

Background: A putative Lon protease has been identified in peroxisomes of various species (Pln).
Results: Pln is an ATP-dependent protease that digests unfolded substrates e.g. oxidatively damaged catalase-peroxidase, and displays chaperone-like activity, circumventing accumulation of protein aggregates in peroxisomes that compromise organelle function.
Conclusion: Pln is a bifunctional protein with chaperone and protease activities.
Significance: Pln is crucial for peroxisome proteostasis.
Keywords: Molecular Chaperone, Oxidative Stress, Peroxisomes, Protease, Protein Aggregation, Lon Protease, Protein Quality Control
Abstract
Proteins are subject to continuous quality control for optimal proteostasis. The knowledge of peroxisome quality control systems is still in its infancy. Here we show that peroxisomes contain a member of the Lon family of proteases (Pln). We show that Pln is a heptameric protein and acts as an ATP-fueled protease and chaperone. Hence, Pln is the first chaperone identified in fungal peroxisomes. In cells of a PLN deletion strain peroxisomes contain protein aggregates, a major component of which is catalase-peroxidase. We show that this enzyme is sensitive to oxidative damage. The oxidatively damaged, but not the native protein, is a substrate of the Pln protease. Cells of the pln strain contain enhanced levels of catalase-peroxidase protein but reduced catalase-peroxidase enzyme activities. Together with the observation that Pln has chaperone activity in vitro, our data suggest that catalase-peroxidase aggregates accumulate in peroxisomes of pln cells due to the combined absence of Pln protease and chaperone activities.
Introduction
Functional proteins generally adapt a defined three-dimensional structure, designated their native fold. Nevertheless, they need to retain conformational flexibility to allow functioning, and as a consequence, they are only marginally thermodynamically stable macromolecules. Hence, preexisting folded proteins are at constant risk of being unfolded, especially at environmental stress conditions (1–3). Aberrantly folded polypeptides as well as folding intermediates expose hydrophobic residues that are normally buried in the native fold and are prone to trigger protein aggregation (4). Aggregation of misfolded protein species is linked with the perturbation of cellular functions, aging, and various human disorders (5). To circumvent this, cells evolved sophisticated protein quality control systems, among which are molecular chaperones that facilitate folding and refolding of polypeptides and proteolytic machines that remove misfolded conformers from the cellular interior (6). These systems are indispensable for protein homeostasis (proteostasis) in cells (7).
Yet little is known of protein quality control in peroxisomes. Despite their simple architecture, peroxisomes are important organelles that display an unprecedented diversity of functions depending on metabolic needs and external stimuli (8). They are crucial in man, manifested by the presence various inherited disorders (i.e. neonatal adrenoleukodystrophy, Zellweger syndrome, infantile Refsum's disease, rhizomelic chondrodysplasia punctata) caused by impairment of one or more peroxisomal functions (9, 10). The hallmark of peroxisomes is the metabolism of hydrogen peroxide that is generated as a byproduct of the organelle oxidative metabolism (11). Hydrogen peroxide can be the source of the most highly reactive and toxic form of reactive oxygen species, the hydroxyl radical (•OH), which is generated in the Fenton reaction (12). To cope with reactive oxygen species formation, peroxisomes contain a set of peroxide decomposing enzymes among which are catalase, peroxiredoxins, or peroxidases (11). Peroxisomes may import folded proteins and even oligomeric protein complexes (13) rendering these organelles attractive models to analyze the mechanisms involved in housekeeping of preexisting, folded proteins.
Recently, a conserved ATP-dependent Lon protease has been identified in peroxisomes (14, 15). The mitochondrial isoform of this protease was shown to represent the main quality control protease of the organelle matrix (16, 17). Mitochondrial Lon selectively degrades misfolded, unassembled, and damaged proteins (18, 19). However, the molecular function of the peroxisomal Lon protease is not yet clear.
In this work we demonstrate that peroxisomal Lon is a multifunctional protein that acts as a protease, selectively degrading aberrant peroxisomal matrix proteins, but also assists in refolding of these components. As such, Lon is the first peroxisomal chaperone identified in fungi so far.
EXPERIMENTAL PROCEDURES
Strains and Growth Conditions
Penicillium chrysogenum strains used in this study are listed in Table 1. Cells were grown at 25 °C on mineral medium plates (20) supplemented with 1% glucose, 1% ethanol, or 1% oleic acid or in batch cultures on YGG medium (21), penicillin (PEN)4 production medium supplemented with 0.05% phenylacetic acid (22), or in mineral medium containing 0.1% oleic acid and 0.05% Tween 80. To induce conidiospore formation, R-agar was used (21). Acetamide-resistant (AmdS+) transformants were selected on plates containing 0.1% acetamide (23).
TABLE 1.
P. chrysogenum strains used in this study
| Strain | Genotype or characteristic | Reference |
|---|---|---|
| DS54465 GFP-SKL | DS54465 with integrated PgpdA-GFP.SKL-TpenDE cassette at niaD locus, AmdS−, chlorate-resistant | (50) |
| pln::GFP-SKL | DS54465 GFP-SKL with deletion of pln gene, chlorate-resistant, AmdS+ | This study |
| DsRed-SKL | DS17690 with integrated PpcbC-DsRed.SKL-TpenDE cassette, AmdS− | (26) |
| DsRed-SKL GFP.Pln | DsRed-SKL with integrated PgpdA-mGFP.pln-TpenDE, AmdS+ | This study |
| DsRed-SKL GFP-catalase-peroxidase | DsRed-SKL with integrated PgpdA-mGFP.catalase-peroxidase-TpenDE, AmdS+ | This study |
| DsRed-SKL GFP-chDEH | DsRed-SKL with integrated PgpdA-mGFP.chDEH-TpenDE, AmdS+ | This study |
To determine fungal autolysis, P. chrysogenum strains were cultivated for 120 h in PEN media. Subsequently, each culture was divided into two halves, and 0.1% oleic acid and 0.05% Tween 80 were added to one-half. After incubation for 24 h, fungal biomass was determined as described previously (21). Escherichia coli DH5α was used for cloning purposes. E. coli Rosetta 2 cells (Novagen) were used for production of heterologous protein. E. coli cells were grown at 37 °C on LB medium supplemented with the appropriate antibiotics.
Molecular Techniques
Plasmids and oligonucleotides used in this study are listed in Tables 2 and 3. Standard DNA manipulations were performed according to established procedures (24). Transformation of P. chrysogenum protoplasts was performed as described previously (23). All PCR fragments were sequenced (Service XS). Restriction enzymes (Fermentas), T4 DNA ligase (Fermentas), and other DNA-modifying enzymes were used as recommended by the suppliers. PCR was carried out using high fidelity Phusion (Fermentas) polymerase. Southern blotting was performed according to the DIG High Prime labeling and detection kit (Roche Applied Science). In silico analysis of DNA sequences and construction of vector maps was carried out using Clone Manager 5 software (Scientific and Educational Software, Durham, NC).
TABLE 2.
Plasmids used in this study
| Name | Description | Source/Reference |
|---|---|---|
| pDONR P4-P1R | Multisite Gateway vector, KanR CmR | Invitrogen |
| pDEST R4-R3 | Multisite Gateway vector, AmpR CmR | Invitrogen |
| pDONR 221 | Multisite Gateway vector, KanR CmR | Invitrogen |
| pDONR P2R-P3 | Multisite Gateway vector, KanR CmR | Invitrogen |
| pENTR221/PniadAMDS | pDONR 221 vector with PgpdA-amdS cassette flanked by niaD sequences, KanR | Gift from J. G. Nijland |
| pENTR2.3-PLNdownstream | pDONR P2R-P3 with 3′-flanking region of pln, KanR | This study |
| pENTR4.1-PLNupstream | pDONR P4-P1R with 5′-flanking region of pln, KanR | This study |
| pDest43_ΔPcPLN | pDEST R4-R3 with pln deletion cassette, AmpR | This study |
| pENTR221_PcPLN | pDONR 221 with pln CDS with stop codon, KanR | This study |
| pENTR41/PpcbCGFP | pDONR P4-P1R with pcbC promoter and eGFP gene lacking a stop codon, KanR | (26) |
| pENTR23-His8.TpenDE | His-tag with C-terminal terminator of penDE gene, KanR | (59) |
| pDEST R4-R3/AMDS | pDESTR4-R3 with PgpdA-amdS cassette, AmpR | (26) |
| pDest43.pIPNS.eGFP.pcPLN.Tat | Expression vector with PpcbC-eGFP.pln-TpenDE cassette, AmpR | This study |
| LMOP41pgpdAmGFP | pDONR P4-P1R with gpdA promoter and mGFP gene lacking a stop codon, KanR | This study |
| pDONR221KatG | pDONR 221 with catalase-peroxidase CDS with stop codon, KanR | This study |
| pGFP.KatG | Expression vector with PgpdA-mGFP-catalase-peroxidase-TpenDE cassette, AmpR | This study |
| pBBK-007 | Plasmid with PgpdA-DsRed.SKL-TpenDE expression cassette, AmpR | (50) |
| pGBRH2-mGFP | pBluescript vector with pcbC promoter, mGFP gene, and penDE terminator, AmpR | (26) |
| LMOP-Pgpda-mGFP | pBluescript vector with gpdA promoter, mGFP gene, and penDE terminator, AmpR | This study |
| pDONR221chDEH | pDONR 221 with oxidoreductase CDS with stop codon, KanR | This study |
| pGFP.chDEH | Expression vector with PgpdA-mGFP.oxidoreductase-TpenDE cassette, AmpR | This study |
| pMAL-c2 | Expression vector for N-terminal MBP-tag fusions, AmpR | New England Biolabs |
| pMDBPlnMBP | Expression vector containing P. chrysogenum Pln fused N-terminally to maltose-binding protein tag, AmpR | This study |
| pMDBPlnHis | Expression vector containing P. chrysogenum Pln fused N-terminally to maltose-binding protein tag and C-terminally to the His tag, AmpR | This study |
| pMDBLONTEV | Expression vector containing P. chrysogenum Pln fused N-terminally to maltose-binding protein tag and C-terminally to the His tag, with TEV protease cleavage site between MBP and Pln, AmpR | This study |
| pMDBPlninMBP | Expression vector containing proteolytically inactive mutant of P. chrysogenum Pln, Plnin, fused N-terminally to maltose-binding protein tag, AmpR | This study |
| pMDBPlninHis | Expression vector containing proteolytically inactive mutant of P. chrysogenum Pln, Plnin, fused N-terminally to maltose-binding protein tag and C-terminally to the His tag, AmpR | This study |
| pMDBLONinTEV | Expression vector containing proteolytically inactive mutant of P. chrysogenum Pln, Plnin, fused N-terminally to maltose-binding protein tag and C-terminally to the His tag, with TEV protease cleavage site between MBP and Plnin, AmpR | This study |
| pGEX-4T-2 | Expression vector for N-terminal GST-tag fusions, AmpR | GE Healthcare |
| pGSTKatG | Expression vector containing P. chrysogenum catalase-peroxidase fused N-terminally to glutathione S-transferase tag, AmpR | This study |
TABLE 3.
Oligonucleotides used in this study
| Name | Sequence (5′-3′) |
|---|---|
| AKR0007.PlnB4 | GGGGACAACTTTGTATAGAAAAGTTGTGATATCTTGAACGGGGACG |
| AKR0008.PlnB1 | GGGGACTGCTTTTTTGTACAAACTTGCAGGCCCTAGTGATGATGGAATGG |
| AKR0009.PlnB2 | GGGGACAGCTTTCTTGTACAAAGTGGTCAGAGACGAGGCTGAAATCC |
| AKR0010.PlnB3 | GGGGACAACTTTGTATAATAAAGTTGTATTGACTGGCATGAACGCATCAC |
| AKR0005.PlnB1 | GGGGACAAGTTTGTACAAAAAAGCAGGCTTGATGGGAACAAACGGTAGAAGTACC |
| AKR0006.PlnB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTTCACAGTCGGCTCTCAACGAAG |
| KatGfor | GGGGACAAGTTTGTACAAAAAAGCAGGCTTTATGAGCGAATGTCCTGTTGCGC |
| KatGrev | GGGGACCACTTTGTACAAGAAAGCTGGGTTTCACAGGCGAGGACGACC |
| LMOp080 | GGGGACAACTTTGTATAGAAAAGTTGTTACCGCGGCCGCTCTGTACAGTGAC |
| LMOp081 | GGGGACTGCTTTTTTGTACAAACTTGCCTTGTACAGCTCGTCCATGCCGAG |
| chDEHfor | GGGGACAAGTTTGTACAAAAAAGCAGGCTTTATG GCCACCACGAACGAGTTC |
| chDEHrev | GGGGACCACTTTGTACAAGAAAGCTGGGTTTTACAGACGGGGCTGGTTGC |
| MDBPlnfor | ATAGGATCCATGGGAACAAACGGTAGAAGTACC |
| MDBPlnrev | AAACTGCAGTCACAGTCGGCTCTCAAC |
| MDBhisfor | CGTTGAGAGCCGACTGCATCACCATCACCATCACTGACTGCAGGCAAGC |
| MDBhisrev | GCTTGCCTGCAGTCAGTGATGGTGATGGTGATGCAGTCGGCTCTCAACG |
| MDB.TEV.for | GGATCGAGGGAAGGGAAAATCTTTACTTTCAAGGTATTTCAGAATTCGG |
| MDB.TEV.rev | CCGAATTCTGAAATACCTTGAAAGTAAAGATTTTCCCTTCCCTCGATCC |
| lonmutfor | CCCAAGGATGGTCCTGCTGCTGGTCTTGCCCACGCC |
| lonmutrev | GGCGTGGGCAAGACCAGCAGCAGGACCATCCTTGGG |
| GST.KatGfor | AAAGGATCCGAAAATCTTTACTTTCAAGGTATGAGCGAATGTCCTGTTGCGC |
| GST.KatGrev | AAAGAATTCTCACAGATCTTCTTCAGAAATAAGTTTTTGTTCCAGGCGAGGACGACCGGT |
Construction of the P. chrysogenum Pln Deletion Strain
The pln deletion cassette was constructed using the Multisite Gateway cloning system. The upstream (1.759 bp) and downstream (1.474 bp) flanking regions of pln gene were amplified by PCR with the primers AKR0007.PlnB4 and AKR0008.PlnB1 and with AKR0009.PlnB2 and AKR0010.PlnB3, respectively, using DS54465 genomic DNA as template. The resulting PCR products were introduced into pDONRP2R-P3 (3′-flanking region) and pDONRP4-P1R (5′-flanking region of pln) by using the Gateway BP clonase reaction, resulting in plasmids pENTR2.3-PLNdownstream and pENTR4.1-PLNupstream, respectively. Next, a Gateway LR reaction was performed using pENTR4.1-PLNupstream, pENTR221/PniadAMDS, pENTR2.3-PLNdownstream, and the destination vector pDEST R4-R3, resulting in the formation of the pln deletion construct pDest43_ΔPcPLN. This plasmid was linearized with AatII and transformed into P. chrysogenum DS54465 GFP-SKL protoplasts. Transformants were selected based on their ability to grow on acetamide as a sole nitrogen source. Disruption of the pln gene was confirmed by Southern blotting.
Construction of P. chrysogenum Strains Producing Various GFP Fusion Proteins
A plasmid enabling production of the Pln protein fused N-terminally with GFP was constructed using Multisite Gateway technology. The Pln coding sequence with a stop codon was amplified by PCR using primers AKR0005.PlnB1 and AKR0006.PlnB2 and a cDNA library as template (25). The obtained PCR product was cloned into the entry vector pDONR221, resulting in pENTR221_PcPLN. Subsequently, plasmids pENTR41/PpcbCGFP, pENTR221_PcPLN, pENTR23-His8.TpenDE, and pDEST R4-R3/AMDS were recombined, resulting in a plasmid designated pDest43.pIPNS.eGFP.pcPLN.Tat. This plasmid was linearized with Acc65I and transformed to a P. chrysogenum strain producing DsRed-SKL.
A plasmid enabling production of a catalase-peroxidase protein fused N-terminally with GFP was constructed using the Multisite Gateway technology. The Pgpda region was cut out from vector pBBK-007 and cloned into pGBRH2-mGFP in the place of PpcbC using Asp718I and BamHI, resulting in vector LMOP-Pgpda-mGFP. Next, the Pgpda-mGFP region was amplified by PCR using primers LMOp080 and LMOp081 and LMOP-Pgpda-mGFP as a template and recombined to pDONR4-1 in a BP recombination reaction, resulting in vector LMOP41-Pgpda-mGFP. The catalase-peroxidase coding sequence with a stop codon was amplified by PCR using primers KatGfor and KatGrev and cDNA library as template (25). The obtained PCR product was cloned into the entry vector pDONR221, resulting in pDONR221KatG. Subsequently, plasmids LMOP41pgpdAmGFP, pDONR221KatG, pENTR23-His8.TpenDE, and pDEST R4-R3/AMDS were recombined, resulting in a plasmid pGFPKatG. This plasmid was linearized with NotI and transformed to a P. chrysogenum strain producing DsRed-SKL.
A plasmid enabling the production of the putative oxidoreductase protein fused N-terminally with GFP was constructed using Multisite Gateway technology. The putative oxidoreductase encoding sequence with a stop codon was amplified by PCR using primers chDEHfor and chDEHrev and a cDNA library as template (25). The obtained PCR product was cloned into the entry vector pDONR221, resulting in pDONR221chDEH. Subsequently, plasmids LMOP41pgpdAmGFP, pDONR221chDEH, pENTR23-His8.TpenDE, and pDEST R4-R3/AMDS were recombined, resulting in a plasmid pGFP-chDEH. This plasmid was linearized with AatII and transformed to P. chrysogenum producing DsRed-SKL. All transformants were selected based on their ability to utilize acetamide as a sole nitrogen source.
Production and Purification of Pln
The coding sequence of Pln was amplified using a P. chrysogenum library as a template (25) and primers MDBPlnfor and MDBPlnrev containing BamHI and PstI sites, respectively. The obtained PCR product and expression vector pMAL-c2 (New England Biolabs) were digested with BamHI and PstI and ligated, resulting in plasmid pMDBPlnMBP, encoding P. chrysogenum Pln protein fused to maltose-binding protein (MBP) at the N terminus. Subsequently, a His6 tag was introduced at the C terminus of the MBP-Pln fusion protein using the Lightening site-directed mutagenesis kit (Stratagene) and primers MDBhisfor and MDBhisrev and plasmid pMDBPlnMBP as a template, resulting in plasmid pMDBPlnHis. To introduce a tobacco etch virus (TEV) cleavage site between MBP and Pln, pMDBPlnHis plasmid, primers MDB.TEV.for, and MDB.TEV.rev and Lightening site-directed mutagenesis kit (Stratagene) were used, resulting in the final construct pMDBLONTEV. Correctness of the plasmid was confirmed by sequencing.
E. coli Rosetta 2 cells containing expression plasmid pMDBLONTEV were grown on LB medium at 37 °C to an optical density at 600 nm of 1. 1 mm isopropyl 1-thio-β-d-galactopyranoside was added followed by 18 h incubation at 16 °C. Cells were harvested by centrifugation, and pellets were suspended in buffer A (50 mm Tris-HCl, 100 mm KCl, 10 mm MgCl2, 5% glycerol, pH 8.0) and disrupted using a French press. The soluble fraction, which was obtained by centrifugation for 1 h at 16,000 × g at 4 °C was incubated with nickel-nitrilotriacetic acid resin (Qiagen) for 45 min at 4 °C. The resin was washed with buffer A containing 10 mm imidazole, and bound proteins were eluted with buffer A containing 300 mm imidazole. Fractions were analyzed by SDS-PAGE and Western blotting using α-His6-tag antibodies (Santa Cruz Biotechnology). Fractions containing the MBP-Pln-His6 fusion protein were pooled and loaded on amylose resin (New England Biolabs) equilibrated with buffer B (50 mm Tris-HCl, 100 mm KCl, 10 mm MgCl2, 5% glycerol, pH 8.0) and incubated for 45 min at 4 °C. The resin was washed with buffer B, and bound Pln fusion protein was eluted with buffer B containing 10 mm maltose.
Generation and Purification of a Proteolytically Inactive Pln Mutant (Plnin)
The Pln mutant, S815A (Plnin), encoded by plasmid pMDBPlninMBP, was generated using the pMDBPlnMBP plasmid, primers lonmutfor and lonmutrev, and the Lightening site-directed mutagenesis kit (Stratagene). To introduce a His6 tag at the C terminus of the MBP-Plnin fusion protein, plasmid pMDBPlninMBP used as the template, primers MDBhisfor and MDBhisrev and Lightening site-directed mutagenesis kit (Stratagene) were applied, resulting in plasmid pMDBPlninHis. To introduce a TEV cleavage site between MBP and Plnin, the pMDBPlninHis plasmid was used as a template together with primers MDB.TEV.for and MDB.TEV.rev and the Lightening site-directed mutagenesis kit (Stratagene), resulting in the final construct pMDBLONinTEV. Correctness of the plasmid was confirmed by sequencing. pMDBLONinTEV plasmid was transformed into the Rosetta 2 E. coli cell strain. The Plnin fusion protein was expressed and purified to homogeneity using the protocol for the Pln fusion protein.
Production and Purification of Catalase-Peroxidase
The coding sequence of catalase-peroxidase was amplified using P. chrysogenum cDNA library (25) as a template and primers GST.KatGfor and GST.KatGrev containing BamHI and EcoRI sites, respectively. The obtained PCR product and expression vector pGEX-4T-2 (GE Healthcare) were digested with BamHI and EcoRI and ligated, resulting in plasmid pGSTKatG, encoding catalase-peroxidase P. chrysogenum protein fused to glutathione S-transferase (GST) at the N terminus. Correctness of the plasmid was confirmed by sequencing.
E. coli Rosetta 2 cells containing expression plasmid pGSTKatG were grown on LB medium to an optical density at 600 nm of 1 and induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside with simultaneous addition of 2.5 μm hemin (Sigma) followed with 18 h of incubation at 16 °C. Cells were harvested by centrifugation at 5200 × g for 10 min at 4 °C. The cell pellet was suspended in column buffer (50 mm NaH2PO4, 150 mm NaCl, 1 mm DTT, and 1 mm EDTA, pH 7.2) and disrupted using a French press. The soluble fraction, which was obtained by centrifugation for 1 h 16,000 × g at 4 °C, was incubated with glutathione Superflow resin (Qiagen) for 45 min at 4 °C. The resin was washed with column buffer, and bound proteins were eluted with elution buffer (50 mm Tris-HCl, 0.1 m NaCl, 50 mm reduced l-glutathione, 1 mm DTT, pH 8.0). Obtained fractions were analyzed by SDS-PAGE and Western blotting using α-c-Myc antibodies (Santa Cruz Biotechnology). Fractions containing GST-catalase-peroxidase were pooled and cleaved using AcTEV protease (Invitrogen). Separation of catalase-peroxidase from GST and TEV protease was accomplished using Superose 6 10/300 GL (GE Healthcare) column equilibrated with 50 mm NaH2PO4, 150 mm NaCl, pH 7.2. Purified GST-catalase-peroxidase was used for immunization of rabbits (Eurogentec, Seraing, Belgium; supplemental Fig. S5).
Biochemical Methods
Crude extract of P. chrysogenum cells were prepared as described previously (26). Protein concentrations were determined using the RC-DC assay system or Bio-Rad assay kit (Bio-Rad) using bovine serum albumin (BSA) as a standard. SDS-PAGE and Western blotting were carried out in accordance with established procedures. P. chrysogenum cell fractionation and peroxisome isolation were performed as described previously (26). Catalase activity was measured spectrophotometrically as described before (14). Peroxidase activity was determined spectrophotometrically as described by Fraaije et al. (27). Catalytic and peroxidatic activities were measured in P. chrysogenum extracts at the optimal pH values as determined for purified catalase-peroxidase (supplemental Fig. S4). The ATPase activity of Pln and Plnin was measured colorimetrically using malachite green according to the instructions of the manufacturer (Innova Biosciences).
Isolation of Triton X-100-insoluble Peroxisomal Proteins
Triton X-100 insoluble proteins were isolated from fractions enriched in peroxisomes essentially as described previously (28). For identification of the proteins, bands of interest were cut from the SDS-PAGE gel and analyzed by mass spectrometry (MALDI-TOF peptide mass fingerprint and MALDI-TOF/TOF peptide sequencing, Alphalyse A/S, Denmark).
Analysis of Penicillin Production
β-Lactam antibiotic bioassays were performed as described previously (21). Quantitative analysis of extracellular penicillin G was determined by HPLC using a Shim-Pack XR-ODS column (3.0-mm internal diameter × 75 mm) and an isocratic flow of 310 g·liter−1 acetonitrile, 640 mg·liter−1 KH2PO4, and 340 mg·liter−1 H3PO4. Peaks of penicillin G were detected at a wavelength of 254 nm.
Measurements of β-Oxidation of Fatty Acids in Intact Cells
Spores of P. chrysogenum strains were inoculated in YGG medium and incubated for 48 h. Next, cells were shifted to mineral medium supplemented with 0.1% oleic acid, 0.05% Tween 20 for 10 h. Subsequently, cells were washed and resuspended in phosphate-buffered saline (PBS). β-Oxidation activities were measured in intact cells as described previously (29). Cell suspensions were incubated in 200 μl of PBS containing 10 μm [1-14C]stearate (C18:0). Reactions were allowed to proceed for 1 h at 28 °C followed by termination of the reactions by the addition of 50 μl of 2.6 m perchloric acid. Radiolabeled CO2 was trapped overnight in 500 μl of 2 m NaOH. The 14C-labeled β-oxidation products were subsequently collected after extracting the acidified material with chloroform/methanol/heptane and quantified in a liquid scintillation counter.
Lipid Peroxidation Analysis
Lipids peroxidation was quantified by determination of thiobarbituric acid-reacting substances (TBARSs) according to the method described by Paraszkiewicz et al. (30) using lysed protoplasts. The TBARS concentration was expressed as nmol of malondialdehyde/mg or protein.
In Vitro Protein Degradation Assays
Purified Pln and the proteolytically inactive mutant Plnin were used for in vitro protease assays. Proteolytic activity against unfolded model proteins was assayed with α-casein (Sigma) or β-casein labeled with resorufin (Universal Protease Substrate, Roche Applied Science). For hydrolysis of α-casein, the assay buffer C consisting of 50 mm Tris-HCl, pH 8.0, 10 mm MgCl2, 1 mm DTT with or without 4 mm ATP and the ATP regeneration system (50 mm creatine phosphate, 80 μg/ml creatine kinase) was used. Pln and the proteolytically inactive mutant (Plnin) (final concentration of 0.26 μm) were incubated at 25 °C with 5.8 μm α-casein overnight. Proteolytic degradation of α-casein was detected using SDS-PAGE and Coomassie Brilliant Blue staining. For hydrolysis of β-casein labeled with resorufin Pln and Plnin, 1.2 μm, were incubated at 25 °C with 0.1% of β-casein labeled with resorufin in buffer C according to the instructions of the manufacturer. Proteolysis was monitored spectrophotometrically at 574 nm.
The proteolytic activity against catalase-peroxidase was assayed in buffer C with or without 4 mm ATP and an ATP regeneration system (50 mm creatine phosphate, 80 μg/ml creatine kinase). Pln (0.46 μm) was incubated at 25 °C with native, thermally denatured or H2O2-treated catalase-peroxidase (0.52 μm). To prepare H2O2-treated catalase-peroxidase, catalase-peroxidase (final concentration 5.2 μm) was incubated with 5 mm H2O2 for 30 min at room temperature. Next H2O2 was removed by buffer exchange using an Amicon Ultra-4 and Microcon YM-3 centrifugation device (Millipore). To prepare thermally denatured catalase-peroxidase, catalase-peroxidase (at a concentration of 5.2 μm) was incubated for 10 min at 80 °C. Proteolytic activity was analyzed using SDS-PAGE electrophoresis and Coomassie Brilliant Blue staining. Quantification of catalase-peroxidase bands was performed with ImageJ software. The results from two independent experiments were averaged.
Catalase-Peroxidase Unfolding and Aggregation
Changes in intrinsic (tryptophan) fluorescence (excitation λ = 295 nm, emission λ = 345 nm) were measured at room temperature using 15 μm purified catalase-peroxidase in 50 mm Tris-HCl, pH 8.0, and 150 mm NaCl upon incubation for 10 min with different concentrations of hydrogen peroxide. Fluorescence measurements were performed using Fluoromax-3 spectrofluorometer (Jobin Yvon-Spex).
Aggregation of catalase-peroxidase (1.3 μm in 50 mm Tris-HCl, pH 8.0, and 150 mm NaCl) upon treatment with 20 mm hydrogen peroxide was monitored over a period of 60 min at room temperature by measuring light scattering at 360 nm in a PerkinElmer Life Sciences Lambda 35 spectrophotometer.
Citrate Synthase Aggregation and Refolding Assays
The capacity of Pln to prevent protein aggregation was determined using chemically denatured citrate synthase as a substrate as described previously (31). Citrate synthase (final concentration 30 μm of native enzyme; Sigma) was chemically denatured by incubation at room temperature in 6 m guanidinium/HCl and 20 mm DTT for at least 2 h. Aggregation was initiated by a 200-fold dilution of the denatured enzyme under vigorous stirring in 50 mm Tris-HCl, pH 8.0, with 5 mm MgCl2 at 25 °C with or without Pln and with or without 1 mm ATP. The kinetic of citrate synthase aggregation was monitored by measuring the intensity of light scattering at 360 nm in a PerkinElmer Life Sciences Lambda 35 spectrophotometer.
To measure refolding activity of Pln, chemically denatured citrate synthase was prepared as described above at a concentration of 15 μm (31). Reactivation of citrate synthase was initiated by a 100-fold dilution of the denatured enzyme under vigorous stirring in 50 mm Tris-HCl, pH 8.0, with 5 mm MgCl2 at 25 °C with or without Pln or Plnin and with or without 1 mm ATP. The recovery of citrate synthase activity was tested as described (32) using oxaloacetic acid, acetyl-CoA, and dithio-bis-[2-nitrobenzoic] acid (all from Sigma).
Static and Dynamic Light Scattering
Before static light scattering (SLS), dynamic light scattering (DLS), and small-angle x-ray scattering (SAXS) analysis, Plnin was subjected to gel filtration using a Superdex S200 16/10 column equilibrated with 50 mm Tris, 150 mm NaCl, 1 mm DTT, 4 mm ATP, pH 8.0. The procedure used for SLS coupled to the ÄKTA system is described in Nettleship et al. (33). DLS measurements were performed on a DynaProTM DLS (Protein solutionsTM) at 10 °C with the protein at a concentration of 1.1 μm.
SAXS Analysis
Synchrotron radiation x-ray scattering data from solutions of Plnin were collected on the X33 camera of the EMBL on the storage ring DORIS III (DESY, Hamburg, Germany) (34). Using a photon counting Pilatus 1 m detector at a sample-detector distance of 2.7 m and a wavelength of λ = 0.15 nm, the range of momentum transfer 0. 1 < s < 6 nm−1 was covered (s = 4πsinθ/λ, where 2θ is the scattering angle). Four solute concentrations in the range 0.5–3.7 mg/ml were measured. To monitor radiation damage, 8 successive 15-s exposures were compared, and no significant changes were observed. The data were normalized to the intensity of the transmitted beam and radially averaged; the scattering of the buffer was subtracted, and the difference curves were scaled for protein concentration. The low angle data collected from Plnin were extrapolated to infinite dilution and merged with the higher concentration high angle data to yield the final composite scattering curve.
The forward scattering I(0) and the radius of gyration Rg were evaluated using the Guinier approximation (35) assuming that at very small angles (s < 1.3/Rg) the intensity is represented as I(s) = I(0) exp(−(sRg)2/3). These parameters were computed using the programs AUTORG and AUTOGNOM (36); the latter program also provided the pair distribution function of the particle p(r) and the maximum dimension Dmax. The molecular mass (MM) of Plnin was evaluated by comparison of the forward scattering with that from a reference solution of bovine serum albumin (MM = 66 kDa).
Ab Initio Shape Determination
The “shape scattering” curve was further used to generate low resolution ab initio shapes of the Plnin with the program DAMMIF (37). This program represents the particle shape with an assembly of densely packed beads and employs simulated annealing to construct a compact interconnected model fitting the experimental data Iexp(s) to minimize discrepancy,
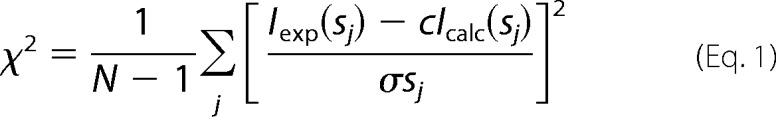 |
where N is the number of experimental points, c is a scaling factor, and Icalc(s) and σ(sj) are the calculated intensity and the experimental error at the momentum transfer sj, respectively. Ten DAMMIF runs were performed to check the stability of solution, and the results were well superimposable with each other. These models were averaged to determine common structural features using the programs DAMAVER (38) and SUPCOMB (39). The latter program aligns two arbitrary low or high resolution models represented by ensembles of points by minimizing a dissimilarity measure called normalized spatial discrepancy. For every point (bead or atom) in the first model, the minimum value among the distances between this point and all points in the second model is found, and the same is done for the points in the second model. These distances are added and normalized against the average distances between the neighboring points for the two models. Generally, normalized spatial discrepancy values close to unity indicate that the two models are similar. The program DAMAVER generates the average model of the set of superimposed structures and also specifies the most typical model (i.e. that having the lowest average normalized spatial discrepancy with all the other models of the set).
Molecular Modeling
The program BUNCH (40) combines rigid body and ab initio modeling of proteins consisting of domains (for which high resolution models are available) linked by flexible loops of unknown structure. A simulated annealing protocol is employed to find the optimal positions and orientations of the domains moved as rigid bodies and probable conformations of the flexible linkers attached to the appropriate terminal residues of domains. These linkers are represented as interconnected chains of dummy residues (41), where protein fragments are substituted by a flexible chain of residues separated by 0.38 nm. The x-ray scattering intensity from the entire model is expressed as,
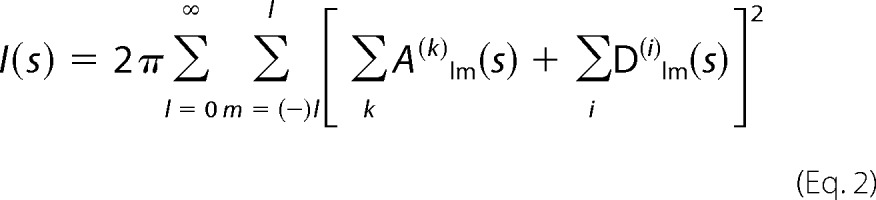 |
Here, A(k)lm(s)H are the partial scattering amplitudes of the rigid domains calculated from their atomic coordinates using CRYSOL (42). The partial scattering amplitudes of the linkers D(i)lm(s) are computed from the positions of the dummy residues using the form factor equal to that of an average residue in water (41). Starting from an arbitrary configuration of domains and linkers, the program performs random modifications of the model maintaining the structures of the domains and connectivity of the linkers.
Microscopy
Confocal laser scanning microscopy images were obtained using a Zeiss LSM510 confocal laser scanning microscope equipped with a Zeiss Plan-Neofluar ×100/1.3 and Zeiss Plan-Apochromatic ×63/1.4 oil objectives. Images were acquired using AIM 4.2 software (Carl Zeiss). GFP fluorescence was analyzed by excitation of the cells with a 488-nm argon-krypton laser, and fluorescence was detected with BP 500–530 Photo Multiplier Tube. DsRed fluorescence was analyzed by excitation of the cells with a 543-nm argon/krypton laser, and fluorescence was detected with a BP 565–615 Photo Multiplier Tube.
Wide-field fluorescence microscopy was performed with a Zeiss Axio Observer Z1 fluorescence microscope. Images were obtained with an EC-Plan-Neofluar ×100/1.3 objective and coolsnap HQ2 Camera (Roper Scientific Inc.). The GFP signal was analyzed with a 470/40-nm bandpass excitation filter, a 495-nm dichromatic mirror, and a 525/50-nm bandpass emission filter. DsRed as well as dihydroethidium (DHE, Invitrogen) fluorescence was visualized with a 545/25-nm bandpass excitation filter, a 570-nm dichromatic mirror, and a 605/70-nm bandpass emission filter. Z-stack images were made with an interval of 1 μm. Images were acquired using AxioVision 4.7 software (Carl Zeiss). P. chrysogenum mycelia were stained with fluorescent probes before microscopy investigation in accordance with manuals provided by the suppliers (Invitrogen). Maximum fluorescence intensities of DHE-stained nuclei were measured using ImageJ software.
For electron microscopy P. chrysogenum cells were prepared as described previously (43). Image analysis was performed using ImageJ (rsbweb.nih.gov), and figures were prepared using Adobe Photoshop CS3.
RESULTS
P. chrysogenum Peroxisomes Contain a Protease of the Lon A Protein Family
Previously, we identified a putative peroxisomal Lon protease (designated Pln) in the fungus P. chrysogenum by in silico identification of proteins that contain a putative peroxisomal targeting signal 1 (PTS1 (26). We confirmed the peroxisomal localization of this protein by fluorescence microscopy using cells producing a GFP-Pln fusion protein in conjunction with DsRed-SKL as a peroxisomal matrix marker (Fig. 1A).
FIGURE 1.

Peroxisomes contain an ATP-dependent Lon protease. A, shown is confocal laser scanning microscopy analysis of the subcellular localization of peroxisomal Lon protease using cells producing GFP-Pln and DsRed-SKL as marker of peroxisomes. GFP fluorescence co-localized with DsRed. Confocal laser scanning microscopy pictures were taken after 40 h of growth in PEN medium. The scale bar represents 5 μm. B, shown is a schematic representation of the domain architecture of P. chrysogenum Pln showing the three conserved domains as well as the C-terminal PTS1 tripeptide, SRL. aa, amino acids. C, P. chrysogenum Pln degrades α-casein in an ATP-dependent manner. Purified Pln or the inactive variant Plnin was incubated with α-casein in the presence (ATP+) or absence (ATP−) of ATP and an ATP regeneration system. After overnight incubation, the samples were subjected to SDS-PAGE, and the gels were stained with Coomassie Brilliant Blue. Upon incubation in the presence of ATP and Pln, the bulk of the α-casein protein was degraded, as evident from the absence of the 35-kDa α-casein band. The bands marked with asterisks represent proteins of the ATP regeneration system. D, Pln degrades β-casein in an ATP-dependent manner. Quantitative analysis of the proteolytic degradation of resorufin-labeled β-casein by Pln (green) and Plnin (blue) in the presence (ATP+) or absence (ATP−) of ATP and an ATP regeneration system is shown. Controls lacking Pln or Plnin (−) are shown in gray. E, ATPase activities of purified Pln and Plnin. n.s., statistically not significant (Student's t test). Error bars in D and E represent S.D.
Inspection of the protein sequence revealed that Pln belongs to the Lon A protein family. These proteins have three functional domains: an N-terminal (Lon N domain), an ATPase (AAA+ module with Walker A and B motifs), and a protease domain (P domain with conserved catalytic dyad) (Fig. 1B).
Pln, purified to homogeneity as MBP fusion protein upon production in E. coli (supplemental Fig. S1), proved to be rather unstable in contrast to its proteolytically inactive variant (Plnin; S815A, see below). However, both MBP fusion proteins (Pln and Plnin) precipitated upon cleavage of MBP by TEV protease. We, therefore, performed all further in vitro studies using MBP fusion proteins. In vitro assays revealed that Pln displayed ATP-driven proteolytic activity toward α-casein and β-casein, both unfolded protein substrates (Fig. 1, C and D). As expected, this activity was fully abolished by the single amino acid substitution, S815A, in the conserved catalytic dyad. Also no proteolytic activity was detected when the assays were performed in the absence of ATP (Fig. 1, C and D). Purified Pln showed ATPase activity, which was not abolished by the mutation in the catalytic dyad (Fig. 1E).
P. chrysogenum Pln Is a Heptameric Protein
Previous reports indicate that Lon proteases form large oligomeric complexes, ranging from tetramers to octamers (44–46). To gain insight into the oligomeric state of P. chrysogenum Pln, we subjected the MBP-Plnin fusion protein to SLS, DLS, and SAXS in the presence of ATP (Table 4, supplemental Fig. S2). SLS and DLS analysis suggested that Plnin consists of 6.7 and 7.2 monomers, respectively, indicative of a heptamer (Table 4). Such an oligomeric state has also been reported for mitochondrial Lon from Saccharomyces cerevisiae (47).
TABLE 4.
SLS, DLS, and SAXS data
| Static light scattering | Molecular weight (Mr) | 984 kDa (± 11%) |
|---|---|---|
| Number weighted mean (Mn) | 982 kDa (± 11%) | |
| Poly-disparity (Mr/Mn) | 1.0 (± 16%) | |
| Dynamic light scattering | Hydrodynamic radius | 11.5 nm (± 31%) |
| Molecular weight | 1060 kDa (± 31%) | |
| SAXS analysis | Radius of gyration | 10.3 nm (± 9%) |
| Molecular weight | 1100 kDa (± 10%) | |
| Theoretical molecular weight of Plnin | 147 kDa (monomer) | |
| 882 kDa (hexamer) | ||
| 1029 kDa (heptamer) | ||
| 1176 kDa (octamer) |
Next, we performed SAXS analysis of Plnin to obtain a low resolution model of the protein (supplemental Fig. S2). The molecular mass of the protein estimated from the forward scattering amounted to 1100 ± 100 kDa comparable to the SLS and DLS estimates of 980 ± 110 and 1060 ± 330 kDa respectively. The experimental radius of gyration Rg and maximum size Dmax (10.3 ± 9 and 34 ± 3 nm, respectively) point to an oblate structure. The smaller angles (up to s = 0.5 nm−1) can be fitted (χ = 2.5) with an oblate ellipsoid of revolution with a diameter of 29 nm and a height 9.4 nm. Assuming that one Plnin monomer is 147 kDa, the oligomeric state of Plnin in solution could be hexameric, heptameric, or octameric. The low resolution shape of Plnin was first reconstructed ab initio using the bead modeling program DAMMIF (37) employing the range of scattering vectors up to s = 0.8 nm−1. Ten ab initio models were constructed using the symmetries P6, P7, and P8 (hexameric, heptameric, and octameric, respectively) and compared; the reconstructions in P6 and P7 were more reliable (normalized spatial discrepancy = 0.82 ± 0.08 and 0.75 ± 0.09, respectively) than in P8 (normalized spatial discrepancy = 2.13 ± 0.25). Using the available crystal structure of MBP (PBD code 1ANF; Ref. 48) as well as two models built with the program SWISS-Model (49) corresponding to amino acids 11–201 and 378–902 in Plnin (no structure of full-length Lon is available), hexamers, heptamers, and octamers were constructed using the program BUNCH (40). P8 models exhibited many domain clashes, effectively ruling out an octameric state for Plnin. The P6 models had a systematically worse fit (χ = 1.4) compared with the P7 models (χ = 1.2), although the difference is not large. However this coupled with the SLS and DLS data inclines us to favor a heptameric state for Plnin in solution. This assumption is supported by our fit to the ellipsoid, as the obtained height also corresponds to a P7 model. A putative P6 model would require a 1.8× bigger height. The most probable ab initio P7 model out of 10 reconstructions (Fig. 2) displays an oblate, ring-shaped, and seven-armed structure. The BUNCH rigid body model, which is comparable with the DAMMIF model in overall appearance, is similar to the transmission electron microscopy model of mitochondrial Lon (47).
FIGURE 2.

Ab initio models of heptameric MBP-Plnin. The DAMMIF bead model is shown in gray. The BUNCH rigid body model, consisting of the crystal structure of MBP (green) and the homology models of residues 11–201 (cyan) and 378–902 (magenta), is shown in stick representation. A, top view. B, side view.
Lon Deficiency Is Associated with a Defect in Growth on Oleic Acid and Enhanced Oxidative Stress
To gain insight into the in vivo function of Pln, a P. chrysogenum knock-out strain was constructed (designated pln). The P. chrysogenum pln strain displayed normal growth on complex media (R-agar) as well as on mineral media containing glucose or ethanol as sole carbon source (Fig. 3A). The cells also grew normally on PEN media (data not shown), which was accompanied by the production of penicillin G levels that were similar to the parental strain (Fig. 3, D and E).
FIGURE 3.

Growth properties and penicillin production. A, growth of pln and wild-type (WT) cells on agar plates containing complex medium (R-agar) or mineral medium supplemented with 1% glucose, 1% ethanol, or 1% oleic acid as sole carbon sources is shown. Plates were incubated at 25 °C for 5–10 days. B, shown is quantification of biomass 24 h after the addition of 0.1% oleic acid to WT or pln cultures grown for 120 h on PEN medium. Error bars represent S.D. C, shown is fatty acid (C18:0) β-oxidation in intact cells of WT (blue) and pln (red) incubated for 10 h in medium containing 0.1% oleate. β-Oxidation is expressed as the sum of [14C]CO2 and water-soluble β-oxidation products produced/μg of protein used in the assay. D, spent media of pln and WT strains cultivated on penicillin G production media for 5 days were diluted and loaded into wells of a bioassay plate overgrown with cells of a Micrococcus luteus strain. Cell densities of the wild-type and pln cultures (in terms of dry weight/liter) were comparable. After 24 h of incubation at 30 °C, the sizes of the growth inhibition zones were similar, indicating that similar concentrations of penicillin G were produced. E, spent medium of pln and wild-type cultures grown for 5 days on PEN medium were used to quantitatively determine penicillin G levels by HPLC. Average values from three independent cultures are presented for each strain. The bars represent S.D. n.s., statistically not significant (Student's t test).
Remarkably, pln cells displayed a severe defect in growth on oleic acid as a sole carbon source (Fig. 3A). Further analysis suggested that oleic acid was toxic for pln cells. This was indicated by the observation that the addition of oleic acid to stationary phase cultures of pln cells resulted in a decrease in biomass (Fig. 3B). The oleic acid growth defect was not due to a block in peroxisomal fatty acid metabolism as C18:0 β-oxidation was not decreased in these cells (Fig. 3C).
Deletion of PLN in Hansenula polymorpha resulted in enhanced oxidative stress (14). We, therefore, also analyzed whether the lack of Pln influences the homeostasis of reactive oxygen species in P. chrysogenum. Cells were stained with DHE, a fluorescent dye that specifically reacts with intracellular 0 2̇̄ and is converted to the red fluorescent compound ethidium, which irreversibly binds to double-stranded DNA and appears as punctate nuclear staining (Fig. 4A). Enhanced nuclear staining with DHE was observed in pln cells grown on PEN medium relative to the wild-type control (supplemental Fig S3). This difference was much more pronounced when peroxisome proliferation and the peroxisomal β-oxidation pathway were induced by the addition of oleic acid (50); Fig. 4, A and B).
FIGURE 4.

Lack of peroxisomal Lon protease results in enhanced oxidative stress. A, shown are fluorescence microscopy images of DHE stained WT and pln cells grown for 120 h in PEN medium after the addition of 0.1% oleic acid and further incubation for 12 h. pln cells show enhanced DHE fluorescence relative to the wild-type control. The scale bars represent 5 μm. DIC, differential interference contrast. B, shown is quantification of DHE fluorescence intensities from cells shown in A. Error bars represent S.E. C, lipid peroxidation was quantified by determination of TBARSs. Measurements were performed using cell homogenates of wild-type and pln cells grown for 120 h on PEN medium. The TBARS concentration is expressed as nmol of malondialdehyde (MDA)/mg of protein. Error bars represent S.D. Statistical analysis was performed with Student's t test.
We also measured the difference in lipid oxidative damage in pln and wild-type cells, which is determined by the measurement of TBARSs. Malondialdehyde is an important byproduct of lipid peroxidation and reacts with thiobarbituric acid. As shown in Fig. 4C, significantly enhanced lipid peroxidation was observed in pln cells grown on PEN media relative to that observed in the wild-type control.
The Absence of Pln Results in the Formation of Protein Aggregates in the Peroxisomal Matrix
Cells of the pln strain showed normal import of peroxisomal matrix proteins, as was evident from the peroxisomal localization of GFP-SKL in pln cells, which was similar to that in the wild-type control (Fig. 5). The absence of Pln was associated with the accumulation of protein aggregates in the peroxisomal matrix that were visualized as electron-dense inclusions in the organelle lumen by electron microscopy (Fig. 6B). These inclusions were confined to peroxisomes and never observed in other cell compartments of cells of the pln strain. Also, peroxisomal protein aggregates were never observed in wild-type control cells (Fig. 6A). The protein content of the aggregates that are present in P. chrysogenum pln cells likely represents substrates of Pln. To identify these proteins, the aggregates (defined as proteins that are not solubilized by Triton X-100) were isolated from Triton X-100-treated peroxisomal fractions by high velocity centrifugation. SDS-PAGE revealed two distinct protein bands that were enhanced in the insoluble fractions obtained from organelles isolated from pln cells relative to identical fractions obtained from wild-type controls (Fig. 6C). Mass spectrometry revealed that these bands consisted of a putative catalase-peroxidase and a putative oxidoreductase from the glucose-methanol-choline (GMC) oxidoreductase family (Fig. 6C). Hex1, which forms a crystalline inclusion of Woronin bodies, is not solubilized by Triton X-100 (51), and was observed in both the insoluble fractions isolated from pln and wild-type control organelles. The peroxisomal localization of catalase-peroxidase and glucose-methanol-choline oxidoreductase was confirmed using P. chrysogenum cells producing N-terminal GFP fusions of these proteins and DsRed-SKL as peroxisomal marker (Fig. 6, D and E).
FIGURE 5.

Localization of GFP containing a PTS1 in WT and pln cells. Fluorescence microscopy images showing GFP-SKL localization in WT and pln cells grown for 120 h in PEN media. Like in the wild-type control, no cytosolic GFP was observed in pln cells. The scale bars represent 5 μm. DIC, differential interference contrast.
FIGURE 6.

The absence of Pln results in the formation of protein aggregates in the peroxisomal matrix. A and B, electron microscopy images of a detail of a wild-type (A) and pln (B) cell show the presence of a protein aggregate (arrow, Fig. 6B) in the peroxisomal matrix of the pln cell. The cells were fixed with KMnO4. P, peroxisome; M, mitochondrion; N, nucleus. The scale bar represents 1 μm. C, shown is a Coomassie Brilliant Blue (CBB)-stained SDS-PAGE gel of Triton-X100-insoluble proteins isolated from peroxisomal fractions obtained from wild-type and pln cells. The indicated protein bands were identified by mass spectrometry. GMC, glucose-methanol-choline. D and E, shown are subcellular localizations of N-terminal GFP fusion proteins of catalase-peroxidase (D) and glucose-methanol-choline oxidoreductase (E), analyzed by fluorescence microscopy in cells cultivated for 40 h in PEN media. DsRed-SKL was used as a marker of peroxisomes. The scale bar represents 5 μm.
Unfolded Catalase-Peroxidase Is a Substrate of Pln in Vitro
Catalase-peroxidase, one of the major Triton X-100 insoluble proteins in peroxisome fractions of pln cells (Fig. 6C), was selected for further analysis. As peroxisomes are oxidative organelles, we studied whether oxidative stress may cause catalase-peroxidase enzyme inactivation or protein aggregation.
Catalase-peroxidase was produced in E. coli and purified to homogeneity (supplemental Fig. S1). Enzyme assays revealed that the catalase-peroxidase enzyme displayed catalase and peroxidase activities with pH optima of 6–7 and 5, respectively (supplemental Fig. S4). Treatment of purified catalase-peroxidase protein with increasing concentrations of H2O2 resulted in a decrease in intrinsic Trp fluorescence and peroxidase enzyme activity, indicative for protein unfolding (Fig. 7A). Relatively low concentrations of H2O2 already caused significant changes in Trp fluorescence and peroxidase activity (enzyme activity decreased by 40% after treatment with 100 μm H2O2). Catalase-peroxidase was also prone to aggregation upon treatment with H2O2 in vitro, as evident from light scattering measurements (Fig. 7B). Finally, we tested whether catalase-peroxidase was a substrate for Pln using an in vitro degradation assay. These studies revealed that the native protein was not degraded by Pln. However, upon pretreatment with H2O2 or preincubation at high temperature, the protein was degraded by Pln (Fig. 7, C and D). These data indicate that unfolded catalase-peroxidase, but not the native protein, is a substrate of the peroxisomal Lon.
FIGURE 7.

Damaged catalase-peroxidase is the substrate of Pln. A, catalase-peroxidase unfolds (blue) and loses peroxidase activity (green) upon hydrogen peroxide treatment. Unfolding of catalase-peroxidase (15 μm) was monitored by measurement of changes in intrinsic tryptophan fluorescence. The protein was incubated for 10 min with the indicated concentrations of hydrogen peroxide before the measurements. Values are expressed as percentages of signal/activity. Error bars represent S.E. B, catalase-peroxidase is prone to aggregation upon hydrogen peroxide treatment as analyzed by light scattering measurements at 360 nm. AU, absorbance units. C, Coomassie Brilliant Blue-stained gel demonstrates that H2O2 or heat-treated catalase-peroxidase is a substrate for Pln in vitro. The amount of catalase-peroxidase protein treated with H2O2 or an elevated temperature, but not untreated (native) protein, decreased upon incubation with Pln relative to the control without Pln. Representative gels are presented. D, quantification of the catalase-peroxidase bands (compare Fig. 7C) from two independent degradation experiments revealed a 57.5% (±12%) decrease in the level of catalase-peroxidase protein treated with H2O2 and a 47% (±5.6) reduction of catalase-peroxidase protein levels when exposed to elevated temperature (80 °C) relative to the control sample without Pln (set to 100%). In contrast, no significant degradation of the untreated, native catalase-peroxidase protein was detected (decrease of 3.3 ± 1.6%). E, a Western blot shows the catalase-peroxidase protein levels in cell extracts of wild-type and pln cells grown for 120 h on PEN medium. Quantification of the bands revealed an ∼2-fold increase in catalase-peroxidase protein in pln cells. Equal protein amounts were loaded per lane. EF-1α was used as loading control. The experiment was performed twice, and a representative blot is presented. The blots were prepared from the same samples as used for enzyme activity measurements (shown in Fig. 7, F and G). Catalase (F) and peroxidase (G) activities in cell extracts of wild-type and pln cells grown for 120 h on PEN medium are shown. Values are expressed as the percentage of the specific enzymatic activity of wild type. The observed differences in enzyme activities between wild-type and pln cells are statistically significant (Student's t test). Error bars represents S.E.
Pln Displays Chaperone-like Activity
The presence of aggregates in peroxisomes of pln cells could be explained by aggregation of unfolded catalase-peroxidase protein. Western blot analysis revealed that pln cells contained enhanced catalase-peroxidase protein levels (Fig. 7E), whereas enzyme activity measurements revealed that the catalytic (Fig. 7F) and peroxidatic activities (Fig. 7G) were reduced. These observations suggest that Pln may display, additional to its proteolytic activity, also chaperone activity in vivo. To test the putative chaperone activity of Pln, citrate synthase (CS) was used as a substrate in in vitro protein refolding experiments. To test if purified Pln can suppress aggregation of this substrate, guanidine hydrochloride-denatured CS was diluted into buffer containing purified Pln. Relative to the control without Pln, a significant decrease in CS aggregation (∼40%) was observed (Fig. 8A). This decrease was independent of the presence of ATP (Fig. 8, A and B). The addition of BSA instead of Pln did not prevent CS aggregation (Fig. 8C). Aggregation of denatured CS was inhibited by Pln in a dose-dependent manner (Fig. 8D) and was the most efficient at a ratio of one or more Pln monomers to two CS monomers. Purified Pln also stimulated reactivation of chemically denatured CS (Fig. 8F), which was also optimal at a ratio of at least 1:2 Pln monomers to CS monomers (Fig. 8E). This stimulatory effect of Pln was not observed in the absence of ATP (Fig. 8G). Interestingly, the proteolytically inactive mutant of Pln (Plnin) facilitated refolding of CS with the same efficiency as the active protein (Fig. 8F). These data suggest that Pln is a multifunctional enzyme that, besides proteolytic activity, displays chaperone-like activity that is independent of the proteolytic catalytic dyad.
FIGURE 8.

Peroxisomal Lon protease displays chaperone-like activity. A and B, the aggregation kinetics of 150 nm denatured CS were monitored in the absence of Pln or presence of 80 nm Pln in the presence (A) or absence (B) of 1 mm ATP. AU, absorbance units. C, a reduction in aggregation was not observed when the reaction was performed in the presence of 80 nm BSA and ATP (red). D, denatured CS was diluted to a final concentration 150 nm in a solution supplemented with different concentrations of Pln (monomer). Aggregation kinetics of CS were measured. Normalized light scattering values obtained after 360 s are shown. The highest measured value was set to 100. E, reactivation of 150 nm denatured CS was determined in the presence of 1 mm ATP and different concentrations of Pln (monomer). CS reactivation was determined after 15 min of incubation. The highest refolding activity of Pln was set to 100%. F, reactivation of 150 nm denatured CS was determined in the presence of 1 mm ATP. Without the addition of Pln, only minor reactivation was observed. However, in the presence of 80 nm Pln or Plnin, ∼25% of the CS was reactivated. G, reactivation of denatured CS (150 nm) was similar in reactions containing Pln or lacking Pln when the reactions were performed in the absence of ATP.
DISCUSSION
In this work we provide evidence that peroxisomes of the fungus P. chrysogenum contain an isoform of the Lon protease, termed Pln, a member of the AAA protein family, that displays a dual function as ATP-stimulated protease in conjunction with a role as molecular chaperone. As such, Pln is the first chaperone that is identified in fungal peroxisomes.
Because proteins are marginally stable macromolecules with a limited lifespan, we imagined that also peroxisomal matrix proteins require constant housekeeping processes. As yet, the fate of luminal proteins within the oxidative milieu of peroxisomes was, however, still an enigma. Recently, a Lon protease was identified in the peroxisomal matrix of rat (15), yeast (14), and plant (52). Studies performed in H. polymorpha indeed suggest that the deletion of peroxisomal Lon protease (pln) affected organelle quality control and had detrimental effects on peroxisome function and cell vitality (14).
The lack of the peroxisomal Lon protease in P. chrysogenum was associated with the accumulation of protein aggregates in the peroxisomal matrix, suggesting that Pln is required to prevent accumulation of misfolded proteins and their subsequent aggregation in the organelle matrix. It is tempting to speculate that aggregate-forming proteins in pln cells represent the preferred substrates of Pln. As major constituents of the aggregates, we identified a heme-containing catalase-peroxidase and a putative FAD-dependent oxidoreductase from the glucose-methanol-choline family. The catalase-peroxidase protein was the major component of the aggregate and was, therefore, analyzed further. Interestingly, our data indicate that purified catalase-peroxidase is prone to oxidative damage and loses activity and forms aggregates upon exposure to hydrogen peroxide.
Western blot analysis revealed that catalase-peroxidase protein levels were elevated in crude extracts of pln cells relative to wild type. This indicates that catalase-peroxidase most likely constitutes a substrate of the Pln protease in vivo. Indeed, damaged catalase-peroxidase (heat denaturated or H2O2 treated), but not the native enzyme, was degraded in vitro by Pln in vitro. pln cells also showed reduced catalase-peroxidase enzyme activities. Together with our in vitro experiments, which indicate that Pln has chaperone activity, this observation suggests that Pln also shows chaperone activity toward catalase-peroxidase. Hence, the increased catalase-peroxidase protein levels together with the reduced enzyme activities most likely is the result of the absence of both the proteolytic and chaperone activities of the Lon protease in P. chrysogenum pln cells.
As yet the bulk of the research on Lon substrate specificity has been carried out using bacterial Pln homologues and artificial substrates. Although some possible substrates have been proposed for mitochondrial Lon in man and bakers' yeast, direct evidence for Lon substrates has not been presented so far (19). Here, we show for the first time a bona fide substrate for Lon protease, namely catalase-peroxidase for Pln from P. chrysogenum. Our data are consistent with the view that catalase peroxidase, which is a crucial component of peroxisomal antioxidant defense system (26), may exemplify a particularly vulnerable peroxisomal protein and, therefore, requires substantial turnover by peroxisomal Lon protease.
In Arabidopsis thaliana, peroxisomal Lon protease was proposed to be involved in sustaining matrix protein import into peroxisomes of older cotyledon (52). For mammalian systems, contradictory results were reported; in cells of a HeLa cell line, peroxisomal matrix import was not affected by knockdown of peroxisomal Lon protease (53), whereas overproduction of a dominant negative variant of this protein in HEK293 cells resulted in mislocalization of the PTS1-containing enzyme catalase (54). In the filamentous fungus P. chrysogenum as well as in the methylotropic yeast H. polymorpha, peroxisomal Lon protease is not involved in sustaining matrix protein import, as mislocalization of GFP-SKL was not observed in these organisms (this study and Ref. 14).
Surprisingly, in P. chrysogenum, various peroxisomal metabolic pathways function normally in the absence of Pln except for the utilization of oleate, a process that requires peroxisomal β-oxidation. Moreover, oleic acid also was shown to be toxic to the cells. Activity measurements of the β-oxidation pathway revealed no major change in pln cells relative to wild-type controls. Hence, oleic acid oxidation can proceed normally in these cells. However, strongly enhanced oxidative stress was observed upon the addition of oleic acid to pln cells. A possible explanation for the failure of the cells to grow on oleate is that enhanced levels of H2O2, produced in the first oxidation step of oleate, inactivates the catalase-peroxidase protein, thereby releasing its co-factor heme, resulting in the generation of oxygen radicals via iron from heme in the Fenton reaction and thus leading to cell death.
Our biophysical data suggest that the P. chrysogenum peroxisomal Lon protease forms heptameric ring-shaped assemblies. Almost all members of the AAA protein family assemble into ring-like higher order structures (55). A stoichiometry of seven subunits was previously reported for mitochondrial Lon from S. cerevisiae (47), whereas bacterial Lon proteases are hexameric (19). Stahlberg et al. (47) demonstrated that significant conformational changes occur in yeast mitochondrial Lon protease depending on the presence of ATP. Because we analyzed Pln only in the presence of ATP, we cannot exclude that peroxisomal Lon protease undergoes similar ATP-dependent structural changes.
The hallmark of the AAA family of proteins is the ATP binding domain (AAA module), which contains conserved Walker A and B motifs, that function as an unfoldase (56). Cycles of ATP binding and hydrolysis drive conformational changes, creating pulses of pulling force that denature the substrate and translocate the unfolded polypeptide through the pore into the proteolytic chamber. Several cycles of protein binding and release are required to accomplish protein degradation by ATP-fueled proteolytic machines (57). Our in vitro experiments indicate that Pln displays ATP-driven refolding activities. Our data suggest that a destabilized protein that is captured by the peroxisomal Lon is able to escape from translocation into the proteolytic chamber and, instead of being degraded, is refolded. This is noteworthy as the observed chaperone activity was independent of the proteolytic activity of Pln. In agreement with these findings, a chaperone-like activity has been suggested before for the mitochondrial isoform of Lon protease (58) based on in vivo experiments.
Taken together, our data suggest that the P. chrysogenum peroxisomal Lon is a multifunctional protein (Fig. 9) and represents the first fungal peroxisomal chaperone identified. Our data are consistent with the view that Pln represents the first example of a conserved peroxisomal protein that functions in peroxisome matrix quality control and actively assists in refolding of peroxisomal matrix proteins.
FIGURE 9.

Model proposing the role of Pln in protein quality control in the peroxisome matrix. Peroxisomal matrix proteins most likely are delivered to the organelle in a folded conformation (1). Peroxisomal matrix proteins encounter stress (e.g. oxidative stress generated by peroxisomal metabolism) that may accelerate protein damage (2) that can be followed by protein aggregation (3). To cope with this, peroxisomes possess a bifunctional ATP-dependent Pln that controls the quality of peroxisomal proteome in two ways, as a chaperone protein (4) that facilitates refolding of destabilized peroxisomal proteins and as a protease that digests misfolded macromolecules (5) to prevent their aggregation. The lack of Pln may lead to accumulation of damaged proteins that may compromise peroxisomal function and cell viability.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr. F. Ulrich Hartl for valuable discussions. We thank Stefan Weber, Katarzyna Łukaszuk, and Annemarie Kralt for assistance in various parts of this study, EMBL Hamburg for providing resources, and the SPC facility (EMBL Hamburg) for technical support.
This work was supported in part by a Rubicon Fellowship (825.08.023) from the Netherlands Organization for Scientific Research (to C. W.).

This article contains supplemental Figs. S1–S5.
- PEN
- penicillin production
- MBP
- maltose-binding protein
- TEV
- tobacco etch virus
- TBARS
- thiobarbituric acid-reacting substance
- SLS
- static light scattering
- DLS
- dynamic light scattering
- SAXS
- small-angle x-ray scattering
- MM
- molecular mass
- DHE
- dihydroethidium
- PTS1
- peroxisomal targeting signal 1
- CS
- citrate synthase
- s
- scattering vector.
REFERENCES
- 1. Dobson C. M. (2003) Protein folding and misfolding. Nature 426, 884–890 [DOI] [PubMed] [Google Scholar]
- 2. Jaenicke R. (1998) Protein self-organization in vitro and in vivo: partitioning between physical biochemistry and cell biology. Biol. Chem. 379, 237–243 [DOI] [PubMed] [Google Scholar]
- 3. Jahn T. R., Radford S. E. (2005) The Yin and Yang of protein folding. FEBS J. 272, 5962–5970 [DOI] [PubMed] [Google Scholar]
- 4. Ellis R. J., Minton A. P. (2006) Protein aggregation in crowded environments. Biol. Chem. 387, 485–497 [DOI] [PubMed] [Google Scholar]
- 5. Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 6. Tyedmers J., Mogk A., Bukau B. (2010) Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 11, 777–788 [DOI] [PubMed] [Google Scholar]
- 7. Powers E. T., Morimoto R. I., Dillin A., Kelly J. W., Balch W. E. (2009) Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78, 959–991 [DOI] [PubMed] [Google Scholar]
- 8. Islinger M., Grille S., Fahimi H. D., Schrader M. (2012) The peroxisome. An update on mysteries. Histochem. Cell Biol. 137, 547–574 [DOI] [PubMed] [Google Scholar]
- 9. Wanders R. J., Waterham H. R. (2006) Peroxisomal disorders. The single peroxisomal enzyme deficiencies. Biochim. Biophys. Acta 1763, 1707–1720 [DOI] [PubMed] [Google Scholar]
- 10. Steinberg S. J., Raymond G. V., Braverman N. E., Moser A. B. (1993) Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum, University of Washington, Seattle, WA [Google Scholar]
- 11. Antonenkov V. D., Grunau S., Ohlmeier S., Hiltunen J. K. (2010) Peroxisomes are oxidative organelles. Antioxid. Redox Signal. 13, 525–537 [DOI] [PubMed] [Google Scholar]
- 12. Reeder B. J., Svistunenko D. A., Cooper C. E., Wilson M. T. (2004) The radical and redox chemistry of myoglobin and hemoglobin. From in vitro studies to human pathology. Antioxid. Redox Signal 6, 954–966 [DOI] [PubMed] [Google Scholar]
- 13. Walton P. A., Hill P. E., Subramani S. (1995) Import of stably folded proteins into peroxisomes. Mol. Biol. Cell 6, 675–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aksam E. B., Koek A., Kiel J. A., Jourdan S., Veenhuis M., van der Klei I. J. (2007) A peroxisomal lon protease and peroxisome degradation by autophagy play key roles in vitality of Hansenula polymorpha cells. Autophagy 3, 96–105 [DOI] [PubMed] [Google Scholar]
- 15. Kikuchi M., Hatano N., Yokota S., Shimozawa N., Imanaka T., Taniguchi H. (2004) Proteomic analysis of rat liver peroxisome. Presence of peroxisome-specific isozyme of Lon protease. J. Biol. Chem. 279, 421–428 [DOI] [PubMed] [Google Scholar]
- 16. Wagner I., Arlt H., van Dyck L., Langer T., Neupert W. (1994) Molecular chaperones cooperate with PIM1 protease in the degradation of misfolded proteins in mitochondria. EMBO J. 13, 5135–5145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. von Janowsky B., Knapp K., Major T., Krayl M., Guiard B., Voos W. (2005) Structural properties of substrate proteins determine their proteolysis by the mitochondrial AAA+ protease Pim1. Biol. Chem. 386, 1307–1317 [DOI] [PubMed] [Google Scholar]
- 18. Bota D. A., Davies K. J. (2002) Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 4, 674–680 [DOI] [PubMed] [Google Scholar]
- 19. Venkatesh S., Lee J., Singh K., Lee I., Suzuki C. K. (2012) Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim. Biophys. Acta 1823, 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Dijken J. P., Otto R., Harder W. (1976) Growth of Hansenula polymorpha in a methanol-limited chemostat. Physiological responses due to the involvement of methanol oxidase as a key enzyme in methanol metabolism. Arch. Microbiol. 111, 137–144 [DOI] [PubMed] [Google Scholar]
- 21. Bartoszewska M., Kiel J. A., Bovenberg R. A., Veenhuis M., van der Klei I. J. (2011) Autophagy deficiency promotes β-lactam production in. Penicillium chrysogenum. Appl. Environ. Microbiol. 77, 1413–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hillenga D. J., Versantvoort H. J., Driessen A. J., Konings W. N. (1994) Structural and functional properties of plasma membranes from the filamentous fungus. Penicillium chrysogenum. Eur. J. Biochem. 224, 581–587 [DOI] [PubMed] [Google Scholar]
- 23. Opaliński L., Kiel J. A., Homan T. G., Veenhuis M., van der Klei I. J. (2010) Penicillium chrysogenum Pex14/17p. A novel component of the peroxisomal membrane that is important for penicillin production. FEBS J. 277, 3203–3218 [DOI] [PubMed] [Google Scholar]
- 24. Sambrook J., Fritsch E. F., Maniatis T., Ford N. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 25. Kiel J. A., Hilbrands R. E., Bovenberg R. A., Veenhuis M. (2000) Isolation of Penicillium chrysogenum PEX1 and PEX6 encoding AAA proteins involved in peroxisome biogenesis. Appl. Microbiol. Biotechnol. 54, 238–242 [DOI] [PubMed] [Google Scholar]
- 26. Kiel J. A., van den Berg M. A., Fusetti F., Poolman B., Bovenberg R. A., Veenhuis M., van der Klei I. J. (2009) Matching the proteome to the genome. The microbody of penicillin-producing Penicillium chrysogenum cells. Funct. Integr. Genomics 9, 167–184 [DOI] [PubMed] [Google Scholar]
- 27. Fraaije M. W., Roubroeks H. P., Hagen W. R., Van Berkel W. J. (1996) Purification and characterization of an intracellular catalase-peroxidase from. Penicillium simplicissimum. Eur. J. Biochem. 235, 192–198 [DOI] [PubMed] [Google Scholar]
- 28. Bender T., Lewrenz I., Franken S., Baitzel C., Voos W. (2011) Mitochondrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol. Biol. Cell 22, 541–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Roermund C. W., Elgersma Y., Singh N., Wanders R. J., Tabak H. F. (1995) The membrane of peroxisomes in Saccharomyces cerevisiae is impermeable to NAD(H) and acetyl-CoA under in vivo conditions. EMBO J. 14, 3480–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paraszkiewicz K., Bernat P., Naliwajski M., Długoński J. (2010) Lipid peroxidation in the fungus Curvularia lunata exposed to nickel. Arch. Microbiol. 192, 135–141 [DOI] [PubMed] [Google Scholar]
- 31. Buchner J., Grallert H., Jakob U. (1998) Analysis of chaperone function using citrate synthase as nonnative substrate protein. Methods Enzymol. 290, 323–338 [DOI] [PubMed] [Google Scholar]
- 32. West S. M., Kelly S. M., Price N. C. (1990) The unfolding and attempted refolding of citrate synthase from pig heart. Biochim. Biophys. Acta 1037, 332–336 [DOI] [PubMed] [Google Scholar]
- 33. Nettleship J. E., Brown J., Groves M. R., Geerlof A. (2008) Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering. Methods Mol. Biol. 426, 299–318 [DOI] [PubMed] [Google Scholar]
- 34. Roessle M., Klaering R., Ristau U., Robrahn B., Jahn D., Gehrmann T., Konarev P., Round A., Fiedler S., Hermes C., Svergun D. (2007) Upgrade of the small-angle X-ray scattering beamline X33 at the European Molecular Biology Laboratory, Hamburg. J. Appl. Crystallogr. 40, 190–194 [Google Scholar]
- 35. Guinier A. (1939) La diffraction des rayons X aux tres petits angles; application a l'etude de phenomenes ultramicroscopiques. Ann. Phys. (Paris) 12, 161–237 [Google Scholar]
- 36. Petoukhov M. V., Konarev P., Kikhney A. G., Svergun D. I. (2007) ATSAS 2.1. Toward automated and web-supported small-angle scattering data analysis. J. Appl. Crystallogr. 40, 223–228 [Google Scholar]
- 37. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kozin M., Svergun D. I. (2001) Automated matching of high and low resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 40. Petoukhov M. V., Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Petoukhov M. V., Eady N. A., Brown K. A., Svergun D. I. (2002) Addition of missing loops and domains to protein models by x-ray solution scattering. Biophys. J. 83, 3113–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Svergun D. I., Barberato C., Koch M. H. J. (1995) CRYSOL. A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 43. Waterham H. R., Titorenko V. I., Haima P., Cregg J. M., Harder W., Veenhuis M. (1994) The Hansenula polymorpha PER1 gene is essential for peroxisome biogenesis and encodes a peroxisomal matrix protein with both carboxyl- and amino-terminal targeting signals. J. Cell Biol. 127, 737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Goldberg A. L., Moerschell R. P., Chung C. H., Maurizi M. R. (1994) ATP-dependent protease La (lon) from Escherichia coli. Methods Enzymol. 244, 350–375 [DOI] [PubMed] [Google Scholar]
- 45. Roudiak S. G., Seth A., Knipfer N., Shrader T. E. (1998) The lon protease from Mycobacterium smegmatis. Molecular cloning, sequence analysis, functional expression, and enzymatic characterization. Biochemistry 37, 377–386 [DOI] [PubMed] [Google Scholar]
- 46. Lee A. Y., Hsu C. H., Wu S. H. (2004) Functional domains of Brevibacillus thermoruber lon protease for oligomerization and DNA binding. Role of N-terminal and sensor and substrate discrimination domains. J. Biol. Chem. 279, 34903–34912 [DOI] [PubMed] [Google Scholar]
- 47. Stahlberg H., Kutejová E., Suda K., Wolpensinger B., Lustig A., Schatz G., Engel A., Suzuki C. K. (1999) Mitochondrial Lon of Saccharomyces cerevisiae is a ring-shaped protease with seven flexible subunits. Proc. Natl. Acad. Sci. U.S.A. 96, 6787–6790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Quiocho F. A., Spurlino J. C., Rodseth L. E. (1997) Extensive features of tight oligosaccharide binding revealed in high resolution structures of the maltodextrin transport/chemosensory receptor. Structure 5, 997–1015 [DOI] [PubMed] [Google Scholar]
- 49. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace. A web-based environment for protein structure homology modeling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 50. Meijer W. H., Gidijala L., Fekken S., Kiel J. A., van den Berg M. A., Lascaris R., Bovenberg R. A., van der Klei I. J. (2010) Peroxisomes are required for efficient penicillin biosynthesis in. Penicillium chrysogenum. Appl. Environ. Microbiol. 76, 5702–5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu F., Lu Y., Pieuchot L., Dhavale T., Jedd G. (2011) Import oligomers induce positive feedback to promote peroxisome differentiation and control organelle abundance. Dev. Cell 21, 457–468 [DOI] [PubMed] [Google Scholar]
- 52. Lingard M. J., Bartel B. (2009) Arabidopsis LON2 is necessary for peroxisomal function and sustained matrix protein import. Plant Physiol. 151, 1354–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Okumoto K., Kametani Y., Fujiki Y. (2011) Two proteases, trypsin domain-containing 1 (Tysnd1) and peroxisomal lon protease (PsLon), cooperatively regulate fatty acid β-oxidation in peroxisomal matrix. J. Biol. Chem. 286, 44367–44379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Omi S., Nakata R., Okamura-Ikeda K., Konishi H., Taniguchi H. (2008) Contribution of peroxisome-specific isoform of Lon protease in sorting PTS1 proteins to peroxisomes. J. Biochem. 143, 649–660 [DOI] [PubMed] [Google Scholar]
- 55. Iyer L. M., Leipe D. D., Koonin E. V., Aravind L. (2004) Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 146, 11–31 [DOI] [PubMed] [Google Scholar]
- 56. Hanson P. I., Whiteheart S. W. (2005) AAA+ proteins. Have engine, will work. Nat. Rev. Mol. Cell Biol. 6, 519–529 [DOI] [PubMed] [Google Scholar]
- 57. Sauer R. T., Bolon D. N., Burton B. M., Burton R. E., Flynn J. M., Grant R. A., Hersch G. L., Joshi S. A., Kenniston J. A., Levchenko I., Neher S. B., Oakes E. S., Siddiqui S. M., Wah D. A., Baker T. A. (2004) Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 119, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rep M., van Dijl J. M., Suda K., Schatz G., Grivell L. A., Suzuki C. K. (1996) Promotion of mitochondrial membrane complex assembly by a proteolytically inactive yeast Lon. Science 274, 103–106 [DOI] [PubMed] [Google Scholar]
- 59. Nijland J. G., Kovalchuk A., van den Berg M. A., Bovenberg R. A., Driessen A. J. (2008) Expression of the transporter encoded by the cefT gene of Acremonium chrysogenum increases cephalosporin production in Penicillium chrysogenum. Fungal Genet. Biol. 45, 1415–1421 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.