

The data presented here suggest that in an asynchronous cell culture, heat shock might affect DNA integrity both directly and via arrest of replication fork progression and that the phosphorylation of histone H2AX has a protective effect on the arrested replication forks in addition to its known DNA damage signaling function.
Abstract
Heat shock (HS) is one of the better-studied exogenous stress factors. However, little is known about its effects on DNA integrity and the DNA replication process. In this study, we show that in G1 and G2 cells, HS induces a countable number of double-stranded breaks (DSBs) in the DNA that are marked by γH2AX. In contrast, in S-phase cells, HS does not induce DSBs but instead causes an arrest or deceleration of the progression of the replication forks in a temperature-dependent manner. This response also provoked phosphorylation of H2AX, which appeared at the sites of replication. Moreover, the phosphorylation of H2AX at or close to the replication fork rescued the fork from total collapse. Collectively our data suggest that in an asynchronous cell culture, HS might affect DNA integrity both directly and via arrest of replication fork progression and that the phosphorylation of H2AX has a protective effect on the arrested replication forks in addition to its known DNA damage signaling function.
INTRODUCTION
Heat shock (HS), or hyperthermia, is one of the better-known exogenous cellular stresses. This phenomenon represents the subjection of a whole organism (or particular cells) to an abnormally high environmental temperature. An increase in temperature can cause protein unfolding and aggregation, which can lead to a variety of cellular pathologies, such as defects of the cytoskeleton (Toivola et al., 2010), fragmentation of the endoplasmic reticulum and Golgi apparatus, a decreasing number of mitochondria and lysosomes (Welch and Suhan, 1985), and poisoning of RNA splicing (Vogel et al., 1995; Boulon et al., 2010), among others (for a review, see Richter et al., 2010). Although the optimal temperature range and corresponding extreme levels of environmental temperature differ between organism classes, the defensive mechanism, known as the HS response, is highly conserved among species. Since its discovery in 1962 (Ritossa, 1962, 1964), the HS response has been studied extensively to understand the molecular mechanisms of this process. On the molecular level, the most studied trait of the HS response is the induction of the expression and subsequent functioning of a set of highly conserved factors, known as HS proteins (HSPs; Welch et al., 1991; Richter et al., 2010). The earliest HSPs to be discovered, such as HSP70 and HSP90 (Welch and Feramisco, 1982), function as molecular chaperones that facilitate protein folding and assembly (Gething and Sambrook, 1992; Hartl, 1996). Recent genome-wide studies of gene expression showed that the transcription of 50–200 genes was upregulated under HS conditions in a variety of model organisms (Gasch et al., 2000; Tabuchi et al., 2008). These stress-induced genes encode not only molecular chaperones but also factors that participate in protein degradation and transport and RNA repair, among other processes (Richter et al., 2010). Hyperthermia-induced up-regulation of HSP transcription is mediated by HS transcription factors (HSFs), which are constitutively expressed in higher eukaryotes and become activated under HS conditions (Sarge et al., 1993; Anckar and Sistonen, 2011). Although the influence of HS on higher eukaryotes has been extensively studied, little is known about its effects on DNA and DNA-associated processes, such as DNA repair and replication. Several works published in the early 1980s focused on the impact of HS on DNA synthesis. It was shown, mainly using radioactive labeling and sucrose gradient sedimentation, that the exposure of Chinese hamster ovary and HeLa cells to heat inhibited DNA replication (Wong and Dewey, 1982; Warters and Stone, 1983, 1984). This inhibitory effect was suggested to be a source of chromosomal aberrations in S-phase cells exposed to HS (Dewey et al., 1978). Since then, only a few papers concretizing the mechanisms of HS-induced inhibition of DNA replication have appeared. It has been suggested that the nucleolus functions as a heat sensor that uses nucleolin as a signaling molecule to initiate inhibitory responses equivalent to a checkpoint (Wang et al., 2001). This effect appears to be mediated by the association of nucleolin with replication factor A (RPA; Wang et al., 2001; Iliakis et al., 2004a). Nevertheless, the exact molecular mechanism of the inhibition of DNA synthesis evoked by hyperthermia in higher eukaryotes is still unclear. We know that this phenomenon does exist, but we do not know whether the inhibition is an arrest or merely a slowing of the replication process, whether it has any epigenetic marks, or whether there are any defensive mechanisms protecting genomic integrity under the conditions of DNA synthesis suppression by HS.
It had been generally accepted for years that HS does not induce DNA double-stranded breaks (DSBs), only single-stranded breaks originate as a result of DNA replication inhibition (Corry et al., 1977; Jorritsma and Konings, 1984; Warters et al., 1985). However, during the past 10 yr, it has been shown by several groups that HS might stimulate the phosphorylation of histone H2AX (Kaneko et al., 2005; Hunt et al., 2007; Takahashi et al., 2008; Laszlo and Fleischer, 2009a, b), which is a variant of the core histone H2A. The phosphorylation of Ser-139 in the C-terminus of H2AX by ATM, ATR, or DNA-PK is believed to be one of the first events in DNA damage response (Rogakou et al., 1998; Paull et al., 2000). Since its discovery, this modification, known as γH2AX, was regarded as a universal marker of DNA DSBs (Rogakou et al., 1998). Phosphorylation induced at a site of a DSB is amplified and can spread along the DNA fiber for kilobases or even megabases from the initial DSB (Rogakou et al., 1999). It was determined that a single γH2AX focus visualized immunocytochemically corresponds to one DSB (Sedelnikova et al., 2002). As for HS-induced γH2AX foci formation, the published data and their interpretation are contradictory; whereas two groups of investigators argue that γH2AX does mark DSBs induced by HS (Kaneko et al., 2005; Takahashi et al., 2008), the third group favors the view that H2AX phosphorylation in heat-stressed cells does not occur due to DNA damage but is a by-product of other cellular processes perturbed by HS (Hunt et al., 2007; Laszlo and Fleischer, 2009a, b). This point of view is indirectly supported by recent evidence suggesting DSB-independent phosphorylation of H2AX (Hammond et al., 2003; Hunt et al., 2007; Shimura et al., 2007; Laszlo and Fleischer, 2009a, b).
In this study, we show that HS affects DNA structure and DNA-associated processes in a cell cycle phase–dependent manner. In G1 and G2 cells, HS induces a countable number of DNA DSBs, which are marked by γH2AX. In contrast, in S-phase cells, HS does not induce DSBs but causes an arrest or deceleration of the progression of the replication forks in a temperature-dependent manner. Interestingly, this response also provoked the phosphorylation of H2AX, which appeared at sites of replication (replication foci). Moreover, the results obtained suggested that the phosphorylation of H2AX at or close to a replication fork rescued the fork from total collapse. Collectively our data suggest HS might have a dual effect on the DNA integrity in an asynchronous cell culture and phosphorylation of H2AX has a directly protective effect on the arrested replication forks in addition to its known DSB signaling function.
RESULTS
HS induces H2AX phosphorylation in human cells
To address the question of whether phosphorylation of H2AX induced by HS has a functional role in higher eukaryotes, we first verified that this phenomenon exists and may be observed in different cells. We used acute (30-min) HS with temperatures ranging between 42 and 45.5ºC. To determine whether the conditions used represented a functional HS, we measured the mRNA levels of several HSPs (hsp70, hsp40, and hsp105/110) in control cells and in cells stressed by cultivation at the above-mentioned temperatures for 30 min. Quantitative reverse transcriptase PCR analysis showed that the expression of the HSPs increased in response to the treatments used (Supplemental Figure S1). Another important question that had to be asked was whether the conditions of HS being used were physiologically relevant and did not affect cell viability. To check this, we analyzed the viability of the cells at different stages (immediately after HS and at different recovery time points) using two approaches: MTT assay (Figure S2) and the trypan blue exclusion method (data not shown). It is evident that that the HS conditions used in our study did not lead to cell death. Moreover, when placed in normal conditions, the HS-treated cells continued to divide and displayed the same proliferation rate as the control cells. After these preparatory experiments, control and heat-shocked mcf-7 cells were immunostained using an antibody against γH2AX (Figure 1A). This experiment clearly demonstrated that the control cells did not contain any γH2AX foci, whereas the heat-stressed ones did. Practically the same results were obtained using another cancer cell line, Jurkat, and primary cultures of human embryonic fibroblasts (data not shown). To verify that the γH2AX foci observed in heat-stressed cells were not an artifact, we performed immunostaining using another antibody against γH2AX and, as expected, obtained similar results (data not shown). Finally, we checked the effect of HS on H2AX phosphorylation using a biochemical approach. Figure 1B shows the appearance of the phosphorylated form of H2AX in the nuclear extracts of heat-stressed cells analyzed by Western blot hybridization. Thus our data, along with the results of Takahashi et al. (2008), strongly suggest that the phosphorylation of H2AX in response to HS is a widespread phenomenon in mammals.
FIGURE 1:
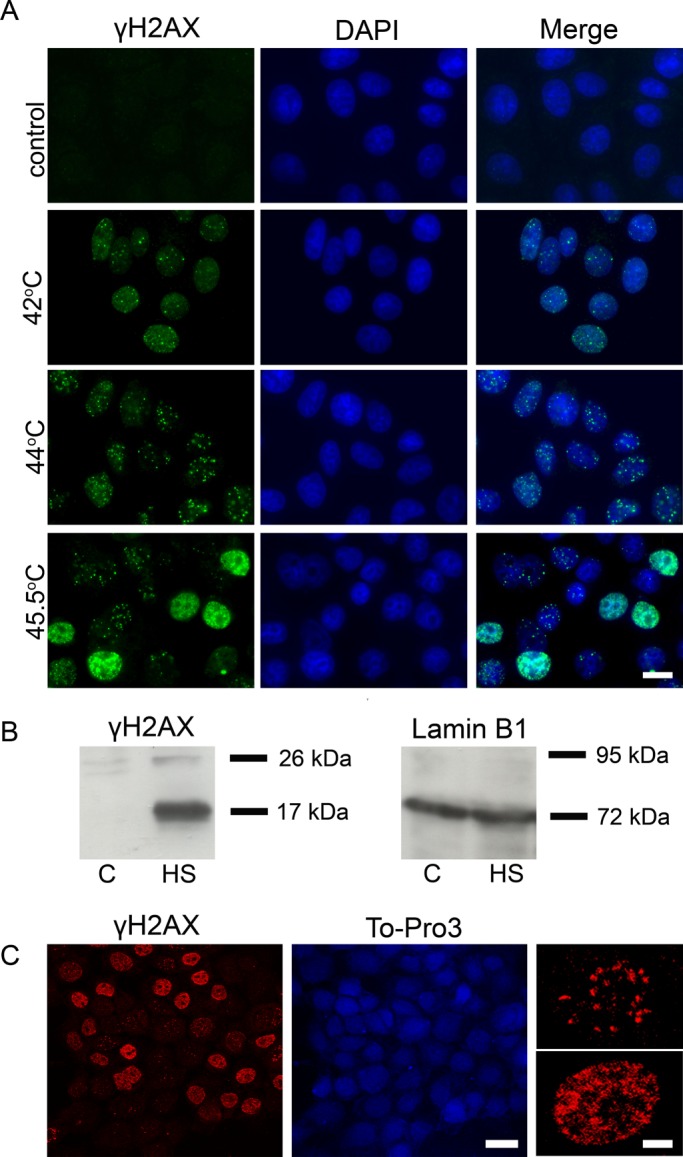
Hyperthermia induces the phosphorylation of histone variant H2AX at Ser-139 in human cells. (A) Immunofluorescence analysis of γH2AX in control (untreated) human mcf-7 cells and cells that were heat-stressed at different temperatures (42, 44, and 45.5°C for 30 min). The DNA was stained with DAPI. Scale bar: 20 μm. (B) Western blot analysis of γH2AX in the nuclei of control (untreated, C) and heat-shocked (45.5°C, 30 min; HS) mcf-7 cells. Lamin B1 was used as a loading control. (C) Immunofluorescence analysis of heat-treated (45.5°C, 30 min) mcf-7 cells performed using a Leica SP2 confocal laser scanning microscope (scale bar: 20 μm). Enlarged cells with two different patterns of γH2AX distribution are shown on the right (scale bar: 5 μm). The DNA was stained with To-Pro 3 iodide fluorescent dye.
It kindled our interest that the immunostained cells could be divided into two distinct groups according to the size/shape and number of γH2AX foci. One group contained a countable number of large-size foci visually similar to well-known irradiation-induced foci (IRIF; Lou et al., 2003; Stewart et al., 2003), and the second contained numerous small-sized γH2AX foci (Figure 1, A and C).
Patterns of HS-induced γH2AX foci are cell cycle phase–dependent
To obtain more information about the distribution of γH2AX foci in HS-treated cells, we determined the percentage of cells containing different numbers of γH2AX foci. The results of this analysis (Figure 2A) clearly demonstrate that two distinct cell populations exist: one containing 5–30 γH2AX foci and another containing 100 or more foci. It is notable that the percentage of cells containing numerous γH2AX foci was similar to the percentage of S-phase cells (45–50%), as determined by flow cytometry (Figure 2B). It was tempting to suppose that the two populations of cells with different patterns of HS-induced γH2AX foci represent cells at different phases of the cycle. To check this supposition, we performed several complex immunostaining experiments. First, we labeled S-phase cells by a short (20-min) pulse of bromodeoxyuridine (BrdU), a nucleotide analogue that is incorporated into DNA during replication. This labeling was followed by HS (45.5ºC, 30 min). Finally, the cells were double-immunostained with antibodies against γH2AX and BrdU. The results shown in Figure 2C demonstrate that the second pattern of γH2AX distribution (small-sized, numerous foci) was typical exclusively of BrdU-positive (i.e., S-phase) cells. Furthermore, the γH2AX foci in these cells virtually colocalized with the sites of BrdU incorporation. In contrast, the first pattern, which represents a countable number of IRIF-like γH2AX foci, was not observed in BrdU-positive cells and thus might be typical for cells in G1 or G2 or both. To distinguish between the above-mentioned possibilities, we stained heat-stressed cells with antibodies against γH2AX, cyclin B1 (a marker of G2 phase), and 5-ethynyl-2′-deoxyuridine (EdU), another nucleotide analogue that had been incorporated into S-phase cells before HS. The results presented in Figure 2D demonstrate that the IRIF-like γH2AX foci were present in both G1 (EdU−, cyclin B1−) and G2 (EdU−, cyclin B1+) cells.
FIGURE 2:

Patterns of HS-induced γH2AX foci are dependent on the cell cycle phase. (A) The percentage of HS-treated (45.5°C, 30 min) mcf-7 cells possessing different numbers of γH2AX foci. More than 200 individual cells immunostained with anti-γH2AX antibody were analyzed. (B) Flow cytofluorometric analysis of the cell cycle distributions of the control (untreated) and heat-stressed cells. Aliquots of asynchronously proliferating control mcf-7 cells and cells that were heat-shocked for 30 min at 45.5°C were stained with propidium iodide and analyzed by flow cytometry. The relative DNA content of 500,000 cells was plotted against the cell number in each histogram. Calculated percentages of S-phase cells in each case are indicated below the corresponding histograms. (C) Human mcf-7 cells were pulse-labeled with BrdU (100 μM, 20 min), heat-stressed (45.5°C, 30 min), and double-immunostained against γH2AX and BrdU. The DNA was stained with DAPI. A superimposition of the green and red channels is shown in the last section (Merge). Scale bar: 20 μm. (D) Human mcf-7 cells were pulse-labeled with EdU (10 μM, 30 min), heat-stressed (45.5°C, 30 min), and immunostained against γH2AX and cyclin B1. EdU was revealed by Click Chemistry. A superimposition of the green, red, and blue channels is shown in the last section (Merge). Scale bar: 20 μm.
HS induces the formation of DNA DSBs in G1 and G2 cells
As was mentioned in the Introduction, γH2AX is regarded as a universal marker of DSBs (Rogakou et al., 1998; Paull et al., 2000). It was therefore reasonable to check whether HS-induced H2AX phosphorylation marks DSBs induced by HS. For this purpose, we used the single-cell gel electrophoresis (SCGE) technique, also known as the “comet assay.” The modification of the method used—neutral SCGE—permitted us to analyze only DNA DSBs (for details, see Materials and Methods). The most meaningful parameter of comet—tail moment, which represents the tail length multiplied by the fraction of DNA in the tail—was chosen as a criterion for the degree of DNA breakage. As a positive control for the existence of DSBs, we used mcf-7 cells treated with the DNA topoisomerase II poison etoposide (also known as VP16; Baldwin and Osheroff, 2005; Montecucco and Biamonti, 2007). The mcf-7 cells, heat-stressed at 45.5ºC for 30 min, were studied in a parallel experiment (Figure 3A). In spite of the massive H2AX phosphorylation, we observed only a minor increase of the tail moment in heat-stressed cells, and even this increase was detected not in the entire population but only in a small group of cells. Keeping in mind that HS induces two types of γH2AX foci depending on the cell cycle phase, we decided to analyze the induction of DSBs under HS conditions separately in S-phase and non–S-phase cells. With this aim, we used a modification of the comet assay that allowed us to discriminate S-phase cells based on the incorporation of BrdU (Figure 3B; McGlynn et al., 1999). The results of the neutral BrdU-comet assay demonstrated that hyperthermia induced DSBs only in non–S-phase cells (Figure 3C). Surprisingly, the tail moment of S-phase cells, which contained many more HS-induced γH2AX foci, remained unchanged (Figure 3C). It is also interesting that the S-phase cells contained a significant number of single-stranded DNA breaks, as shown by an alkaline BrdU-comet assay (Figure 3D). To verify the observed cell cycle phase–specific DSB induction under HS conditions, we used in situ DNA end-labeling assays utilizing two different enzymes, bacterial DNA polymerase I (PolI) and terminal deoxynucleotidyl transferase (TdT). The difference between the two is that PolI requires a DNA template and can incorporate nucleotides only at the site of a single-stranded nick or gap, whereas TdT can work without a template and simply adds nucleotides to the end of the DNA. These experiments demonstrated that both enzymes incorporated nucleotides (fluor-dUTP) in cells with the S-phase γH2AX foci distribution pattern (Figure 4, A and B), but, in contrast to PolI, TdT incorporated fluor-dUTP in the nuclei of non–S-phase cells as well (Figure 4B). Most importantly, TdT incorporated nucleotides exactly at the sites of H2AX phosphorylation (Figure 4B, lower line). Together, the results of SCGE and in situ assays strongly suggest that HS induces double-stranded DNA break formation during the G1 and G2 phases of the cell cycle. Consequently, we decided to term the HS-induced γH2AX foci that appeared in G1 and G2 cells and did mark DSBs as DF (damage-associated foci).
FIGURE 3:
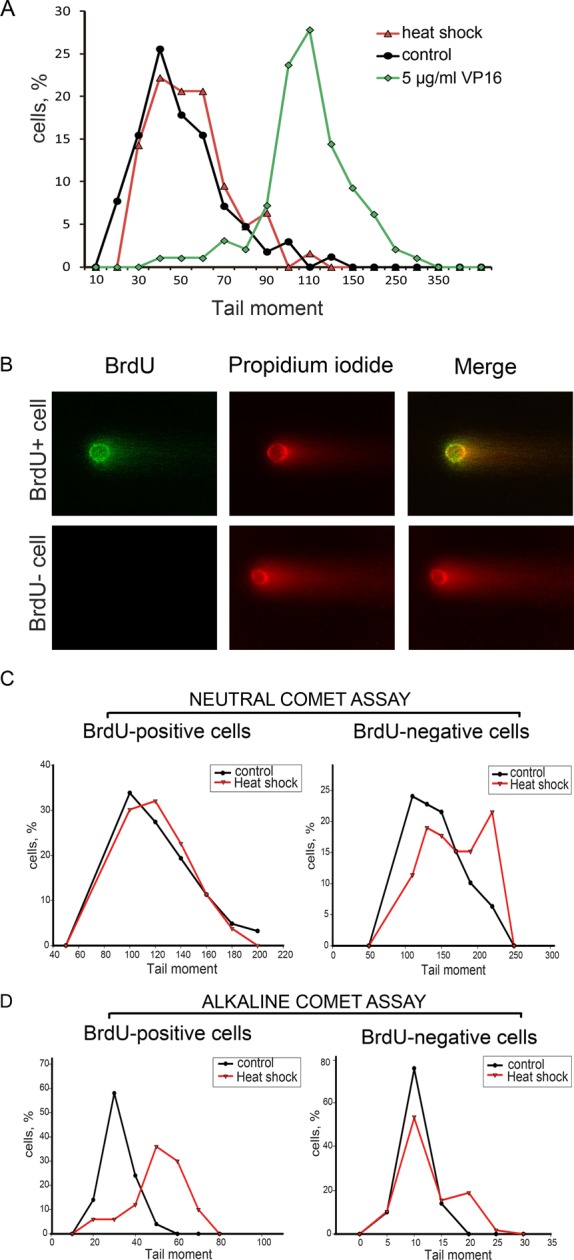
Analysis of HS-induced DNA damage by SCGE (comet assay). (A) Asynchronous mcf-7 cells (control) were treated with HS (45.5°C, 30 min) or etoposide (VP16; 5 μg/ml, 30 min) and then subjected to neutral SCGE analysis. The percentage of cells with different tail moments is shown (the results of one representative experiment out of three reproducibly repeated experiments). (B) A representative image of a BrdU-positive comet prepared by neutral BrdU-SCGE from heat-stressed mcf-7 cells. (C and D) Asynchronous mcf-7 cells (control) were pulse-labeled with BrdU (100 μM, 60 min), treated with HS (45.5°C, 30 min), and subjected to either neutral (C) or alkaline (D) BrdU-SCGE analysis (for details, see Materials and Methods). BrdU-positive and BrdU-negative cells were analyzed separately. The designations are the same as in (A).
FIGURE 4:

Analysis of HS-induced DNA damage by TdT and PolI nucleotide analogue incorporation assays. Control (untreated) and heat-stressed (45.5°C, 30 min) mcf-7 cells were fixed, permeabilized, and subjected to a fluorescein-labeled nucleotide analogue (green channel) incorporation assay using either E. coli DNA polymerase I (A) or terminal deoxynucleotidyl transferase (B). As a positive control, mcf-7 cells fixed and then treated with DNase I (0.1 U/ml, 30 min) were used. After incorporation of a nucleotide analogue, the cells were immunostained with an antibody against γH2AX (red). The DNA was stained with DAPI (blue). A superimposition of the green and red channels is shown as “Merge.” Scale bars: 20 and 5 μm (for last row).
HS has a gradual effect on DNA replication
The presence of a significant number of single-stranded DNA breaks in S-phase heat-stressed cells, as indicated by the comet assay and the in situ end labeling (Figures 3C and 4), might be due to a disruption of the DNA replication process. We found that the γH2AX foci colocalized with the replication foci in S-phase cells (see above, Figure 2). This overlap is especially evident when the analyzed cell contains a relatively small number of replication foci (Figure 5A). We use the abbreviation RF (replication-associated foci) to designate γH2AX foci that appear at sites of replication fork progression in response to HS. It is known that H2AX phosphorylation might be induced by replication arrest caused, for example, by aphidicolin or hydroxyurea (Figure 5B; Hammond et al., 2003; Kurose et al., 2006). It was thus reasonable to suggest that HS causes the arrest of replication forks, which, in turn, triggers the phosphorylation of H2AX. To verify this possibility, we checked whether mcf-7 cells that were heat-stressed at different temperatures (42, 44, and 45.5ºC) retain the ability to incorporate BrdU into replication foci. As demonstrated by the immunostaining of incorporated BrdU, the 45.5ºC HS arrested DNA replication, while the 42 and 44ºC HS did not (data not shown). To investigate these effects more precisely, we used a DNA fiber analysis (molecular combing; Jackson and Pombo, 1998; Chastain et al., 2006). The mcf-7 cells heat shocked for 30 min at 42, 44, or 45.5ºC were pulsed with BrdU (15 min) either directly after HS or after recovery for 0.5, 3, 6, and 24 h at 37ºC and were subjected to DNA fiber analysis. The tracks of incorporated BrdU were revealed by immunostaining (Figure 5C). For each of the above-mentioned time points, the length of at least 150 tracks was measured, and the average replication velocity was calculated (Figure 5C). The results obtained show that HS affects DNA replication gradually: only heating to 45.5ºC caused the complete arrest of replication fork progression, whereas heating to 42 or 44ºC merely decelerated replication in a temperature-dependent manner (Figure 5C). To check what actually happened under mild HS conditions— stochastic arrest of a portion of replication forks or a slowing down of all replication fork progression—we performed double-labeling experiments using BrdU and EdU incorporation. The results obtained clearly demonstrated that 1) under mild HS conditions (42 and 44ºC) virtually all replication forks were slowing down (not stopped); and 2) during the recovery after 45ºC, HS replication was mostly restarted and not initiated at new origins (Figure S3). At all temperature points, the recovery of replication started as early as 30 min and reached the control level within 6 h (Figures 5C and S3). Interestingly, after 6 h of recovery, the apparent speed of replication fork progression actually exceeded the speed in control cells (Figure 5C), which may represent a compensatory mechanism aimed to finish the DNA replication in time.
FIGURE 5:
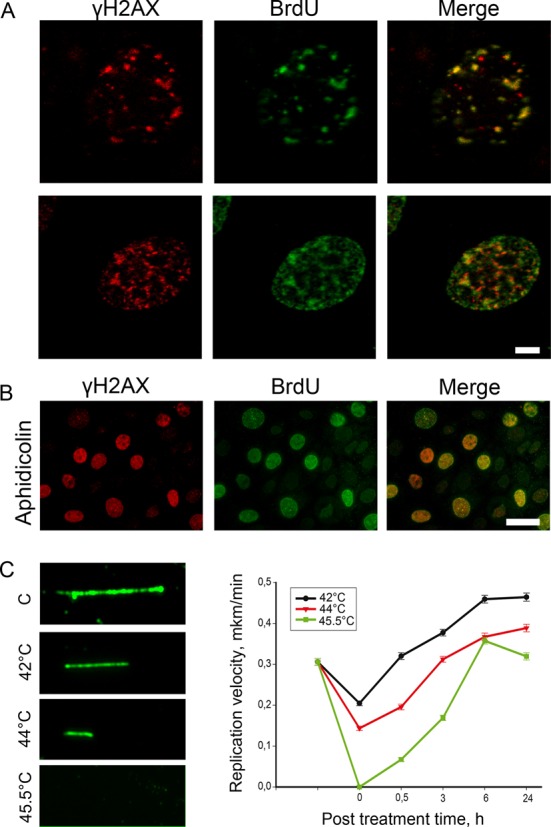
HS has a gradual effect on DNA replication. (A) Human mcf-7 cells were pulse-labeled with BrdU (100 μM, 20 min), heat stressed (45.5°C, 30 min), and double-immunostained against γH2AX (red) and BrdU (green). Representative images are shown. Scale bar: 5 μm. (B) Human mcf-7 cells were pulse-labeled with BrdU (100 μM, 20 min), treated with aphidicolin (6 mM, 60 min), and double-immunostained against γH2AX (red) and BrdU (green). Scale bar: 20 μm. (C) A DNA fiber analysis (molecular combing) of the DNA replication speed under HS conditions. Human mcf-7 cells were heat-stressed for 30 min at different temperatures (42, 44, and 45.5°C) and allowed to recover at 37°C for 0, 0.5, 3, 6, and 24 h. After these time intervals, the cells were pulse-labeled with BrdU (100 μM, 20 min) and subjected to DNA fiber analysis. The tracks of incorporated BrdU were revealed by immunostaining. For each time point, the lengths of at least 150 tracks were measured, and the average replication velocity was calculated. The error bars represent the SEM. Representative images of the BrdU tracks are shown to the left of the graphs.
H2AX molecules forming DF and RF are phosphorylated by different phosphatidylinositol 3-kinase–related kinases (PIKKs)
It is well established that H2AX is phosphorylated by phosphatidylinositol 3-kinase-related kinases: ATM, ATR, or DNA-PK (Burma et al., 2001; Durocher and Jackson, 2001; Stiff et al., 2004). There are no cell cycle–dependent differences in the expression levels of these kinases; all three are expressed during G1, S, and G2 phases (Lee et al., 1997; Gately et al., 1998). However, it is possible that the H2AX molecules forming DF and RF are phosphorylated by different PIKKs. To address this question, we performed an inhibitory analysis using caffeine, a specific inhibitor of ATM/ATR (Sarkaria et al., 1999), and NU7026, a specific inhibitor of DNA-PK (Willmore et al., 2004). The mcf-7 cells were treated with these inhibitors for 3–6 h, pulsed with BrdU, heated to 45.5ºC for 30 min, and then subjected to double immunostaining with antibodies against γH2AX and BrdU. The results obtained showed that only RF were formed in the caffeine-treated cells, and only DF were formed in the NU7026-treated cells (Figure 6A). Thus the phosphorylation of H2AX associated with DSBs in heat-stressed cells is mediated by ATM or ATR, while the replication arrest/delay–associated phosphorylation is mediated by a DNA-dependent protein kinase. To verify these data, we down-regulated expression levels of ATM, ATR, or catalytic subunit of DNA-PK using small interfering RNA (siRNA) technology. The mcf-7 cells knocked down for ATM, ATR, or DNA-PKcs were pulsed with EdU, heated to 45.5ºC for 30 min, and then immunostained for γH2AX and EdU (Figure S4). The results obtained fully confirmed the conclusions made on the basis of inhibitory analysis. Moreover, we managed to distinguish the roles of ATM and ATR in forming damage-associated γH2AX foci—ATM, but not ATR, mediated DNA damage–induced phosphorylation of H2AX in heat-stressed cells (Figure S4).
FIGURE 6:

Characterization of HS-induced γH2AX foci in human mcf-7 cells. (A) HS-induced H2AX phosphorylation is mediated by different PIKKs. Human mcf-7 cells were treated with either an ATM/ATR inhibitor (caffeine; 10 mM, 3 h) or a DNA-PKcs inhibitor (NU7026; 10 μM, 6 h), pulse-labeled with BrdU (100 μM, 20 min), heat stressed (45.5°C, 30 min), and double-immunostained against γH2AX (red) and BrdU (green). The DNA was stained with DAPI (blue). A superimposition of the green and red channels is shown as the “Merge.” Scale bar: 20 μm. (B) The kinetics of γH2AX focus recovery after HS in mcf-7 cells. Human mcf-7 cells that were either untreated, treated with HS (45.5°C, 30 min), or treated with HS and allowed to recover for the indicated time intervals (1 and 6 h) were pulse-labeled with EdU (10 μM, 30 min) and then immunostained against γH2AX (red). EdU (green) was revealed by Click Chemistry. The DNA was stained with DAPI (blue). A superimposition of the green and red channels is shown as the “Merge.” Scale bars: 20 μm.
Another difference between DF and RF is their recovery time, which is the time required for the disappearance of the γH2AX foci (γH2AX dephosphorylation and/or displacement). We demonstrated that DF practically disappeared during the first 6 h of recovery at 37ºC, which was similar to typical DSB repair time (Iliakis et al., 2004b; Kantidze et al., 2006), whereas RF remained visible for 24 or more hours. The restart of replication after 30 min of recovery of the cells at 37ºC (see above, Figure 5) resulted in the physical separation of the replication foci and the γH2AX foci (Figures 6B and S5).
H2AX phosphorylation rescues DNA replication fork from total collapse
The most important question concerning the phosphorylation of H2AX in S-phase cells is whether this phosphorylation has a functional role. To approach this problem, we analyzed the effect of HS on DNA replication in the heat-stressed mcf-7cells that had been pretreated with the DNA-PK inhibitor NU7026 to prevent the replication-coupled phosphorylation of H2AX. HS was performed for 30 min at 42, 44, or 45.5ºC. The cells were pulsed with BrdU (20 min) and then subjected to DNA fiber analysis. Surprisingly, the suppression of H2AX phosphorylation led to an approximately twofold decrease in replication speed, as determined by the decrease in length of the BrdU tracks, in the cells heated to 42ºC and to replication arrest in the cells heated to 44ºC (Figure 7A). The recently published results of Shimura et al. (2007) suggested that DNA-PK had a crucial role in preventing DSB formation in response to aphidicolin treatment, which inhibits the DNA replication process. It is plausible that DSBs formed at the sites of replication fork movement may cause the above-described replication-associated effects of HS. To test this possibility, we performed a BrdU-neutral comet analysis on mcf-7 cells pretreated with NU7026 and then subjected to HS. Figure 7B demonstrates the significant tail-moment increase in the cells subjected to this treatment. These results allowed us to conclude that H2AX phosphorylation at replication sites prevented the formation of DSBs and, therefore, rescued the DNA replication forks from total collapse.
FIGURE 7:
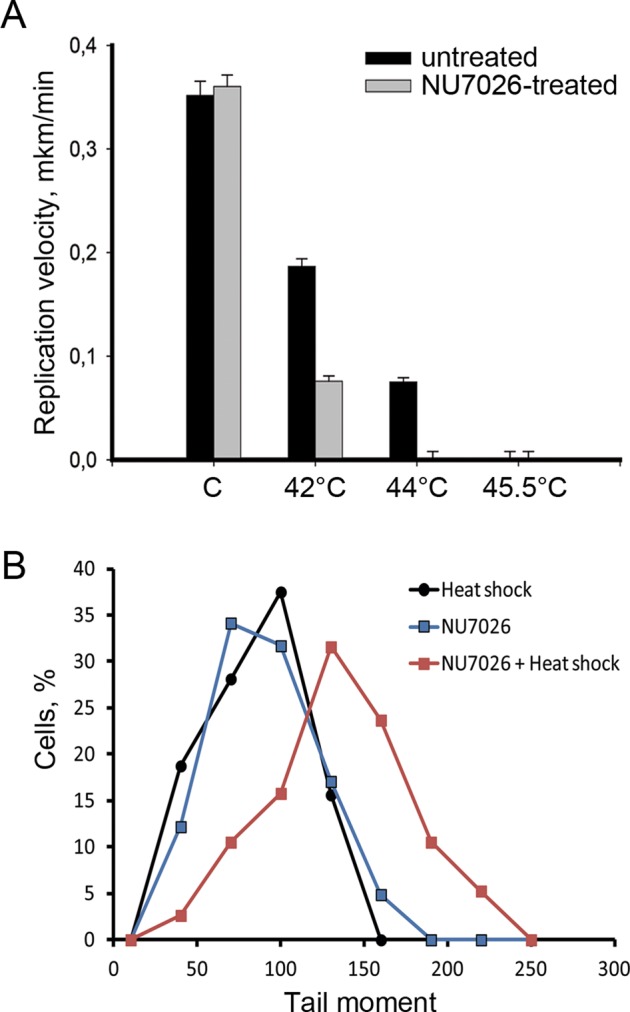
H2AX phosphorylation preserves the DNA replication fork from total collapse. (A) DNA fiber analysis (molecular combing) of replication speed under HS conditions in cells treated with NU7026. Human mcf-7 cells were treated with a DNA-PKcs inhibitor (NU7026; 10 μM, 6 h), heat stressed for 30 min at different temperatures (42, 44, and 45.5°C), pulse-labeled with BrdU (100 μM, 20 min), and subjected to DNA fiber analysis. The tracks of incorporated BrdU were revealed by immunostaining. For each treatment point, the lengths of 80–100 tracks were measured, and the average replication velocity was calculated. The error bars represent the SEM. (B) Neutral BrdU-SCGE analysis of the heat-stressed cells pretreated with the DNA-PKcs inhibitor. Human mcf-7 cells were treated either with HS (45.5°C, 30 min), NU7026 (10 μM, 6 h), or both (NU7026 and HS), and were then subjected to neutral BrdU-SCGE (BrdU incorporation was performed just before heating, as described in the legend to Figure 3). Only BrdU-positive cells were analyzed. The percentage of cells demonstrating different tail moments is shown. The results of one representative experiment out of three reproducibly repeated experiments are shown.
DISCUSSION
DSB formation under HS conditions
The literature concerning the possibility of DSB induction by HS is rather controversial. Most authors agree that by itself HS does not introduce DSB (Hunt et al., 2007; Laszlo and Fleischer, 2009a, b). Nevertheless, some published results suggest that HS induces DSB, although to a lesser extent than do x-rays (Wong et al., 1995). The present study demonstrates that HS did actually induce the formation of DSBs. This conclusion is strongly supported by the results of the neutral SCGE analysis and by the TdT incorporation assay. Unexpectedly, the formation of DSBs under HS conditions turned out to be restricted to non–S-phase cells. The HS-induced DSBs were marked by H2AX phosphorylation, which was ATM-dependent (Figure 8). The mechanism of DSB induction by HS is not clear. It is also not clear why the DSBs are induced only in non–S-phase cells. The G1 and G2 phases are fundamentally different. It is therefore not easy to suggest a mechanism that operates in both of these phases but does not operate in the S phase. One possibility is that DSBs are induced in the S phase as well but are very quickly repaired. It might also be hypothesized that HS-induced DSBs were not formed in the cells de novo, but were unmasked only for DNA damage sensors. This assumption is based on recently published data suggesting the existence in eukaryotic cells of unrepaired lesions that might be formed during S phase, transmitted to G2 and G1 (through mitosis) phases, and sequestered in 53BP1-containing nuclear compartments (Harrigan et al., 2011; Lukas et al., 2011). One would expect that these compartments are degraded under HS conditions and that DSBs became accessible to DNA damage sensor systems. This might be the case, because we also managed to observe such 53BP1-containing compartments, which were disrupted in response to HS (Figure S6). However, the fact that we could not observe considerable existence of DSBs in untreated G1 and G2 cells (in comparison with S-phase cells) by neutral comet assay did not allow us to suggest this mechanism as the only possible one. These DSBs are likely to be introduced de novo, rather than unmasked by HS treatment. It has been proposed that HS-induced DSBs originate as a result of conversion of single-stranded DNA breaks formed due to replication fork collapse and/or the generation of reactive oxygen species (Bruskov et al., 2002; Takahashi and Ohnishi, 2005). Our present results do not support the idea that the collapse of replication forks under HS conditions contributes to the formation of DSB. Indeed, if that notion were true, the DSBs would be generated in the S phase rather than in the G1 and G2 phases. As to reactive oxygen species, they might be considered as a possible, but not exclusive, cause of HS-induced DSBs. This problem requires further investigation.
FIGURE 8:
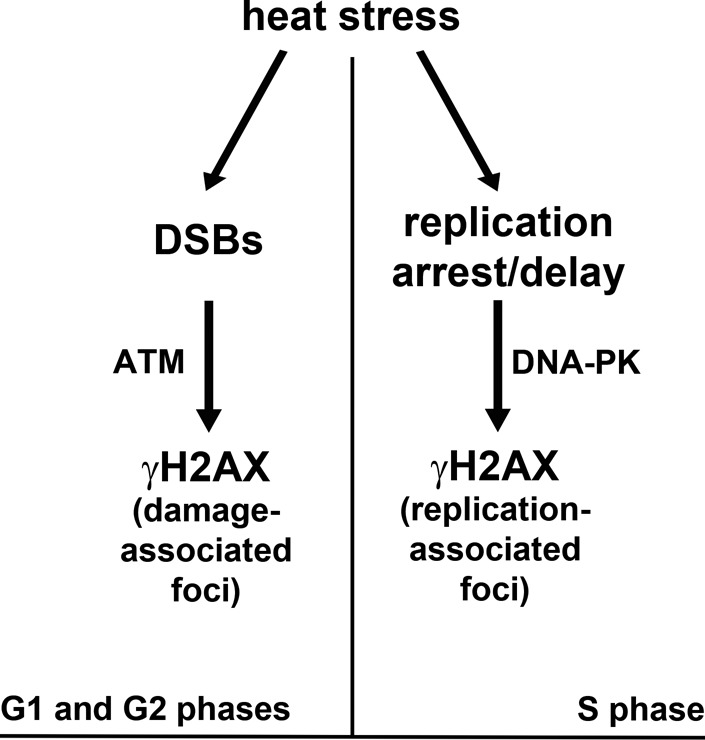
Model of the HS-induced, cell cycle phase–dependent histone H2AX phosphorylation.
Suppression of DNA replication under HS conditions
Although the effect of HS on DNA synthesis has been known for ∼30 yr (Wong and Dewey, 1982; Warters and Stone, 1983, 1984), it has never been studied in detail using contemporary molecular and cellular biology techniques. Our results clearly show that HS induces replication arrest/delay in human cells. The most interesting trait of this effect was its gradualism; the different temperature conditions used led to a progressive decrease of the apparent speed of replication fork progression (42 and 44ºC) and finally to a total arrest of replication at 45.5ºC. In accordance with previous observations (Hammond et al., 2003; Kurose et al., 2006; Conti et al., 2010; Gagou et al., 2010), we found that under HS conditions, H2AX was phosphorylated at BrdU incorporation (replication) sites. In contrast to DSB-associated H2AX phosphorylation, the replication-associated phosphorylation appeared to be mediated by DNA-dependent protein kinase (Figure 8). The exclusive role of DNA-PK in γH2AX formation at replication sites might be due to its association with RPA at replication forks (Shao et al., 1999). Most importantly, the results of the present study suggest that the formation of γH2AX domains at or close to replication forks prevented replication arrest under mild HS conditions (42 and 44ºC) and that it is likely to protect stalled replication forks from a collapse that would result in the formation of DSBs. In this regard, it is noteworthy that a mechanism based on the DNA-PK– and γH2AX–dependent prevention of DSB formation at replication sites was proposed to protect the integrity of genomes under conditions of replication arrest by aphidicolin (Shimura et al., 2007).
Dual role of γH2AX
Since its discovery, γH2AX has been considered to be a marker of DSBs, recruiting repair factors to the sites of DNA damage (Gately et al., 1998; Rogakou et al., 1998; Paull et al., 2000; Celeste et al., 2002). However, this interpretation had been brought into question by the results obtained during the last few years by several independent groups. In particular, it was shown that H2AX might be phosphorylated in a DSB-independent manner under hypoxic, HS, and replication-arrest conditions (Hammond et al., 2003; Hunt et al., 2007; Shimura et al., 2007; Laszlo and Fleischer, 2009a, b). Nevertheless, it should be mentioned that the data regarding H2AX phosphorylations outside the DNA DSBs remain controversial, and the idea that γH2AX marks not only DSBs is not universally accepted. In this paper, we accurately showed that H2AX might be phosphorylated both in DSB-dependent and DSB-independent modes in one cellular population. Moreover, the results of our inhibitor analysis suggested that γH2AX formation was mediated by different PIKKs: ATM triggered the DSB-associated phosphorylation, while DNA-PK triggered the replication arrest/delay–associated phosphorylation (Figure 8). In connection with these results, it should be noted that both enzymes (ATM and DNA-PK) have been previously claimed to phosphorylate H2AX in different situations (Stiff et al., 2004; Shrivastav et al., 2009). In this study, for the first time, we clearly discriminated the role of these enzymes in the DSB-dependent and replication arrest–dependent phosphorylation of H2AX. As to DSB-associated γH2AX, its functional role seems to be clear: it acts as a DNA damage secondary sensor, as described elsewhere (for a review, see Hammond et al., 2003; Kinner et al., 2008). In the case of replication arrest, it appears that phosphorylation of H2AX directly protects replication forks rather than participating in a signaling cascade.
It should be noted that under conditions of replication arrest, the majority of visible H2AX foci colocalize with replication foci. Such massive DSB-independent formation of γH2AX foci imposes serious restrictions on the utilization of γH2AX as a marker of DSB.
MATERIALS AND METHODS
Antibodies
The primary antibodies used for immunofluorescence and/or Western blot hybridization were lamin B1 (rabbit; cat. #ab16048; Abcam, Cambridge, MA), γH2AX (rabbit; cat. #39117; Active Motif, Carlsbad, CA), γH2AX (rabbit; cat. #ab2893; Abcam), γH2AX (mouse; clone JBW301; cat. #05-636; Upstate/Millipore, Billerica, MA), BrdU (mouse; clone 131-14871; cat. #MAB4072; Chemicon/Millipore), cyclin B1 (rabbit; cat. #sc-752; Santa Cruz Biotechnology, Santa Cruz, CA), and 53BP1 (rabbit; cat. # sc-22760; Santa Cruz Biotechnology). The secondary antibodies conjugated to either Alexa Fluor 488 or Alexa Fluor 555 were purchased from Molecular Probes/Invitrogen (Carlsbad, CA); the horseradish peroxidase–conjugated, anti–mouse and anti–rabbit immunoglobulin G (IgG) were purchased from Amersham/GE Healthcare (Waukesha, WI).
Cell culture and treatments
The human breast cancer cells (mcf-7) and the primary human fibroblasts were cultured in DMEM (PanEco, Moscow, Russia) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT). The human lymphoid cells (Jurkat) were grown in RPMI-1640 medium (PanEco) supplemented with 10% FBS. The cells were cultured at 37ºC in a conventional humidified CO2 incubator. The inhibitor of DNA-PK, NU7026 (Sigma-Aldrich, St. Louis, MO), was diluted in dimethyl sulfoxide (DMSO) and added to the culture media 6 h before heating at a final concentration of 10 μM. The inhibitor of topoisomerase II, VP16 (Sigma-Aldrich), was diluted in DMSO and added at a final concentration of 10 μg/ml for 30 min. For ATM/ATR inhibition, caffeine-sodium benzoate (Sigma-Aldrich) was diluted in phosphate-buffered saline (PBS) and added to supplemented medium for 3 h at a concentration of 10 mM. Replication arrest was induced by treating the cells with aphidicolin (Sigma-Aldrich) for 1 h at a concentration of 6 mM.
Hyperthermia
Exponentially growing cells were immersed in a precision-controlled water bath at 42, 44, or 45.5°C (± 0.05°C) for 30 min. For the recovery experiments, the HS-treated cells were maintained at 37°C for 1, 3, 6, 12, or 24 h. Under the experimental conditions used, no marked change of pH was detected in the medium during the treatment.
BrdU and EdU incorporation
Exponentially growing cells were incubated with 100 μM BrdU (Sigma-Aldrich) or 10 μM EdU (Invitrogen) for 20 or 30 min, respectively, at 37°C. After incubation, the cells were washed three times with PBS and fixed in either CSK buffer (10 mM PIPES, pH 7.0, 100 mM NaCl, 1.5 mM MgCl2, 300 mM sucrose, and 1.2 mM phenylmethylsulfonylfluoride [PMSF]) supplemented with 1% paraformaldehyde (PFA) and 2.5% Triton X-100 for 15 min at room temperature or 100% cold methanol (−20ºC) for 10 min before staining. The samples were then processed using a Click−iT EdU Imaging Kit (Invitrogen) according to the manufacturer's recommendations or were immunostained as described below.
Immunofluorescence microscopy
For immunostaining, MCF-7 cells were grown on microscope slides; an aliquot of the suspension of Jurkat cells was centrifuged onto silane-coated microscope slides (Sigma-Aldrich) at 1500 rpm for 5 min in a Cytospin 4 cytocentrifuge (Thermo Electron, Waltham, MA). All of the samples were fixed and permeabilized in either CSK buffer supplemented with 1% PFA and 2.5% Triton X-100 for 15 min at room temperature or 100% cold methanol (−20ºC) for 10 min. The fixed cells were washed three times (5 min each) in PBS (7 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.4, 137 mM NaCl, and 2.7 mM KCl). For the BrdU immunostaining, DNA was denatured by 2 N HCl at 37ºC for 1 h and then neutralized in 0.1 M borate buffer. After being washed, the cells were preincubated with 1% bovine serum albumin (BSA) in PBS for 30 min and were then incubated with antibodies in PBS supplemented with 1% BSA for 1 h at room temperature. After incubation, the cells were washed three times (5 min each time) with PBS supplemented with 0.2% BSA and 0.05% Tween 20. Subsequently, primary antibodies bound to antigens were visualized using Alexa Fluor 488– or Alexa Fluor 555–conjugated secondary antibodies. The DNA was counterstained with the fluorescent dyes 4,6-diamino-2-phenylindole (DAPI) or To-Pro 3 iodide for 10 min at room temperature. The samples were mounted using Dako fluorescent mounting medium (Dako/Invitrogen).
The immunostained samples were analyzed using a Leica DRMB fluorescence microscope (objectives: Leica N Plan 10×/0.25 and Leica 100× PH3 oil; camera: Leica DC 350F; acquisition software: Leica DCTwain version 5.1.1, Wetzlar, Germany), a Zeiss AxioScope A.1 fluorescence microscope (objectives: Zeiss N-Achroplan 40×/0.65 and Zeiss EC Plan-Neofluar 100×/1,3 oil; camera: Zeiss AxioCam MRm; acquisition software: Zeiss AxioVision Rel. 4.8.2; Jena, Germany) or a Leica TCS SP2 laser-scanning confocal microscope (objective: PL FLUOTAR 40.0×/1.0 oil; acquisition software: LCS). The images were processed using ImageJ software (version 1.44) and Adobe Photoshop CS5 (San Jose, CA).
Nuclear extract preparation and immunoblotting
Exponentially growing mcf-7 cells were lysed by incubation in cell lysis buffer (10 mM NaCl, 20 mM HEPES, pH 7.6, 1.5 mM MgCl2, 1 mM ZnSO4, 20% glycerol, 0.1% Triton X-100, 1 mM dithiothreitol, and 1 mM PMSF) supplemented with Protease Inhibitor Cocktail (Roche, Indianapolis, IN) for 2 min on ice. After centrifugation, the pelleted nuclei were collected and shaken for 60 min on ice in nuclear extraction buffer (0.5 M NaCl, 20 mM HEPES, pH 7.6, 1.5 mM MgCl2, 1 mM ZnSO4, 20% glycerol, 0.1% Triton X-100, 1 mM dithiothreitol [DTT], and 1 mM PMSF) supplemented with Protease Inhibitor Cocktail (Roche). After centrifugation (7000 × g for 10 min), the nuclear extracts were stored at −70°C. The protein concentration was measured on a Qubit Fluorometer (Invitrogen).
Aliquots of each sample were separated by 12% SDS–PAGE and blotted onto polyvinylidene difluoride membranes (Hybond-P; Amersham/GE Healthcare, Fairfield, CT). The membranes were blocked overnight in 2% ECL Advance blocking reagent (GE Healthcare) in PBS containing 0.1% Tween 20 (PBS-T) and were then incubated for 1 h with a primary antibody diluted in PBS containing 0.1% Tween 20 and 2% blocking reagent. After three washes with PBS-T, the membranes were incubated for 1 h with secondary antibodies (horseradish peroxidase–conjugated anti–rabbit or anti–mouse IgG) in PBS containing 0.1% Tween 20 and 2% blocking agent. The immunoblots were visualized using an Amersham ECL kit. For data presentation, the films were scanned and processed with Adobe Photoshop CS5 software.
Flow cytometry
For the flow cytometry analysis, adherent cells were trypsinized with 0.25% trypsin for several minutes at 37ºC. The trypsin was inactivated with a fourfold volume of DMEM medium. Next the cells were filtered through a 40-μm nylon mesh and fixed with 70% ice-cold ethanol for 1 h. After fixation, the cells were washed three times with PBS and then incubated for 10 min in permeabilization buffer (PBS containing 0.1% Triton X-100). After being washed, the cells were incubated for 30 min at room temperature with 1 mg/ml RNase and 50 μg/ml propidium iodide. The samples were analyzed using a Beckman Coulter Epics Altra flow cytometer.
TdT labeling
The cultured cells were fixed in CSK buffer for 15 min at room temperature. Following washing in PBS, the cells were preincubated at room temperature for 30 min with a 50-μl volume of TdT equilibration buffer containing 25 mM Tris-HCl (pH 7.2), 0.2 M potassium cacodylate, 1 mM CoCl2, and 0.01% Triton X-100. Next the cells were incubated at 37ºC for 1 h with equilibration buffer supplemented with 15 U terminal deoxynucleotidyl transferase (Fermentas, Vilnius, Lithuania) and 40 μM fluorescein-labeled dTTP. The reaction was terminated by washing the slides with PBS; the slides were then used for immunostaining. For a positive control, fixed cells were treated with RNase-free DNase I (1 U/ml; Fermentas) for 30 min at room temperature in PBS.
DNA PolI dUTP labeling (in situ nick translation)
Cells were fixed and permeabilized in CSK buffer (10 mM PIPES, pH 7.0, 100 mM NaCl, 1.5 mM MgCl2, 300 mM sucrose, and 1.2 mM PMSF) supplemented with 1% PFA and 2.5% Triton X-100 for 15 min at room temperature. After being washed, the cells were incubated at room temperature for 30 min with a 50 μl nick translation mixture containing 50 mM Tris-HCI (pH 7.5), 10 mM MgCl2, 1 mM DTT, 1 U of Escherichia coli DNA polymerase I (Fermentas), 10 μM each of dATP, dGTP, dCTP, and dTTP (Sileks, Moscow, Russia), and 3 μM fluorescein-labeled dTTP. The reaction was terminated by washing the slides with PBS; the slides were then used for immunostaining. For a positive control, fixed cells were treated with RNase-free DNase I (1 U/ml; Fermentas) for 30 min at room temperature in PBS.
Neutral SCGE (comet assay)
After treatment, cells were trypsinized with 0.25% trypsin for several minutes at 37ºC. The trypsin was inactivated with a fourfold volume of DMEM medium. Cells were filtered through a 40-μm nylon mesh and resuspended immediately in PBS at a concentration of 105 cells/ml. A 50-μl aliquot of this suspension was mixed with 50 μl of prewarmed 1% low–melting point agarose type VII (Sigma-Aldrich), and 75 μl was pipetted immediately onto an agarose-coated (dried) electrophoresis glass slide. The slide was kept at 4°C for 10 min. The slide was then immersed in lysis solution (30 mM EDTA, 0.5% SDS, pH 8.0, supplemented with 10 μg/ml proteinase K) at 37°C for 1 h. After being washed with PBS, the slide was subjected to electrophoresis for 20 min at 1.5 V/cm. After electrophoresis, the slide was stained with a 1:10,000 dilution of SYBR Green. The comets were visualized under a Leica DRMB fluorescence microscope. The images of comets were analyzed with the CometScore software. For each sample, 100 randomly chosen cells were scored for the tail moment.
Alkaline SCGE
The cells were combined with low–melting point agarose at 37°C and spread onto a standard comet assay slide. The slides were incubated in cold alkaline lysis buffer (10 mM Tris-HCl, 2.5 M NaCl, 100 mM EDTA, pH 10, supplemented with 1% Triton X-100 and 10% DMSO) at 4°C for 1 h in the dark. The slides were removed from the lysis buffer, washed for 10 min in PBS, and transferred to an electrophoresis chamber. After equilibration for 20 min in electrophoresis buffer (300 mM NaOH, 1 mM EDTA, pH 13), electrophoresis was conducted at 25 V and 300 mA for 20 min. The slides were then washed in a neutralization buffer (0.4 M Tris, pH 7.5) three times for 5 min and stained as described for the BrdU-comet assay.
BrdU-SCGE (BrdU-comet assay)
For the BrdU-comet assay, after DNA labeling with 100 μM BrdU (Sigma-Aldrich), the cells were processed as described above. After neutral or alkaline electrophoresis, the gels were incubated in 2 N HCl for 1 h at 37°C and then neutralized in 0.1 M borate buffer. After being washed with PBS, the slides were preincubated with 1% BSA in PBS for 30 min and were then incubated with an anti-BrdU antibody (Chemicon/Millipore) in PBS supplemented with 0.2% BSA at 37°C for 1 h in a humid chamber. After incubation, the slides were washed three times (5 min each) with PBS supplemented with 0.2% BSA and 0.05% Tween 20. Subsequently, primary antibodies bound to antigens were visualized using Alexa 488–conjugated anti–mouse IgG (Molecular Probes/Invitrogen). The comets were counterstained with propidium iodide.
DNA fiber analysis
The cells were single-labeled with 10 μM BrdU for 20 min. For double labeling, the cells were pulsed with 10 μM EdU for 30 min before HS, and were then kept in fresh medium with 50 μM BrdU for 30 min. After treatment, cells were trypsinized with 0.25% trypsin for several minutes at 37ºC. The trypsin was inactivated with a fourfold volume of DMEM medium. The cells were resuspended immediately in PBS at a concentration of 20,000 cells/ml and centrifuged onto silane-coated microscope slides (Sigma-Aldrich) at 1500 rpm for 5 min in a Cytospin 4 cytocentrifuge. Next slides were placed in a Coplin jar with lysis buffer (0.5% SDS, 50 mM EDTA, pH8.0) in an upright position for 5 min at room temperature. To obtain more extended DNA fibers, we fixed the slides at a 70° angle, and lysis solution was dripped onto the top for 2 min. The slides were lifted up slowly and were fixed in 1% formaldehyde for 10 min. For detection of incorporated nucleotide analogues (BrdU and EdU), the DNA fibers were denatured in 2 M HCl for 1 h at 37ºC and neutralized in 0.1 M borate buffer (pH 8.5). The slides were rinsed in PBS and were then used for immunostaining. Incorporated EdU was revealed using Click Chemistry (Invitrogen).
Supplementary Material
Acknowledgments
This work was supported by the Presidium of the Russian Academy of Sciences (MCB grant) and the Ministry of Science and Education of the Russian Federation (grant to O.L.K.).
Abbreviations used:
- BrdU
bromodeoxyuridine
- BSA
bovine serum albumin
- DAPI
4,6-diamino-2-phenylindole
- DF
damage-associated foci
- DMSO
dimethyl sulfoxide
- DSB
double-stranded breaks
- DTT
dithiothreitol
- EdU
5-ethynyl-2′-deoxyuridine
- FBS
fetal bovine serum
- HS
heat shock
- HSP
heat shock protein
- IgG
immunoglobulin G
- IRIF
irradiation-induced foci
- PBS
phosphate-buffered saline
- PBS-T
PBS containing 0.1% Tween 20
- PFA
paraformaldehyde
- PIKK
phosphatidylinositol 3-kinase–related kinase
- PMSF
phenylmethylsulfonylfluoride
- PolI
DNA polymerase I
- RF
replication-associated foci
- RPA
replication factor A
- SCGE
single-cell gel electrophoresis
- siRNA
small interfering RNA
- TdT
terminal deoxynucleotidyl transferase
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-12-1009) on July 11, 2012.
REFERENCES
- Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents. 2005;5:363–372. doi: 10.2174/1568011054222364. [DOI] [PubMed] [Google Scholar]
- Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell. 2010;40:216–227. doi: 10.1016/j.molcel.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruskov VI, Malakhova LV, Masalimov ZK, Chernikov AV. Heat-induced formation of reactive oxygen species and 8-oxoguanine, a biomarker of damage to DNA. Nucleic Acids Res. 2002;30:1354–1363. doi: 10.1093/nar/30.6.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- Celeste A, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chastain PD, II, Heffernan TP, Nevis KR, Lin L, Kaufmann WK, Kaufman DG, Cordeiro-Stone M. Checkpoint regulation of replication dynamics in UV-irradiated human cells. Cell Cycle. 2006;5:2160–2167. doi: 10.4161/cc.5.18.3236. [DOI] [PubMed] [Google Scholar]
- Conti C, Leo E, Eichler GS, Sordet O, Martin MM, Fan A, Aladjem MI, Pommier Y. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 2010;70:4470–4480. doi: 10.1158/0008-5472.CAN-09-3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corry PM, Robinson S, Getz S. Hyperthermic effects on DNA repair mechanisms. Radiology. 1977;123:475–482. doi: 10.1148/123.2.475. [DOI] [PubMed] [Google Scholar]
- Dewey WC, Sapareto SA, Betten DA. Hyperthermic radiosensitization of synchronous Chinese hamster cells: relationship between lethality and chromosomal aberrations. Radiat Res. 1978;76:48–59. [PubMed] [Google Scholar]
- Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol. 2001;13:225–231. doi: 10.1016/s0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- Gagou ME, Zuazua-Villar P, Meuth M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol Biol Cell. 2010;21:739–752. doi: 10.1091/mbc.E09-07-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gately DP, Hittle JC, Chan GK, Yen TJ. Characterization of ATM expression, localization, and associated DNA-dependent protein kinase activity. Mol Biol Cell. 1998;9:2361–2374. doi: 10.1091/mbc.9.9.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething MJ, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Hammond EM, Green SL, Giaccia AJ. Comparison of hypoxia-induced replication arrest with hydroxyurea and aphidicolin-induced arrest. Mutat Res. 2003;532:205–213. doi: 10.1016/j.mrfmmm.2003.08.017. [DOI] [PubMed] [Google Scholar]
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol. 2011;193:97–108. doi: 10.1083/jcb.201011083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- Hunt CR, et al. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res. 2007;67:3010–3017. doi: 10.1158/0008-5472.CAN-06-4328. [DOI] [PubMed] [Google Scholar]
- Iliakis G, Krieg T, Guan J, Wang Y, Leeper D. Evidence for an S-phase checkpoint regulating DNA replication after heat shock: a review. Int J Hyperthermia. 2004a;20:240–249. doi: 10.1080/02656730310001656379. [DOI] [PubMed] [Google Scholar]
- Iliakis G, Wang H, Perrault AR, Boecker W, Rosidi B, Windhofer F, Wu W, Guan J, Terzoudi G, Pantelias G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet Genome Res. 2004b;104:14–20. doi: 10.1159/000077461. [DOI] [PubMed] [Google Scholar]
- Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorritsma JB, Konings AW. The occurrence of DNA strand breaks after hyperthermic treatments of mammalian cells with and without radiation. Radiat Res. 1984;98:198–208. [PubMed] [Google Scholar]
- Kaneko H, Igarashi K, Kataoka K, Miura M. Heat shock induces phosphorylation of histone H2AX in mammalian cells. Biochem Biophys Res Commun. 2005;328:1101–1106. doi: 10.1016/j.bbrc.2005.01.073. [DOI] [PubMed] [Google Scholar]
- Kantidze OL, Iarovaia OV, Razin SV. Assembly of nuclear matrix-bound protein complexes involved in non-homologous end joining is induced by inhibition of DNA topoisomerase II. J Cell Physiol. 2006;207:660–667. doi: 10.1002/jcp.20597. [DOI] [PubMed] [Google Scholar]
- Kinner A, Wu W, Staudt C, Iliakis G. γ-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurose A, Tanaka T, Huang X, Traganos F, Dai W, Darzynkiewicz Z. Effects of hydroxyurea and aphidicolin on phosphorylation of ataxia telangiectasia mutated on Ser 1981 and histone H2AX on Ser 139 in relation to cell cycle phase and induction of apoptosis. Cytometry A. 2006;69:212–221. doi: 10.1002/cyto.a.20241. [DOI] [PubMed] [Google Scholar]
- Laszlo A, Fleischer I. The heat-induced γ-H2AX response does not play a role in hyperthermic cell killing. Int J Hyperthermia. 2009a;25:199–209. doi: 10.1080/02656730802631775. [DOI] [PubMed] [Google Scholar]
- Laszlo A, Fleischer I. Heat-induced perturbations of DNA damage signaling pathways are modulated by molecular chaperones. Cancer Res. 2009b;69:2042–2049. doi: 10.1158/0008-5472.CAN-08-1639. [DOI] [PubMed] [Google Scholar]
- Lee SE, Mitchell RA, Cheng A, Hendrickson EA. Evidence for DNA-PK-dependent and -independent DNA double-strand break repair pathways in mammalian cells as a function of the cell cycle. Mol Cell Biol. 1997;17:1425–1433. doi: 10.1128/mcb.17.3.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957–961. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- Lukas C, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol. 2011;13:243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- McGlynn AP, Wasson G, O'Connor J, McKerr G, McKelvey-Martin VJ, Downes CS. The bromodeoxyuridine comet assay: detection of maturation of recently replicated DNA in individual cells. Cancer Res. 1999;59:5912–5916. [PubMed] [Google Scholar]
- Montecucco A, Biamonti G. Cellular response to etoposide treatment. Cancer Lett. 2007;252:9–18. doi: 10.1016/j.canlet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. 2010;40:253–266. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Ritossa FM. A new puffing pattern induced by temperature shock and DNP in Drosophila. Experientia. 1962;18:571–573. [Google Scholar]
- Ritossa FM. Experimental activation of specific loci in polytene chromosomes of Drosophila. Exp Cell Res. 1964;35:601–607. doi: 10.1016/0014-4827(64)90147-8. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Sarge KD, Murphy SP, Morimoto RI. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol. 1993;13:1392–1407. doi: 10.1128/mcb.13.3.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- Sedelnikova OA, Rogakou EP, Panyutin IG, Bonner WM. Quantitative detection of (125)IdU-induced DNA double-strand breaks with γ-H2AX antibody. Radiat Res. 2002;158:486–492. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999;18:1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura T, Martin MM, Torres MJ, Gu C, Pluth JM, DeBernardi MA, McDonald JS, Aladjem MI. DNA-PK is involved in repairing a transient surge of DNA breaks induced by deceleration of DNA replication. J Mol Biol. 2007;367:665–680. doi: 10.1016/j.jmb.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastav M, Miller CA, De Haro LP, Durant ST, Chen BP, Chen DJ, Nickoloff JA. DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair (Amst) 2009;8:920–929. doi: 10.1016/j.dnarep.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- Tabuchi Y, Takasaki I, Wada S, Zhao QL, Hori T, Nomura T, Ohtsuka K, Kondo T. Genes and genetic networks responsive to mild hyperthermia in human lymphoma U937 cells. Int J Hyperthermia. 2008;24:613–622. doi: 10.1080/02656730802140777. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Mori E, Somakos GI, Ohnishi K, Ohnishi T. Heat induces γH2AX foci formation in mammalian cells. Mutat Res. 2008;656:88–92. doi: 10.1016/j.mrgentox.2008.07.012. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Ohnishi T. Does γH2AX foci formation depend on the presence of DNA double strand breaks? Cancer Lett. 2005;229:171–179. doi: 10.1016/j.canlet.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Toivola DM, Strnad P, Habtezion A, Omary MB. Intermediate filaments take the heat as stress proteins. Trends Cell Biol. 2010;20:79–91. doi: 10.1016/j.tcb.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JL, Parsell DA, Lindquist S. Heat-shock proteins Hsp104 and Hsp70 reactivate mRNA splicing after heat inactivation. Curr Biol. 1995;5:306–317. doi: 10.1016/s0960-9822(95)00061-3. [DOI] [PubMed] [Google Scholar]
- Wang Y, Guan J, Wang H, Leeper D, Iliakis G. Regulation of DNA replication after heat shock by replication protein a-nucleolin interactions. J Biol Chem. 2001;276:20579–20588. doi: 10.1074/jbc.M100874200. [DOI] [PubMed] [Google Scholar]
- Warters RL, Brizgys LM, Axtell-Bartlett J. DNA damage production in CHO cells at elevated temperatures. J Cell Physiol. 1985;124:481–486. doi: 10.1002/jcp.1041240318. [DOI] [PubMed] [Google Scholar]
- Warters RL, Stone OL. The effects of hyperthermia on DNA replication in HeLa cells. Radiat Res. 1983;93:71–84. [PubMed] [Google Scholar]
- Warters RL, Stone OL. Histone protein and DNA synthesis in HeLa cells after thermal shock. J Cell Physiol. 1984;118:153–160. doi: 10.1002/jcp.1041180207. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Feramisco JR. Purification of the major mammalian heat shock proteins. J Biol Chem. 1982;257:14949–14959. [PubMed] [Google Scholar]
- Welch WJ, Kang HS, Beckmann RP, Mizzen LA. Response of mammalian cells to metabolic stress; changes in cell physiology and structure/function of stress proteins. Curr Top Microbiol Immunol. 1991;167:31–55. doi: 10.1007/978-3-642-75875-1_2. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Suhan JP. Morphological study of the mammalian stress response: characterization of changes in cytoplasmic organelles, cytoskeleton, and nucleoli, and appearance of intranuclear actin filaments in rat fibroblasts after heat-shock treatment. J Cell Biol. 1985;101:1198–1211. doi: 10.1083/jcb.101.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmore E, de Caux S, Sunter NJ, Tilby MJ, Jackson GH, Austin CA, Durkacz BW. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood. 2004;103:4659–4665. doi: 10.1182/blood-2003-07-2527. [DOI] [PubMed] [Google Scholar]
- Wong RS, Dewey WC. Molecular studies on the hyperthermic inhibition of DNA synthesis in Chinese hamster ovary cells. Radiat Res. 1982;92:370–395. [PubMed] [Google Scholar]
- Wong RS, Dynlacht JR, Cedervall B, Dewey WC. Analysis by pulsed-field gel electrophoresis of DNA double-strand breaks induced by heat and/or X-irradiation in bulk and replicating DNA of CHO cells. Int J Radiat Biol. 1995;68:141–152. doi: 10.1080/09553009514551041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.