

Background: Dimethyl Fumarate (DMF) treatment leads to a reduction in IFN-γ+CD4+ T (Th1) cells in patients with multiple sclerosis.
Results: DMF reduces the antigen-presenting capacity of dendritic cells (DCs) via suppression of NF-κB and ERK1/2-MSK1 signaling.
Conclusion: DMF impaired DC maturation, resulting in decreased Th1 and Th17 cell differentiation.
Significance: This study defined one of the molecular mechanisms of DMF in inflammatory diseases.
Keywords: Antigen Presentation, Cytokine, Dendritic Cells, MAP Kinases (MAPKs), Multiple Sclerosis, NF-Kappa B (NF-KB), Dimethyl Fumarate
Abstract
Dimethyl fumarate (DMF) is an effective novel treatment for multiple sclerosis in clinical trials. A reduction of IFN-γ-producing CD4+ T cells is observed in DMF-treated patients and may contribute to its clinical efficacy. However, the cellular and molecular mechanisms behind this clinical observation are unclear. In this study, we investigated the effects of DMF on dendritic cell (DC) maturation and subsequent DC-mediated T cell responses. We show that DMF inhibits DC maturation by reducing inflammatory cytokine production (IL-12 and IL-6) and the expression of MHC class II, CD80, and CD86. Importantly, this immature DC phenotype generated fewer activated T cells that were characterized by decreased IFN-γ and IL-17 production. Further molecular studies demonstrated that DMF impaired nuclear factor κB (NF-κB) signaling via reduced p65 nuclear translocalization and phosphorylation. NF-κB signaling was further decreased by DMF-mediated suppression of extracellular signal-regulated kinase 1 and 2 (ERK1/2) and its downstream kinase mitogen stress-activated kinase 1 (MSK1). MSK1 suppression resulted in decreased p65 phosphorylation at serine 276 and reduced histone phosphorylation at serine 10. As a consequence, DMF appears to reduce p65 transcriptional activity both directly and indirectly by promoting a silent chromatin environment. Finally, treatment of DCs with the MSK1 inhibitor H89 partially mimicked the effects of DMF on the DC signaling pathway and impaired DC maturation. Taken together, these studies indicate that by suppression of both NF-κB and ERK1/2-MSK1 signaling, DMF inhibits maturation of DCs and subsequently Th1 and Th17 cell differentiation.
Introduction
Multiple sclerosis (MS)3 is an inflammatory demyelinating disease of the central nervous system (CNS). Although the cause of MS is unknown, accumulating evidence suggests that it is an autoimmune disorder mediated by autoreactive myelin-specific CD4+ T cells (1). Much effort has been invested to better understand the phenotypes of these pathogenic CD4+ T cells, and both IFN-γ-producing Th1 cells and IL-17-secreting Th17 cells have been identified as critical mediators in MS (2). Dendritic cells play a critical role in instructing the development of these pathogenic T cells. In the steady state, immature DCs lack the capacity to initiate a strong T cell response due to low expression of MHC class II, CD80, and CD86 (3, 4). Activation of DCs through Toll-like receptor signaling or encounter with antigen leads to maturation and increased expression of MHC class II, CD80, and CD86. These modifications allow DCs to become efficient at presenting antigen to naive T cells, which is critical for launching the adaptive immune response. Cytokine production is also initiated during the maturation of DCs. This is important as unique cytokine microenvironments influence naive T cell differentiation (3, 4). IL-12 instructs T cells toward IFN-γ-producing CD4+ cells (Th1), whereas IL-6 in the absence of IL-12 favors IL-17-producing cell (Th17) differentiation (2).
Given the important role of DCs in instructing CD4+ T cell differentiation, the association of DCs with MS has been investigated. Peripheral DCs from patients with MS are more mature and polarize naive T cells to secrete higher levels of cytokines when compared with DCs from healthy individuals (5). In addition to their key role in priming naive T cells in the periphery, CD11c+ DCs are also found in the cerebrospinal fluid and inflammatory MS lesions in the brain (6). This observation attracted the interest of many scientists to study the phenotypes and functions of DCs retained in the CNS. Evidence suggests that these CD11c+ DCs are capable of acquiring a mature phenotype in MS lesions to stimulate autoreactive T cells locally (7). In addition, these DCs may migrate to cervical draining lymph nodes and initiate new waves of autoreactive T cells. Based on these studies, DCs are emerging as a critical player in the priming of autoreactive T cells in the periphery and the CNS.
Dimethyl fumarate (DMF), in combination with monomethyl fumarate, was first approved for the treatment of psoriasis in Germany in 1994 (8). Two phase III clinical trials have demonstrated that oral administration of DMF significantly reduces gadolinium-enhancing lesions, annual relapse rate, and disease progression in relapsing-remitting MS (9, 10). The safety record, efficacy, and oral availability of DMF make it potentially more beneficial to MS patients than currently available therapies. Although immunological studies show that DMF treatment leads to a reduction in IFN-γ-producing CD4+ T cells in both MS and psoriasis patients (11, 12), the cellular and molecular mechanisms of DMF are not fully understood. Understanding the mechanisms by which DMF exerts its therapeutic effects will not only benefit drug development, but will also enhance our understanding of the molecular pathways that are critical to the pathophysiology of MS.
In the present study, we focus on the effect of DMF on DC maturation and subsequent DC-mediated T cell responses because DCs are the key element in the interface of innate and adaptive immune responses. We show that DMF inhibits maturation of DCs by significantly reducing cytokine production (IL-12 and IL-6) and expression of MHC class II, CD80, and CD86. These immature DCs generate fewer activated T cells, namely IFN-γ- and IL-17-producing CD4+ T cells. Further, our molecular studies demonstrate that DMF hinders the maturation of DCs via suppression of both p65 and ERK1/2-MSK1 signaling.
EXPERIMENTAL PROCEDURES
Bone Marrow-derived Dendritic Cells (BMDCs) Generated in Vitro
BMDCs were generated according to standard procedures (12). In brief, bone marrow cells were removed from the femurs and tibias of B10.PL or C57BL/6 mice and cultured in RPMI 1640 (Invitrogen) supplemented with l-glutamine and penicillin/streptomycin at 2 mm, β-mercaptoethanol (5 × 10−5 m), 10% FBS, and 2% J558L medium. J558L cells (kindly provided by Dr. Miguel Stadecker) secrete granulocyte-macrophage colony-stimulating factor (GM-CSF), which is needed to promote differentiation of bone marrow cells into DCs. Cells were cultured at 2 × 106 cells/well for 10 days with 5% CO2 and 95% air at 37 °C. Fresh medium supplemented with 2% J558L medium were added to the cell culture on days 3, 6, and 8. Cells were harvested on day 10 for in vitro assays. For mitogen-activated protein kinase (MAPK) and phospho-histone-3 analysis, BMDCs were harvested on day 10 and serum-starved for 3–4 h before LPS or DMF addition. This procedure allows us to 1) exclude the effects of the serum factors (e.g. growth factors) that may consistently activate MAPK and p-histone-3 and 2) distinguish between specific agonist-induced signaling and that mediated by DMF.
Dendritic Cells Generated in Vivo
DCs were expanded in vivo by subcutaneously administering 4 × 106 B16 melanoma cells, which overexpress murine Flt3L, to C57BL/6 female mice. DCs were isolated 3–4 weeks later. Whole spleens were processed into a single cell suspension, and CD11c+ DCs were then purified using CD11c magnetic bead positive selection (Miltenyi Biotec, Auburn, CA) on an AUTOMACS separator (Miltenyi Biotec) (13). Purity of DCs was verified by flow cytometry to be greater than 95%.
Dendritic Cells and MOG35–55-specific T Cell Co-culture
In vivo-generated DCs were purified and plated onto 24-well plates at 1 × 106 cells/well. Cells were stimulated with LPS (100 ng/ml, Sigma-Aldrich) in the presence or absence of DMF (70 μm, Sigma-Aldrich) for 24 h. Supernatants were then removed, and DCs were washed twice in warm PBS. The myelin oligodendrocyte glycoprotein (MOG)-specific 2D2 T cell receptor transgenic mouse generated by Dr. Vijay Kuchroo (14) was a generous gift from Dr. Caroline Whitacre. MOG35–55-specific T cells (4 × 106/well) were added to the conditioned DC culture in the presence of MOG35–55 (2 μg/ml). T cell activation and cytokine production were determined by flow cytometry and ELISA, respectively.
Thymidine Incorporation Assay
DCs generated in vivo were purified and plated on a 96-well plate at 1 × 105 cells/well. Cells were stimulated with LPS (100 ng/ml) in the presence or absence of DMF (70 μm) for 24 h. Supernatants were then removed, and DCs were washed twice in warm PBS. MOG35–55-specific T cells (4 × 105 cells/well) were added to the conditioned DCs in 96-well plates for 48 h. Cells were pulsed with 50 μl of medium containing 0.5 μCi of [3H]thymidine for 18 h. Cells were then harvested with the FilterMate harvester (PerkinElmer Life Sciences) onto UniFilter plates (PerkinElmer Life Sciences) and incubated overnight at room temperature to dry. A total of 30 μl of MicroScint 20 (PerkinElmer Life Sciences) were added to each well, and plates were sealed. [3H]Thymidine incorporation was read using a TopCount NXT machine (PerkinElmer Life Sciences).
Cytokine ELISA
Supernatants were collected for each culture condition and analyzed for IL-12p70, IL-6, IFN-γ, and IL-17 as described previously (15). ELISA was performed using purified anti-mouse capture antibodies and biotinylated rat anti-mouse detection antibodies (BD Biosciences). Cytokine concentrations were calculated by generating a standard curve using recombinant proteins (R&D Systems) and analyzed using SoftMax Pro software (Molecular Devices, Sunnyvale, CA).
Flow Cytometry and Intracellular Cytokine Staining
Flow cytometric analysis was performed to evaluate CD44 expression on T cells as described previously (15). In the co-culture, GolgiPlug (1 mg/ml; BD Biosciences) was used to block cytokine secretion for the last 4 h prior to staining. In addition, the expression of CD11c, CD80, CD86, and MHC class II on DCs was also examined under different conditions. About 105 live cell events were acquired on a FACSCanto II (BD Biosciences) and analyzed using FlowJo software (Tree Star). The following antibodies were purchased from BD Biosciences: fluorescein (FITC)-conjugated anti-CD44, peridinin-chlorophyll protein complex (PerCP)-conjugated anti-CD4, allophycocyanin (APC)-conjugated CD11c, fluorescein (FITC)-conjugated MHC class II, phycoerythrin (PE)-conjugated CD86, and V450-conjugated anti-CD80.
Immunohistochemistry
BMDCs were plated onto coverslips at 50,000 cells/coverslip and stimulated by LPS (100 ng/ml) in the presence or absence of DMF (70 μm). At 5 and 60 min after stimulation, cells were washed by 0.1 m PBS and fixed by 2% paraformaldehyde for 30 min at 4 °C. Cells were then stained by primary monoclonal rabbit anti-mouse p65 antibody (1:500, Cell Signaling). The location and distribution of p65 were visualized by incubation with Alexa Fluor 568 (1:500, Invitrogen)-conjugated goat anti-rabbit secondary antibody. DAPI (4′,6-diamidino-2-phenylindole, Sigma) was used to stain cell nuclei. To analyze the p65 distribution and reduce human bias, an automated scanning program was developed using the MetaMorph image analysis system (16). The intensity of p65 labeling in whole cell and nucleus was respectively collected by the program. Nuclear p65 distribution is expressed by the ratio of nuclear p65 and whole cell p65.
Western Blot
Whole cell protein was extracted using 1% Triton X-100 lysis buffer (20 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride (PMSF), 1 mm sodium orthovanadate, and 1:100 protease inhibitor (Sigma-Aldrich)) for phospho-p65, total p65, and IκBα detection. For MAPKs detection, whole cell protein was extracted using 1× SDS buffer with 1 mm PMSF, 1 mm sodium orthovanadate, and 1:100 protease inhibitor. Whole cell lysates were electrophoretically separated on Criterion Tris-HCl gel (10%) (Bio-Rad), transferred to nitrocellulose membranes (GE Healthcare), and then probed with the antibody of interest. Detections were performed using HRP-conjugated secondary antibodies followed by development with ECL Western blotting substrate (Cell Signaling), as described previously (17). The density of the bands was determined by using an AlphaImager (Alpha Innotech Corp.) p65 antibody, and pSer536-p65 antibody, IκBα antibody, pThr581-MSK1 antibody, phospho-ERK1/2 antibody, phospho-JNK antibody, and phospho-p38 antibody were purchased from Cell Signaling. pSer276-p65 antibody was from Abcam. β-Actin was from Sigma. U0126 was from Cell Signaling, and H89 was purchased from EMD Millipore.
Statistical Analysis
GraphPad Prism software (GraphPad, La Jolla, CA) was used to perform all statistical analyses. All statistical analyses were performed using a two-tailed Student's t test. A 0.05 p value was considered significant.
RESULTS
DMF Inhibits DC Maturation
As a promising novel medication, the beneficial effects of DMF correlate with a reduction in IFN-γ+ CD4+ T cells in patients (10). Given the important role of DCs in instructing T cell differentiation, we sought to determine whether DMF impairs DC maturation and subsequent DC-mediated T cell responses. BMDCs were stimulated by LPS, a TLR4 agonist, in the presence or absence of DMF, and cytokine secretion was determined by ELISA at 6 and 24 h. DMF treatment significantly inhibited production of IL-12 and IL-6, proinflammatory cytokines that mediate Th1 and Th17 differentiation (2) (Fig. 1A). The inhibitory effect of DMF was dose-dependent, with maximal inhibition achieved at the 70 μm concentration. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay indicated no toxicity of DMF at the dosages used (data not shown). Our cytokine data are in agreement with previous work from both our laboratory and others (12, 18).
FIGURE 1.
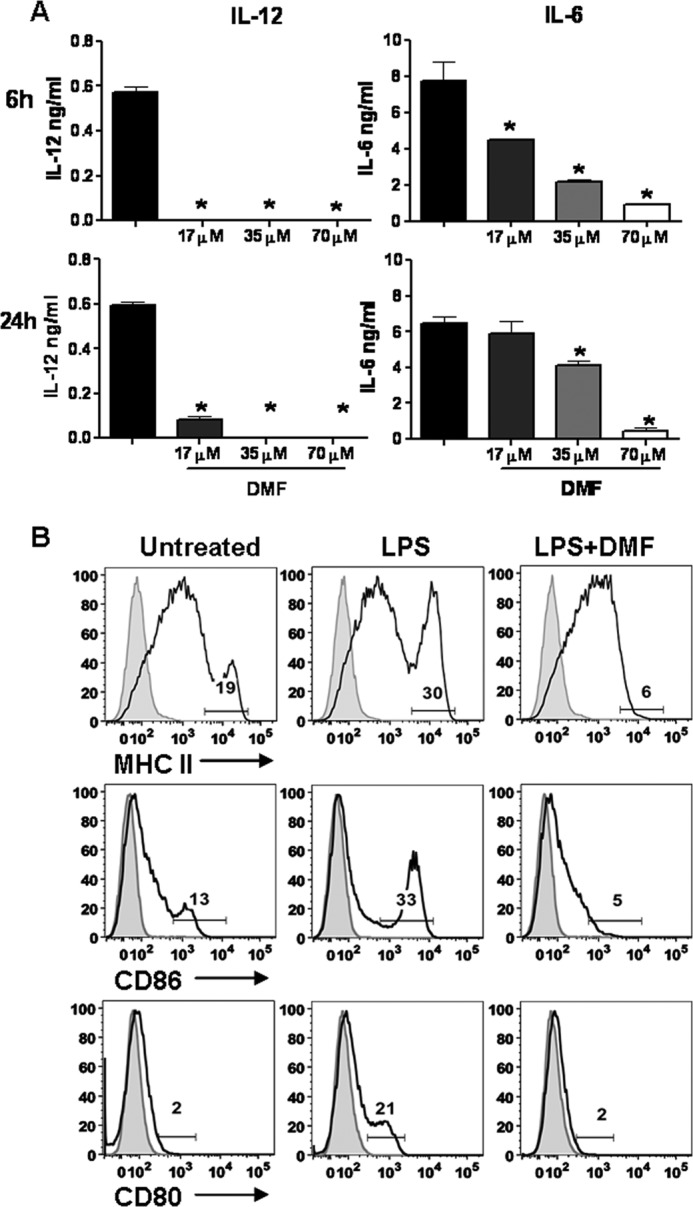
DMF inhibits dendritic cell maturation. A, BMDCs were stimulated by LPS (100 ng/ml) in the presence or absence of different dosages of DMF (17, 35, and 70 μm). Cytokine secretion was determined by ELISA at 6 and 24 h. Error bars indicate S.E. B, DCs were generated in vivo, purified, and stimulated by LPS (100 ng/ml) in the presence or absence of DMF (70 μm) for 24 h. MHC class II, CD80, and CD86 were then stained for flow cytometric analysis. Numbers indicate the percentages of MHC class IIhigh, CD80-positive, or CD86high. *, p < 0.01. Data are derived from one experiment representative of three independent experiments.
To further characterize DC responses to DMF, the expression of DC maturation markers was evaluated. Although the GM-CSF-differentiated DCs method is reproducible, easy, and inexpensive, GM-CSF-differentiated DCs display an inflammatory phenotype (19). Therefore, for the DC maturation and co-culture studies, we utilized DCs generated in vivo that are phenotypically equivalent to steady-state DCs in vivo (19). Our results suggest that LPS stimulation led to enhanced expression of MHC class IIhigh, CD80, and CD86high, whereas the addition of DMF completely abrogated the up-regulation of these surface molecules (Fig. 1B). Taken together, these data indicate that DMF inhibits DC maturation by suppressing both pro-inflammatory cytokine production and the expression of cell surface molecules required for effective antigen presentation to T cells.
DMF-treated DCs Are Less Effective Antigen-presenting Cells
As potent antigen-presenting cells, DCs present peptides on MHC molecules to naive T cells and instruct T cell activation and differentiation. Because DMF treatment inhibited DC maturation and the production of cytokines that mediate the differentiation of Th1 and Th17 cells, we next wanted to test whether DMF subsequently influenced DC-mediated T cell activation. In vivo-generated DCs were stimulated by LPS in the presence (LPS/DMF-DCs) or absence (LPS-DCs) of DMF for 24 h, washed to remove DMF and LPS, and then co-cultured with T cell receptor transgenic T cells specific for MOG35–55 in the presence of MOG35–55 peptide for 48–72 h. This experimental set-up allowed us to rule out direct effects of DMF on T cells because only DCs are exposed to DMF. T cell activation and proliferation were evaluated by flow cytometry and [3H]thymidine assay, respectively. As demonstrated in Fig. 2A, T cells generated by LPS/DMF-DCs had a markedly lower percentage of CD4+ T cells expressing CD44, a marker of effector and memory T cells, when compared with those generated by LPS-DCs. To determine whether this reduction in cellular activation also resulted in decreased cytokine production, namely the hallmark Th1 and Th17 cytokines IFN-γ and IL-17, cytokine production was evaluated by ELISA. Consistently, both IFN-γ and IL-17 in the LPS/DMF-DC+MOG35–55-specific T cell co-culture were significantly reduced (Fig. 2B). In addition, T cells generated by LPS-DCs had a high proliferation, whereas T cells from LPS/DMF-DCs co-culture had a significantly decreased proliferation (Fig. 2C). Thus, DMF inhibits the maturation and function of DCs, as well as the subsequent DC-mediated T cell response, resulting in reduced activation, cytokine production, and proliferation of MOG35–55-specific T cells.
FIGURE 2.

LPS/DMF-DCs generate less T cell activation and proliferation characterized by decreased IFN-γ and IL-17 production. Purified DCs were stimulated by LPS (100 ng/ml) in the presence or absence of DMF (70 μm) for 24 h, supernatant was then removed, and DCs were washed twice by warm PBS. MOG35–55-specific T cells were then added to the conditioned DC culture in the presence of MOG35–55 peptides (2 μg/ml). A, T cell activation was determined by flow cytometric analysis of CD44, an effector T cell marker, at both 48 h and 72 h after co-culture was set up. The number above the gate indicates the percentage of activated CD4+CD44+ T cells. B, cytokine production including IFN-γ and IL-17 in the co-culture system supernatant was evaluated by ELISA. C, proliferation was assessed by [3H]thymidine incorporation. Cells were pulsed at 48 h with [3H]thymidine and harvested 18 h later. [3H]Thymidine incorporation was measured using a TopCount NXT (PerkinElmer Life Sciences). *, p < 0.01. Data are derived from one experiment, which is representative of at least three independent experiments. Error bars indicate S.E.
DMF Inhibits the NF-κB Pathway in DCs
To explore the molecular mechanisms through which DMF impairs the maturation of DCs, the NF-κB pathway was analyzed because it is critical for TLR-induced DC maturation (see Fig. 7, Point 1). In the resting state, p65 is sequestered by IκBα in the cytoplasm. Upon stimulation with agents such as LPS, IκBα is rapidly phosphorylated and degraded. As a consequence, p65 is freed and translocates to the nucleus, where it binds to promoters of inflammatory genes and drives gene transcription. During this process, p65 is also phosphorylated at different sites to allow for optimal transcriptional activity (20). To test whether DMF treatment influences p65 activity, BMDCs were stimulated by LPS in the presence or absence of DMF. Cells were fixed at 5 and 60 min after LPS stimulation. Total p65 distribution was then evaluated by histology at each time point. As shown in Fig. 3A, the majority of p65 resides in the cytoplasm at 5 min after LPS activation, and as expected, a robust translocation of p65 to the nucleus was observed at 60 min. In contrast, DMF treatment retains p65 in the cytoplasm, resulting in significantly reduced p65 translocation to the nucleus (Fig. 3, A and B).
FIGURE 7.
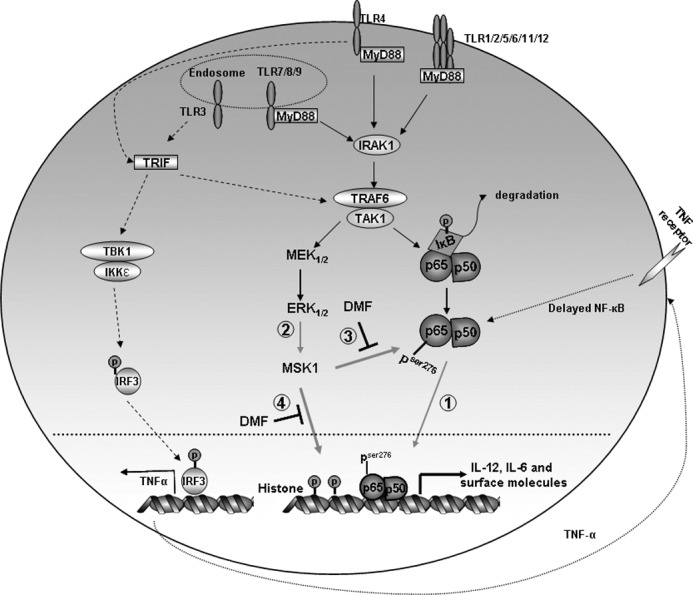
TLR signaling and proposed model for DMF-mediated impairment of DC maturation. All TLR signaling (except TLR3) events recruit the adaptor MyD88, followed by the interleukin-1 receptor-associated kinase (IRAK) family of protein kinases, leading to TNF receptor-associated factor-6 (TRAF6) activation followed by NF-κB activation. In addition to MyD88-dependent pathway, MyD88-independent (TIR domain-containing adapter-inducing interferon-β (TRIF)-dependent) pathway is utilized in TLR3/4 signaling. TLR3/4 recruit the adaptor TRIF, which can subsequently lead to activation of interferon regulatory factor 3 (IRF3). IRF3 then translocates into the nucleus and transcribes target genes, e.g. TNF-α. TNF-α binds to the TNF receptor and induces delayed NF-κB activation. In our study, we show that DMF suppresses p65 phosphorylation and nuclear localization (Point 1). p65 activation is further reduced by DMF-mediated MSK1 reduction (Point 2), which results in decreased phosphorylation of p65 at serine 276 (Point 3) and histone-3 at serine 10 (Point 4). IKKϵ, IκB kinase ϵ. TAK1, TGF-β activated kinase-1, TBK1, TANK-binding kinase-1.
FIGURE 3.

DMF inhibits p65 phosphorylation and nuclear translocation. A and B, BMDCs were stimulated by LPS (100 ng/ml) in the presence or absence of DMF (70 μm). A, cells were fixed by 2% paraformaldehyde at 5 and 60 min after LPS stimulation and then labeled with p65 and DAPI, revealing increased cytoplasmic retention of p65 in DMF conditions. The immunohistochemical results are quantified in B, which shows the ratio of nuclear p65 over whole cell p65 in LPS (black bar) and LPS+DMF (white bar) conditions. Data are derived from one experiment representative of two independent experiments. *, p < 0.01. Error bars indicate S.E. C, time course of phospho-p65 and IκBα expression in LPS and LPS+DMF conditions. BMDCs were stimulated by LPS (500 ng/ml) in the presence or absence of DMF (70 μm). Whole cell protein was then extracted at 0, 5, 20, 30, 40, and 60 min, and phospho-p65, p65, IκBα, and β-actin levels were determined by Western blot and quantified in D as the ratio of phospho-p65/p65 or IκBα/β-actin. Data are derived from one experiment representative of three independent experiments.
It has been well described that degradation of IκBα plays a critical role in altering the dynamic balance of p65 between cytoplasm and nucleus, favoring nuclear entry of p65 (21). To determine whether decreased degradation of IκBα was responsible for the reduced translocation of p65 to the nucleus after DMF treatment, IκBα protein levels were determined by Western blot. As demonstrated in Fig. 3C, DMF does not prevent IκBα degradation, suggesting that DMF does not inhibit p65 liberation. Phosphorylation of p65 enhances its nuclear stability and its capacity to recruit other co-activators to the promoter. To determine whether DMF inhibits phosphorylation of p65 at specific serine residues, Western blotting was performed on whole protein extracts using antibodies specific for phospho-Ser-276 and phospho-Ser-536. As demonstrated in Fig. 3, C and D, DMF reduces p65 phosphorylation, which may result in reduced capacity of p65 nuclear localization and decreased p65 transcriptional activity. These analyses of p65 localization and phosphorylation provide molecular evidence that DMF modulates signaling mechanisms that lead to DC maturation and cytokine production.
DMF Suppresses ERK1/2-MSK1 Signaling
In addition to NF-κB activation, MAPK family signaling is also involved and plays different roles in DC maturation (22). Suppression of p38 or extracellular signal-regulated kinase 1 and 2 (ERK1/2) activation by inhibitors prevents DC maturation, whereas activation of c-Jun N-terminal kinase (JNK) may limit DC maturation (23–25). To determine whether DMF alters MAPK signaling (see Fig. 7, Point 2), BMDCs were serum-starved for 3–4 h and then treated with DMF 1 h prior to LPS stimulation. U0126, an ERK1/2 inhibitor, was included as a positive control. Whole cell proteins were extracted at 5, 10, 20, and 30 min after LPS stimulation, and MAPK activity was determined by Western blot. Fig. 4 demonstrates that DMF treatment specifically inhibited phosphorylation of ERK1/2 (Fig. 4, A and B) but did not influence phosho-p38 or phospho-JNK activity (Fig. 4C). Previous studies suggest that MSK1, a downstream kinase of ERK1/2, positively regulates p65 signaling pathways. MSK1 enhances p65 transcriptional capacity by promoting phosphorylation of p65 at serine 276 (see Fig. 7, Point 3). In addition, MSK1 phosphorylates histone-3 at serine 10, which in turn promotes a permissive chromatin structure for transcriptional activity induced by p65 (see Fig. 7, Point 4). To address this issue, MSK1 activity was determined by Western blot. As shown in Fig. 4, A and B, LPS stimulation results in enhanced phosphorylation of MSK1, and this enhancement was impaired by DMF treatment.
FIGURE 4.

DMF inhibits ERK1/2-MSK1 signaling. Serum-starved-BMDCs were treated with DMF (70 μm) or ERK1/2 inhibitor U0126 (10 μm) 1 h before the addition of LPS (100 ng/ml). Whole cell protein was then extracted at 5, 10, 20, and 30 min, and p-ERK1/2, ERK1/2, p-JNK, JNK, p-p38, p38, p-MSK1, and β-actin were determined by Western blot. A, time course of expression of p-ERK1/2 and p-MSK1. B, quantification of Western blot images. Values are expressed as the ratio of p-ERK1/2/ERK1/2, and p-MSK1/β-actin. C, time course of expression of p-JNK, JNK, p-p38, and p38 in each condition. Data are derived from one experiment representative of three independent experiments.
DMF-mediated ERK1/2-MSK1 Reduction Contributes to the Compromised NF-κB Activity
Although the MAPK and NF-κB pathways are independent, they can interact via MSK1, where MSK1 phosphorylates p65 and enhances its transcriptional activity (see Fig. 7, Point 3). To dissect the contribution of ERK1/2-MSK1 signaling reduction to NF-κB inhibition caused by DMF, the MSK1 inhibitor H89 was used (20, 26–28). The effect of DMF-mediated MSK1 reduction on different levels of the p65 pathway was examined and compared with that of H89 (Fig. 5). BMDCs were stimulated by LPS (500 ng/ml), LPS+DMF, or LPS+H89 (10 μm), and whole cell proteins were analyzed for pSer10-H3 and pSer276-p65. As expected, H89 did not affect IκBα degradation. Interestingly, H89 successfully mimicked the inhibitory effect of DMF on pSer276-p65 and pSer10-H3, suggesting that DMF-induced-MSK1 reduction contributes to the compromised NF-κB activity.
FIGURE 5.

DMF-mediated-MSK1 reduction interferes with p65 signaling. A, serum-starved-BMDCs were treated with DMF (70 μm) or MSK1 inhibitor H89 (10 μm) 1 h before the addition of LPS (100 or 500 ng/ml). Whole cell protein was then extracted at 5, 20, 30, and 40 min, and pSer10-histone-3, histone-3, pSer276-p65, p65, IκBα, and β-actin were determined by Western blot. B, quantification of Western blot images. Values are expressed as the ratio of pSer276-p65/p65, pSer10-histone-3/histone-3, and IκBα/β-actin. Data are derived from one experiment representative of three independent experiments.
H89 Mimics the Inhibitory Effect of DMF on DC Maturation
To further determine whether DMF-induced MSK1 reduction is functionally important, the effect of H89 on DC maturation was determined. We hypothesized that H89 is able to mimic the inhibitory effect of DMF on DC maturation. To test this, purified DCs generated in vivo were stimulated by LPS, LPS+H89, or LPS+DMF for 24 h. The expression of MHC class II, CD80, and CD86 was evaluated by flow cytometric analysis (Fig. 6A). To determine whether the MHC class IIhigh, CD80, and CD86high cells are the same population, cells were first gated on MHC class IIhigh and then on CD80 and CD86high (Fig. 6B). In the untreated condition, about 45% of MHC class II high cells are CD86high, and only about 5% are CD80 positive. In contrast, after LPS stimulation, the percentage of MHC class IIhigh cells increased, virtually all MHC IIhigh cells were also CD86high, and about 73% of MHC class IIhigh CD86high cells were also CD80-positive. This demonstrates that LPS induces a mature population expressing MHC class IIhigh CD86high and CD80, which is expected to be highly efficient at driving T cell responses. Importantly, DMF completely blocked the development of this mature population. H89 partially mimicked the effect of DMF on DC maturation by decreasing the expression of MHC class IIhigh from 38 to 8%, expression of CD80 from 31 to 4%, and expression of CD86high from 44% to 11% (Fig. 6A). In addition, cytokine production was evaluated in different conditions. DMF reduced the secretion of both IL-12 (undetectable) and IL-6 (95% reduction). Similarly, H89 treatment led to a 85% reduction in IL-12 and a 60% reduction in IL-6 (Fig. 6C) Taken together, our data suggest that MSK1 reduction is critical for the inhibitory effect of DMF on DC maturation.
FIGURE 6.

DMF-mediated MSK1 reduction is essential for the inhibitory effect of DMF on DC maturation. Purified DCs were stimulated by LPS (100 ng/ml), LPS+H89 (10 μm), or LPS+DMF (70 μm) for 24 h. Untreated condition was included as control. A, MHC class IIhigh, CD80, and CD86high expression was determined by flow cytometry. B, cells were first gated on MHC class IIhigh and then gated on CD80 and CD86high. C, cytokine secretion in each condition was evaluated by ELISA. *, p < 0.01. Data are derived from one experiment representative of two independent experiments. Error bars indicate S.E.
DISCUSSION
Our data show that the expression of DC maturation markers including MHC class IIhigh, CD80, and CD86high is substantially inhibited by DMF. In addition, IL-12 and IL-6 production is significantly reduced. These cytokines are essential for Th1 and Th17 cell differentiation (12, 29). TLR4-mediated NF-κB activation involves both myeloid differentiation factor 88 (MyD88)-dependent and MyD88-independent pathways (Fig. 7). Although proinflammatory cytokine production is mainly via MyD88-dependent signaling, these cytokines are still inducible in MyD88/TNF receptor-associated factor-6 (TRAF6) double knock-out mice, indicating the involvement of MyD88-independent pathway in cytokine production after TLR4 stimulation (30, 31) Thus, it is possible that the inhibitory effect of DMF on DC maturation is via both pathways. Additionally, DMF may also block signaling of other TLRs because MyD88-dependent pathway is involved in signaling of all TLRs (with the exception of TLR3), and MyD88-independent pathway is involved in TLR3/TLR4 signaling (31–33).
Our data further demonstrate that these immature DCs have impaired capacity in driving T cell activation. T cells generated by LPS/DMF-DCs activate and proliferate less and produce decreased levels of Th1 and Th17 hallmark cytokines. This is in agreement with the clinical observation that DMF treatment leads to pathogenic Th1 (IFN-γ+CD4+ T cell) cell reduction (11, 12). Thus, our study provides an important mechanism by which DMF suppresses T cell activation and proliferation. Clinical data in regard to the effect of DMF on Th17 cells, another pathogenic cell population, are not yet available. However, IL-17 production in the LPS/DMF-DCs+MOG35–55-specific T cell co-culture was also significantly reduced, suggesting that Th17 differentiation is affected by DMF treatment as well. It would be of interest to determine whether the Th17 population in MS patients is decreased during the course of DMF treatment. It should be noted that some studies suggest that DMF leads to direct enhancement in CD4+ T cell apoptotic rate. However, the increases in apoptotic rate were not sustained, and by 12 weeks of treatment, the rate of apoptosis had returned to base-line levels, yet the IFN-γ-producing CD4+ T cell count remains low during the entire treatment course (11, 12). Because of its immunosuppressive effects, concerns may arise on potential adverse effects of DMF treatment. However, based on the MS clinical trial data and experiences from psoriasis patients, there is no evidence of increased risk of opportunistic infection or malignancy in DMF-treated individuals (8–11).
To enhance our understanding of the effects of DMF on DCs, we began to explore the molecular targets of DMF in DCs. Previous studies showed that both NF-κB and MAPK activation contribute to LPS-mediated DC maturation (34, 35). In the resting state, p65 is bound to IκBα in the cytosol. Upon LPS stimulation, IκBα is phosphorylated, ubiquitinated, and degraded by the proteasome machinery, releasing p65 to translocate to the nucleus and transactivate genes (Fig. 7, Point 1). In addition, site-specific phosphorylation also occurs to increase p65 transcriptional activity either by enhancing binding to coactivators and basal transcription factors or by increasing p65 nuclear localization and stability (21, 36). Phosphorylation at the same site can be achieved by multiple kinases, often in a cell-specific manner. For instance, Ser-276 of p65 is phosphorylated by protein kinase A (PKA) and MSK1, whereas phosphorylation at Ser-536 is mediated by different IκB kinases (37, 38). Phosphorylation of both Ser-536 and Ser-276 plays critical roles in regulating p65 activity (27, 36, 38–41).
In the current study, we demonstrate that DMF inhibits p65 signaling by decreasing its phosphorylation and its entry to the nucleus. However, reduced p65 nuclear localization is independent of IκBα as DMF does not prevent its degradation. Our finding is in agreement with others where in TNF-α stimulated endothelial cells, DMF inhibits translocation of p65 to the nucleus without inhibiting IκBα degradation (42). What causes reduced nuclear localization of p65 by DMF treatment? One possible explanation might be the reduced phosphorylation of p65 caused by DMF. Phosphorylation of p65 at serine 276 was suggested to promote acetylation and retention of p65 in the nucleus (43). In addition, evidence also suggests that pSer536-p65 does not exist in the classical NF-κB complex of p50 and p65. Instead of being controlled by IκB, its nuclear localization is probably facilitated by another protein that is not involved in the classical NF-κB pathway (44). In our study, the fact that the reduction in pSer536-p65 is independent of IκBα degradation indicates that DMF regulates phospho-p65 nuclear translocation through an alternative mechanism (44). We hope to address this question once these phospho-p65 nuclear translocation regulators are identified.
MSK1, a downstream protein of ERK1/2 or p38, potentiates p65 transcriptional activity (Fig. 7, Point 3). Initially identified as a nuclear kinase, MSK1 was later shown to present in the cytosol as well (26). MSK1 phosphorylates p65 at serine 276, and this phosphorylation was much reduced in MSK1/2-deficient mouse embryonic fibroblasts (36). It has been well documented that once phosphorylated at serine 276, p65 undergoes configuration change, allowing its enhanced capacity of recruiting other co-factors to the gene promoters (20). In addition, through phosphorylation of histone-3 at serine 10, MSK1 promotes local relaxation of DNA and enhances the accessibility of p65 to gene promoters including IL-6, IL-12p40, and IL-8 (28, 45). In fact, by chromatin immunoprecipitation (ChIP) analysis, MSK1 and its substrates including both pSer276-p65 and pSer10-histone-3 were shown to localize to the endogenous NF-κB containing IL-6 promoter upon TNF-α stimulation (27, 46).
In our study, we demonstrate that DMF inhibits MSK1 activation through its upstream protein ERK1/2 (Fig. 7, Point 2). Our observation is in conflict with a study published by Gesser et al. (47) where DMF inhibits MSK1 without affecting p38 or ERK1/2. However, different cell types and different stimuli may explain the difference we observed. Further, the importance of MSK1 as a target of DMF was supported by the mimicry experiments. We showed that without decreasing IκBα degradation, H89, an MSK1 inhibitor, reduces phosphorylation of p65 at serine 276 and of histone-3 at serine 10. Moreover, this reduction in pSer276-p65 and pSer10-histone-3 is functionally important because H89 successfully inhibits DC maturation by reducing IL-6/IL-12 and MHC class IIhigh, CD80, and CD86high expression. It should be noted that H89 partially mimics the function of DMF, which indicates the involvement of other phospho-p65 (e.g., pSer536-p65) as targets by DMF.
In conclusion, we demonstrate that DMF inhibits DC maturation and function, and subsequently, suppresses Th1 and Th17 cell differentiation. Further, our molecular data demonstrate that DMF exerts its effect via suppression of both p65 and ERK1/2-MSK1 signaling. Our study provides an important mechanism for the action of DMF in modulating inflammation and provides insight into pathways that are effective targets for therapeutic intervention in inflammatory diseases with unknown etiology such as multiple sclerosis.
Acknowledgments
We thank Todd Shawler for flow cytometry expertise, Dr. John Gensel for developing the imaging program for p65 histological quantification, Zachary Vangundy for AUTOMACS expertise, and Curtis Pannell for crucial support of mouse studies. We also thank Dr. Paul Drew, Dr. Susheela Tridandapani, and Dr. Denis Guttridge for the critical reading of our manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 NS067441-01 to A. E. L. R.) and RO1 NS037513-10 (to M. K. R.). This work was also supported by National Multiple Sclerosis Society Grant NMSS 4358-A-13 (to M. K. R.).
- MS
- multiple sclerosis
- DMF
- dimethyl fumarate
- DC
- dendritic cell
- BMDC
- bone marrow-derived dendritic cell
- MOG
- myelin oligodendrocyte glycoprotein
- MSK1
- mitogen stress-activated kinase 1
- MyD88
- myeloid differentiation factor 88
- TLR
- Toll-like receptor
- p
- phospho.
REFERENCES
- 1. Frohman E. M., Racke M. K., Raine C. S. (2006) Multiple sclerosis–the plaque and its pathogenesis. N. Engl. J. Med. 354, 942–955 [DOI] [PubMed] [Google Scholar]
- 2. Lovett-Racke A. E., Yang Y., Racke M. K. (2011) Th1 versus Th17: are T cell cytokines relevant in multiple sclerosis? Biochim. Biophys. Acta 1812, 246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chastain E. M., Duncan D. S., Rodgers J. M., Miller S. D. (2011) The role of antigen-presenting cells in multiple sclerosis. Biochim. Biophys. Acta 1812, 265–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steinman R. M., Banchereau J. (2007) Taking dendritic cells into medicine. Nature 449, 419–426 [DOI] [PubMed] [Google Scholar]
- 5. Karni A., Abraham M., Monsonego A., Cai G., Freeman G. J., Hafler D., Khoury S. J., Weiner H. L. (2006) Innate immunity in multiple sclerosis: myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J. Immunol. 177, 4196–4202 [DOI] [PubMed] [Google Scholar]
- 6. Kivisäkk P., Mahad D. J., Callahan M. K., Sikora K., Trebst C., Tucky B., Wujek J., Ravid R., Staugaitis S. M., Lassmann H., Ransohoff R. M. (2004) Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann. Neurol. 55, 627–638 [DOI] [PubMed] [Google Scholar]
- 7. Serafini B., Rosicarelli B., Magliozzi R., Stigliano E., Capello E., Mancardi G. L., Aloisi F. (2006) Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J. Neuropathol. Exp. Neurol. 65, 124–141 [DOI] [PubMed] [Google Scholar]
- 8. Mrowietz U., Asadullah K. (2005) Dimethylfumarate for psoriasis: more than a dietary curiosity. Trends Mol Med. 11, 43–48 [DOI] [PubMed] [Google Scholar]
- 9.(2011) Trial watch: Phase III success for Biogen's oral multiple sclerosis therapy. Nat. Rev. Drug Discov. 10, 404. [DOI] [PubMed] [Google Scholar]
- 10. Kappos L., Gold R., Miller D. H., Macmanus D. G., Havrdova E., Limmroth V., Polman C. H., Schmierer K., Yousry T. A., Yang M., Eraksoy M., Meluzinova E., Rektor I., Dawson K. T., Sandrock A. W., O'Neill G. N. (2008) Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomized, double-blind, placebo-controlled phase IIb study. Lancet 372, 1463–1472 [DOI] [PubMed] [Google Scholar]
- 11. Schimrigk S., Brune N., Hellwig K., Lukas C., Bellenberg B., Rieks M., Hoffmann V., Pöhlau D., Przuntek H. (2006) Oral fumaric acid esters for the treatment of active multiple sclerosis: an open-label, base line-controlled pilot study. Eur. J. Neurol. 13, 604–610 [DOI] [PubMed] [Google Scholar]
- 12. Ghoreschi K., Brück J., Kellerer C., Deng C., Peng H., Rothfuss O., Hussain R. Z., Gocke A. R., Respa A., Glocova I., Valtcheva N., Alexander E., Feil S., Feil R., Schulze-Osthoff K., Rupec R. A., Lovett-Racke A. E., Dringen R., Racke M. K., Röcken M. (2011) Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J. Exp. Med. 208, 2291–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Papenfuss T. L., Powell N. D., McClain M. A., Bedarf A., Singh A., Gienapp I. E., Shawler T., Whitacre C. C. (2011) Estriol generates tolerogenic dendritic cells in vivo that protect against autoimmunity. J. Immunol. 186, 3346–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bettelli E., Pagany M., Weiner H. L., Linington C., Sobel R. A., Kuchroo V. K.(2003) Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 197, 1073–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huss D. J., Winger R. C., Cox G. M., Guerau-de-Arellano M., Yang Y., Racke M. K., Lovett-Racke A. E. (2011) TGF-β signaling via Smad4 drives IL-10 production in effector Th1 cells and reduces T cell trafficking in EAE. Eur. J. Immunol. 41, 2987–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fuseler J. W., Merrill D. M., Rogers J. A., Grisham M. B., Wolf R. E. (2006) Analysis and quantitation of NF-κB nuclear translocation in tumor necrosis factor α (TNF-α) activated vascular endothelial cells. Microsc. Microanal. 12, 269–276 [DOI] [PubMed] [Google Scholar]
- 17. Lovett-Racke A. E., Rocchini A. E., Choy J., Northrop S. C., Hussain R. Z., Ratts R. B., Sikder D., Racke M. K. (2004) Silencing T-bet defines a critical role in the differentiation of autoreactive T lymphocytes. Immunity 21, 719–731 [DOI] [PubMed] [Google Scholar]
- 18. Lehmann J. C., Listopad J. J., Rentzsch C. U., Igney F. H., von Bonin A., Hennekes H. H., Asadullah K., Docke W. D. (2007) Dimethylfumarate induces immunosuppression via glutathione depletion and subsequent induction of heme oxygenase 1. J. Invest. Dermatol. 127, 835–845 [DOI] [PubMed] [Google Scholar]
- 19. Xu Y., Zhan Y., Lew A. M., Naik S. H., Kershaw M. H. (2007) Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J. Immunol. 179, 7577–7584 [DOI] [PubMed] [Google Scholar]
- 20. Chen L. F., Greene W. C. (2004) Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 5, 392–401 [DOI] [PubMed] [Google Scholar]
- 21. Bakkar N., Guttridge D. C. (2010) NF-κB signaling: a tale of two pathways in skeletal myogenesis. Physiol. Rev. 90, 495–511 [DOI] [PubMed] [Google Scholar]
- 22. Hazzalin C. A., Mahadevan L. C. (2002) MAPK-regulated transcription: a continuously variable gene switch? Nat. Rev. Mol. Cell Biol. 3, 30–40 [DOI] [PubMed] [Google Scholar]
- 23. Svajger U., Obermajer N., Anderluh M., Kos J., Jeras M. (2011) DC-SIGN ligation greatly affects dendritic cell differentiation from monocytes compromising their normal function. J. Leukoc Biol. 89, 893–905 [DOI] [PubMed] [Google Scholar]
- 24. Mou H. B., Lin M. F., Huang H., Cai Z. (2011) Transforming growth factor-β1 modulates lipopolysaccharide-induced cytokine/chemokine production and inhibits nuclear factor-κB, extracellular signal-regulated kinases, and p38 activation in dendritic cells in mice. Transplant Proc. 43, 2049–2052 [DOI] [PubMed] [Google Scholar]
- 25. Handley M. E., Thakker M., Pollara G., Chain B. M., Katz D. R. (2005) JNK activation limits dendritic cell maturation in response to reactive oxygen species by the induction of apoptosis. Free Radic. Biol. Med. 38, 1637–1652 [DOI] [PubMed] [Google Scholar]
- 26. Beck I. M., Vanden Berghe W., Vermeulen L., Bougarne N., Vander Cruyssen B., Haegeman G., De Bosscher K. (2008) Altered subcellular distribution of MSK1 induced by glucocorticoids contributes to NF-κB inhibition. EMBO J. 27, 1682–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vermeulen L., De Wilde G., Van Damme P., Vanden Berghe W., Haegeman G. (2003) Transcriptional activation of the NF-κB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 22, 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arthur J. S. (2008) MSK activation and physiological roles. Front. Biosci. 13, 5866–5879 [DOI] [PubMed] [Google Scholar]
- 29. Yang Y., Weiner J., Liu Y., Smith A. J., Huss D. J., Winger R., Peng H., Cravens P. D., Racke M. K., Lovett-Racke A. E. (2009) T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J. Exp. Med. 206, 1549–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kawai T., Takeuchi O., Fujita T., Inoue J., Mühlradt P. F., Sato S., Hoshino K., Akira S. (2001) Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167, 5887–5894 [DOI] [PubMed] [Google Scholar]
- 31. Hansen B. S., Hussain R. Z., Lovett-Racke A. E., Thomas J. A., Racke M. K. (2006) Multiple Toll-like receptor agonists act as potent adjuvants in the induction of autoimmunity. J. Neuroimmunol. 172, 94–103 [DOI] [PubMed] [Google Scholar]
- 32. Covert M. W., Leung T. H., Gaston J. E., Baltimore D. (2005) Achieving stability of lipopolysaccharide-induced NF-κB activation. Science 309, 1854–1857 [DOI] [PubMed] [Google Scholar]
- 33. Doyle S. L., O'Neill L. A. (2006) Toll-like receptors: from the discovery of NF-κB to new insights into transcriptional regulations in innate immunity. Biochem. Pharmacol. 72, 1102–1113 [DOI] [PubMed] [Google Scholar]
- 34. Guha M., Mackman N. (2001) LPS induction of gene expression in human monocytes. Cell. Signal. 13, 85–94 [DOI] [PubMed] [Google Scholar]
- 35. Rescigno M., Martino M., Sutherland C. L., Gold M. R., Ricciardi-Castagnoli P. (1998) Dendritic cell survival and maturation are regulated by different signaling pathways. J. Exp. Med. 188, 2175–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen L. F., Williams S. A., Mu Y., Nakano H., Duerr J. M., Buckbinder L., Greene W. C. (2005) NF-κB RelA phosphorylation regulates RelA acetylation. Mol. Cell. Biol. 25, 7966–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. O'Mahony A. M., Montano M., Van Beneden K., Chen L. F., Greene W. C. (2004) Human T cell lymphotropic virus type 1 Tax induction of biologically Active NF-κB requires IκB kinase-1-mediated phosphorylation of RelA/p65. J. Biol. Chem. 279, 18137–18145 [DOI] [PubMed] [Google Scholar]
- 38. Joo J. H., Jetten A. M. (2008) NF-κB-dependent transcriptional activation in lung carcinoma cells by farnesol involves p65/RelA(Ser-276) phosphorylation via the MEK-MSK1 signaling pathway. J. Biol. Chem. 283, 16391–16399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reber L., Vermeulen L., Haegeman G., Frossard N. (2009) Ser-276 phosphorylation of NF-κB p65 by MSK1 controls SCF expression in inflammation. PLoS One 4, e4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arun P., Brown M. S., Ehsanian R., Chen Z., Van Waes C. (2009) Nuclear NF-κB p65 phosphorylation at serine 276 by protein kinase A contributes to the malignant phenotype of head and neck cancer. Clin. Cancer Res. 15, 5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang X., Takahashi N., Matsui N., Tetsuka T., Okamoto T. (2003) The NF-κB activation in lymphotoxin β receptor signaling depends on the phosphorylation of p65 at serine 536. J. Biol. Chem. 278, 919–926 [DOI] [PubMed] [Google Scholar]
- 42. Loewe R., Holnthoner W., Gröger M., Pillinger M., Gruber F., Mechtcheriakova D., Hofer E., Wolff K., Petzelbauer P. (2002) Dimethylfumarate inhibits TNF-induced nuclear entry of NF-κB/p65 in human endothelial cells. J. Immunol. 168, 4781–4787 [DOI] [PubMed] [Google Scholar]
- 43. Gao N., Asamitsu K., Hibi Y., Ueno T., Okamoto T. (2008) AKIP1 enhances NF-κB-dependent gene expression by promoting the nuclear retention and phosphorylation of p65. J. Biol. Chem. 283, 7834–7843 [DOI] [PubMed] [Google Scholar]
- 44. Sasaki C. Y., Barberi T. J., Ghosh P., Longo D. L. (2005) Phosphorylation of RelA/p65 on serine 536 defines an IκBα-independent NF-κB pathway. J. Biol. Chem. 280, 34538–34547 [DOI] [PubMed] [Google Scholar]
- 45. Saccani S., Pantano S., Natoli G. (2002) p38-dependent marking of inflammatory genes for increased NF-κB recruitment. Nat. Immunol. 3, 69–75 [DOI] [PubMed] [Google Scholar]
- 46. Pathak S. K., Basu S., Bhattacharyya A., Pathak S., Banerjee A., Basu J., Kundu M. (2006) TLR4-dependent NF-κB activation and mitogen- and stress-activated protein kinase 1-triggered phosphorylation events are central to Helicobacter pylori peptidyl prolyl cis-, trans-isomerase (HP0175)-mediated induction of IL-6 release from macrophages. J. Immunol. 177, 7950–7958 [DOI] [PubMed] [Google Scholar]
- 47. Gesser B., Johansen C., Rasmussen M. K., Funding A. T., Otkjaer K., Kjellerup R. B., Kragballe K., Iversen L. (2007) Dimethylfumarate specifically inhibits the mitogen and stress-activated kinases 1 and 2 (MSK1/2): possible role for its anti-psoriatic effect. J. Invest. Dermatol. 127, 2129–2137 [DOI] [PubMed] [Google Scholar]