CT Secondary Structure
The Cx32CT structure was determined to be intrinsically disordered; however, CaM binding induced the Cx32CT to form α-helical structure. The ability to form the α-helical structure as observed for the CT domains from the α (Cx43) (12), β (Cx32), and γ (Cx45)3 subdivisions suggests this transitioning from a intrinsically disordered to α-helical structure is a general mechanism for connexins to facilitate a biological reaction, such as modulating protein-protein interactions. However, we did not observe a change in the Cx32CT structure when bound to the SAP97 GUK domain. The crystal structure of the SAP97 SH3-HOOK-GUK domains in complex with a peptide reveals that when the GUK domain binds to certain binding partners (e.g. mitotic spindle regulatory protein LGN) the interaction is inducing a one turn α-helix in the LGN protein (38). Therefore, the Cx32CT might also be induced to form the α-helical structure upon binding the GUK domain; however, this minimal structural change would be difficult to observe by differential CD.
Functional Significance of the Cx32CT/CaM Interaction
Cx32 gap junction channels mediate a pathway for Ca2+ signaling across the myelin sheath from the adaxonal cytoplasm to the perinuclear region of Schwann cells (7–9, 64). Under normal conditions, neuron activity results in ATP release that causes a rise in the intracellular Ca2+ levels in Schwann cells (20, 21). In response to increased intracellular levels of Ca2+, Cx32 channel closure is facilitated by the binding of activated CaM and Ca2+ is extruded via channels on the plasma membrane to maintain [Ca2+]i homeostasis (6, 18–20). However, if [Ca2+]i is maintained at elevated concentrations, the resulting effect is Schwann cell demyelination and proliferation (1, 20, 59). The Ca2+-mediated demyelination affects the paranodes and Schmidt-Lanterman incisures first and then progresses to disrupt the compact myelin (59). This observation emphasizes the importance of the Cx32 gap junctions because the localization of these channels is in the same area initially affected by increased Ca2+. The elevated Ca2+ would inhibit the Cx32/SAP97 interaction because CaM binds the same Cx32CT residues with higher affinity. However, if Ca2+ levels returned to normal, SAP97 could once again bind Cx32 to restore myelin homeostasis. This is consistent with the observation that after elevated [Ca2+]i, Schwann cells can recover and remyelinate axons (1, 59).
Our data revealed that all three Cx32CT CMTX mutations (R219H, R230C, and F235C) had reduced binding affinities for CaM suggesting that elevated Ca2+ levels would no longer close the Cx32 channels. Therefore, if the Cx32/CaM interaction is inhibited, Ca2+ would permeate throughout the layers of the Schwann cell ultimately leading to demyelination. Consistent with this hypothesis are the functional studies of the Cx32-F235C channels that were predominantly open (57) and our scrape-loading assay, which demonstrated that all three CMTX mutations form functional channels, albeit to different degrees.
Previous studies showing that CaM interacts directly with two small Cx32CT peptides (Ala216-Arg230 and Glu208-Asn226) suggested that only one lobe of CaM binds at a time with the C-lobe having a higher affinity than the N-lobe (13, 41). Using a longer Cx32CT construct (amino acids Cys217-Cys283), we confirmed that CaM interacts with Cx32CT residues Cys217-Arg230; however, we also observed additional affects extending to residue Ser266. This observation combined with CaM inducing a Cx32CT α-helical structure with a 1:1 stoichiometry, indicates that both lobes of CaM bind the Cx32CT at the same time. Consistent with this observation is that the Cx32CT domain affected both the N- and C-terminal lobes of CaM, indicating that a global conformational change occurred when bound to the Cx32CT domain. Previous reports of other CaM complexes, such as Ca2+-CaM/CaMKI, also showed such global changes in the amide chemical shifts (60).
Functional Significance of the Cx32CT/SAP97 GUK Interaction
The interaction between Cx32 and SAP97 was shown to be important for the proper localization of SAP97 to the cell membrane suggesting a role for Cx32 in the regulation of cell proliferation (25). SAP97 is enriched at Schwann cell paranodes where it coordinates membrane homeostasis during Schwann cell myelination via interacting with Mtmr2 to negatively regulate membrane addition and the exocyst protein Sec8 to promote membrane formation (5). Additionally, the loss of SAP97 expression and mutations in Mtmr2 individually both lead to Schwann cell demyelination (5, 61).
Our studies have identified that the SAP97 GUK domain is involved in the interaction with Cx32CT. This is important because Mtmr2 and Sec8 would be able to interact with the SAP97 PDZ2 and PDZ1 + 2 domains, respectively, when SAP97 is bound to Cx32 at the membrane (5). Our studies demonstrating that Cx32CT CMTX mutations cause a decrease in the binding affinity for the Cx32/SAP97 interaction strongly suggest that in Schwann cells these mutations disrupt the proper localization of SAP97, thus inhibiting Mtmr2- and Sec8-mediated myelin homeostasis.
Structural and functional studies of SAP97 and its homolog PSD-95 revealed that the intramolecular SH3 domain interaction with the GUK domain can regulate the binding of intermolecular partners (37, 38, 62). Using NMR we showed that the Cx32CT domain binds to SAP97 with a similar KD regardless of the presence of the SAP97 SH3 domain. Although the GKAP/SAP97 GUK interaction is inhibited by the SH3 domain (37), our data show that the presence of the SH3 domain does not interfere with Cx32CT binding, similar to SAP97 binding to the kinesin-3 motor protein GAKIN (63).
Relationship between Cx32/SAP97/CaM
The finding that SAP97 and CaM affect the same region of the Cx32CT suggests that both proteins cannot bind Cx32 at the same time. In general, the preference for binding could be regulated via several different mechanisms such as changes in cellular conditions and phosphorylation state of Cx32. Changes in intracellular Ca2+ levels could regulate which partner binds to the Cx32CT such that upon an increase in [Ca2+]i, SAP97 would be displaced by CaM. If this shift in partner preference occurred near the axon, the Ca2+-dependent closure of Cx32 channels by CaM would inhibit the diffusion of Ca2+ to the outer membrane thus maintaining the SAP97/Cx32 interaction at the site where Mtmr2 and Sec8 tethering is required for myelin homeostasis.
The phosphorylation of connexin proteins is involved in regulating gap junctional communication at several stages including trafficking, assembly/disassembly, degradation, as well as gating of the channels (66). The role that phosphorylation plays with regards to Cx32 has been less well characterized; however, some serine kinases have been identified such as cAMP-PK, Ca2+-CaM PK II, and PKC (67, 68). The Cx32CT contains several serine residues within the SAP97 and CaM binding sites that are suggested sites for phosphorylation, Ser229 and Ser240 as well as Ser233 (68). We observed that Cx32CT is differentially phosphorylated on tyrosine and serine residues in RT4-D6P2T rat schwannoma cells. The Cx32CT contains only one tyrosine residue (Tyr243), which is located within the SAP97 and CaM binding domain, and can be phosphorylated by the epidermal growth factor receptor (67). This suggests that alterations in the phosphorylation state of the Cx32CT could also play a role in regulating myelin homeostasis. Consistent with this possibility are studies showing that tyrosine phosphorylation of Cx32 is inhibited in the presence of CaM and CaM binding can be disrupted when its targets are phosphorylated (65, 67, 69). Therefore, any increased tyrosine kinase activity leading to Cx32CT phosphorylation could also decrease CaM binding leading to the same CMTX phenotype as the Cx32CT mutations. Consistent with this possibility is the observation that ErbB tyrosine kinase receptors (related to epidermal growth factor receptor) are up-regulated in CMT patients that exhibit demyelinated axons (70). Future studies will address if any of the serine residues also alter the binding affinity for the Cx32/CaM (and SAP97 GUK domain) interaction.
In summary, we describe here the biophysical characterization of the Cx32CT domain alone and in complex with two molecular partners, SAP97 and CaM. Our studies support the importance of protein-protein interactions in the regulation of Cx32 gap junction channels and for myelin homeostasis. The significance of these findings is enhanced in the context of CMTX, where mutations in the Cx32CT could cause demyelination by disrupting the binding of protein partners such as SAP97 and CaM.
Acknowledgments
We are grateful to Dr. Greg Taylor (Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center) for the RT4-D6P2T cells, Dr. Luis Marky (Department of Pharmaceutical Sciences, University of Nebraska Medical Center) for the use of the iTC200, Dr. Steve Caplan (Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center) for the HeLa cells, and Dr. Parmender Mehta and Kristen Johnson (Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center) for help with the scrape-loading assay.
This work was supported, in whole or in part, by National Institutes of Health Grant 2R01GM072631, American Heart Association Grant 11GRNT7820017, and Eppley Cancer Center Support Grant 2P30CA036727.
J. Kopanic and P. L. Sorgen, unpublished data.
- CaM
- calmodulin
- CL
- Cx32, connexin 32
- CT
- carboxyl-terminal
- SAP97
- synapse-associated protein 97
- GUK
- guanylate kinase domain
- ITC
- isothermal titration calorimetry
- TFE
- trifluoroethanol
- CMTX
- X-linked Charcot-Marie-Tooth disease
- Mtmr2
- myotubularin-related protein 2
- Y2H
- yeast two-hybrid
- IP
- immunoprecipitation
- SH3
- Src homology domain 3
- bd
- binding domain
- ad
- activation domain
- HSQC
- heteronuclear single quantum coherence.
REFERENCES
- 1. Fansa H., Keilhoff G., Horn T., Altmann S., Wolf G., Schneider W. (2000) Stimulation of Schwann cell proliferation and axonal regeneration by FK 506. Restor. Neurol. Neurosci. 16, 77–86 [PubMed] [Google Scholar]
- 2. Jessen K. R., Mirsky R. (2010) Control of Schwann cell myelination. F1000 Biol. Rep. 2:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maurel P., Salzer J. L. (2000) Axonal regulation of Schwann cell proliferation and survival and the initial events of myelination requires PI 3-kinase activity. J. Neurosci. 20, 4635–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Waegh S., Brady S. T. (1990) Altered slow axonal transport and regeneration in a myelin-deficient mutant mouse. The trembler as an in vivo model for Schwann cell-axon interactions. J. Neurosci. 10, 1855–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bolis A., Coviello S., Visigalli I., Taveggia C., Bachi A., Chishti A. H., Hanada T., Quattrini A., Previtali S. C., Biffi A., Bolino A. (2009) Dlg1, Sec8, and Mtmr2 regulate membrane homeostasis in Schwann cell myelination. J. Neurosci. 29, 8858–8870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mata M., Fink D. J. (1988) Calmodulin distribution in peripheral nerve. An EM immunocytochemical study. Brain Res. 475, 297–304 [DOI] [PubMed] [Google Scholar]
- 7. Niemann A., Berger P., Suter U. (2006) Pathomechanisms of mutant proteins in Charcot-Marie-Tooth disease. Neuromolecular Med. 8, 217–242 [DOI] [PubMed] [Google Scholar]
- 8. Nagy J. I., Dudek F. E., Rash J. E. (2004) Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Res. Brain Res. Rev. 47, 191–215 [DOI] [PubMed] [Google Scholar]
- 9. Ressot C., Bruzzone R. (2000) Connexin channels in Schwann cells and the development of the X-linked form of Charcot-Marie-Tooth disease. Brain Res. Brain Res. Rev. 32, 192–202 [DOI] [PubMed] [Google Scholar]
- 10. Scherer S. S., Xu Y. T., Messing A., Willecke K., Fischbeck K. H., Jeng L. J. (2005) Transgenic expression of human connexin 32 in myelinating Schwann cells prevents demyelination in connexin 32-null mice. J. Neurosci. 25, 1550–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scherer S. S., Xu Y. T., Nelles E., Fischbeck K., Willecke K., Bone L. J. (1998) Connexin 32-null mice develop demyelinating peripheral neuropathy. Glia 24, 8–20 [DOI] [PubMed] [Google Scholar]
- 12. Bouvier D., Spagnol G., Chenavas S., Kieken F., Vitrac H., Brownell S., Kellezi A., Forge V., Sorgen P. L. (2009) Characterization of the structure and intermolecular interactions between the connexin 40 and connexin 43 carboxyl-terminal and cytoplasmic loop domains. J. Biol. Chem. 284, 34257–34271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Török K., Stauffer K., Evans W. H. (1997) Connexin 32 of gap junctions contains two cytoplasmic calmodulin-binding domains. Biochem. J. 326, 479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ponsaerts R., De Vuyst E., Retamal M., D'hondt C., Vermeire D., Wang N., De Smedt H., Zimmermann P., Himpens B., Vereecke J., Leybaert L., Bultynck G. (2010) Intramolecular loop/tail interactions are essential for connexin 43-hemichannel activity. FASEB J. 24, 4378–4395 [DOI] [PubMed] [Google Scholar]
- 15. Dyson H. J., Wright P. E. (2002) Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 12, 54–60 [DOI] [PubMed] [Google Scholar]
- 16. Receveur-Bréchot V., Bourhis J. M., Uversky V. N., Canard B., Longhi S. (2006) Assessing protein disorder and induced folding. Proteins 62, 24–45 [DOI] [PubMed] [Google Scholar]
- 17. Verkhivker G. M., Bouzida D., Gehlhaar D. K., Rejto P. A., Freer S. T., Rose P. W. (2003) Simulating disorder-order transitions in molecular recognition of unstructured proteins. Where folding meets binding. Proc. Natl. Acad. Sci. U.S.A. 100, 5148–5153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen Y., Zhou Y., Lin X., Wong H. C., Xu Q., Jiang J., Wang S., Lurtz M. M., Louis C. F., Veenstra R. D., Yang J. J. (2011) Molecular interaction and functional regulation of connexin 50 gap junctions by calmodulin. Biochem. J. 435, 711–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lurtz M. M., Louis C. F. (2003) Calmodulin and protein kinase C regulate gap junctional coupling in lens epithelial cells. Am. J. Physiol. Cell Physiol. 285, C1475–1482 [DOI] [PubMed] [Google Scholar]
- 20. Nobbio L., Sturla L., Fiorese F., Usai C., Basile G., Moreschi I., Benvenuto F., Zocchi E., De Flora A., Schenone A., Bruzzone S. (2009) P2X7-mediated increased intracellular calcium causes functional derangement in Schwann cells from rats with CMT1A neuropathy. J. Biol. Chem. 284, 23146–23158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stevens B., Fields R. D. (2000) Response of Schwann cells to action potentials in development. Science 287, 2267–2271 [DOI] [PubMed] [Google Scholar]
- 22. Dimitratos S. D., Woods D. F., Stathakis D. G., Bryant P. J. (1999) Signaling pathways are focused at specialized regions of the plasma membrane by scaffolding proteins of the MAGUK family. Bioessays 21, 912–921 [DOI] [PubMed] [Google Scholar]
- 23. Fanning A. S., Anderson J. M. (1999) Protein modules as organizers of membrane structure. Curr. Opin. Cell Biol. 11, 432–439 [DOI] [PubMed] [Google Scholar]
- 24. Hibino H., Inanobe A., Tanemoto M., Fujita A., Doi K., Kubo T., Hata Y., Takai Y., Kurachi Y. (2000) Anchoring proteins confer G protein sensitivity to an inward-rectifier K+ channel through the GK domain. EMBO J. 19, 78–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Duffy H. S., Iacobas I., Hotchkiss K., Hirst-Jensen B. J., Bosco A., Dandachi N., Dermietzel R., Sorgen P. L., Spray D. C. (2007) The gap junction protein connexin32 interacts with the Src homology 3/hook domain of discs large homolog 1. J. Biol. Chem. 282, 9789–9796 [DOI] [PubMed] [Google Scholar]
- 26. Duffy H. S., Sorgen P. L., Girvin M. E., O'Donnell P., Coombs W., Taffet S. M., Delmar M., Spray D. C. (2002) pH-dependent intramolecular binding and structure involving Cx43 cytoplasmic domains. J. Biol. Chem. 277, 36706–36714 [DOI] [PubMed] [Google Scholar]
- 27. Török K., Tzortzopoulos A., Grabarek Z., Best S. L., Thorogate R. (2001) Dual effect of ATP in the activation mechanism of brain Ca2+/calmodulin-dependent protein kinase II by Ca2+/calmodulin. Biochemistry 40, 14878–14890 [DOI] [PubMed] [Google Scholar]
- 28. Lees J. G., Smith B. R., Wien F., Miles A. J., Wallace B. A. (2004) CDtool-an integrated software package for circular dichroism spectroscopic data processing, analysis, and archiving. Anal. Biochem. 332, 285–289 [DOI] [PubMed] [Google Scholar]
- 29. Bouvier D., Kieken F., Sorgen P. L. (2007) 1H, 13C, and 15N backbone resonance assignments of the carboxyl-terminal domain of connexin 40. Biomol. NMR Assign. 1, 155–157 [DOI] [PubMed] [Google Scholar]
- 30. Kay E., Kiefer P., Saarinen T. (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114, 10663–10665 [Google Scholar]
- 31. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 32. Johnson B., Blevins R. (1994) NMRView: a program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 33. Chakraborty S., Mitra S., Falk M. M., Caplan S. H., Wheelock M. J., Johnson K. R., Mehta P. P. (2010) E-cadherin differentially regulates the assembly of connexin 43 and connexin 32 into gap junctions in human squamous carcinoma cells. J. Biol. Chem. 285, 10761–10776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Govindarajan R., Zhao S., Song X. H., Guo R. J., Wheelock M., Johnson K. R., Mehta P. P. (2002) Impaired trafficking of connexins in androgen-independent human prostate cancer cell lines and its mitigation by α-catenin. J. Biol. Chem. 277, 50087–50097 [DOI] [PubMed] [Google Scholar]
- 35. Yang J. J., Buck M., Pitkeathly M., Kotik M., Haynie D. T., Dobson C. M., Radford S. E. (1995) Conformational properties of four peptides spanning the sequence of hen lysozyme. J. Mol. Biol. 252, 483–491 [DOI] [PubMed] [Google Scholar]
- 36. Brokx R. D., Scheek R. M., Weljie A. M., Vogel H. J. (2004) Backbone dynamic properties of the central linker region of calcium-calmodulin in 35% trifluoroethanol. J. Struct. Biol. 146, 272–280 [DOI] [PubMed] [Google Scholar]
- 37. Wu H., Reissner C., Kuhlendahl S., Coblentz B., Reuver S., Kindler S., Gundelfinger E. D., Garner C. C. (2000) Intramolecular interactions regulate SAP97 binding to GKAP. EMBO J. 19, 5740–5751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhu J., Shang Y., Xia C., Wang W., Wen W., Zhang M. (2011) Guanylate kinase domains of the MAGUK family scaffold proteins as specific phosphoprotein-binding modules. EMBO J. 30, 4986–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tavares G. A., Panepucci E. H., Brunger A. T. (2001) Structural characterization of the intramolecular interaction between the SH3 and guanylate kinase domains of PSD-95. Mol. Cell 8, 1313–1325 [DOI] [PubMed] [Google Scholar]
- 40. Whitmore L., Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dodd R., Peracchia C., Stolady D., Török K. (2008) Calmodulin association with connexin 32-derived peptides suggests trans-domain interaction in chemical gating of gap junction channels. J. Biol. Chem. 283, 26911–26920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chou J. J., Li S., Klee C. B., Bax A. (2001) Solution structure of Ca2+-calmodulin reveals flexible hand-like properties of its domains. Nat. Struct. Biol. 8, 990–997 [DOI] [PubMed] [Google Scholar]
- 43. Ikura M., Clore G. M., Gronenborn A. M., Zhu G., Klee C. B., Bax A. (1992) Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science 256, 632–638 [DOI] [PubMed] [Google Scholar]
- 44. Ikura M., Spera S., Barbato G., Kay L. E., Krinks M., Bax A. (1991) Secondary structure and side chain 1H and 13C resonance assignments of calmodulin in solution by heteronuclear multidimensional NMR spectroscopy. Biochemistry 30, 9216–9228 [DOI] [PubMed] [Google Scholar]
- 45. Zhou Y., Yang W., Lurtz M. M., Ye Y., Huang Y., Lee H. W., Chen Y., Louis C. F., Yang J. J. (2007) Identification of the calmodulin binding domain of connexin 43. J. Biol. Chem. 282, 35005–35017 [DOI] [PubMed] [Google Scholar]
- 46. Zhou Y., Yang W., Lurtz M. M., Chen Y., Jiang J., Huang Y., Louis C. F., Yang J. J. (2009) Calmodulin mediates the Ca2+-dependent regulation of Cx44 gap junctions. Biophys. J. 96, 2832–2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gifford J. L., Ishida H., Vogel H. J. (2011) Fast methionine-based solution structure determination of calcium-calmodulin complexes. J. Biomol. NMR. 50, 71–81 [DOI] [PubMed] [Google Scholar]
- 48. Ogura K., Kumeta H., Takahasi K., Kobashigawa Y., Yoshida R., Itoh H., Yazawa M., Inagaki F. (2012) Solution structures of yeast Saccharomyces cerevisiae calmodulin in calcium- and target peptide-bound states reveal similarities and differences to vertebrate calmodulin. Genes Cells. 17, 159–172 [DOI] [PubMed] [Google Scholar]
- 49. Bergoffen J., Scherer S. S., Wang S., Scott M. O., Bone L. J., Paul D. L., Chen K., Lensch M. W., Chance P. F., Fischbeck K. H. (1993) Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262, 2039–2042 [DOI] [PubMed] [Google Scholar]
- 50. Hahn A. F., Bolton C. F., White C. M., Brown W. F., Tuuha S. E., Tan C. C., Ainsworth P. J. (1999) Genotype/phenotype correlations in X-linked dominant Charcot-Marie-Tooth disease. Ann. N.Y. Acad. Sci. 883, 366–382 [PubMed] [Google Scholar]
- 51. Yum S. W., Kleopa K. A., Shumas S., Scherer S. S. (2002) Diverse trafficking abnormalities of connexin 32 mutants causing CMTX. Neurobiol. Dis. 11, 43–52 [DOI] [PubMed] [Google Scholar]
- 52. Musil L. S., Cunningham B. A., Edelman G. M., Goodenough D. A. (1990) Differential phosphorylation of the gap junction protein connexin 43 in junctional communication-competent and -deficient cell lines. J. Cell Biol. 111, 2077–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Elvira M., Díez J. A., Wang K. K., Villalobo A. (1993) Phosphorylation of connexin 32 by protein kinase C prevents its proteolysis by μ-calpain and m-calpain. J. Biol. Chem. 268, 14294–14300 [PubMed] [Google Scholar]
- 54. Deschênes S. M., Walcott J. L., Wexler T. L., Scherer S. S., Fischbeck K. H. (1997) Altered trafficking of mutant connexin 32. J. Neurosci. 17, 9077–9084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Castro C., Gómez-Hernandez J. M., Silander K., Barrio L. C. (1999) Altered formation of hemichannels and gap junction channels caused by C-terminal connexin 32 mutations. J. Neurosci. 19, 3752–3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huang Y., Sirkowski E. E., Stickney J. T., Scherer S. S. (2005) Prenylation-defective human connexin32 mutants are normally localized and function equivalently to wild-type connexin 32 in myelinating Schwann cells. J. Neurosci. 25, 7111–7120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liang G. S., de Miguel M., Gómez-Hernández J. M., Glass J. D., Scherer S. S., Mintz M., Barrio L. C., Fischbeck K. H. (2005) Severe neuropathy with leaky connexin32 hemichannels. Ann. Neurol. 57, 749–754 [DOI] [PubMed] [Google Scholar]
- 58. Peracchia C. (2004) Chemical gating of gap junction channels. Roles of calcium, pH, and calmodulin. Biochim. Biophys. Acta 1662, 61–80 [DOI] [PubMed] [Google Scholar]
- 59. Smith K. J., Hall S. M. (1988) Peripheral demyelination and remyelination initiated by the calcium-selective ionophore ionomycin. In vivo observations. J. Neurol. Sci. 83, 37–53 [DOI] [PubMed] [Google Scholar]
- 60. Mal T. K., Skrynnikov N. R., Yap K. L., Kay L. E., Ikura M. (2002) Detecting protein kinase recognition modes of calmodulin by residual dipolar couplings in solution NMR. Biochemistry 41, 12899–12906 [DOI] [PubMed] [Google Scholar]
- 61. Bolino A., Muglia M., Conforti F. L., LeGuern E., Salih M. A., Georgiou D. M., Christodoulou K., Hausmanowa-Petrusewicz I., Mandich P., Schenone A., Gambardella A., Bono F., Quattrone A., Devoto M., Monaco A. P. (2000) Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat. Genet. 25, 17–19 [DOI] [PubMed] [Google Scholar]
- 62. Nix S. L., Chishti A. H., Anderson J. M., Walther Z. (2000) hCASK and hDlg associate in epithelia, and their src homology 3 and guanylate kinase domains participate in both intramolecular and intermolecular interactions. J. Biol. Chem. 275, 41192–41200 [DOI] [PubMed] [Google Scholar]
- 63. Yamada K. H., Hanada T., Chishti A. H. (2007) The effector domain of human Dlg tumor suppressor acts as a switch that relieves autoinhibition of kinesin-3 motor GAKIN/KIF13B. Biochemistry 46, 10039–10045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meier C., Dermietzel R., Davidson K. G., Yasumura T., Rash J. E. (2004) Connexin32-containing gap junctions in Schwann cells at the internodal zone of partial myelin compaction and in Schmidt-Lanterman incisures. J. Neurosci. 24, 3186–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sotkis A., Wang X. G., Yasumura T., Peracchia L. L., Persechini A., Rash J. E., Peracchia C. (2001) Calmodulin colocalizes with connexins and plays a direct role in gap junction channel gating. Cell Commun. Adhes. 8, 277–281 [DOI] [PubMed] [Google Scholar]
- 66. Lampe P. D., Lau A. F. (2004) The effects of connexin phosphorylation on gap junctional communication. Int. J. Biochem. Cell Biol. 36, 1171–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Díez J. A., Elvira M., Villalobo A. (1995) Phosphorylation of connexin 32 by the epidermal growth factor receptor tyrosine kinase. Ann. N.Y. Acad. Sci. 766, 477–480 [DOI] [PubMed] [Google Scholar]
- 68. Sáez J. C., Nairn A. C., Czernik A. J., Spray D. C., Hertzberg E. L., Greengard P., Bennett M. V. (1990) Phosphorylation of connexin 32, a hepatocyte gap-junction protein, by cAMP-dependent protein kinase, protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Eur. J. Biochem. 192, 263–273 [DOI] [PubMed] [Google Scholar]
- 69. McIlroy B. K., Walters J. D., Blackshear P. J., Johnson J. D. (1991) Phosphorylation-dependent binding of a synthetic MARCKS peptide to calmodulin. J. Biol. Chem. 266, 4959–4964 [PubMed] [Google Scholar]
- 70. Massa R., Palumbo C., Cavallaro T., Panico M. B., Bei R., Terracciano C., Rizzuto N., Bernardi G., Modesti A. (2006) Overexpression of ErbB2 and ErbB3 receptors in Schwann cells of patients with Charcot-Marie-tooth disease type 1A. Muscle Nerve 33, 342–349 [DOI] [PubMed] [Google Scholar]