

Abstract
The complete 15,413-bp mitochondrial genome (mitogenome) of Sesamia inferens (Walker) (Lepidoptera: Noctuidae) was sequenced and compared with those of four other noctuid moths. All of the mitogenomes analyzed displayed similar characteristics with respect to gene content, genome organization, nucleotide comparison, and codon usages. Twelve-one protein-coding genes (PCGs) utilized the standard ATN, but the cox1 gene used CGA as the initiation codon; cox1, cox2, and nad4 genes had the truncated termination codon T in the S. inferens mitogenome. All of the tRNA genes had typical cloverleaf secondary structures except for trnS1(AGN), in which the dihydrouridine (DHU) arm did not form a stable stem-loop structure. Both the secondary structures of rrnL and rrnS genes inferred from the S. inferens mitogenome closely resembled those of other noctuid moths. In the A+T-rich region, the conserved motif “ATAGA” followed by a long T-stretch was observed in all noctuid moths, but other specific tandem-repeat elements were more variable. Additionally, the S. inferens mitogenome contained a potential stem-loop structure, a duplicated 17-bp repeat element, a decuplicated segment, and a microsatellite “(AT)7”, without a poly-A element upstream of the trnM in the A+T-rich region. Finally, the phylogenetic relationships were reconstructed based on amino acid sequences of mitochondrial 13 PCGs, which support the traditional morphologically based view of relationships within the Noctuidae.
Keywords: Sesamia inferens, mitochondrial genome, lepidoptera, Noctuidae, phylogenetic relationship
1. Introduction
The pink stem borer, Sesamia inferens (Walker), is an important rice pest that is widely distributed in China, Japan, India and other countries, and causes severe damage to rice crops in rice planting areas [1–3]. Since 1990, with the expansion of hybrid rice planting areas and changes in climate and rice cultivation systems, the S. inferens population has been gradually increasing in China, and has become an important rice pest next to Chilo suppressalis (Walker), especially in the Lower–Middle Reaches of the Yangze River [3]. At present, the mitochondrial genomes (mitogenomes) from more than 393 species of arthropods have been completely or partially determined, including 241 species of insects. However, only 36 mitogenomes from six lepidopteran superfamilies have been sequenced, with seven species of Bombycoidea, one species of Geometroidea, 14 species of Papilionoidea, six species of Pyraloidea, three species of Tortricoidea, and five species of Noctuoidea, including Helicoverpa armigera, Hyphantria cunea, Lymantria dispar, Ochrogaster lunifer, and S. inferens [4–7].
The insect mitogenome is normally a closed-circular duplex molecule, ranging from 14 to 20 kb in length. The mitogenome usually contains 13 protein-coding genes (PCGs: atp6, atp8, cox1, cox2, cox3, cob, nad1, nad2, nad3, nad4, nad5, nad6, and nad4L), large and small ribosomal RNA genes (rrnL and rrnS), 22 tRNA genes, which are involved in energy production, electron transport and oxidative phosphorylation [8,9]. In addition, at least one segment of the most variable A+T-rich region is present in the insect mitogenome, including initiation sites for the transcription and replication of the genome [10,11].
Compared with the nuclear genome, the animal mitogenome—except for lice [12]—usually has a smaller size, a stable and relatively short circular structure. In addition, it has other characteristics such as a higher rate of base substitution and a presumed lack of intermolecular recombination. Mitogenome has been widely used as an informative molecular marker to reveal fundamental information for phylogenetic inference, the identification of species, phylogeography, the analysis of population structure and dynamics, and molecular evolution at the genomic level. This has been especially true for the past several decades, especially since the human mitogenome was sequenced [13]. More and more insect mitogenomes will increase the richness of information for phylogenetic analyses and evolutionary biology.
In this study, we report the completed mitogenome of S. inferens and provide a thorough description of its genomic features, including gene order, nucleotide composition of PCGs, secondary structures of tRNA and rRNA genes, and A+T-rich region. In addition, we compare the S. inferens mitogenome with those of four other noctuid moths, which can provide further insights into the relationships among Noctuidae species. Detailed genetic information on this important rice pest may help in the development of methods for its control or prevention.
2. Results and Discussion
2.1. Genome Structure, Organization and Composition
The complete mitogenome of S. inferens was found to be a 15,413 bp long circular molecule, and its sequence was deposited into GenBank (Accession number: JN039362) (Figure 1 and Table 1). The size was shorter than that of H. cunea (15,481 bp), L. dispar (15,569 bp), and O. lunifer (15,593 bp) [4,5,7]. All of the 37 typical animal mitochondrial genes (13 PCGs, 22 tRNA and 2 rRNA genes) were present, and the gene order was the same as in other sequenced noctuid moths, in an order of trnM, trnI, and trnQ. The position of the trnM gene was usually translocated to the 5′ upstream position of trnI in the lepidopteran species, which was different from those of other order species [14]. This indicated that the mitochondrial gene arrangement in lepidopteran species evolved independently after splitting from its stem lineage [15]. In addition, a total of 257 bp of intergenic spacer sequences were present in 18 locations with the exception of the A+T-rich region, and a total of 25 bp of overlapping nucleotides was scattered over six locations in the S. inferens mitogenome.
Figure 1.

A map of the mitogenome of Sesamia inferens. Transfer RNA genes are designated by single-letter amino acid codes. CR represents the A+T-rich region. The gene name without underline indicates the direction of transcription from left to right, and with underline indicates right to left. s1–s4, intergenic spacers.
Table 1.
Annotation of the mitogenome of Sesamia inferens.
| Name | Direction | Site | Size (bp) | Intergenic spacer | Anticodon | Start codon | Stop codon |
|---|---|---|---|---|---|---|---|
| trnM | F | 1..68 | 68 | 0 | 32..34 CAT | ||
| trnI | F | 69..133 | 65 | −3 | 98..100 GAT | ||
| trnQ | R | 131..199 | 69 | 68 | 167..169 TTG | ||
| nad2 | F | 268..1284 | 1017 | 6 | ATT | TAA | |
| trnW | F | 1291..1358 | 68 | −8 | 1322..1324 TCA | ||
| trnC | R | 1351..1423 | 73 | 4 | 1391..1393 GCA | ||
| trnY | R | 1428..1494 | 67 | 5 | 1460..1462 GTA | ||
| cox1 | F | 1500..3030 | 1531 | 3 | CGA | T- | |
| trnL2(UUR) | F | 3034..3100 | 67 | 0 | 3064..3066 TAA | ||
| cox2 | F | 3101..3782 | 682 | 0 | ATT | T- | |
| trnK | F | 3783..3853 | 71 | 0 | 3813..3815 CTT | ||
| trnD | F | 3854..3919 | 66 | 0 | 3884..3886 GTC | ||
| atp8 | F | 3920..4087 | 168 | −11 | ATT | TAA | |
| atp6 | F | 4077..4754 | 696 | −1 | ATG | TAA | |
| cox3 | F | 4754..5542 | 789 | 2 | ATG | TAA | |
| trnG | F | 5545..5609 | 65 | 0 | 5575..5577 TCC | ||
| nad3 | F | 5610..5963 | 354 | 16 | ATT | TAA | |
| trnA) | F | 5980..6048 | 69 | 2 | 6010..6012 TGC | ||
| trnR | F | 6051..6114 | 64 | 9 | 6078..6080 TCG | ||
| trnN | F | 6124..6188 | 65 | 2 | 6154..6156 GTT | ||
| trnS1(AGN) | F | 6191..6256 | 66 | 0 | 6216..6218 GCT | ||
| trnE | F | 6257..6324 | 68 | 8 | 6287..6289 TTC | ||
| trnF | R | 6333..6399 | 67 | −1 | 6364..6366 GAA | ||
| nad5 | R | 6399..8150 | 1752 | 0 | ATT | TAA | |
| trnH | R | 8151..8215 | 65 | 0 | 8183..8185 GTG | ||
| nad4 | R | 8216..9554 | 1339 | 44 | ATG | T- | |
| nad4L | R | 9599..9892 | 294 | 7 | ATG | TAA | |
| trnT | F | 9900..9969 | 70 | 0 | 9929..9931 TGT | ||
| trnP | R | 9970..10034 | 65 | 7 | 10002..10004 TGG | ||
| nad6 | F | 10042..10575 | 534 | 44 | ATC | TAA | |
| cob | F | 10620..11768 | 1149 | 1 | ATG | TAA | |
| trnS2(UCN) | F | 11770..11836 | 67 | 18 | 11802..11804 TGA | ||
| nad1 | R | 11855..12799 | 945 | 1 | ATG | TAA | |
| trnL1(CUN) | R | 12801..12867 | 67 | −1 | 12836..12838 TAG | ||
| rrnL | R | 12867..14251 | 1385 | 0 | - | ||
| trnV | R | 14253..14318 | 66 | 0 | 14284..14286 TAC | ||
| rrnS | R | 14319..15102 | 784 | 0 | |||
| A+T-rich region | 15103..15413 | 311 | 0 |
Negative numbers indicate that adjacent genes overlap.
The nucleotide composition of the S. inferens mitogenome was also biased toward AT nucleotides (80.3%), which was slightly higher than those of L. dispar (79.9%) and O. lunifer (77.8%), but was slightly lower than those of H. armigera (81.0%) and H. cunea (80.4%) [4–7] (Table 2). The AT skew in the S. inferens mitogenome was slightly negative (−0.001), indicating the occurrence of more Ts than As. This value was different from those of other Noctuidae species, such as H. armigera (0.002), H. cunea (0.010), L. dispar (0.016), O. lunifer (0.030) [4–7]. Meanwhile, the GC skew in the S. inferens mitogenome was negative (−0.228), demonstrating the occurrence of more Cs than Gs. This was also true for H. armigera (−0.184), H. cunea (−0.230), L. dispar (−0.247), O. lunifer (−0.318) [4–7].
Table 2.
Skewed nucleotide composition in regions of noctuid moths’ mitogenomes.
| Region | Whole mitogenome | PCGs | |||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Species | Length (bp) | AT (%) | Length (bp) | AT (%) | 1st position (%) | 2nd position (%) | 3rd position (%) |
| H. armigera | 15347 | 81.0 | 11,133 | 79.4 | 70.2 | 94.1 | 73.8 |
| H. cunea | 15481 | 80.4 | 11,136 | 78.5 | 73.2 | 70.3 | 92.0 |
| L. dispar | 15569 | 79.9 | 11,169 | 77.7 | 90.2 | 72.9 | 70.1 |
| O. lunifer | 15593 | 77.8 | 11,196 | 75.7 | 72.0 | 70.0 | 85.1 |
| S. inferens | 15413 | 80.3 | 11,160 | 78.6 | 87.7 | 72.9 | 75.2 |
2.2. Protein-Coding Genes
The mitochondrial sequence of S. inferens, as in most insects, contained 13 PCGs (Table 2). Most of the PCGs utilized typical ATN start codons (ATG, ATT, and ATC), among which six genes started with codon ATG (atp6, cox3, nad4, nad4L, cob, and nad1), five with ATT (nad2, cox2, atp8, nad3, and nad5), and the nad6 gene with ATC. However, the cox1 gene in the S. inferens mitogenome had the start codon CGA as observed in most other lepidopteran species sequenced to date [16]. Notably, the start codon of the cox1 gene is usually found to use nonstandard putative codons, as previously reported in arthropod species [17–20].
The conventional termination codon TAA, likely resulting from post-transcriptional polyadenylation [21], was observed in 10 PCGs. However, the cox1, cox2, and nad4 genes utilized truncated termination codons, which are commonly observed in lepidopteran species.
The average AT content of the 13 PCGs in the S. inferens mitogenome was 78.6%, with a negative AT skew of −0.148 and a slightly positive GC skew of 0.037 (Table 2). The AT and GC skews were similar to those of H. armigera (−0.139, 0.029), H. cunea (−0.146, 0.023), and L. dispar (−0.148, 0.013), indicating that the contents of T and G were higher than those of A and C, respectively [5–7]. It is to be noted that both the AT skew (−0.141) and the GC skew (−0.004) were slightly negative in the 13 PCGs of O. lunifer [4]. Also, the AT contents at different codon positions was analyzed. In the S. inferens mitogenome, the AT contents was 87.7%, 72.9% and 75.2% at the first, second and third codon positions, respectively, with the highest AT contents at the first codon position, which was similar to the result from L. dispar mitogenome (90.2%, 72.9%, 70.1%), but was different from that in H. armigera (70.2%, 94.1%, 73.8%), H. cunea (73.2%, 70.3%, 92.0%) and O. lunifer mitogenomes (72.0%, 70.0%, 85.1%) [4–7] (Table 2).
Excluding the start and termination codons, the 13 PCGs in the S. inferens mitogenome consisted of 3702 codons in total, consistent with the observations in four other noctuid moths, which ranged from 3687 in the H. armigera to 3718 codons in the O. lunifer mitogenome [4–7] (Figure 2). The codon families exhibited a very similar behavior among the five species (Figures 2 and 3). There were at least six codon families with at least 50 codons (CDs) (Asn, Ile, Leu2, Met, Phe, Tyr), and two families with at least 100 CDs per thousand CDs (Leu2 and Phe) as observed in five insects. In addition, the AT-rich CDs favor synonymous CDs with a lower AT content, as revealed by the Relative Synonymous Codon Usage (RSCU) results, and exemplified by the Leu2 family, in which the TTA codon accounted for the large majority of CDs (Figure 3). The codon families with high CDs had A and T predominantly in the third position, which might reflect selection for optimal tRNA usage, genome bias, and the speed and efficacy of genome/DNA repair [22].
Figure 2.

Codon distributions in five noctuid moths’ mitogenomes as indicated. The total number of codons per thousand codons (designated as CDsp T) is shown on the y-axis and the codon families on the x-axis.
Figure 3.

The relative synonymous codon usage (RSCU) in five noctuid moths’ mitogenomes as a function of codon families. Red codons indicate that the codons are missing in the mitogenome.
2.3. Transfer RNA Genes
The mitogenome of S. inferens had the characteristic 22 tRNAs sets interspersed by rRNAs or PCGs, with an AT content of 81.5% and a total of 1478 bp in size ranging from 64 to 73 bp/tRNA. All tRNA genes had the typical cloverleaf secondary structures except for the trnS1(AGN) gene, in which a stable stem-loop structure of the dihydrouridine (DHU) arm was missing (Figure 4), as observed in many metazoan mitogenomes [23–25]. Eight tRNAs were encoded by the L-strand and the remaining 14 by the H-strand, which was identical in all the species. All tRNA genes usually contained a 7-bp amino acid acceptor (AA) stem, where most nucleotide substitutions were compensatory. The anticodon (AC) stem and the loop (7 bp) were both conserved in all tRNAs, whereas two U–U pairs were usually located at the second and third couplets in the anticodon stem of trnS2(UCN). The length of DHU was 3–4 bp, except for trnS1(AGN). The TΨC arm was usually 3–6 bp in length. Except for trnD, trnE, and trnY, a total of 29 unmatched base pairs were detected in the S. inferens tRNAs, but 18 of them were G-U pairs, which form a weak bond in the tRNAs, and are well-known non-canonical pairs in tRNA secondary structures [26]. The remaining 11 were mismatched base pairs including seven U-U, one C-U, and three A-C pairs. Notably, the numbers of mismatches varied in different noctuid moths, with 4, 7, 23 and 10 mismatches in H. armigera, H. cunea, L. dispar, and O. lunifer, respectively [4–7].
Figure 4.



Inferred secondary structures for 22 typical tRNAs of the Sesamia inferens mitogenome. The tRNAs are labeled with the abbreviations of their corresponding amino acids. Base-pairing is indicated as follows: Watson–Crick pairs by lines, wobble GU pairs by dots and mismatched pairs by circles.
2.4. Ribosomal RNA Genes
As in other mitochondrial sequences from Insecta species, there were two rRNAs in S. inferens with a total length of 2169 bp and an AT content of 84.1%. The large ribosomal gene (rrnL) had a length of 1385 bp, located between trnL1(CUN) and trnV, whereas the small one (rrnS) had a length of 784 bp between trnV and the A+T-rich region. The gene sizes and locations were typical as in other noctuid moths’ mitogenomes. Both tRNA and rRNA genes of Noctuidae species predominantly contained A and T [4–7] (Table 2). Furthermore, both the AT and GC skews were slightly positive in the tRNA and rRNA genes, consistent with the results in H. armigera and H. cunea mitogenomes [5,6].
Both the rrnL and rrnS secondary structures were predicted according to models proposed for these genes in other insects [27,28]. Although the length of individual helices varied in insect species, the secondary structures of both rrnL and rrnS in S. inferens were quite similar to their counterparts in Apis mellifera, Manduca sexta, and other insect species [27,28]. In S. inferens, the rrnL gene contained five domains (labeled I, II, IV, V, and VI) with 49 helices, as observed in other noctuid moths (Figure 5). Each domain was separated by a single-stranded region, and domain III was absent in arthropod mitogenomes. Also, S. inferens had the rrnS with 33 helices in three domains (labeled I, II, III) (Figure 6).
Figure 5.

The predicted rrnL secondary structure from the Sesamia inferens mitogenome. The 5′ half of rrnL is displayed in A and the remaining 3′ half in B. Base-pairings are indicated as follows: Watson-Crick pairs by lines, wobble GU pairs by dots and mismatched pairs by circles.
Figure 6.

The predicted rrnS secondary structure in the Sesamia inferens mitogenome. Tertiary interactions and base triples are shown connected by continuous lines. Base-pairings are indicated in Figure 5.
2.5. Non-Coding and Overlapping Regions
In the S. inferens mitogenome, there was a total of 257 bp of intergenic spacer sequences, which resided at 18 locations (except for the A+T-rich region) and varied in size from 1 to 68 bp. It is to be noted that there were four major intergenic spacers (s1–s4) spanning at least 18 bp, all of which were rich in As and Ts (Figure 7). The s1 spacer (68 bp) located between trnQ and nad2 had a higher AT content compared with the counterpart in the O. lunifer mitogenome. And so did the other three spacers. The s3 spacer (44 bp), inserted between nad6 and cob, is not conserved in five noctuid moths, which was mostly a 4 bp repeat in the S. inferens mitogenome while the other had a stretch of TA dinucleotides in the L. dispar mitogenome. However, the s2 spacer (44 bp), placed between nad4 and nad4L, was longer than its counterparts in H. armigera and L. dispar mitogenomes, but it was twice the length of the counterpart in the L. dispar mitogenome (44 vs. 22) and about the same size as the counterpart in the H. armigera mitogenome (44 vs. 43). In addition, the s4 spacer (18 bp), located between trnS2(UCN) and nad1, did not contain the motif “ATACTAA” that was conserved across four other noctuid moths and other lepidopteran species mitogenomes [28,29].
Figure 7.

Intergenic spacer sequences in the mitogenomes of Sesamia inferens and other noctuid moths.
Also, a total of 25 bp overlapping nucleotides was scattered over six locations and ranged in sizes from 1 to 11 bp in the S. inferens mitogenome, with the longest one (11 bp) located between atp8 and atp6. Interestingly, an 8-bp motif “AAGCCTTA” conserved in the four other noctuid moths’ mitogenomes [4–7] was also detected between trnW and trnC in the present study.
2.6. A+T-rich Region
In mitochondrial genomes, the A+T-rich region had been reported to possess essential elements involved in the initiation of replication and transcription of mitogenome [30]. The A+T-rich region of the S. inferens mitogenome was 311 bp in length with an AT content of 95.8%. Similarly, the sizes and AT contents in the A+T-rich regions of four other noctuid moths were 328 bp and 95.1% AT content (H. armigera), 357 bp and 95.0% AT content (H. cunea), 369 bp and 96.1% AT content (L. dispar), and 319 bp and 93.4% AT content (O. lunifer) [4–7]. Notably, the S. inferens A+T-rich region displayed a higher AT content (95.8%) than all the other locations in the mitogenome (Table 2).
There was a conserved structure that combined the motif “ATAGA” and a 19-bp poly-T stretch in S. inferens A+T-rich region (Figure 8). A very similar pattern occurred in four other noctuid moths, and it was widely conserved in lepidopteran mitogenomes and might be the origin of light-strand replication [4–7,30]. There was also a stem-and-loop structure in S. inferens A+T-rich region containing a 5′ flanking “TATA” and 3′ flanking “G(A)nT” sequence, which were considered to be the site of initiation of secondary strand synthesis [31,32] (Figures 8 and 9). The stem-and loop structure might be the characteristic of insect mitogenomes, and it had been found in several insect orders including Orthoptera, Lepidoptera, Diptera, Plecoptera, and Hymenoptera, but it was not observed in four other noctuid moths [30,33–36]. Also, the conserved 3′ flanking “G(A)nT” sequence might not be present in all insects or the sequence might take on different forms [35,37].
Figure 8.

The structure of the A+T-rich region of the Sesamia inferens mitogenome. The origin of light strand replication, the potential stem loop structure, the duplicated 17-bp repeat element, the decuplicated 8 bp segment (derived from a consensus sequence “ATATTAAT”) and the microsatellite “(AT)n” elements are displayed as indicated in the figure.
Figure 9.
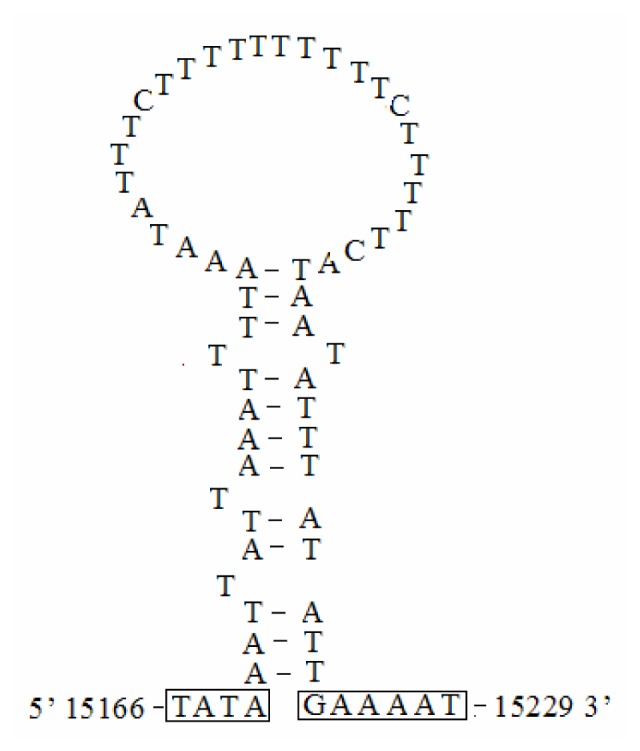
The potential stem-loop structure found in the Sesamia inferens A+T-rich region. The motifs (“GAAAAT” and “TATA”) in the flanking region of the stem-loop structures are indicated by boxing [32].
The presence of varying copy numbers of tandemly repeated elements was one of the characteristics in insect mitogenomes [30]. The S. inferens A+T-rich region contained a duplicated 17-bp repeat element with minor variations, and a decuplicated highly divergent 8-bp segment “ATATTAAT” (Figure 8), which resembled the octuplicated 8-bp repeat element “TATATATT” in the L. dispar A+T-rich region [7]. Additionally, there was a microsatellite “(AT)7” in S. inferens A+T-rich region (Figure 8), and similar patterns were observed in H. cunea “(AT)8” and O. lunifer “(AT)7” mitogenomes [4,5].
A conserved 9-bp poly-A element was observed upstream of trnM in H. cunea, L. dispar, and O. lunifer mitogenomes, which might be involved in controlling transcription and replication initiation [4,5,7,38]. However, this structure was not detected in S. inferens and H. armigera mitogenomes [6].
2.7. Phylogenetic Relationships
In this study, the amino acid sequences of the 13 PCGs were concatenated, rather than analyzed separately, to construct the phylogenetic relationships, which may result in a more complete analysis [39]. Based on morphological and genomic analyses, the relationship among six superfamilies in lepidopteran species has been reported in other articles [29,40,41].
In the present study, the NJ and MP trees showed similar topologies except for some node confidence values (Figure 10). The S. inferens mitogenome together with mitogenomes of H. armigera, H. cunea, L. dispar, and O. lunifer clustered with other superfamilies, which was consistent with the morphological classification [40]. Geometroidea (Phthonandria atrilineata) was closely related to Noctuoidea and Bombycoidea (Bombyx mandarina, Bombyx mori, Eriogyna pyretorum, M. sexta) [28,42–44], but Papilionoidea (Calinaga davidis and Coreana raphaelis) [15,45] was the sister to Pyraloidea (C. suppressalis and Ostrinia furnacalis) [29,46], Tortricoidea (Adoxophyes honmai and Grapholita molesta) [16,38] and other superfamilies, in disagreement with the tree topology [29,40,41]. The results indicated that more mitogenomes could elaborate on phylogenetic relationships among lepidopteran superfamilies in greater detail to some extent.
Figure 10.

Phylogeny of the lepidopteran species. Phylogenetic tree inferred from amino acid sequences of 13 PCGs of the mitogenome by using Neighbor Joining (NJ) and Maximum parsimony (MP). Numbers at each node indicate bootstrap support; percentages of NJ probabilities (first value) and MP bootstrap support values (second value), respectively. Drosophila melanogaster [47] and Locusta migratoria manilensis were used as outgroup.
3. Experimental Section
3.1. DNA Extraction
Adult S. inferens were collected from rice paddies surrounding Yangzhou (32°40.025N, 119°44.017E), Jiangsu Province, China. Samples were preserved in 100% ethanol and stored at −70 °C until DNA extraction was performed. Whole genomic DNA was extracted from a single sample using the protocol of DNAVzol (Bioteke, Beijing, China) and then used for the PCR amplification.
3.2. Primers Design, PCR Amplification, Cloning and Sequencing
Two pairs of LA-PCR primers were used for amplification of two overlapping fragments of S. inferens mitogenome, which were 16SAA (5′-ATGCTWCCTTTGCACRGTCAAGATACYGCGGC-3′), 16SBB (5′-CTTATCGAYAAAAAAGWTTGCGACCTCRATGTTG-3′), LP01 (5′-TGATTAGCTCCACAAATTTCTGAACATTGACC-3′), and LP02 (5′-WACACCAGTTCATATTDAACCAGAATGATATT-3′) [48]. Sub-PCR primers were designed from comparisons of 35 lepidopteran sequences available in GenBank and referenced to the universal primers of insect mitogenomes [49].
The conditions for amplification of two long fragments were as follows: An initial denaturation for 2 min at 96 °C followed by 35 cycles of 10 s at 98 °C, 10 min at 58 °C, and a subsequent 10 min final extension at 72 °C. PCR conditions for amplification of other fragments were as follows: An initial denaturation for 5 min at 95 °C, followed by 35 cycles of denaturation for 1 min at 94 °C, annealing for 1 min at 45–50 °C, elongation for 1–3 min (depending on putative length of the fragments) at 68 °C, and a final extension step of 72 °C for 10 min. For most fragments, LA Taq polymerase (TaKaRa, Kyoto, Japan) was used for the PCR amplification, but for fragments less than 1.3 kb, LA Taq polymerase was replaced by Taq polymerase (TaKaRa, Kyoto, Japan) in the PCR reaction. All PCR reactions were performed in an ABI thermal cycler (PE Applied Biosystems, CA, USA).
PCR products were separated by electrophoresis in a 1.0% agarose gel and purified using a DNA Gel Extraction Kit (Bioteke, Beijing, China). Purified PCR products were ligated into the T-vector (TaKaRa, Kyoto, Japan) and then transformed into XL-1 blue competent bacteria, according to the method of Wei et al. [50]. The positive recombinant clone was sequenced using upstream and downstream primers from bi-directions on ABI 3730XL Automated DNA Sequencer (PE Applied Biosystems, CA, USA) at least three times.
3.3. Sequence Analysis
The Staden package was used for sequence assembly and annotation [51]. The PCGs and rRNA genes in S. inferens mitogenome were identified by sequence alignment with other lepidopteran species, especially the Noctuidae by using Clustal X version 2.0 [4–7,52]. The PCG nucleotide sequences without start and termination codons were translated on the basis of the Invertebrate Mitochondrial Genetic Code. The AT content was calculated using MEGA version 5.0 [35]. Composition skew analysis was carried out with formulas AT skew = [A − T]/[A + T] and GC skew = [G − C]/[G + C] [53].
Transfer RNA genes were identified using tRNAscan-SE software available online [54], and the software of XRNA 1.2.0b was used to draw the secondary structure of RNA genes. The secondary structures of the rrnL and rrnS genes were inferred based on models developed for other insect species [27,55]. To infer the rRNA gene secondary structures, we used a commonly accepted comparative approach to correct for unusual pairings with RNA-editing mechanisms that are well known in arthropod mitogenomes [56,57]. The entire A+T-rich region was subjected to a search for tandem repeats using the Tandem Repeats Finder program [58].
3.4. Phylogenetic Analysis
To illustrate the phylogenetic relationship of Lepidoptera, the other sixteen complete lepidopteran mitogenomes were downloaded from Genbank, including A. honmai (DQ073916), B. mori (AY048187), B. mandarina (FJ384796), C. davidis (HQ658143), C. raphaelis (DQ102703), C. suppressalis (JF339041), E. pyretorum (FJ685653), G. molesta (HQ392511), H. armigera (GU188273), H. cunea (GU592049), L. dispar (FJ617240), M. sexta (EU286785), O. furnacalis (AF467260), O. lunifer (AM946601), P. atrilineata (EU569764), Drosophila melanogaster (U37541) and Locusta migratoria manilensis (GU344101) were used as outgroup.
The alignment of the concatenated amino acid sequences of each 13 mitochondrial PCGs was aligned with Clustal X [52] using default settings. Then, based on the concatenated amino acid data set from the 13 PCGs, phylogenetic analyses were constructed using MEGA version 5.0 software with Neighbor Joining (NJ) and Maximum Parsimony (MP) methods [35]. Bootstrap analysis was done with 1000 replications, and bootstrap values were calculated using the 50% majority rule.
4. Conclusions
In the present study, the mitogenome for S. inferens was one of five sequenced mitogenomes of the Noctuidae species, which includes about 10% of all recorded lepidopteran species. Compared to four other noctuid moths, the newly determined mitogenome also shared the gene order, nucleotide composition of protein-coding genes, the presence of intergenic spacers, and other features [4–7]. The order of trnM, trnI, and trnQ in the mitogenome might be one of the features that characterize the whole order Lepidoptera. The S. inferens mitogenome was biased to use T rather than A, which was different from other noctuid moths. The s4 spacer did not contain the motif “ATACTAA” that was one potential characteristic feature conserved across four other noctuid moths, the reason may be related to genetic mutation [59]. The great difference was that the stem-and-loop structure was found in the A+T-rich region of S. inferens mitogenome, but it was not observed in the four other noctuid moths. Although the S. inferens mitogenome was added, the position of Noctuidae is not improved. More insect mitogenomes will be helpful to fully resolve the phylogeny of Noctuoidea.
Acknowledgements
We thank Guo (Harvard University) for improving the text of the manuscript. This work was supported by the Special Fund for the National Basic Research and Development Program (973) of China (No. 2012CB114100).
References
- 1.Savary S., Willocquet L., Elazegui F.S., Castilla N.P., Teng P.S. Rice pest constraints in tropical Asia: Quantification of yield losses due to rice pests in a range of production situations. Plant Dis. 2000;84:357–369. doi: 10.1094/PDIS.2000.84.3.357. [DOI] [PubMed] [Google Scholar]
- 2.Singh J., Shera P.S. Relative abundance of different species of rice stem borers in punjab, India. Presented at the national conference on plant protection—New horizons in the millennium. rajasthan college of agriculture; Udaipur, India. 2001. [Google Scholar]
- 3.Sheng C.F., Wang H.T., Sheng S.Y., Gao L.D., Xuan W.J. Pest status and loss assessment of crop damage caused by the rice borers, Chilo suppressalis and Tryporyza incertulas in China. Entomol. Knowl. 2003;40:289–294. [Google Scholar]
- 4.Salvato P., Simonato M., Battisti A., Negrisolo E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae) BMC Genomics. 2008;9 doi: 10.1186/1471-2164-9-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liao F., Wang L., Wu S., Li Y.P., Zhao L., Huang G.M., Niu C.J., Liu Y.Q., Li M.G. The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae) Int. J. Biol. Sci. 2010;6:172–186. doi: 10.7150/ijbs.6.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin J., Hong G.Y., Wang A.M., Cao Y.Z., Wei Z.J. Mitochondrial genome of the cotton bollworm Helicoverpa armigera (Lepidoptera: Noctuidae) and comparison with other Lepidopteran. Mitochondrial DNA. 2010;21:160–169. doi: 10.3109/19401736.2010.503242. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Y.J., Zhou G.L., Fang R., Ye J., Yi J.P. The complete sequence determination and analysis of Lymantria dispar (Lepidoptera: Lymantriidae) mitochondrial genome. Plant Quar. 2010;4:6–11. [Google Scholar]
- 8.Wolstenholme D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992;141:173–216. doi: 10.1016/s0074-7696(08)62066-5. [DOI] [PubMed] [Google Scholar]
- 9.Boore J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1726–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Da Silva N.M., de Souza Dias A., da Silva Valente V.L., Victor H.V. Characterization of mitochondrial control region, two intergenic spacers and tRNAs of Zaprionus indianus (Diptera: Drosophilidae) Genetica. 2009;137:325–332. doi: 10.1007/s10709-009-9396-5. [DOI] [PubMed] [Google Scholar]
- 11.Wei S.J., Shi M., He J.H., Sharkey M.J., Achterberg C.V., Chen X.X. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genomics. 2010;11 doi: 10.1186/1471-2164-11-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cameron S.L., Yoshizawa K., Mizukoshi A., Whiting M.F., Johnson K.P. Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera) BMC Genomics. 2011;12 doi: 10.1186/1471-2164-12-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson S., Bankier A.T., Barrell B.G., de Bruijn M.H., Coulson A.R., Drouin J., Eperon I.C., Nierlich D.P., Roe B.A., Sanger F., et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 14.Boore J.L., Lavrov D., Brown W.M. Gene translocation links insects and crustaceans. Nature. 1998;393:667–668. doi: 10.1038/33577. [DOI] [PubMed] [Google Scholar]
- 15.Kim I., Lee E.M., Seol K.Y., Yun E.Y., Lee Y.B., Hwang J.S., Jin B.R. The mitochondrial genome of the Korean hairsteak, Coreana raphaelis (Lepidoptera: Lycaenidae) Insect Mol. Biol. 2006;15:217–225. doi: 10.1111/j.1365-2583.2006.00630.x. [DOI] [PubMed] [Google Scholar]
- 16.Gong Y.J., Shi B.C., Kang Z.J., Zhang F., Wei S.J. The complete mitochondrial genome of the oriental fruit moth Grapholita molesta (Busck) (Lepidoptera: Tortricidae) Mol. Biol. Rep. 2012;39:2893–2900. doi: 10.1007/s11033-011-1049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Bruijn M.H. Drosophila melanogaster mitochondrial DNA, a novel organization and genetic code. Nature. 1983;304:234–241. doi: 10.1038/304234a0. [DOI] [PubMed] [Google Scholar]
- 18.Beard C.B., Hamm D.M., Collins F.H. The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol. Biol. 1993;2:103–124. doi: 10.1111/j.1365-2583.1993.tb00131.x. [DOI] [PubMed] [Google Scholar]
- 19.Junqueira A.C., Lessinger A.C., Torres T.T., da Silva F.R., Vettore A.L., Arruda P., Azeredo Espin A.M. The mitochondrial genome of the blowfly Chrysomya chloropyga (Diptera: Calliphoridae) Gene. 2004;339:7–15. doi: 10.1016/j.gene.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 20.Krzywinski J., Grushko O.G., Besansky N.J. Analysis of the complete mitochondrial DNA from Anopheles funestus: An improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution. Mol. Phylogenet. Evol. 2006;39:417–423. doi: 10.1016/j.ympev.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 21.Masta S.E., Boore J.L. The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol. Biol. Evol. 2004;21:893–902. doi: 10.1093/molbev/msh096. [DOI] [PubMed] [Google Scholar]
- 22.Crozier R.H., Crozier Y.C. The mitochondrial genome of the honeybee Apis mellifera: Complete sequence and genome organization. Genetics. 1993;133:97–117. doi: 10.1093/genetics/133.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia X. Maximizing transcription efficiency causes codon usage bias. Genetics. 1996;144:1309–1320. doi: 10.1093/genetics/144.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shao R.F., Barker S.C. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): Convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol. Biol. Evol. 2003;20:362–370. doi: 10.1093/molbev/msg045. [DOI] [PubMed] [Google Scholar]
- 25.Yamauchi M.M., Miya M.U., Nishida M. Complete mitochondrial DNA sequence of the swimming crab, Portunus trituberculatus (Crustacea: Decapoda: Brachyura) Gene. 2003;311:129–135. doi: 10.1016/s0378-1119(03)00582-1. [DOI] [PubMed] [Google Scholar]
- 26.Gutell R.R., Lee J.C., Cannone J.J. The accuracy of ribosomal RNA comparative structure models. Curr. Opin. Struct. Biol. 2002;12:301–310. doi: 10.1016/s0959-440x(02)00339-1. [DOI] [PubMed] [Google Scholar]
- 27.Gillespie J.J., Johnston J.S., Cannone J.J., Gutell R.R. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): Structure, organization, and retrotransposable elements. Insect Mol. Biol. 2006;15:657–686. doi: 10.1111/j.1365-2583.2006.00689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cameron S.L., Whiting M.F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008;408:112–123. doi: 10.1016/j.gene.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 29.Chai H.N., Du Y.Z., Zhai B.P. Characterization of the complete mitochondrial genomes of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae) Int. J. Biol. Sci. 2012;8:561–579. doi: 10.7150/ijbs.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang D.X., Hewitt G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997;25:99–120. [Google Scholar]
- 31.Clary D.O., Wolstenholme D.R. Drosophila mitochondrial DNA: Conserved sequences in the A+T-rich region and supporting evidence for a secondary structure model of the small ribosomal RNA. J. Mol. Evol. 1987;25:116–125. doi: 10.1007/BF02101753. [DOI] [PubMed] [Google Scholar]
- 32.Zhang D., Szymura J.M., Hewitt G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995;40:382–391. doi: 10.1007/BF00164024. [DOI] [PubMed] [Google Scholar]
- 33.Rand D.M., Harrison R.G. Molecular population genetics of mtDNA size variation in crickets. Genetics. 1989;121:551–569. doi: 10.1093/genetics/121.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schultheis A.S., Weigt L.A., Hendricks A.C. Arrangement and structural conservation of the mitochondrial control region of two species of Plecoptera: Utility of tandem repeat-containing regions in studies of population genetics and evolutionary history. Insect Mol. Biol. 2002;11:605–610. doi: 10.1046/j.1365-2583.2002.00371.x. [DOI] [PubMed] [Google Scholar]
- 35.Hong M.Y., Lee E.M., Jo Y.H., Park H.C., Kim S.R., Hwang J.S., Jin B.R., Kang P.D., Kim K.G., Han Y.S., Kim I. Complete nucleotide sequence and organization of the mitogenome of the silk moth Caligula boisduvalii (Lepidoptera: Saturniidae) and comparison with other lepidopteran insects. Gene. 2008;413:49–57. doi: 10.1016/j.gene.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 36.Kim S.R., Kim M.I., Hong M.Y., Kim K.Y., Kang P.D., Hwang J.S., Han Y.S., Jin B.R., Kim I. The complete mitogenome sequence of the Japanese oak silkmoth, Antheraea yamamai (Lepidoptera: Saturniidae) Mol. Biol. Rep. 2009;36:1871–1880. doi: 10.1007/s11033-008-9393-2. [DOI] [PubMed] [Google Scholar]
- 37.Kim I., Cha S.Y., Yoon M.H., Hwang J.S., Lee S.M., Sohn H.D., Jin B. The complete nucleotide sequence and gene organization of the mitochondrial genome of the oriental mole cricket, Gryllotalpa orientalis (Orthoptera: Gryllotalpidae) Gene. 2005;353:155–168. doi: 10.1016/j.gene.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 38.Lee E.S., Shin K.S., Kim M.S., Park H., Cho S., Kim C.B. The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae) Gene. 2006;373:52–57. doi: 10.1016/j.gene.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Hassanin A. Phylogeny of Arthropoda inferred from mitochondrial sequences: Strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol. Phylogenet. Evol. 2006;38:100–116. doi: 10.1016/j.ympev.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 40.Kristensen N.P., Skalski A.W. Phylogeny and Paleontology. In: Kristensen N.P., editor. Lepidoptera: Moths and Butterflies. 1. Evolution, Systematics, and Biogeography. Handbook of Zoology. Part 35. IV. De Gruyter; Berlin, Germany and New York, NY, USA: 1999. pp. 7–25. [Google Scholar]
- 41.Yang L., Wei Z.J., Hong G.Y., Jiang S.T., Wen L.P. The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae) Mol. Biol. Rep. 2009;36:1441–1449. doi: 10.1007/s11033-008-9334-0. [DOI] [PubMed] [Google Scholar]
- 42.Hu X.L., Cao G.L., Xue R.Y., Zheng X.J., Zhang X., Duan H.R., Gong C.L. The complete mitogenome and phylogenetic analysis of Bombyx mandarina strain Qingzhou. Mol. Biol. Rep. 2010;37:2599–2608. doi: 10.1007/s11033-009-9781-2. [DOI] [PubMed] [Google Scholar]
- 43.Lu C., Liu Y.Q., Liao X.S., Li B., Xiang Z.X., Han H., Wang X.G. Complete sequence determination and analysis of Bombyx mori mitochondrial genome. J. Agric. Biotechnol. 2002;10:163–170. [Google Scholar]
- 44.Jiang S.T., Hong G.Y., Yu M., Li N., Yang Y., Liu Y.Q., Wei Z.J. Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogyna pyretorum (Lepidoptera: Saturniidae) Int. J. Biol. Sci. 2009;5:351–365. doi: 10.7150/ijbs.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xia J., Hu J., Zhu G.P., Zhu C.D., Hao J.S. Sequencing and analysis of the complete mitochondrial genome of Calinaga davidis Oberthür (Lepidoptera: Nymphalidae) Acta Entomol. Sin. 2011;54:555–565. [Google Scholar]
- 46.Coates B.S., Sumerford D.V., Hellmich R.L., Lewis L.C. Partial mitochondrial genome sequences of Ostrinia nubilalis and Ostrinia furnicalis. Int. J. Biol. Sci. 2005;1:13–18. doi: 10.7150/ijbs.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewis D.L., Farr C.L., Kaguni L.S. Drosophila melanogaster mitochondrial DNA: Completion of the nucleotide sequence and evolutionary comparisons. Insect Mol. Biol. 1995;4:263–278. doi: 10.1111/j.1365-2583.1995.tb00032.x. [DOI] [PubMed] [Google Scholar]
- 48.Hwang U.W., Park C.J., Yong T.S., Kim W. One-step PCR amplification of complete arthropod mitochondrial genomes. Mol. Phylogenet. Evol. 2001;19:345–352. doi: 10.1006/mpev.2001.0940. [DOI] [PubMed] [Google Scholar]
- 49.Simon C., Frati F., Beckenbach A.T., Crespi B., Liu H., Flook P. Evolution, weighting and phylogenetics utility of mitochondrial gene sequences and compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994;87:651–701. [Google Scholar]
- 50.Wei Z.J., Hong G.Y., Jiang S.T., Tong Z.X., Lu C. Characters and expression of the gene encoding DH, PBAN and other FXPRLamide family neuropeptides in Antheraea pernyi. J. Appl. Entomol. 2008;132:59–67. [Google Scholar]
- 51.Staden Package. [accessed on 5 December 2011]. Available online: http://staden.sourceforge.net.
- 52.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 53.Perna N.T., Kocher T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995;41:353–358. doi: 10.1007/BF00186547. [DOI] [PubMed] [Google Scholar]
- 54.Lowe T.M., Eddy S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cannone J.J., Subramanian S., Schnare M.N., Collett J.R., D’Souza L.M., Du Y., Feng B., Lin N., Madabusi L.V., Müller K.M., et al. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform. 2002;3 doi: 10.1186/1471-2105-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gutell R.R., Larsen N., Woese C.R. Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol. Rev. 1994;58:10–26. doi: 10.1128/mr.58.1.10-26.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buckley T.R., Simon C., Flook P.K., Misof B. Secondary structure and conserved motifs of the frequently sequenced domains IV and V of the insect mitochondrial large subunit rRNA gene. Insect Mol. Biol. 2000;9:565–580. doi: 10.1046/j.1365-2583.2000.00220.x. [DOI] [PubMed] [Google Scholar]
- 58.Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bachtrog D. Reduced selection for codon usage bias in Drosophila Miranda. J. Mol. Evol. 2007;64:586–590. doi: 10.1007/s00239-006-0257-x. [DOI] [PubMed] [Google Scholar]