

Abstract
Two cationic, amphipathic peptoids (poly-N-substituted glycines) were developed as new molecular transporters, which have extensive cellullar uptake and utilize different internalization mechanisms from purely cationic polyguanidine comparators.
The advantages of cell-penetrating peptides (CPPs) as molecular transporters lie in their high efficiency in delivering cargo into various cell lines in vitro, and their promising in vivo applications1, 2.
Like polyarginines and other guanidinium-rich transporters, many CPPs comprise just cationic residues; however, cationic, amphipathic CPPs with both basic and hydrophobic residues, such as pAntp-(43–58), Transportan, and pVEC-(615–632), are also common3, 4. Although the mechanisms by which CPPs enter mammalian cells have not been elucidated in detail, it is generally accepted that both energy-dependent endocytosis and energy-independent direct translocation are involved2. In addition, structural properties of CPPs as well as properties of the associated cargo have a significant impact on the adopted internalization mechanisms3.
Host-defense peptides typically have cationic, amphipathic structures. They are strongly active on negatively charged membranes and/or zwitterionic membranes5. Several host-defense peptides, such as magainin 2, buforin 2, and α-helical model amphipathic peptides (MAPs), have been reported to show extensive cellular uptake without requiring permanent membrane permeabilization6. This has led to interest in employing membrane-crossing host-defense peptides as transporters or as templates to design novel vectors5
One limitation of the peptide-based transporters is their rapid degradation in vivo. Peptoids, or poly-N-substituted glycines, are an emerging class of protease-resistant peptidomimetics in designing molecular transporters. Previously, polyguanidine peptoids, lysine-like polypeptoids, α-peptide/β-peptoid chimeras were developed as efficient transporters7. In addition, Tan and coworkers demonstrated that peptoids were anywhere from 3- to 30-fold more permeable than structurally analogous peptides8.
In this work, we designed and synthesized a library of cationic, amphipathic peptoids as molecular transporters and investigated how peptoid structures influenced their cellular uptake and cytotoxicity. Unlike previous studies, we focused on the cationic, amphipathic peptoids as transporters. In a previous study of host-defense peptide mimics, we serendipitously found an antimicrobial and anticancer peptoid (4, Table 1) that translocated into mammalian cells efficiently at low concentrations without causing observable cytotoxicity9. Hence, several cationic, amphipathic variants (3–11, Table 1) were designed with different lengths, charges and hydrophobicity. Arg8 (1) and a polyguanidine peptoid (Nbtg8, 2) were tested as polyguanidine comparators with the charge-to-length ratio (CTLR) of 1. The peptoids were synthesized employing a two-step submonomer method (Fig. 1A)10. With a periodic incorporation of α-chiral side chains, peptoids can form polyproline type-I-like helices which allows the cationic, amphipathic structure to be easily recapitulated into three-faced helices in peptoids comprising trimer repeats, (X-Y-Z)n11. To quantify cellular uptake, all constructs were labeled with 5(6)-carboxyfluorescein (CF) on the N-terminus without a spacer (Fig. 1C). Thus, all the studies described herein were about the cellular uptake of transporters conjugated to small molecules (MW ca. <3000), and the results do not preclude different phenomena in delivering larger cargos.
Table 1. Fluorescently labeled cell-penetrating constructs.
HPLC elution is reported as the percentage of compound elution in water/acetonitrile (0.1% trifluoroacetic acid) on a C18 column in analytic HPLC. Net charge indicates molecular charges at neutral pH. CTLR stands for charge-to-length ratio. LC50 means lethal concentrations causing 50% death of MCF-7 cells, quantified via MTS assays for 24h treatment. Peptoids labeled with asterisk have been reported with antimicrobial or anticancer activities9,11.
| Group | Construct | Sequence | MW (Da) | HPLC elution(% ACN) | Net Charge | CTLR | LC50 (μM)(MCF-7) |
|---|---|---|---|---|---|---|---|
| Polyguanidine comparators | 1 | CF-(Arg)8-NH2 | 1624 | 43.8 | +8 | 1 | 80 |
| 2 | CF-(Nbtg)8 -NH2 | 1737 | 44.9 | +8 | 1 | >100 | |
| NLys(l/3) variants | 3* | CF-(NLys-Nspe-Nspe)3-NH2 | 1727 | 73.2 | +3 | 0.33 | 67 |
| 4* | CF-(NLys-Nspe-Nspe)4-NH2 | 2177 | 76.0 | +4 | 0.33 | 12 | |
| 5* | CF-(NLys-Npm-Npm)4-NH2 | 2065 | 69.2 | +4 | 0.33 | 38 | |
| 6* | CF-(NLys-Nssb-Nssb)4-NH2 | 1905 | 73.0 | +4 | 0.33 | >100 | |
| NLys(2/3) variants | 7 | CF-(NLys-NYLys-Nspe)4-NH2 | 2045 | 52.5 | +8 | 0.66 | >100 |
| 8 | CF-(NLys)8-(Nspe)4-NH2 | 2045 | 60.0 | +8 | 0.66 | 48 | |
| 9 | CF-(NLys)8-(NLeu)4-NH2 | 1853 | 53.5 | +8 | 0.66 | >100 | |
| Guanidylated variants | 10* | CF-(Nbtg-Nspe-Nspe)4-NH2 | 2345 | 75.5 | +4 | 0.33 | 11 |
| 11 | CF-(Nbtg-Nbtg-Nspe)4-NH2 | 2381 | 54.8 | +8 | 0.66 | 50 |
Fig. 1. The construct synthesis.

(A), the submonomer synthesis scheme of peptoids; (B), peptoid monomer structures with shorthand names; (C), chemical structures of cell penetrating constructs (X) labeled with 5(6)-Carboxyfluorescein (CF).
The sequences of fluorescently labeled variants, the solvent composition at RP-HPLC elution as a measure of their hydrophobicity, net charges, CTLR and the lethal concentration (LC50) in MCF-7 (human breast cancer cell line) are summarized in Table 1. LC50 of constructs in MCF-7 cells were derived from the cell viability curve for 24h treatment measured via MTS assays. Uptake of 1–11 in MCF-7 cells was quantified in Guava Easycyte Plus® flow cytometry system (Fig. 2).
Fig. 2. The cellular uptake of construct 1 to 11.

MCF-7 cells were incubated with 8μM of each construct diluted in 10% FBS media for 1h at 37°C. Cells were washed thoroughly and trypsinized. Cellular fluorescence was quantified on Guava Easycyte Plus® flow cytometry system.
Polyguanidine comparators (1 and 2) displayed efficient uptake (Fig. 2) and minor cytotoxicity (Table 1). The NLys(1/3) variants, 3–6 with CTLR at 0.33, have been reported in our previous antimicrobial and anticancer studies. Compared to 4 (with Nspe), 5 with less hydrophobic, achiral Npm residues and 6 with less bulky and hydrophobic, chiral Nssb both showed lower cellular uptake (Fig. 2) and lower cytotoxicity as indicated by their increased LC50. This indicated the importance of chiral, aromatic hydrophobic residues for the high uptake of those peptoid transporters. Moreover, the cytotoxicity and cellular uptake of 3 (a 9mer) was much lower than those of the 12mer analogue 4 (Table 1). 4 had the most efficient uptake yet with the highest cytotoxicity among the NLys(1/3) variants.
The NLys(2/3) variants, 7–9, have 8 NLys per molecule with CTLR at 0.66. Compared to 4, 7 with reduced hydrophobicity displayed dramatically decreased uptake and cytotoxicity. Interestingly, 8 with the same compositon as 7 but with segregated NLys and Nspe compartments had higher cytotoxicity and uptake. 9 with nonaromatic NLeu were basically inactive.
Guanidinium heads are known to enhance the membrane interactions of cell-penetrating peptides via hydrogen bonding with phospholipids12. 10 is the guanidinylated analogue of 4, and 11 is the guanidinylated analogue of 7. For peptoids with CTLR at 0.33, guanidylation did not have obvious effects, suggested by similar uptake and cytotoxicity of 4 and 10. For peptoids with CTLR at 0.66, guanidylation dramatically increased cellular uptake. The uptake efficiency of 11 was much higher than that of 7 (Fig. 2) which indicated stronger membrane interactions contributed by guanidinium heads. Moreover, 11 displayed lower cytotoxicity than 4 and 10 (Table 1). To summarize, both guanidinium heads and chiral, aromatic hydrophobic residues can make the cationic, amphipathic constructs more permeable. 4 with CTLR at 0.33 is a good candidate with ease of synthesis, the most efficient uptake but with high cytotoxicity. 11 with guanidinium heads and CTLR at 0.66 had strong uptake as well but with lower cytotoxicity.
To compare the uptakes of cationic, amphipathic 4 and 11 with polyguanidine comparators 1 and 2, the time dependence of their uptakes was studied as shown in Fig. 3. A longer incubation gradually increased the uptake of peptoid constructs, 4, 11 and 2, up to 18h tested. The time course of 1 (Arg8) was different: its initial uptake was slower than peptoid constructs indicated by low cellular fluorescent signals at 30min, the maximum uptake was reached around 4h, and then a decreased uptake was observed with very low fluorescent signals present in cells 18h later. The decrease is likely to be caused by peptide degradation. Moreover, the intracellular distributions of these two groups were distinct, as shown by live cell confocal imaging (Fig. 4). Cationic, amphipathic peptoids 4 and 11 had a punctuated distribution. 4 was mainly present in the cytoplasm (Fig. 4A), and 11 had some nuclear as well as cytoplasmic distribution (Fig. 4B). In contrast, 1 (Fig. 4C) and 2 (Fig. 4D) had a diffusive distribution throughout the cells. Different concentrations were used to achieve similar imaging brightness. Similar phenomenon has been shown about pAntp and Arg9 which displayed distinct intracellular patterns13. Furthermore, in a temperature study, we showed that the cellular uptake of 4 was significantly reduced at 4°C compared to 37°C (Fig. 5). The uptake of 11 was also greatly reduced at 4°C but it still had considerable uptake. This temperature dependency indicated that translocation of 4 and 11 were either energy-dependent or very sensitive to the membrane fluidity which would have greatly changed at 4°C. In contrast, at 4°C, the uptake of 1 and 2 was not reduced, indicating an energy-independent internalization for polyguanidine comparators. The uptake of 1 was even increased at 4°C. The intracellular distributions and the temperature study indicated that distinct internalization mechanisms were used by polyguanidine constructs and cationic, amphipathic constructs.
Fig. 3. The time-dependent cellular uptake of construct 4, 11, 1 and 2.
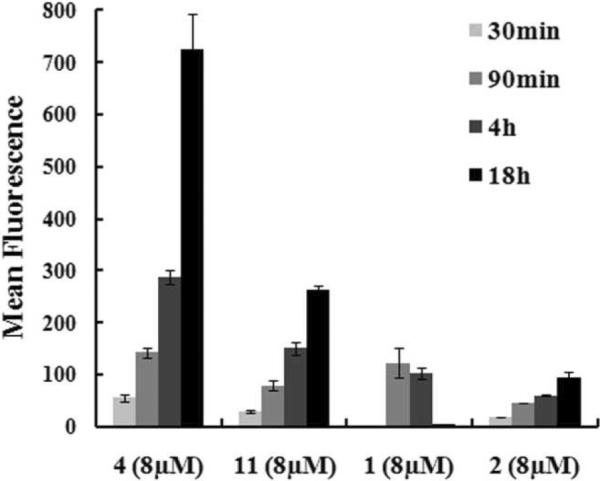
MCF-7 cells were incubated at 37°C with 8μM of each construct in 10% FBS media for the indicated time. The cellular uptake was quantified on Guava Easycyte Plus® system.
Fig. 4. Intracellular distributions of construct 4, 11, 1 and 2--the live-cell confocal imaging.

MCF 7 cells were incubated at 37°C with 8μM of 4 (A)9, 16μM of 11 (B), 50μM of 1 (C) and 2 (D) in 10% FBS media for 1h. Different concentrations were used to achieve similar image brightness. Cells were washed thoroughly and imaged on Leica confocal laser microscope (×63 oil lens)
Fig. 5. The cellular uptake of 4, 11, 1 and 2 at 37°C and 4°C.
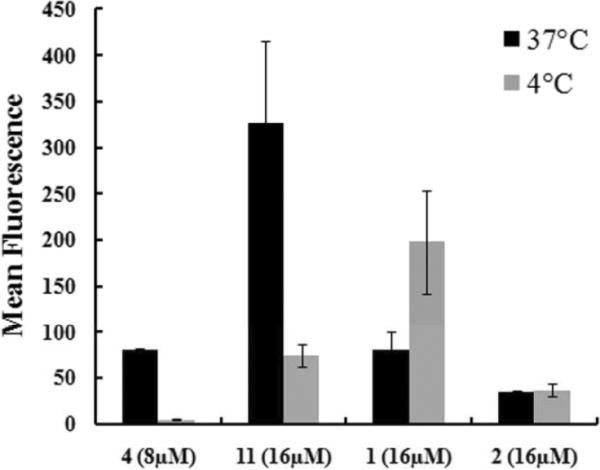
MCF-7 cells were incubated with 8μM of 4, 16μM of 11, 1, and 2 in 10% FBS media for 1h at 37°C and 4°C. For 4°C experiments, cells and reagents were pre-cooled 30min at 4°C. The cellular uptake was quantified on Guava Easycyte system.
In summary, two cationic, amphipathic peptoid transporters with good water solubility (4 and 11) were developed with efficient cellular uptake, which employ distinct uptake mechanisms from polyguanidine constructs. Our findings showed how structures can determine the cellular uptake efficiency and mechanisms of cell-penetrating peptides, and should provide useful guidance in designing novel molecular transporters.
Supplementary Material
Acknowledgments
This work was supported by NIH R01 AI072666-04.
Footnotes
† Electronic Supplementary Information (ESI) available: [Experimental details, HPLC and ESI data of presented compounds, Figure S1–2].
Notes and references
- 1.Dowdy SF, Snyder EL. Pharmaceutical Research. 2004;21:389. doi: 10.1023/B:PHAM.0000019289.61978.f5. [DOI] [PubMed] [Google Scholar]; Wender PA, Galliher WC, et al. Advanced Drug Delivery Reviews. 2008;60:452. doi: 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelley SO, Stewart KM, et al. Organic & Biomolecular Chemistry. 2008;6:2242. doi: 10.1039/b719950c. [DOI] [PubMed] [Google Scholar]
- 3.Ferrer M, Maiolo JR, et al. Biochimica Et Biophysica Acta-Biomembranes. 2005;1712:161. doi: 10.1016/j.bbamem.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Langel U, Elmquist A, et al. Experimental Cell Research. 2001;269:237. doi: 10.1006/excr.2001.5316. [DOI] [PubMed] [Google Scholar]; Langel U, Pooga M, et al. Faseb Journal. 1998;12:67. doi: 10.1096/fasebj.12.1.67. [DOI] [PubMed] [Google Scholar]
- 5.Castanho MARB, Henriques ST, et al. Biochemical Journal. 2006;399:1. doi: 10.1042/BJ20061100. [DOI] [PMC free article] [PubMed] [Google Scholar]; Giralt E, Fernandez-Carneado J, et al. Biopolymers. 2004;76:196. doi: 10.1002/bip.10585. [DOI] [PubMed] [Google Scholar]; Neundorf I, Splith K. European Biophysics Journal with Biophysics Letters. 2011;40:387. doi: 10.1007/s00249-011-0682-7. [DOI] [PubMed] [Google Scholar]
- 6.Matsuzaki K, Takeshima K, et al. Journal of Biological Chemistry. 2003;278:1310. doi: 10.1074/jbc.M208762200. [DOI] [PubMed] [Google Scholar]; Oehlke J, Scheller A, et al. Biochimica Et Biophysica Acta-Biomembranes. 1998;1414:127. doi: 10.1016/s0005-2736(98)00161-8. [DOI] [PubMed] [Google Scholar]
- 7.Bradley M, Unciti-Broceta A, et al. Bioorganic & Medicinal Chemistry. 2009;17:959. doi: 10.1016/j.bmc.2008.02.068. [DOI] [PubMed] [Google Scholar]; Brase S, Schroder T, et al. Journal of Medicinal Chemistry. 2008;51:376. doi: 10.1021/jm070603m. [DOI] [PubMed] [Google Scholar]; Foged C, Franzyk H, et al. Biochimica Et Biophysica Acta-Biomembranes. 2008;1778:2487. doi: 10.1016/j.bbamem.2008.06.020. [DOI] [PubMed] [Google Scholar]; Schepers U, Schroder T, et al. Bioconjugate Chemistry. 2007;18:342. doi: 10.1021/bc0602073. [DOI] [PubMed] [Google Scholar]; Wender PA, Mitchell DJ, et al. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:13003. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kodadek T, Tan NC, et al. Bioorganic & Medicinal Chemistry. 2008;16:5853. doi: 10.1016/j.bmc.2008.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang W, Jiwon S, et al. in submission. 2011 [Google Scholar]
- 10.Zuckermann RN, Kerr JM, et al. Journal of the American Chemical Society. 1992;114:10646. [Google Scholar]
- 11.Chongsiriwatana NP, Patch JA, et al. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2794. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]; Czyzewski AM, Kapoor R, et al. Journal of antimicrobial agents and chemotherapy. In review. [Google Scholar]; Wu CW, Sanborn TJ, et al. Abstracts of Papers of the American Chemical Society. 2001;222:U420. [Google Scholar]
- 12.Rothbard JB, Jessop TC, et al. Journal of the American Chemical Society. 2004;126:9506. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 13.Duchardt F, Fotin-Mleczek M, et al. Traffic. 2007;8:848. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.