

Abstract
Pheochromocytomas are rare tumours originating from the chromaffin tissue. The clinical manifestations are variable and are not specific; as a result, pheochromocytomas often imitate other diseases. The diagnosis is usually established by biochemical studies, i.e., the measurement of catecholamines or their metabolites in urine or plasma, followed by radiographic and scintigraphic studies for localisation. Surgical removal of the tumour is the preferred treatment. We report a 30-year-old woman presenting with an adrenal incidentaloma that was 7.6 × 5.3 × 4.8 cm in size on an abdominal computed tomography scan. Investigations for adrenal hormones, including a low-dose dexamethasone suppression test, plasma aldosterone level, 24-hour urinary metanephrine and vanillylmandelic acid levels, and plasma metanephrine level were all within the normal ranges. During the surgical resection, the patient had a hypertensive spell. Surgery was postponed, and the blood pressure was adequately controlled with α blockers, followed by β blockers. After 2 weeks, the surgery was followed by a pathological biopsy that confirmed the pheochromocytoma diagnosis.
Keywords: adrenal incidentaloma, catecholamines, hypertension, pheochromocytoma, scintigraphy
Introduction
Pheochromocytomas are catecholamine-producing tumours arising from chromaffin cells in the sympathoadrenal system. Their prevalence is estimated at 0.1% to 0.6%, and 80% to 85% of them arise from the adrenal medulla; however, tumours of the adrenal medulla predominantly secrete norepinephrine over epinephrine, whereas the adrenal medulla normally secretes 80% epinephrine (1). The various catecholamines give rise to sustained or paroxysmal hypertension, along with symptoms of headaches, palpitations, profuse sweating, breathlessness, anxiety, a sense of dread, chest pain, nausea, vomiting, tremors, and paraesthesia. Hyperglycaemia due to the anti-insulinaemic actions of catecholamines can produce polyuria and polydipsia. In severe cases, a patient can present with myocardial infarction, heart failure, pulmonary oedema, arrhythmias, or intracranial haemorrhage (2). A diagnosis is established by measuring the levels of metanephrines in the urine or blood (2). Localisation of the tumour is performed by computed tomography (CT) or magnetic resonance imaging (MRI). Occasionally, the tumour can present as an asymptomatic adrenal incidentaloma after radiography. Here, we report one such interesting case.
Case Report
A 30-year-old woman was referred to the department of endocrinology for an evaluation of an adrenal incidentaloma that was detected on a CT scan. She did not have any complaints of episodic headaches, palpitations, sweating, chest pain, or hypertension. There was no history of weight gain, excess hair growth on the body, acne, proximal muscle weakness, or menstrual irregularity. No previous history of hypertension, weakness, anorexia, vomiting, abdominal pain, fever, diarrhoea, polyuria, or polydipsia could be elucidated.
On examination, patient’s pulse rate was 76 beats/min, blood pressure (BP) was 110/70 mmHg, without any postural drop, and respiratory rate was 20 breaths/min. Her height was 152 cm, and her weight was 47.2 kg. She was pale and without oedema. There was no evidence of moon facies, hirsutism, acne, purple abdominal striae, acanthosis nigricans, or knuckle or mucosal pigmentation. Her systematic examination, which included a fundoscopy, was normal.
Routine haemogram, urine examination, liver and kidney function tests, and other laboratory parameters including serum electrolytes, arterial blood gas analysis, and plasma calcium and phosphorous levels were normal. No abnormalities were noted in the electrocardiography or chest X-ray. Biochemical tests to determine the functional hormone secretion were found to be normal and are depicted in Table 1.
Table 1:
The baseline biochemical parameters of the patient
| Parameter | Value | Normal range |
|---|---|---|
| Plasma cortisol at 8:00 AM (μg/dL) | 17.2 | 5–25 |
| Plasma cortisol after ONDST (μg/dL) | 2.4 | < 5 |
| Plasma aldosterone (ng/dL) | 5.4 | 2–20 |
| 24-hour urine fractionated metanephrines (mg/24 h) | 0.7 | < 1.3 |
| 24-hour urine VMA (mg/24 h) | 5.3 | 1–8 |
| Plasma metanephrines (μg/dL) | 34 | < 60 |
Abbreviation: ONDST = overnight 1-mg dexamethasone suppression test, VMA = vanillylmandelic acid
CT of the abdomen revealed a well-defined, heterogeneous enhancing mass lesion that was 7.6 × 5.3 × 4.8 cm in size, with an attenuation score of 35 HU, and without any calcifications at the upper pole of the right kidney. The left adrenal gland appeared to be normal (Figure 1). Considering the possibility of a non-functional adrenal incidentaloma that was more than 6 cm in size and with a CT attenuation score of 35 HU, neither of which suggest a benign lesion, the patient was subjected to a surgical resection. Intra-operatively, the patient had a spell of hypertensive emergency with a BP of 230/120 mmHg. The surgery was postponed, and the patient was prescribed 5 mg of prazosin twice daily, followed by 50 mg of metoprolol twice daily after testing for the adequacy of the α blockade. Two weeks later, with a BP of 140/80 mmHg, the patient underwent a laparoscopic tumour resection. The pathological study revealed an encapsulated tumour that had a nested arrangement of large polygonal cells with moderate granular cytoplasms and round to oval nuclei, which confirmed the pheochromocytoma diagnosis (Figure 2). Post-operatively, the patient required 5 mg of prazosin twice daily. One month later, the patient was treated with 5 mg of prazosin once daily without any recurrence of hypertension.
Figure 1:
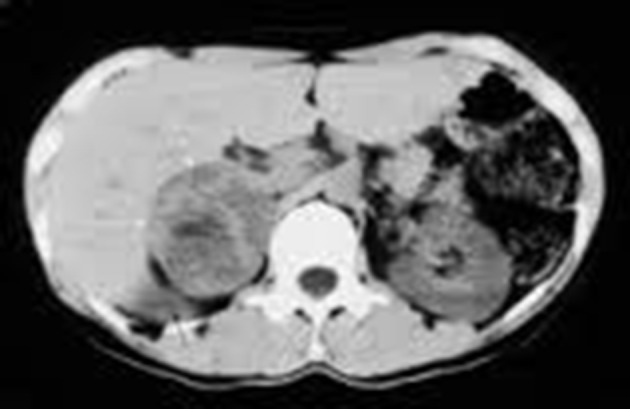
A computed tomography of the abdomen showing a well-defined, heterogeneous enhancing mass lesion that was 7.6 × 5.3 × 4.8 cm in size without any calcifications at the upper pole of right kidney. The left adrenal gland appears to be normal.
Figure 2:
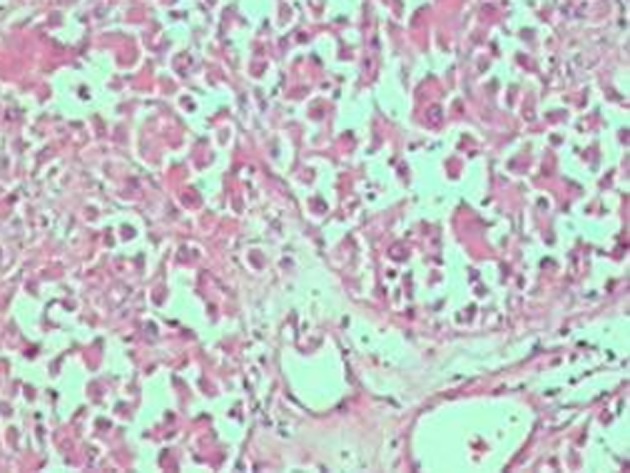
The histopathology of the biopsy specimen, showing a nested arrangement of large polygonal cells with moderate granular cytoplasms and round to oval nuclei.
Discussion
Here, we discuss the case of a biochemically negative pheochromocytoma that presented with an intra-operative hypertensive crisis. Approximately 5.0%–6.5% of adrenal incidentalomas are pheochromocytomas, and 8% of the patients with a pheochromocytoma are completely asymptomatic and usually have a familial form. In approximately 8%–9% of patients with sporadic paragangliomas and 21%–31% with hereditary paragangliomas, plasma and urinary concentrations of the catecholamines are normal (2). Timmers et al. (3) have reported on biochemically silent abdominal paragangliomas in 4 patients with a mutation in the succinate dehydrogenase subunit B (SDHB) gene. The reason provided was a defective catecholamine synthesis that resulted from the absence of tyrosine hydroxylase. In a recent case series of 20 patients by Vanderveen et al. (4), 2 patients with false negative biochemical tests were found.
The widespread use of radiological investigations has increased the detection of adrenal incidentalomas in individuals over 30 years of age. The clinical parameters that need to be considered include functionality and aggressiveness (5). An identification of the functionality of cortisol, aldosterone, and catecholamine and androgen secretion is essential, and in our patient, all of these parameters were within normal limits. Some imaging features favour a malignancy: these include a large size (more than 6 cm), irregular margins, heterogeneity, high unenhanced CT attenuation values (more than 10 HU), calcification, a delayed wash out of contrast on an enhanced CT, and a high to intermediate signal intensity on a T2-weighted MRI. A biopsy would confirm the nature of the lesion, which in our case was a benign pheochromocytoma.
A CT scan with contrast-enhanced images and an MRI scan are used to localise adrenal pheochromocytomas (6). Metaiodobenzylguanidine (MIBG) and positron emission tomography (PET) scanning are largely reserved for an extra-adrenal paraganglioma or particularly large tumours to rule out metastasis. In PET scanning, the Gallium-DOTA-TOC/NOC and DOPA-PET were found to perform better than FDG-PET in detecting the paragangliomas. Heterogeneity, a high Hounsfield density on a CT (more than 10 HU), a marked enhancement with intravenous contrast and delayed contrast washout (less than 60% at 10 minutes), a high signal intensity on a T2-weighted MRI, and cystic or haemorrhagic changes indicate a pheochromocytoma, adrenocortical carcinoma, or metastasis. However, a pheochromocytoma with lipid degeneration can result in low attenuation scores (less than 10 HU) and more than 60% washout in delayed CT scanning (7). Motta-Ramirez et al. (8) have elucidated that, although there was an increasing trend towards calcification in the CT scan findings of symptomatic patients, no significant differences were found in asymptomatic patients with respect to the age at presentation, the volume/ diameter of the mass, the necrosis score, or the attenuation (all had scores of more than 10 HU). Benign adrenal incidentalomas are characterised by having a size less than 5 cm, sharp margins, smooth contours, a lack of demonstrable growth on serial examinations, attenuation scores less than 10 HU, and more than 60% washout by delayed CT scanning (6,9). In our patient, the CT scan revealed a nonhomogeneous mass of 35 HU without any calcification.
It has been reported that an incidental pheochromocytoma of less than 1 cm in size has no symptoms (7). Our patient, who had a much larger mass, had no symptoms or signs of the disease. Possible reasons for this finding include the following: (i) the presence of a smaller piece of functional tissue, (ii) the release of a small amount of unmetabolised catecholamines due to a rapid intratumoural turnover rate, (iii) episodically secreting tumours, (iv) silent stress-activated tumours, and (v) false negative results due to the high-temperature handling of the laboratory specimen and an ingestion of caffeine 24 hours prior to the testing.
The biochemical diagnosis of a pheochromocytoma is made by measuring the 24-hour urinary fractionated metanephrines (98% sensitivity and 98% specificity) and fractionated catecholamines (10). There is a high specificity for the 24-hour urinary vanillylmandelic acid (95%) and the 24-hour urinary total metanephrines (99%). The plasma metanephrines are the preferred screening test for patients who are considered to have a high risk for a pheochromocytoma and for those suspected of having a familial form. Because the free metanephrines are formed extraneuronally, and to a large extent within the chromaffin tissues (e.g., the adrenal medulla and pheochromocytomas), these metabolites are also the more sensitive markers for a pheochromocytoma than the other catecholamine metabolites that are derived mainly from neuronal sources. Other tests, including plasma catecholamines and 24-hour urinary catecholamines, have a poor diagnostic accuracy. Though biochemically silent, the characteristic findings and biopsy diagnosis of a pheochromocytoma in our patient justified the use of an α blocker.
Histologically, paragangliomas are capsulated and are composed of round or polygonal epithelioid/chief cells arranged in the characteristically compact cell nests (zellballen) or trabecular patterns. The chief cells have centrally located nuclei with finely clumped chromatin and a moderate amount of eosinophilic, granular cytoplasm. Spindle-shaped sustentacular or supporting cells are located peripherally. Tumours of a higher grade are characterised by a progressive loss in the relationship between the chief cells and the sustentacular cells and a decrease in the number of sustentacular cells. In our patient, although the typical zellballen pattern was not found, the presence of an encapsulated tumour with a nested arrangement of large polygonal cells with moderate granular cytoplasms and round to oval nuclei confirmed the pheochromocytoma diagnosis. The presence of markers such as chromogranin A (CGA), neuron-specific enolase, and synaptophysin would confirm the neuroendocrine nature of the chief cells (11). Algeciras-Schimnich et al. (12) have determined that the likelihood ratio for having a pheochromocytoma/paraganglioma was 7.9 with CGA levels greater than 270 ng/mL; however, for CGA levels less than 270 ng/mL, this ratio was 0.15. Therefore, a patient in whom both the fractionated plasma metanephrines and CGA are positive is more than 50 times more likely to have a chromaffin tumour than a patient with only positive plasma fractionated metanephrines. For a combination of plasma and urine fractionated metanephrines, the corresponding likelihood ratios were 4.6 and 0.11 (12). Our patient did not undergo any of the marker-based investigations.
Malignant pheochromocytomas are histologically and biochemically similar to benign ones. The only reliable clue to the presence of a malignant pheochromocytoma is a local invasion or distant metastases, which may occur as long as 20 years after a resection (13). Thus, even when pheochromocytomas or paragangliomas are considered benign on a pathological examination, a long-term follow-up is indicated for all patients to confirm that diagnosis. Other markers for a malignancy are an absent or weak expression of the inhibin/activin βB subunit (14) and the presence of the SDHB subunit. In the absence of any invasion, we considered the mass in our patient to be benign.
Approximately 15%–20% of patients with catecholamine secreting tumours have a germ-line mutation in genes such as SDHB, SDHC, SDHB (familial paraganglioma), RET (MEN 2 A & B), MENIN (MEN-1), NF-1 (neurofibromatosis), and VHL (von Hippel– Lindau syndrome). Suspicious circumstances, including bilaterality, a family history of pheochromocytoma, younger age (20 years or below), or the presence of co-phenotypes, call for genetic testing (15,16). In our patient, the factors of age of presentation at 30 years, a unilateral pheochromocytoma, and the absence of a family history and other co-phenotypes supported the decision against a familial origin. Karasek et al. (17) describes the appropriate genetic testing pattern for nonsyndromic, nonfamilial cases that is based on a histological evaluation, the localisation of the tumour, and a biochemical phenotype of pheochromocytomas/paragangliomas, e.g., “the rule of three”.
Pre-operative optimisation of blood pressure requires an adequate α blockade with increasing doses followed by a β blockade. The Roizen’s criteria (18) for a significant α blockade are used (Table 2). A surgical resection is the appropriate treatment and cures 90% of patients. A laparoscopic removal is commonly performed. A laparotomy is reserved for tumours that are more than 8 cm in size and that show local invasion (19). Both of these approaches are equally successful in terms of overall survival. The greatest intra-operative concern is a release of catecholamines leading to life-threatening hypertension. Hypertensive crises can cause myocardial infarction, heart failure, dysrhythmia, and cerebral haemorrhage. Severe hypertension can occur at any time during the surgery, but the induction, intubation, and tumour palpation tend to lead to the greatest catecholamine release. In our patient, the presence of a mass that was 7.6 × 5.3 × 4.8 cm in size called for a laparoscopic resection.
Table 2:
The Roizen’s criteria (18) for an appropriate pre-operative α blockade and surgical optimisation
|
|
|
|
Induction agents should be titrated slowly to maintain normotension. A short-acting narcotic, such as fentanyl, with its minimal myocardial depression, in combination with a sedative/ hypnotic is preferable. An adequate depth of anaesthesia is required to prevent the patient’s response to the stimulus of intubation (20). Vecuronium or rocuronium, which have few cardiovascular effects, may be used for muscle relaxation; however, pancuronium should be avoided because of its sympathomimetic effects. Inhalational agents (isoflurane, sevoflurane, or desflurane) may be used with or without intravenous agents. Other medications known to trigger the release of catecholamines, including metoclopramide, pentazocine, droperidol, atracurium, succinylcholine, selective serotonin reuptake inhibitors, monoamine oxidase inhibitors, imipramine, opioids, and curare, should be avoided (21).
Intra-operatively, the α blockade is continued with phentolamine. Its most common side effect is a reflex tachycardia due to the baroreceptor reflex following an α2 blockade. Labetalol should be used to control the tachycardia. Calcium channel blockers and nitroprusside can be used a second line of therapy (21). In our case, intravenous phentolamine was used.
Following early ligation of the vein that drains the pheochromocytoma, intravenous fluid administration is essential for volume expansion. The sudden drop in catecholamines can lead to significant hypotension, which requires aggressive fluid replacement with a combination of crystalloids and colloids. Pressors may be necessary to maintain blood pressure in severe hypotension, but they are best avoided and are contraindicated if the patient is hypovolaemic. Often, the hypotension of a pheochromocytoma is refractory to agents such as norepinephrine, epinephrine, and dopamine because of the desensitisation of the sympathetic receptors to the previous persistently high levels of catecholamines (20).
The 24-hour urinary fractionated metanephrines should be measured 1 to 2 weeks after surgery; normal values indicate a complete resection. A periodic follow-up for blood pressure is needed. Furthermore, annual biochemical testing to assess metastatic disease, tumour recurrence, or a delayed appearance of a primary tumour is recommended, and this was advised for our patient (16).
Conclusion
Pheochromocytoma is a clinically important disorder because it leads to high morbidity and mortality rates if untreated. Fractionated metanephrines and catecholamines in a 24-hour urine analysis is the preferred biochemical test. In a biochemically silent pheochromocytoma, characteristic CT findings should identify its possible presence. A combination of anatomical imaging studies (CT or MRI) with functional imaging studies (MIBG and PET) is used to locate adrenal, extra-adrenal, recurrent, and metastatic tumours. An adequate α and β blockade should be ensured before proceeding to tumour removal. For individuals with adrenal tumours larger than 4–6 cm, a surgical resection is appropriate. However, for a pheochromocytoma, surgery is recommended irrespective of the size and is even recommended in the context of normal biochemical study results to prevent future complications.
Acknowledgments
All the authors would like to thank Ms Sruti Jammula, a PhD research scholar in the department of Pharmaceutics, Roland Institute of Pharmaceutical Sciences, Berhampur, India, for her sincere help in preparing the manuscript, formulating the basic intellectual content in its basic format, and giving us valuable inputs regarding treatment of the condition mentioned in our case report.
Footnotes
Authors’ Contribution
Conception and design, critical revision of the article: SUKK, SIKK, SP, KDM
Analysis and interpretation of the data: SUKK, SIKK
Drafting of the article: SUKK, SIKK, SP
Final approval of the article: SUKK, KDM
References
- 1.Anderson GH Jr, Blakeman N, Streeten DH. The effect of age on prevalence of secondary forms of hypertension in 4429 consecutively referred patients. J Hypertens. 1994;12(5):609–615. doi: 10.1097/00004872-199405000-00015. [DOI] [PubMed] [Google Scholar]
- 2.Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, et al. Biochemical diagnosis of pheochromocytoma: Which test is best? JAMA. 2002;287(11):1427–1434. doi: 10.1001/jama.287.11.1427. [DOI] [PubMed] [Google Scholar]
- 3.Timmers HJLM, Pacak K, Huynh TT, Abu-Asab M, Tsokos M, Merino MJ, et al. Biochemically silent abdominal paragangliomas in patients with mutations in the succinate dehydrogenase subunit B gene. J Clin Endocrinol Metab. 2008;93(12):4826–4832. doi: 10.1210/jc.2008-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vanderveen KA, Thompson SM, Callstrom MR, Young WF Jr, Grant CS, Farley Dr, et al. Biopsy of pheochromocytomas and paragangliomas: Potential for disaster. Surgery. 2009;146(6):1158–1166. doi: 10.1016/j.surg.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 5.Chen YY. Pheochromocytoma presenting with normal urinary catecholamines and metabolites: A case report. Formos J Endocrin Metab. 2009;1(1):33–37. [Google Scholar]
- 6.Szolar DH, Korobkin M, Reittner P, Berghold A, Bauernhofer T, Trummer H, et al. Adrenocortical carcinomas and adrenal pheochromocytomas: Mass and enhancement loss evaluation at delayed contrast enhanced CT. Radiology. 2005;234(2):479–485. doi: 10.1148/radiol.2342031876. [DOI] [PubMed] [Google Scholar]
- 7.Blake MA, Krishnamoorthy SK, Boland GW, Sweeney AT, Pitman MB, Harisinghani M, et al. Low-density pheochromocytoma on CT: A mimicker of adrenal adenoma. AJR Am J Roentgenol. 2003;181(6):1663–1668. doi: 10.2214/ajr.181.6.1811663. [DOI] [PubMed] [Google Scholar]
- 8.Motta-Ramirez GA, Remer EM, Herts BR, Gill IS, Hamrahian AH. Comparison of CT findings in symptomatic and incidentally discovered pheochromocytomas. AJR Am J Roentgenol. 2005;185(3):684–688. doi: 10.2214/ajr.185.3.01850684. [DOI] [PubMed] [Google Scholar]
- 9.Boraschi P, Braccini G, Grassi L, Campatelli A, Di Vito A, Mosca F, et al. Incidentally discovered adrenal masses: Evaluation with gadolinium enhancement and fat-suppressed MR imaging at 0.5 T. Eur J Radiol. 1997;24(3):245–252. doi: 10.1016/s0720-048x(97)01046-2. [DOI] [PubMed] [Google Scholar]
- 10.Kudva YC, Sawka AM, Young WF Jr. Clinical review 164: The laboratory diagnosis of adrenal pheochromocytoma: The Mayo Clinic experience. J Clin Endocrinol Metab. 2003;88(10):4533–4539. doi: 10.1210/jc.2003-030720. [DOI] [PubMed] [Google Scholar]
- 11.Kliewer KE, Wen DR, Cancilla PA, Cochran AJ. Paragangliomas: Assessment of prognosis by histologic, immunohistochemical, and ultrastructural techniques. Hum Pathol. 1989;20(1):29–39. doi: 10.1016/0046-8177(89)90199-8. [DOI] [PubMed] [Google Scholar]
- 12.Algeciras-Schimnich A, Preissner CM, Young WF Jr, Singh RJ, Grebe SKG. Plasma chromoganin A or urinary fractionated metanephrines follow-up testing improves the diagnostic accuracy of plasma fractionated metanephrines for pheochromocytoma. J Clin Endocrinol Metab. 2008;93(1):91–95. doi: 10.1210/jc.2007-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldstein RE, O’Neill JA Jr, Holcomb GW 3rd, Morgan WM 3rd, Neblett WW 3rd, Oates JA, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755–766. doi: 10.1097/00000658-199906000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salmenkivi K, Arola J, Voutilainen R, Ilvesmaki V, Haglund C, Kahri AI. Inhibin/activin betaB-subunit expression in pheochromocytomas favors benign diagnosis. J Clin Endocrinol Metab. 2001;86(5):2231–2235. doi: 10.1210/jcem.86.5.7446. [DOI] [PubMed] [Google Scholar]
- 15.Gimm O, Koch CA, Januszewicz A, Opocher G, Neumann HP. The genetic basis of pheochromocytoma. Front Horm Res. 2004;31:45–60. doi: 10.1159/000074657. [DOI] [PubMed] [Google Scholar]
- 16.Young WF Jr. In: Williams textbook of endocrinology. 11th ed. Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, editors. Philadelphia (PA): Saunders Elsevier; 2008. Endocrine hypertension; pp. 505–537. [Google Scholar]
- 17.Karasek D, Frysak Z, Pacak K. Genetic testing for pheochromocytoma. Curr Hypertens Rep. 2010;12(6):456–464. doi: 10.1007/s11906-010-0151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roizen MF, Horrigan RW, Koike M, Eger IE 2nd, Mulroy MF, Frazer B. A prospective randomized trial of four anesthetic techniques for resection of pheochromocytoma. Anesthesiology. 1982;57:A43. [Google Scholar]
- 19.Assalia A, Gagner M. Laparoscopic adrenalectomy. Br J Surg. 2004;91(10):1259–1274. doi: 10.1002/bjs.4738. [DOI] [PubMed] [Google Scholar]
- 20.Grant F. Anesthetic considerations in the multiple endocrine neoplasia syndromes. Curr Opin Anaesthesiol. 2005;18(3):345–352. doi: 10.1097/01.aco.0000169245.88506.13. [DOI] [PubMed] [Google Scholar]
- 21.Dortzbach K, Gainsburg DM, Frost EA. Variants of pheochromocytoma and their anaesthetic implications—A case report and literature review. Middle East J Anesth. 2010;20(6):897–905. [PubMed] [Google Scholar]