

ILK is identified as a new partner for ADAM12L cell signaling functions. ADAM12L colocalizes with ILK at focal adhesions and induces the Akt-dependent survival pathway via stimulation of β1 integrins and activation of PI3K. This effect is independent of ADAM12L proteolytic activity and involves its cytoplasmic domain.
Abstract
Based on its shedding and binding activities, the disintegrin and metalloprotease 12 (ADAM12) has been implicated in cell signaling. Here we investigate the intracellular protein interaction network of the transmembrane ADAM12L variant using an integrative approach. We identify the integrin-linked kinase (ILK) as a new partner for ADAM12L cellular functions. We demonstrate that ADAM12L coimmunoprecipitates with ILK in cells and that its cytoplasmic tail is required for this interaction. In human cultured hepatic stellate cells (HSCs), which express high levels of endogenous ADAM12L and ILK, the two proteins are redistributed to focal adhesions upon stimulation of a β1 integrin–dependent pathway. We show that down-regulation of ADAM12L in HSCs leads to cytoskeletal disorganization and loss of adhesion. Conversely, up-regulation of ADAM12L induces the Akt Ser-473 phosphorylation-dependent survival pathway via stimulation of β1 integrins and activation of phosphoinositide 3-kinase (PI3K). Depletion of ILK inhibits this effect, which is independent of ADAM12L proteolytic activity and involves its cytoplasmic domain. We further demonstrate that overexpression of ADAM12L promotes kinase activity from ILK immunoprecipitates. Our data suggest a new role for ADAM12L in mediating the functional association of ILK with β1 integrin to regulate cell adhesion/survival through a PI3K/Akt signaling pathway.
INTRODUCTION
ADAM12 is a member of a disintegrin and metalloprotease (ADAM) protein family whose members are cell-surface multidomain proteins involved in ectodomain shedding, adhesion, and signaling activities (Edwards et al., 2008). ADAM archetypes are transmembrane proteins that contain propeptide, extracellular metalloprotease, disintegrin-like, cysteine-rich, and epidermal growth factor (EGF)–like transmembrane and cytoplasmic domains.
In humans, the expression of ADAM12 is mainly associated with adult skeletal, cardiac, and smooth muscle cells and is dramatically increased in several pathologies, including cancer (Kveiborg et al., 2008). Within tissues, ADAM12 up-regulation has been described in both cancer and stromal cells, and increased urine protein levels were also reported in patients with breast or bladder tumors. Genome-wide analyses of human tumors identified ADAM12 as a new candidate cancer gene (Sjoblom et al., 2006), and it is now considered as a prognosis marker for human bladder cancer (Roy et al., 2004; Fröhlich et al., 2006) and breast cancer (Pories et al., 2008; Narita et al., 2010). In support of its involvement in cancer, ADAM12 was shown to regulate tumor progression in genetically modified mouse models. However, the mechanism involved remains unclear (Kveiborg et al., 2005; Peduto et al., 2006).
The human ADAM12 gene is expressed as two alternatively spliced transcripts that give rise to transmembrane ADAM12L and secreted ADAM12S forms, the latter lacking the transmembrane and cytoplasmic domains. Both ADAM12 variants can act as proteases and have been shown to cleave IGFBP3 and IGFBP5 (Loechel et al., 2000; Shi et al., 2000), EGF receptor ligands (Asakura et al., 2002; Horiuchi et al., 2007), Notch ligand Delta-like 1 (Dyczynska et al., 2007), and placental oxytocinase (Ito et al., 2004). Besides its metalloprotease activity, the membrane-anchored ADAM12L long form is involved in cell–cell and cell–matrix interaction via its binding to cell-surface molecules that include integrins (Eto et al., 2000; Kawaguchi et al., 2003), syndecans (Iba et al., 2000), and type II transforming growth factor-β (TGF-β) receptor (Atfi et al., 2007). However ADAM12L is not constitutively addressed to the membrane, and its distribution at the cell surface is a dynamic process that requires PKCɛ activation (Sundberg et al., 2004), recruitment of RACK1 (Bourd-Boittin et al., 2008), and c-Src activity (Stautz et al., 2010). All these events take place in response to external cell stimulation such as integrin engagement and depend on the intracellular domain of ADAM12L, since its truncation prevents translocation of the protein (Hougaard et al., 2000). The cytoplasmic tail of ADAM12 has been shown to physically interact with several signaling proteins, including the Src nonreceptor tyrosine kinases c-Src and YES (Suzuki et al., 2000), the adapter proteins Grb2 (Suzuki et al., 2000) and Fish (Abram et al., 2003), the regulatory subunit of phosphatidylinositol 3-kinase, p85α (Asakura et al., 2002), the cytoplasmic PACSIN3 phosphoprotein (Mori et al., 2003), eve-1, an EEN-binding protein implicated in shedding activity (Tanaka et al., 2004), and two actin-related proteins, α-actinin-1 and -2 (Galliano et al., 2000; Cao et al., 2001). This complex protein interaction network contributes to the regulation of ADAM12L translocation and function and might in turn play a pivotal role in the coordination of cell signaling and responses to the microenvironment.
In liver cancer, we previously demonstrated that up-regulation of ADAM12 expression was associated with the activation of hepatic stellate cells (HSCs; Le Pabic et al., 2003) and the regulation of the TGF-β signaling pathway (Atfi et al., 2007). In addition, our data showed that type I collagen—the major collagen in fibrosis tissue—promoted the localization of ADAM12 to the surface of HSCs through a β1 integrin–dependent mechanism (Bourd-Boittin et al., 2008). In this study, we explored the ADAM12L protein interaction network using an integrative data-mining approach. We identify the integrin-linked kinase (ILK)—a kinase associated with human malignancies—as a new potential partner for ADAM12L. Our results demonstrate that ADAM12L coimmunoprecipitates with ILK and that stimulation of HSCs by type I collagen induces the recruitment of both proteins to focal adhesion-like structures. We show that down-regulation of ADAM12L leads to a loss of cell adhesion, whereas overexpression induces the Akt-dependent survival pathway and increases the kinase activity of ILK immunoprecipitates. We propose that ADAM12 plays a major role in adhesion-cell survival processes through enhancement of the β1 integrin/ILK/Akt signaling pathway.
RESULTS
An ADAM12 protein interaction network as part of the integrin signaling network
The interaction of ADAM12L with numerous receptors and intracellular signal mediators (listed in Table 1) suggests that it could act as a docking/signaling protein. Using the STRING database to extract direct (physical) and indirect (functional) associations between proteins, we built the ADAM12L protein interaction network shown in Figure 1. The high level of interconnections suggests strong associations with the same biological functions. The connectivity of the graph was next enriched by adding five known interactions not yet incorporated in the database, between ADAM12 and TGFBR2, GNB2L1, PRKCE, and PRKCD, and between Src and TGFBR2. Of interest, all known proteins identified as ADAM12-binding proteins were previously shown to be associated with Src. These include the four SH3 domain–containing proteins SH3D19, SH3PXD2A, YES1, and PACSIN3. In addition ADAM12 and Src both connect to the same subnetwork in which the integrin signaling pathway stands out. Because engagement of β1 integrin (ITGB1) by external stimuli has been recently associated with the Src-mediated modulation of ADAM12 subcellular distribution (Stautz et al., 2010), we sought to investigate a possible interaction between ADAM12 and a major functional partner of integrins, ILK. Our rationale was that ILK directly binds to integrins in response to stimulation by the microenvironment, suggesting a pivotal role in signal transduction that might implicate ADAM12.
TABLE 1:
ADAM12L-interacting proteins used as input for STRING application (http://string-db.org).
| Gene name | Description | Experiment type |
|---|---|---|
| ACTN2 | Actinin α2 | In vivo; in vitro; yeast two-hybrid |
| ACTN1 | Actinin α1 | In vivo; in vitro |
| GRB2 | Growth factor receptor–bound protein 2 | In vivo |
| SRC | V-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homologue (avian) | In vivo; in vitro |
| YES1 | V-yes-1 Yamaguchi sarcoma viral oncogene homologue 1 | In vitro |
| PIK3R1 | Phosphoinositide-3-kinase regulatory subunit 1 (α) | In vivo; in vitro |
| PRKCE | Protein kinase C ε | In vitro |
| PRKCD | Protein kinase C δ | Yeast two-hybrid |
| ITGB1 | Integrin β1 (fibronectin receptor β polypeptide, antigen CD29) | In vitro |
| SH3PXD2A | SH3 and PX domains 2A | In vivo; in vitro |
| PACSIN3 | Protein kinase C and casein kinase substrate in neurons 3 | In vivo; in vitro; yeast two-hybrid |
| SH3D19 | SH3 domain containing 19 | In vivo; in vitro; yeast two-hybrid |
| TGFBR2 | Transforming growth factor β receptor II (70/80 kDa) | In vivo; in vitro; yeast two-hybrid |
| GNB2L1 | Guanine nucleotide binding protein (G protein) β polypeptide 2–like 1 | In vivo; in vitro; yeast two-hybrid |
FIGURE 1:
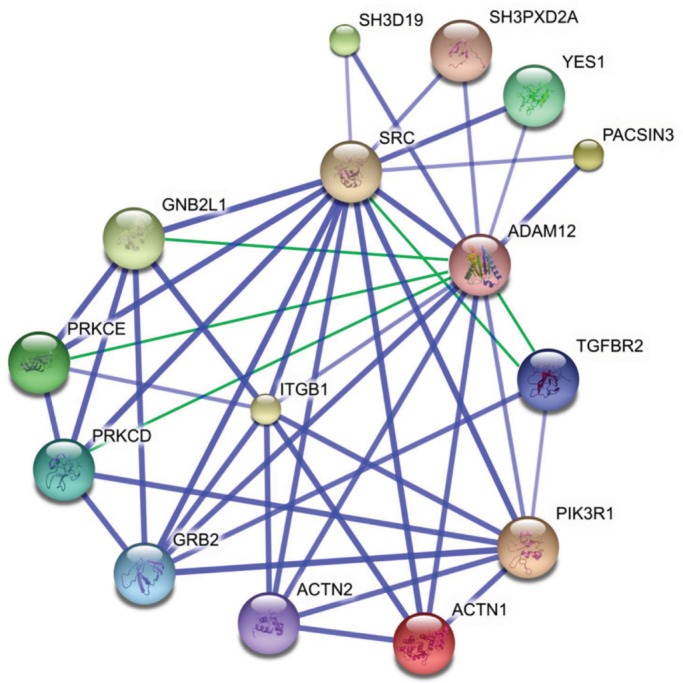
The ADAM12L protein interaction network. The list of the 14 proteins that physically bind ADAM12L (Table 1) was used as input in the STRING application, and the graph was generated using default parameters. The interactions between ADAM12 and PRKCE (Sundberg et al., 2004), PRKCD (Asakura et al., 2002), GNB2L1 (Bourd-Boittin et al., 2008), and TGFBR2 (Atfi et al., 2007) and between TGFBR2 and Src (Galliher and Schiemann, 2007) have not been entered in the database yet and were added manually (green connecting lines).
ADAM12 interacts with the ILK integrin-linked kinase
To test this hypothesis, we first investigated whether ADAM12 interacts with ILK in Cos7 cells, which do not express endogenous ADAM12. Endogenous ILK was immunoprecipitated from extracts of cells transfected with ADAM12L. As shown in Figure 2A, both pro and processed ADAM12—the 110- and 90-kDa forms, respectively—were expressed in Cos7 cells (left) and coimmunoprecipitated with ILK (middle). The reverse immunoprecipitation was performed using a mix of ADAM12 monoclonal antibodies and blotted for ILK (Figure 2A, right). The coimmunoprecipitation of ILK and ADAM12 was confirmed using transfection of V5-tagged ILK in either Cos7 cells transfected with ADAM12 or in the LX2 HSC line, which expresses low endogenous ADAM12 levels (Supplemental Figure S1). To show whether the interaction between ADAM12 and ILK occurs under physiological conditions, we used primary cultures of HSCs isolated from human liver tissues. Cell culture mimics the HSC activation process, which consists in the transition from a quiescent to a fibrogenic state. This is accompanied by increased expression of ADAM12 and ILK, as previously described in chronic liver diseases (Le Pabic et al., 2003; Zhang et al., 2006). The expression of endogenous ADAM12L in activated HSCs is shown in Figure 2B (left). When immunoprecipitation was performed using ILK antibodies, ADAM12/ILK complexes were clearly detected in cell extracts (Figure 2B, middle). The reverse immunoprecipitation using ADAM12 antibodies confirmed the endogenous interaction of ADAM12 and ILK (Figure 2B, right).
FIGURE 2:

ADAM12L interacts with ILK. Cell extracts from (A) Cos7 cells transfected with the long form of ADAM12, ADAM12L, or (B) HSCs were immunoprecipitated with anti-ADAM12 or anti-ILK (IP) and immunoblotted with anti-ILK or anti-ADAM12 antibodies (IB). (C) Cos7 cells were transfected either with ADAM12L or the ADAM12-Δcyto cytoplasmic tail deletion or the short ADAM12S secreted form. Cell extracts were immunoprecipitated with anti-ILK and immunoblotted with anti-ADAM12 (right). Left, immunoblots performed on crude extracts as controls. (D) Cos7 cells transfected with ADAM12L were immunoprecipitated with anti-ILK and immunoblotted (IB) with anti-PINCH, anti-parvin, or anti-ADAM12 antibodies. (E) ADAM12L, ADAM12S, and ADAM12-Δcyto were expressed as His-tagged fusion proteins, and binding assays were performed by incubating His-ADAM12 bound to nickel-agarose with 1 μg of purified ILK. Interacting proteins were immunoblotted with anti-ILK and anti-ADAM12 antibodies.
To identify the domain of ADAM12 that is required for interaction with ILK, we transfected Cos7 cells with ADAM12L, with a truncated form lacking the cytoplasmic domain (ADAM12-Δcyt) or with the ADAM12S short form. As shown in Figure 2C, immunoprecipitation of ILK led to the recovery of high amounts of ADAM12L. In contrast, neither ADAM12L-Δcyt (90 kDa) nor ADAM12S (90 and 68 kDa for the precursor and processed forms, respectively) were detected in immunoprecipitates, implicating the ADAM12 cytoplasmic domain in the interaction with ILK. Because disintegrin and cysteine-rich domains are present in ADAM12L-Δcyt and ADAM12S and have been reported to interact with integrins (Eto et al., 2000; Kawaguchi et al., 2003; Thodeti et al., 2003; Zhao et al., 2004; Huang et al., 2005; Lafuste et al., 2005), the lack of recovery of ADAM12L-Δcyt and ADAM12S in ILK immunoprecipitates suggests that integrins do not mediate the interaction of ADAM12 and ILK. In addition, we investigated the presence of PINCH and parvin in ILK-ADAM12 immune complexes isolated from cell extracts, as ILK has been linked to PINCH and parvin proteins in forming the IPP complex (Legate et al., 2006). As expected, both proteins were detected in ILK immunoprecipitates (Figure 2D), suggesting that the interaction of ADAM12 and ILK takes place within IPP complexes.
To rule out the possible implication of additional proteins, we next investigated ADAM12/ILK interactions using recombinant proteins in in vitro binding assays. ILK was expressed as a glutathione S-transferase (GST) fusion protein and purified on glutathione-agarose beads before removal of the GST tag (recILK). We then performed pull-down assays by incubating recILK with purified histidine (His)-tagged ADAM12 variants. As shown in Figure 2E, recILK only bound full-length ADAM12L, demonstrating an association that is both direct and specific. These results provide strong evidence for a direct interaction of the cytoplasmic domain of ADAM12L with ILK in cells. To investigate the functional relevance of the interaction between ADAM12 and ILK, we next explored the localization of both proteins in HSCs.
ADAM12 and ILK are recruited to focal adhesions upon β1 integrin stimulation
We previously showed that ADAM12 is stored in the cytoplasm and translocated to the membrane of HSCs upon stimulation by type I collagen (Bourd-Boittin et al., 2008). To investigate the dynamics of the interaction between ADAM12 and ILK, we immunolocalized ADAM12 and ILK in HSCs stimulated or not by type I collagen. As shown in Figure 3A, immunostained ILK and ADAM12 were strongly redistributed to the cell surface when HSCs were plated on type I collagen. Similarly, in stimulated HSCs, ADAM12 colocalized with β1 integrin (Figure 3B) and vinculin (Figure 3C), a cytoskeletal focal adhesion protein involved in linkage of integrin adhesion molecules to the actin cytoskeleton. In agreement with these observations, the colocalization of ILK with β1 integrin was increased upon stimulation by type I collagen (Figure 3D). We further demonstrated the involvement of β1 integrin in the redistribution of ILK and ADAM12 to focal adhesions by using anti–β1 integrin blocking antibodies. As shown in Figure 3E, preincubation of HSCs with blocking antibodies inhibited the translocation of ADAM12 to the focal adhesions. These results suggest that activation of the integrin pathway by type I collagen induces the recruitment of both ADAM12 and ILK to focal adhesions.
FIGURE 3:

ADAM12L and ILK are recruited to focal adhesion structures upon β1 integrin stimulation. HSCs were cultured on plastic dishes (–) or on dishes coated with type I collagen (+) overnight in medium containing 2% FBS. Cell were immunostained with antibodies against anti-ADAM12, anti-ILK, anti-vinculin, and anti-ITGB1 as indicated, followed by tetramethylrhodamine isothiocyanate– or fluorescein isothiocyanate–labeled secondary IgG. Representative fields are shown. Colocalization results in yellow cellular staining. (A) Localization of ADAM12 and ILK. (B) Localization of β1 integrin (ITGB1) and ADAM12. (C) Localization of vinculin and ADAM12. (D) Localization of ILK and β1 integrin. (E) HSCs were preincubated with anti–β1 integrin blocking antibodies or control mouse IgG1 before seeding on type I collagen. Scale bars, 10 μm.
ADAM12 modulates hepatic stellate cell adhesion
Because ILK and ADAM12 are recruited to focal adhesions in stimulated HSCs (Figure 3) and ILK has been previously associated with the fibrogenic phenotype of HSCs through its implication in cell adhesion (Shafiei and Rockey, 2006), we next asked whether expression for ADAM12 could affect cell adhesion. We performed ADAM12 small interfering RNA (siRNA) knockdown in human HSCs. After 48 h of treatment, the steady-state levels of ADAM12 mRNA and proteins were significantly reduced in HSCs, indicating robust RNA interference efficiency (Figure 4, A and B). Note that both the long and the short variants of ADAM12 were equally silenced, whereas expression of type I collagen and TGF-β, the two fibrogenic markers of HSCs used as controls, was not modified. During the course of this analysis, we observed changes in cell spreading, and we sought to examine the effect of ADAM12 on the cytoskeleton of HSCs by performing microscopy analysis of actin stress fibers (Figure 4C). Type I collagen induced prominent stress compared with control cultures on plastic dishes. In addition, we found that inhibition of ADAM12 expression led to a striking disorganization and retraction of stress fibers, suggesting major changes in cell spreading and adhesion processes. In line with this conclusion, ADAM12 silencing in HSCs led to a significant decrease in adherent cells cultured on plastic dishes (26 ± 2.5 and 40 ± 2.7% decrease at 24 and 48 h, respectively; Figure 4D, left). Cultivating HSCs on type I collagen delayed this effect, probably by mobilizing more integrins. However, a significant reduction in adherent cells (35.6 ± 2.8%) was observed at 48 h under these growth conditions. In agreement with this observation, ADAM12-silenced cells plated onto type I collagen, fibronectin, or laminin, which engage different integrins, showed a reduced loss of cell adhesion compared with an albumin plating control (Figure 4D, right).
FIGURE 4:

RNA interference–mediated decreased expression of ADAM12 in hepatic stellate cells induces actin cytoskeleton reorganization and loss of adhesion. HSCs were transfected with 2 nM nontargeted siRNA (Scr) or ADAM12L siRNA (siADAM12). After 48 h, Western blotting (A) and real-time PCR analyses (B) were used to confirm the efficiency and specificity of RNA interference in cell extracts. PCR controls included ADAM12S, type I collagen (Col1), and TGF-β. (C) Transfected cells were analyzed by phase-contrast microscopy (left) and stained with rhodamine-conjugated phalloidin to monitor actin stress fibers (right). Nuclei were counterstained using 4′,6-diamidino-2-phenylindole (blue). (D) Cell adhesion was evaluated by counting adherent cells vs. cells released in the medium at 24 and 48 h posttransfection. Results are expressed as the percentage of adherent cells and represented as the mean ± SD of three independent experiments (*p < 0.05; **, ##p < 0.01). Left, Cell adhesion on plastic and collagen type I compared 24 and 48 h posttransfection. Right, cell adhesion on different substrates compared 24 h posttransfection.
The effect of ADAM12 silencing on cell adhesion was maximal at 48 h. Over longer times, expression of ADAM12 resumed in HSCs transiently transfected with siADAM12 (Supplemental Figure S2A). Note that all cells recovered in the culture supernatants after transfection with ADAM12 siRNAs no longer expressed ADAM12. In addition, cell death was unaffected in HSCs transfected with siADAM12 (Supplemental Figure S2B). This conclusion is further supported by the demonstration that overexpression of ADAM12 in CHO cells treated with staurosporine or tumor necrosis factor–related, apoptosis-inducing ligand did not significantly protect cells against death (Supplemental Figure S3). Because ILK plays an important role in the regulation of cell proliferation and migration (McDonald et al., 2008), we also sought to explore how ADAM12 overexpression could modulate these effects. As shown in Supplemental Figure S4, ADAM12 induced only a slight but significant increase in cell proliferation (Supplemental Figure S4A) and migration (Supplemental Figure S4B).
On the basis of the physical and functional association of endogenous ADAM12 and ILK in HSCs, we next explored which signaling pathway(s) might be involved in this effect.
Expression of ADAM12 reinforces the Akt signaling pathway
Cell adhesion to the extracellular matrix is mediated by the integrin-stimulated, ILK-dependent pathway, which involves protein kinase B (Akt) and GSK-3. To examine whether expression of ADAM12 modulates Akt and GSK-3 activation, we transfected Cos7 cells with wild-type and mutant ADAM12 constructs for 48 h and further plated them on type I collagen–coated dishes. Phosphorylation of Akt and GSK-3 was examined by Western blot analysis at the times indicated in Figure 5. Overexpression of ADAM12L significantly enhanced Akt and GSK-3 phosphorylation compared with cells transfected with an empty vector (Figure 5A). Of interest, this induction was independent of the proteolytic activity of ADAM12L, since the catalytically deficient ADAM12-E351Q mutant induced Akt and GSK-3 phosphorylation to an extent similar to that of wild-type ADAM12L (Figure 5B). Neither the ADAM12-Δcyt truncated form nor the ADAM12S secreted form, neither of which binds ILK (Figure 1D), could induce Akt and GSK-3 phosphorylation (Figure 5C), establishing that the effect of ADAM12L on the Akt signaling pathway is specific.
FIGURE 5:
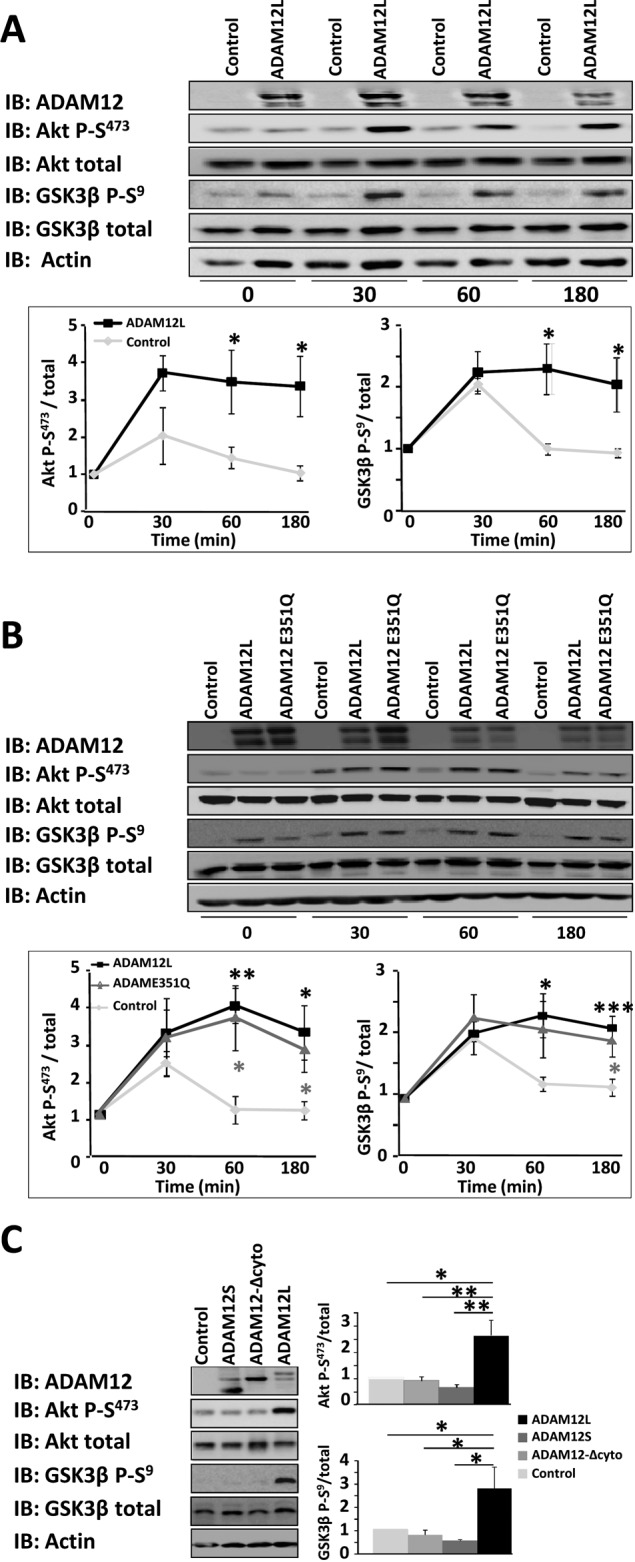
Expression of ADAM12L augments phosphorylation of Akt and GSK-3. Cos7 cells were transfected with empty vector (control) or ADAM12 constructs and cultured for 48 h. The cells were then plated on dishes coated with type I collagen for the indicated times, and the levels of phosphorylated Akt (Akt P-S473), phosphorylated GSK-3β (GSK3β P-S9), total Akt, and GSK-3β were determined by Western blot analysis (IB). Results are expressed as the mean ± SD of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.01). (A) ADAM12L vs. control. (B) ADAM12L vs. the catalytically deficient ADAM12-E351Q mutant. (C) ADAM12L vs. ADAM12S and ADAM12-Δcyto (time, 60 min).
To determine whether this stimulating effect was dependent on ILK and β1 integrin, we assessed the phosphorylation status of Akt in cells either silenced for ILK or preincubated with anti–β1 integrin blocking antibodies. Down-regulation of ILK expression (Figure 6A) or inhibition of β1 integrin (Figure 6B) both diminished Akt phosphorylation induced by overexpression of ADAM12L, implicating β1 integrin and ILK in mediating the effect of ADAM12L. In line with this conclusion, adhesion on type I collagen of HSCs, which express high basal levels of ADAM12L, induced a similar phosphorylation of Akt and GSK3 (Figure 6C).
FIGURE 6:

Induction of Akt phosphorylation by ADAM12L expression requires ILK and stimulation of integrin β1 and PI3K activity. (A) Cos7 cells were cotransfected with 2 nM nontargeted siRNA (Scr) or ILK siRNA (siILK) and empty vector (control) or the ADAM12 construct. Cells were cultured for 48 h, serum starved, and then plated onto dishes coated with type I collagen for 1 h. Levels of phosphorylated Akt (Akt-P-S473) were determined by Western blot analysis (IB); immunoblots for ILK, ADAM12, and actin are shown as controls. (B) Cos7 cells were transfected with ADAM12 constructs, cultured for 48 h, and incubated with anti–β1 integrin blocking antibodies for 30 min before plating on plastic dishes or dishes coated with type I collagen for 1 h. Levels of phosphorylated Akt (Akt-P-S473) were determined by Western blot analysis (IB); immunoblots for ILK, ADAM12, and actin are shown as controls. (C) Adhesion of HSCs on type I collagen augments phosphorylation of Akt and GSK-3. HSC cells were plated onto dishes coated with type I collagen for the indicated times, and the levels of phosphorylated Akt (Akt P-S473), phosphorylated GSK-3β (GSK3β P-S9), total Akt, and GSK-3β were determined by Western blot analysis (IB). Immunoblots for endogenous ADAM12 and actin are shown as controls. (D) Cos7 cells were transfected with empty vector (control) or ADAM12L constructs and cultured for 48 h. The cells were then incubated with the PI3K inhibitor wortmannin (100 nM) or LY294002 (10 μM) and further plated onto type I collagen–coated dishes for 1 h. Levels of phosphorylated Akt (Akt-P-S473) were determined by Western blot analysis (IB); immunoblots for ILK, ADAM12, and actin are shown as controls.
To further explore the molecular mechanisms underlying this novel modulation of the Akt pathway by ADAM12L, we next analyzed the involvement of phosphoinositide 3-kinase (PI3K), an upstream-acting kinase that regulates ILK activity (Delcommenne et al., 1998). For this purpose, we evaluated the effects of two PI3K inhibitors, wortmannin and LY294002, on Akt phosphorylation. Transfected cells were preincubated with the inhibitors before plating on type I collagen for 60 min, and Akt phosphorylation was analyzed by Western blotting. As shown in Figure 6D, wortmannin and LY294002 both blocked the ADAM12L-dependent induction of Akt phosphorylation, implicating PI3K activity in this phenomenon.
Providing additional support for the role of ADAM12L in the promotion of the ILK/Akt-dependent pathway, ILK immunoprecipitates from extracts of cells overexpressing ADAM12L directly phosphorylated the downstream GSK-3 Crosstide peptide substrate (CT-GSK-3), whereas phosphorylation of CT-GSK-3 was very low in the absence of ADAM12 (Figure 7). This effect was correlated with the concentration of ADAM12L in cells (Figure 7A) and was specific to ADAM12L and dependent on ILK, since the phosphorylation of CT-GSK-3 in cells overexpressing ADAM12S was similar to that in control and in ILK-silenced cells (Figure 7B). Taken together, the results of these experiments demonstrate that ADAM12L expression augments the Akt/GSK3 signaling pathway in response to microenvironment stimulation via a β1 integrin/PI3K/ILK–dependent pathway.
FIGURE 7:

ADAM12L expression induces GSK3 phosphorylation in ILK immunoprecipitates. Cos7 cells were transfected with (A) increasing amounts of ADAM12L or (B) empty vector (control), ADAM12L, ADAM12S, nontargeted siRNA (Scr), or ILK siRNA (siILK) in the presence of ADAM12L, as indicated. In vitro kinase assays were carried out by incubating ILK immunoprecipitated from the corresponding cell extracts with the GSK-3 Crosstide peptide substrate (CT-GSK-3). Representative autoradiographs show the amount of 32P incorporation, and results are expressed as the mean ± SD of three independent experiments.
DISCUSSION
Members of the ADAM protein family have emerged as important regulators of cell signaling through shedding activities of membrane-anchored cytokines or growth factors. Besides ligand processing, ADAMs have been proposed to function as signal transmitters. In the present study, we used an in silico approach based on protein integrative annotation to explore the intracellular protein interaction network of the transmembrane variant of ADAM12, ADAM12L. The resulting highly connected graph suggests that integrin signaling plays a critical role in the ADAM12L network. In agreement with this observation, the dynamic association of ADAM12 with Src upon integrin engagement was recently suggested to exert cooperative signals for cell proliferation, migration, and invasion (Stautz et al., 2010). In addition, a functional association of ADAM12 with integrin signaling was implicated in the mesenchymal differentiation of adipocytes (Kawaguchi et al., 2003) and myogenic cells (Lafuste et al., 2005), in cell adhesion and spreading (Thodeti et al., 2003; Zhao et al., 2004), migration (Huang et al., 2005), and invadopodia formation (Albrechtsen et al., 2011). However, the molecular mechanism linking ADAM12 to integrin signaling has remained unclear. We now provide evidence for the interaction of ADAM12 with the integrin-linked kinase ILK and demonstrate the role of ADAM12 in enhancing the functionality of the ILK/Akt signaling pathway upon integrin stimulation. ILK is found in most cells within the IPP complex formed together with PINCH and parvin (Legate et al., 2006). Our coimmunoprecipitation experiments show that the ILK/ADAM12 interaction we report can occur within these complexes and without disrupting them, highlighting the acquisition of a new functionality for IPP as a function of ADAM12 recruitment.
Dynamic translocation of ADAM12 with ILK to focal adhesions
In response to binding of integrins to matrix components, ILK associates with adaptor proteins and is recruited to nascent focal complexes enriched in docking and signaling proteins, including vinculin (Legate and Fassler, 2009). In agreement with this dynamic translocation of ILK, we previously reported that stimulation of integrin pathways by type I collagen induces the recruitment of ADAM12 to the membrane of hepatic stellate cells (Bourd-Boittin et al., 2008). We now demonstrate that, similar to ILK, ADAM12 is redistributed within vinculin-rich structures at the cell surface of HSCs stimulated by type I collagen. This suggests that ADAM12 contributes to the dynamics of protein-complex formation at focal adhesions in HSCs. Transiently increased levels of ADAM12/β1 integrin–containing complexes have been reported at the cell surface of preadipocytes (Kawaguchi et al., 2003). Of interest, this study showed that forced expression of the ADAM12-Δcyto truncated protein, which spontaneously translocates to the membrane, also led to alterations of the actin cytoskeleton, cell adhesion, and survival. These observations suggest that the cytoplasmic tail of ADAM12 is required to sustain these functions. In support of this hypothesis, our data show that ADAM12 silencing induces loss of HSC adhesion and that the cytoplasmic tail of ADAM12 is required for the ILK-dependent survival pathway that correlates with cell adhesion.
Functional association of ADAM12 and ILK for Akt-dependent survival signaling
Over the past several years, the in vivo and in vitro kinase activity of ILK has been widely debated, and ILK has been suggested to function as an adaptor/scaffold protein (Lange et al., 2009; Maydan et al., 2010; Wickstrom et al., 2010; Fukuda et al., 2011; Hannigan et al., 2011). Nevertheless, the classic ILK-dependent pathway involves a PI3K-dependent integrin engagement that leads to the phosphorylation of downstream Akt and GSK-3 targets. In addition, ILK properties can be modulated in vivo by complex protein interactions that result in activities triggered by the contextual environment, such as that found in cancer cells (Troussard et al., 2006). In line with this notion, our data demonstrate that ADAM12, a protein specifically expressed in activated HSCs during chronic liver disease, enhances the activity of the ILK/Akt pathway in response to β1 integrin activation by type I collagen, the main matrix component of liver fibrosis. These results are in agreement with the reported implication of ILK pathway activity in the Akt-dependent regulation of fibroblast survival upon integrin stimulation (Nho et al., 2005).
The contextual association between ADAM12 and the ILK/Akt pathway is further supported by the correlation between ADAM12 and ILK expression. ADAM12 expression is associated with the undifferentiated proliferating state of mesenchymal cells such as chondrocytes in osteoarthritis (Okada et al., 2008), preadipocytes during high-fat diet–induced obesity (Masaki et al., 2005), hepatic stellate cells in liver fibrosis (Le Pabic et al., 2003), myoblasts during myogenic differentiation (Cao et al., 2003), and proliferating astroglial cells in injured brain (Baertling et al., 2010). It is also involved in the initial steps of prekeratinocyte differentiation, where the absence of ADAM12 promotes migration, a differentiated feature of keratinocytes (Harsha et al., 2008; Oh et al., 2009). Similar to ADAM12, increased activation of the ILK/Akt pathway has been associated with cell proliferation (Persad and Dedhar, 2003). In contrast, ILK knockdown in chondrocytes (Grashoff et al., 2003), keratinocytes (Lorenz et al., 2007), fibroblasts (Sakai et al., 2003), and cortical cells (Niewmierzycka et al., 2005) is not associated with a decrease in Akt phosphorylation, suggesting that, in differentiated cells, ILK could signal through an Akt-independent pathway. Taken together, these observations suggest that the specific expression of ADAM12 in proliferating undifferentiated mesenchymal-type cells might favor ILK/Akt pathway activity, thereby promoting the survival signaling pathway that is classically associated with cell adhesion.
Cooperative effects of ILK and ADAM12 in cancer
The ubiquitously expressed ILK is involved in tissue homeostasis. Elevated expression of ILK has been observed in tumors with poor prognosis and has been proposed as a new therapeutic target (Hannigan et al., 2005). Similarly, ADAM12 expression is markedly increased in numerous human cancers (Kveiborg et al., 2008), and we previously reported the association of ADAM12 expression with liver fibrosis and cancer (Le Pabic et al., 2003). In agreement with our results, ILK overexpression has also been shown to be associated with the activation of HSCs in liver fibrosis (Shafiei and Rockey, 2006; Zhang et al., 2006), and induction of ILK activity has been correlated with an increased activation of Akt in hepatocellular carcinomas (Peroukides et al., 2008). In line with these observations, the expression of ADAM12 in mammary-gland tumor cells was recently associated with increased cell proliferation and Akt phosphorylation levels (Fröhlich et al., 2011). Similar to results reported in the present study, this effect was independent of the proteolytic activity of ADAM12, suggesting that the protumorigenic effect of ADAM12 is mediated by the disintegrin- and cysteine-rich domains that interact with integrins (Eto et al., 2000; Kawaguchi et al., 2003; Thodeti et al., 2003; Zhao et al., 2004; Huang et al., 2005; Lafuste et al., 2005). Of interest, we previously showed that these same domains also interact with the TβRII receptor of TGF-β (Atfi et al., 2007), which affects cell survival and proliferation through a PI3K/Akt–dependent pathway (Shin et al., 2001; Wilkes et al., 2005). These data reinforce the notion that ADAM12 might act as a scaffolding protein for integrin- and TGF-β receptor–dependent pathways, leading to the activation of PI3K/ILK/Akt signals and thereby affecting cancer cell survival.
In conclusion, results of this study, which identify ILK as a new ADAM12 partner, unravel a major aspect of the biological significance of the functional association of ADAM12 with the integrin signaling pathway. In addition, induction of the ILK/Akt pathway by ADAM12 suggests a potential role for this protein in cell adhesion and survival. According to this view, ADAM12 would act as a pivotal sensor implicated in the regulation of a microenvironment-dependent equilibrium between differentiation and proliferation.
MATERIALS AND METHODS
Cell culture and transfection
HSCs were isolated from histologically normal specimens of partial hepatectomies from patients undergoing hepatic resection for liver metastases, as previously described (Le Pabic et al., 2003). HSC and Cos7 cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS). For all experiments, cultured HSCs were used between passages 4 and 10. When specified, cells were plated on collagen type I–coated dishes (BD Biosciences, San Diego, CA) or Permanox chamber slides (Lab-Tek, Nalge Nunc International, Rochester, NY). HSCs were transfected using the Amaxa protocols and kits (Amaxa Biosystems, Cologne, Germany) according to the manufacturer's instructions. In some experiments, cells were incubated with PI3K inhibitors, 100 nM wortmannin (Calbiochem, Darmstadt, Germany), or 10 μM LY294002 (Calbiochem) for 30 min or anti-ITBG1 blocking antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) for 30 min at 37°C under agitation without FBS. To knock down ADAM12 and ILK expression, we used synthetic double-stranded ADAM12 siRNAs from Sigma-Aldrich (Saint-Quentin, France). The expression vectors for ADAM12 and mutants thereof, including the complete deletion of the cytoplasmic tail (ADAM12-Δcyt) and the protease-inactive mutant ADAM12-E351Q, were a gift from U. Wewer (Biotech Research and Innovation Center, University of Copenhagen, Copenhagen, Denmark). The expression vector for ILK was a gift from S. Dedhar (Department of Integrative Oncology, BC Cancer Research Center, Vancouver, Canada).
Adhesion assays
Cell adhesion was measured using a trypsinization assay. Briefly, HSCs transfected with 2 nM nontargeted siRNA (Scr) or ADAM12L siRNA (siADAM12) were plated onto collagen type I–, laminin-, and fibronectin-coated dishes or multiwell plates (BD Biosciences) for the indicated times in the absence of serum. Cells were then treated with 0.5 ml of 0.05% trypsin-EDTA for 4 min. Detached cells were collected in medium containing 10% serum and manually counted. Numbers of detached cells expressed as percentage of total were determined from three independent experiments. Controls included uncoated dishes and albumin-coated dishes (10 mg/ml in phosphate-buffered saline [PBS]).
Cell proliferation and migration assays
Cos7 cells were transfected with ADAM12 or control vectors and cultured for 48 h. Cells were then trypsinized, equilibrated in medium for 1 h at room temperature, and plated on collagen type I–coated dishes. For cell proliferation assay, the medium was replaced with DMEM containing 2% FCS in the presence of [3H]thymidine and incubated for the indicated times. Cells were scraped in PBS, sonicated for 10 min, and treated overnight with 30% trichloroacetic acid (TCA). Cell extracts were centrifuged, and the pellets homogenized in formic acid were used for counting TCA-precipitable radioactivity. For migration assays, cells seeded on type I collagen were incubated for 1 h at 37°C before addition of 2.5 μg/ml mitomycin C. After 24 h, the confluent monolayer was scratched using a pipette tip, and images were captured at the indicated times.
Western blotting and immunoprecipitation
Cell lysates were subjected to SDS–PAGE and transferred to nitrocellulose membranes (GE Healthcare, Little Chalfont, United Kingdom). Blots were incubated for 30 min in Tris-buffered saline containing 0.1% Tween 20 and 5% nonfat dry milk and further incubated for 2 h (or overnight at 4°C) with the following antibodies: rabbit anti-ADAM12 (a gift from U. Wewer), rabbit anti-ILK (Cell Signaling Technology, Boston, MA), and monoclonal anti–β-actin-peroxidase (Sigma-Aldrich). Antibodies against phospho-Akt (Ser-473), Phospho-GSK-3β (Ser-9), total Akt, total GSK-3 and α-parvin (D7F9) were from Cell Signaling Technology. PINCH antibodies were from BD Transduction Laboratories (Heidelberg, Germany). Bound antibodies were visualized with horseradish peroxidase–conjugated antibodies using an enhanced chemiluminescence system (Millipore, Billerica, MA). For immunoprecipitation, cell extracts were incubated with cross-linked ILK beads for 2 h at 4°C or preincubated for 1 h with protein G–Sepharose beads (GE Healthcare, Freiberg, Germany) alone to reduce nonspecific protein binding, followed by adsorption overnight to protein G–Sepharose beads prebound with 2 μg of specific or control ADAM12 rabbit immunoglobulin G (IgG). The beads were washed five times, and samples were analyzed by SDS–PAGE and immunoblotting.
Expression of recombinant proteins
pET102D-His-ADAM12 and pGEX-GST-ILK constructs were transformed in BL21-DE3 (Novagen, EMD4Biosciences, Gibbstown, NJ), and expression was induced by 0.25 mM isopropyl 1-thio-α-d-galactopyranoside overnight at 18°C. Cells were harvested by centrifugation at 6000 × g for 15 min and lysed either in ice-cold PBS, pH 7.4 (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4) for GST-ILK or IMAC5 (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 5 mM imidazole) for His-ADAM12, and 0.1 mM 4-(2-aminoethyl-benzensulfonyl fluoride hydrochloride (AEBSF), complete EDTA-free protease inhibitor cocktail (Roche, Indianapolis, IN), and 1 mg/ml lysozyme. After 1 h of incubation on ice and two freeze/thaw cycles, the lysate was sonicated and clarified by centrifugation at 23,000 × g for 1 h at 4°C. His-ADAM12 was bound to Ni-charged Mag Beads (GenScript, Piscataway, NJ) for 1 h at 4°C. After washing with 10 bed volumes of IMAC20 (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 20 mM imidazole), the protein was eluted with IMAC 250 (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 250 mM imidazole) and dialyzed against PBS. GST-ILK was bound to GST MagBeads (GenScript) for 1 h at 4°C. After washing with 10 bed volumes of WB 300 (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 0.02% Tween 20, 0.1 mM AEBSF), the protein was eluted with 25 mM glutathione (Sigma-Aldrich) in PBS. The GST tag was cleaved with thrombin (1 U/10 μg in 25 mM Tris-HCl, pH 7.5, 1 mM dithiothreitol, 50 mM NaCl, 2.5 mM CaCl2) for 2 h at 30°C and immediately removed by chromatography on a GST MagBeads column. All protein fractions were concentrated on Ultracel YM 30 columns (Amicon, Millipore) and stored at −80°C.
Pull-down assays
His-ADAM12 in IMAC5 was bound to Ni-charged Mag-Beads (Qiagen, Hilden, Germany), for 90 min at 4°C, followed by three washes with IMAC5 buffer containing 5 mM imidazole, and incubated with ILK (wt/wt) for a 90 min incubation at 4°C. The nickel-agarose beads were further washed five times with 10 bed volumes of IMAC5 before elution with SDS–PAGE sample buffer. Samples were used for immunoblotting.
Immunostaining and imaging
To detect ADAM12, ILK, and ITGB1 in HSCs, we plated cells on Permanox Lab-Tek chamber slides for 48 h with 2% FBS and fixed them with 4% paraformaldehyde for 15 min. Cells were permeabilized with 0.1% Triton X-100 before incubation with rabbit anti-ADAM12 (rb122), mouse anti-ILK (Santa Cruz Biotechnology), rabbit anti-ILK1 (4G9; Cell Signaling Technology), mouse β1 integrin (Santa Cruz Biotechnology), or mouse anti-vinculin (Sigma-Aldrich) antibodies, followed by Alexa Fluor 488–conjugated anti-mouse and Alexa Fluor 555–conjugated anti-goat antibodies (Invitrogen, Karlsruhe, Germany), respectively. The slides were washed, mounted, and viewed using an automated DMRXA2 microscope (Leica, Wetzlar, Germany).
Relative quantifications of mRNA
Reverse transcription-quantitative PCR was performed using the fluorescent dye SYBR Green methodology with the SybrTM Green I qPCRTM Core Kit (Eurogentec, Seraing, Belgium) and the ABI Prism 7300 thermocycler (PerkinElmer-Cetus, Foster City, CA) as previously described (Le Pabic et al., 2003). The following primer pairs were used for the indicated target genes: ADAM12L, sense, 5′-CACCATTGAAAAACTAAGGTGTGTG-3′, and antisense, 5′-GAGCCTGACAGGGTTGGAAG-3′; ADAM12S, sense, 5′-CTGGGCACCTCCCTTCTGT-3′ and antisense, 5′-TGCTTCTGCTTGCCGGA-3′; type I collagen, sense, 5′-GGTCCTGATGGAAACAATGG-3′, and antisense, 5′-TTCCACCTTGAACACCCTGT-3′; TGF-β, sense, 5′-TGCGCTTGAGATCTTCAAACA-3′, and antisense, 5′-GGGCTAGTCGCACAGACCTC-3′; and 18S rRNA, sense, 5′-CGCCGCTAGAGGTGAAATTC-3′, and antisense, 5′-TTGGCAAATGCTTTCGCTC-3′.
In vitro kinase assays
ILK immunoprecipitates were prepared using ILK antibodies cross-linked to magnetic protein G Dynabeads (Invitrogen). Cell extracts were incubated with cross-linked ILK beads for 2 h at 4°C and successively washed with extraction buffer and kinase assay buffer. Kinase assays were performed by incubating ILK immunoprecipitates with 1 μg/ml GSK-3 Crosstide peptide (Cell Signaling Technology) for 30 min at 30°C in reaction buffer (1 mM dithiothreitol, 10 μM ATP, 1 μCi of 32P-ATP, 0.01% Triton X-100, 25 mM Tris-HCl, pH 7.4, 50 mM NaCl, 10 mM MgCl2). The reaction was stopped by addition of SDS–PAGE sample buffer, and samples were used for immunoblotting and autoradiography analysis.
Supplementary Material
Acknowledgments
This work was supported by the Institut National de la Santé et de la Recherche Médicale, the Ligue Nationale Contre le Cancer, the Association pour la Recherche Contre le Cancer, and the Région Bretagne (PRIR 3193). A.L. and A.F. were recipients of PhD fellowships from the Ligue Nationale Contre le Cancer and the Association pour la Recherche Contre le Cancer, respectively, and K.B. was supported by a postdoctoral fellowship from the Région Bretagne. We thank U. Wewer for providing the ADAM12 constructs and the ADAM12 antibodies and S. Dedhar for providing ILK constructs. We thank Emmanuel Käs (Laboratoire de Biologie Moléculaire Eucaryote, Centre National de la Recherche Scientifique/Université Paul Sabatier, Toulouse, France) for his help in editing and for critical reading of the manuscript.
Abbreviations used:
- ADAM
a disintegrin and metalloprotease
- HSC
hepatic stellate cell
- ILK
integrin-linked kinase
- TGF-β
transforming growth factor-β
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-11-0918) on July 5, 2012.
REFERENCES
- Abram CL, Seals DF, Pass I, Salinsky D, Maurer L, Roth TM, Courtneidge SA. The adaptor protein fish associates with members of the ADAMs family and localizes to podosomes of Src-transformed cells. J Biol Chem. 2003;278:16844–16851. doi: 10.1074/jbc.M300267200. [DOI] [PubMed] [Google Scholar]
- Albrechtsen R, Stautz D, Sanjay A, Kveiborg M, Wewer UM. Extracellular engagement of ADAM12 induces clusters of invadopodia with localized ectodomain shedding activity. Exp Cell Res. 2011;317:195–209. doi: 10.1016/j.yexcr.2010.10.003. [DOI] [PubMed] [Google Scholar]
- Asakura M, et al. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat Med. 2002;8:35–40. doi: 10.1038/nm0102-35. [DOI] [PubMed] [Google Scholar]
- Atfi A, Dumont E, Colland F, Bonnier D, L'Helgoualc'h A, Prunier C, Ferrand N, Clement B, Wewer UM, Theret N. The disintegrin and metalloproteinase ADAM12 contributes to TGF-beta signaling through interaction with the type II receptor. J Cell Biol. 2007;178:201–208. doi: 10.1083/jcb.200612046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baertling F, Kokozidou M, Pufe T, Clarner T, Windoffer R, Wruck CJ, Brandenburg LO, Beyer C, Kipp M. ADAM12 is expressed by astrocytes during experimental demyelination. Brain Res. 2010;1326:1–14. doi: 10.1016/j.brainres.2010.02.049. [DOI] [PubMed] [Google Scholar]
- Bourd-Boittin K, Le Pabic H, Bonnier D, L'Helgoualc'h A, Theret N. RACK1, a new ADAM12 interacting protein. Contribution to liver fibrogenesis. J Biol Chem. 2008;283:26000–26009. doi: 10.1074/jbc.M709829200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Kang Q, Zolkiewska A. Metalloprotease-disintegrin ADAM 12 interacts with alpha-actinin-1. Biochem J. 2001;357:353–361. doi: 10.1042/0264-6021:3570353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Zhao Z, Gruszczynska-Biegala J, Zolkiewska A. Role of metalloprotease disintegrin ADAM12 in determination of quiescent reserve cells during myogenic differentiation in vitro. Mol Cell Biol. 2003;23:6725–6738. doi: 10.1128/MCB.23.19.6725-6738.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyczynska E, Sun D, Yi H, Sehara-Fujisawa A, Blobel CP, Zolkiewska A. Proteolytic processing of delta-like 1 by ADAM proteases. J Biol Chem. 2007;282:436–444. doi: 10.1074/jbc.M605451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008;29:258–289. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto K, Puzon-McLaughlin W, Sheppard D, Sehara-Fujisawa A, Zhang XP, Takada Y. RGD-independent binding of integrin alpha9beta1 to the ADAM-12 and -15 disintegrin domains mediates cell-cell interaction. J Biol Chem. 2000;275:34922–34930. doi: 10.1074/jbc.M001953200. [DOI] [PubMed] [Google Scholar]
- Fröhlich C, Albrechtsen R, Dyrskjot L, Rudkjaer L, Orntoft TF, Wewer UM. Molecular profiling of ADAM12 in human bladder cancer. Clin Cancer Res. 2006;12:7359–7368. doi: 10.1158/1078-0432.CCR-06-1066. [DOI] [PubMed] [Google Scholar]
- Fröhlich C, Nehammer C, Albrechtsen R, Kronqvist P, Kveiborg M, Sehara-Fujisawa A, Mercurio AM, Wewer UM. ADAM12 produced by tumor cells rather than stromal cells accelerates breast tumor progression. Mol Cancer Res. 2011;9:1449–1461. doi: 10.1158/1541-7786.MCR-11-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Knight JD, Piszczek G, Kothary R, Qin J. Biochemical, proteomic, structural, and thermodynamic characterizations of integrin-linked kinase (ILK): cross-validation of the pseudokinase. J Biol Chem. 2011;286:21886–21895. doi: 10.1074/jbc.M111.240093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliano MF, Huet C, Frygelius J, Polgren A, Wewer UM, Engvall E. Binding of ADAM12, a marker of skeletal muscle regeneration, to the muscle-specific actin-binding protein, alpha-actinin-2, is required for myoblast fusion. J Biol Chem. 2000;275:13933–13939. doi: 10.1074/jbc.275.18.13933. [DOI] [PubMed] [Google Scholar]
- Galliher AJ, Schiemann WP. Src phosphorylates Tyr284 in TGF-beta type II receptor and regulates TGF-beta stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res. 2007;67:3752–3758. doi: 10.1158/0008-5472.CAN-06-3851. [DOI] [PubMed] [Google Scholar]
- Grashoff C, Aszodi A, Sakai T, Hunziker EB, Fassler R. Integrin-linked kinase regulates chondrocyte shape and proliferation. EMBO Rep. 2003;4:432–438. doi: 10.1038/sj.embor.embor801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer. 2005;5:51–63. doi: 10.1038/nrc1524. [DOI] [PubMed] [Google Scholar]
- Hannigan GE, McDonald PC, Walsh MP, Dedhar S. Integrin-linked kinase: not so “pseudo” after all. Oncogene. 2011;30:4375–4385. doi: 10.1038/onc.2011.177. [DOI] [PubMed] [Google Scholar]
- Harsha A, Stojadinovic O, Brem H, Sehara-Fujisawa A, Wewer U, Loomis CA, Blobel CP, Tomic-Canic M. ADAM12: a potential target for the treatment of chronic wounds. J Mol Med. 2008;86:961–969. doi: 10.1007/s00109-008-0353-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig P, Blobel CP. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell. 2007;18:176–188. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hougaard S, Loechel F, Xu X, Tajima R, Albrechtsen R, Wewer UM. Trafficking of human ADAM 12-L: retention in the trans-Golgi network. Biochem Biophys Res Commun. 2000;275:261–267. doi: 10.1006/bbrc.2000.3295. [DOI] [PubMed] [Google Scholar]
- Huang J, Bridges LC, White JM. Selective modulation of integrin-mediated cell migration by distinct ADAM family members. Mol Biol Cell. 2005;16:4982–4991. doi: 10.1091/mbc.E05-03-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba K, et al. The cysteine-rich domain of human ADAM 12 supports cell adhesion through syndecans and triggers signaling events that lead to beta1 integrin-dependent cell spreading. J Cell Biol. 2000;149:1143–1156. doi: 10.1083/jcb.149.5.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito N, Nomura S, Iwase A, Ito T, Kikkawa F, Tsujimoto M, Ishiura S, Mizutani S. ADAMs, a disintegrin and metalloproteinases, mediate shedding of oxytocinase. Biochem Biophys Res Commun. 2004;314:1008–1013. doi: 10.1016/j.bbrc.2003.12.183. [DOI] [PubMed] [Google Scholar]
- Kawaguchi N, et al. ADAM12 induces actin cytoskeleton and extracellular matrix reorganization during early adipocyte differentiation by regulating beta1 integrin function. J Cell Sci. 2003;116:3893–3904. doi: 10.1242/jcs.00699. [DOI] [PubMed] [Google Scholar]
- Kveiborg M, Albrechtsen R, Couchman JR, Wewer UM. Cellular roles of ADAM12 in health and disease. Int J Biochem Cell Biol. 2008;40:1685–1702. doi: 10.1016/j.biocel.2008.01.025. [DOI] [PubMed] [Google Scholar]
- Kveiborg M, Fröhlich C, Albrechtsen R, Tischler V, Dietrich N, Holck P, Kronqvist P, Rank F, Mercurio AM, Wewer UM. A role for ADAM12 in breast tumor progression and stromal cell apoptosis. Cancer Res. 2005;65:4754–4761. doi: 10.1158/0008-5472.CAN-05-0262. [DOI] [PubMed] [Google Scholar]
- Lafuste P, Sonnet C, Chazaud B, Dreyfus PA, Gherardi RK, Wewer UM, Authier FJ. ADAM12 and alpha9beta1 integrin are instrumental in human myogenic cell differentiation. Mol Biol Cell. 2005;16:861–870. doi: 10.1091/mbc.E04-03-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange A, Wickstrom SA, Jakobson M, Zent R, Sainio K, Fassler R. Integrin-linked kinase is an adaptor with essential functions during mouse development. Nature. 2009;461:1002–1006. doi: 10.1038/nature08468. [DOI] [PubMed] [Google Scholar]
- Le Pabic H, Bonnier D, Wewer UM, Coutand A, Musso O, Baffet G, Clement B, Theret N. ADAM12 in human liver cancers: TGF-beta-regulated expression in stellate cells is associated with matrix remodeling. Hepatology. 2003;37:1056–1066. doi: 10.1053/jhep.2003.50205. [DOI] [PubMed] [Google Scholar]
- Legate KR, Fassler R. Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J Cell Sci. 2009;122:187–198. doi: 10.1242/jcs.041624. [DOI] [PubMed] [Google Scholar]
- Legate KR, Montanez E, Kudlacek O, Fassler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol. 2006;7:20–31. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- Loechel F, Fox JW, Murphy G, Albrechtsen R, Wewer UM. ADAM 12-S cleaves IGFBP-3 and IGFBP-5 and is inhibited by TIMP-3. Biochem Biophys Res Commun. 2000;278:511–515. doi: 10.1006/bbrc.2000.3835. [DOI] [PubMed] [Google Scholar]
- Lorenz K, Grashoff C, Torka R, Sakai T, Langbein L, Bloch W, Aumailley M, Fassler R. Integrin-linked kinase is required for epidermal and hair follicle morphogenesis. J Cell Biol. 2007;177:501–513. doi: 10.1083/jcb.200608125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki M, Kurisaki T, Shirakawa K, Sehara-Fujisawa A. Role of meltrin {alpha} (ADAM12) in obesity induced by high- fat diet. Endocrinology. 2005;146:1752–1763. doi: 10.1210/en.2004-1082. [DOI] [PubMed] [Google Scholar]
- Maydan M, et al. Integrin-linked kinase is a functional Mn2+-dependent protein kinase that regulates glycogen synthase kinase-3beta (GSK-3beta) phosphorylation. PLoS One. 2010;5:e12356. doi: 10.1371/journal.pone.0012356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald PC, Fielding AB, Dedhar S. Integrin-linked kinase–essential roles in physiology and cancer biology. J Cell Sci. 2008;121:3121–3132. doi: 10.1242/jcs.017996. [DOI] [PubMed] [Google Scholar]
- Mori S, Tanaka M, Nanba D, Nishiwaki E, Ishiguro H, Higashiyama S, Matsuura N. PACSIN3 binds ADAM12/meltrin alpha and up-regulates ectodomain shedding of heparin-binding epidermal growth factor-like growth factor. J Biol Chem. 2003;278:46029–46034. doi: 10.1074/jbc.M306393200. [DOI] [PubMed] [Google Scholar]
- Narita D, Anghel A, Seclaman E, Ilina R, Cireap N, Ursoniu S. Molecular profiling of ADAM12 gene in breast cancers. Rom J Morphol Embryol. 2010;51:669–676. [PubMed] [Google Scholar]
- Nho RS, Xia H, Kahm J, Kleidon J, Diebold D, Henke CA. Role of integrin-linked kinase in regulating phosphorylation of Akt and fibroblast survival in type I collagen matrices through a beta1 integrin viability signaling pathway. J Biol Chem. 2005;280:26630–26639. doi: 10.1074/jbc.M411798200. [DOI] [PubMed] [Google Scholar]
- Niewmierzycka A, Mills J, St-Arnaud R, Dedhar S, Reichardt LF. Integrin-linked kinase deletion from mouse cortex results in cortical lamination defects resembling cobblestone lissencephaly. J Neurosci. 2005;25:7022–7031. doi: 10.1523/JNEUROSCI.1695-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh ST, Cho BK, Schramme A, Gutwein P, Tilgen W, Reichrath J. Hair-cycle dependent differential expression of ADAM 10 and ADAM 12: an immunohistochemical analysis in human hair follicles in situ. Dermatoendocrinology. 2009;1:46–53. doi: 10.4161/derm.1.1.7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada A, Mochizuki S, Yatabe T, Kimura T, Shiomi T, Fujita Y, Matsumoto H, Sehara-Fujisawa A, Iwamoto Y, Okada Y. ADAM-12 (meltrin alpha) is involved in chondrocyte proliferation via cleavage of insulin-like growth factor binding protein 5 in osteoarthritic cartilage. Arthritis Rheum. 2008;58:778–789. doi: 10.1002/art.23262. [DOI] [PubMed] [Google Scholar]
- Peduto L, Reuter VE, Sehara-Fujisawa A, Shaffer DR, Scher HI, Blobel CP. ADAM12 is highly expressed in carcinoma-associated stroma and is required for mouse prostate tumor progression. Oncogene. 2006;25:5462–5466. doi: 10.1038/sj.onc.1209536. [DOI] [PubMed] [Google Scholar]
- Peroukides S, Bravou V, Varakis J, Alexopoulos A, Kalofonos H, Papadaki H. ILK overexpression in human hepatocellular carcinoma and liver cirrhosis correlates with activation of Akt. Oncol Rep. 2008;20:1337–1344. [PubMed] [Google Scholar]
- Persad S, Dedhar S. The role of integrin-linked kinase (ILK) in cancer progression. Cancer Metastasis Rev. 2003;22:375–384. doi: 10.1023/a:1023777013659. [DOI] [PubMed] [Google Scholar]
- Pories SE, et al. Urinary metalloproteinases: noninvasive biomarkers for breast cancer risk assessment. Cancer Epidemiol Biomarkers Prev. 2008;17:1034–1042. doi: 10.1158/1055-9965.EPI-07-0365. [DOI] [PubMed] [Google Scholar]
- Roy R, Wewer UM, Zurakowski D, Pories SE, Moses MA. ADAM 12 cleaves extracellular matrix proteins and correlates with cancer status and stage. J Biol Chem. 2004;279:51323–51330. doi: 10.1074/jbc.M409565200. [DOI] [PubMed] [Google Scholar]
- Sakai T, Li S, Docheva D, Grashoff C, Sakai K, Kostka G, Braun A, Pfeifer A, Yurchenco PD, Fassler R. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 2003;17:926–940. doi: 10.1101/gad.255603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafiei MS, Rockey DC. The role of integrin-linked kinase in liver wound healing. J Biol Chem. 2006;281:24863–24872. doi: 10.1074/jbc.M513544200. [DOI] [PubMed] [Google Scholar]
- Shi Z, Xu W, Loechel F, Wewer UM, Murphy LJ. ADAM 12, a disintegrin metalloprotease, interacts with insulin-like growth factor-binding protein-3. J Biol Chem. 2000;275:18574–18580. doi: 10.1074/jbc.M002172200. [DOI] [PubMed] [Google Scholar]
- Shin I, Bakin AV, Rodeck U, Brunet A, Arteaga CL. Transforming growth factor beta enhances epithelial cell survival via Akt-dependent regulation of FKHRL1. Mol Biol Cell. 2001;12:3328–3339. doi: 10.1091/mbc.12.11.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Stautz D, Sanjay A, Hansen MT, Albrechtsen R, Wewer UM, Kveiborg M. ADAM12 localizes with c-Src to actin-rich structures at the cell periphery and regulates Src kinase activity. Exp Cell Res. 2010;316:55–67. doi: 10.1016/j.yexcr.2009.09.017. [DOI] [PubMed] [Google Scholar]
- Sundberg C, Thodeti CK, Kveiborg M, Larsson C, Parker P, Albrechtsen R, Wewer UM. Regulation of ADAM12 cell-surface expression by protein kinase C epsilon. J Biol Chem. 2004;279:51601–51611. doi: 10.1074/jbc.M403753200. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Kadota N, Hara T, Nakagami Y, Izumi T, Takenawa T, Sabe H, Endo T. Meltrin alpha cytoplasmic domain interacts with SH3 domains of Src and Grb2 and is phosphorylated by v-Src. Oncogene. 2000;19:5842–5850. doi: 10.1038/sj.onc.1203986. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Nanba D, Mori S, Shiba F, Ishiguro H, Yoshino K, Matsuura N, Higashiyama S. ADAM binding protein Eve-1 is required for ectodomain shedding of epidermal growth factor receptor ligands. J Biol Chem. 2004;279:41950–41959. doi: 10.1074/jbc.M400086200. [DOI] [PubMed] [Google Scholar]
- Thodeti CK, Albrechtsen R, Grauslund M, Asmar M, Larsson C, Takada Y, Mercurio AM, Couchman JR, Wewer UM. ADAM12/syndecan-4 signaling promotes beta 1 integrin-dependent cell spreading through protein kinase Calpha and RhoA. J Biol Chem. 2003;278:9576–9584. doi: 10.1074/jbc.M208937200. [DOI] [PubMed] [Google Scholar]
- Troussard AA, et al. Preferential dependence of breast cancer cells versus normal cells on integrin-linked kinase for protein kinase B/Akt activation and cell survival. Cancer Res. 2006;66:393–403. doi: 10.1158/0008-5472.CAN-05-2304. [DOI] [PubMed] [Google Scholar]
- Wickstrom SA, Lange A, Montanez E, Fassler R. The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase! EMBO J. 2010;29:281–291. doi: 10.1038/emboj.2009.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkes MC, Mitchell H, Penheiter SG, Dore JJ, Suzuki K, Edens M, Sharma DK, Pagano RE, Leof EB. Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. 2005;65:10431–10440. doi: 10.1158/0008-5472.CAN-05-1522. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ikegami T, Honda A, Miyazaki T, Bouscarel B, Rojkind M, Hyodo I, Matsuzaki Y. Involvement of integrin-linked kinase in carbon tetrachloride-induced hepatic fibrosis in rats. Hepatology. 2006;44:612–622. doi: 10.1002/hep.21315. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Gruszczynska-Biegala J, Cheuvront T, Yi H, von der Mark H, von der Mark K, Kaufman SJ, Zolkiewska A. Interaction of the disintegrin and cysteine-rich domains of ADAM12 with integrin alpha7beta1. Exp Cell Res. 2004;298:28–37. doi: 10.1016/j.yexcr.2004.04.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.