

This study identifies a motif within the first transmembrane domain of Cx26, from amino acids Val-37 through Ala-40, that is critical for oligomerization and function. The impacts of deafness-associated mutations within this motif upon gap junction channel and hemichannel functions correlate with the severity of disease that they cause.
Abstract
To identify motifs involved in oligomerization of the gap junction protein Cx26, we studied individual transmembrane (TM) domains and the full-length protein. Using the TOXCAT assay for interactions of isolated TM α-helices, we found that TM1, a Cx26 pore domain, had a strong propensity to homodimerize. We identified amino acids Val-37–Ala-40 (VVAA) as the TM1 motif required for homodimerization. Two deafness-associated Cx26 mutations localized in this region, Cx26V37I and Cx26A40G, differentially affected dimerization. TM1-V37I dimerized only weakly, whereas TM1-A40G did not dimerize. When the full-length mutants were expressed in HeLa cells, both Cx26V37I and Cx26A40G formed oligomers less efficiently than wild-type Cx26. A Cx26 cysteine substitution mutant, Cx26V37C formed dithiothreitol-sensitive dimers. Substitution mutants of Val-37 formed intercellular channels with reduced function, while mutants of Ala-40 did not form functional gap junction channels. Unlike wild-type Cx26, neither Cx26V37I nor Cx26A40G formed functional hemichannels in low extracellular calcium. Thus the VVAA motif of Cx26 is critical for TM1 dimerization, hexamer formation, and channel function. The differential effects of VVAA mutants on hemichannels and gap junction channels imply that inter-TM interactions can differ in unapposed and docked hemichannels. Moreover, Cx26 oligomerization appears dependent on transient TM1 dimerization as an intermediate step.
INTRODUCTION
Gap junction channels constitute major pathways for direct cellular interactions because they allow the passage of metabolites, second messengers, and ions between neighboring cells. Opening of hemichannels can allow exchange of such small molecules between the cytoplasm and the extracellular compartment. Gap junction function is critical for many cellular processes; indeed, mutations in gap junction proteins are causally associated with a large spectrum of diseases, including deafness, skin disease, arrhythmias, neuropathies, and cataracts (reviewed by Harris, 2001; Willecke et al., 2002; Saez et al., 2003; Martinez et al., 2009).
A gap junction channel is formed by the docking of two hemichannels or connexons. Each connexon is formed by the oligomerization of six subunit proteins called connexins (Cx). Each member of the connexin family has a similar topology, containing four transmembrane α-helices (TM1–TM4), cytoplasmic amino and carboxyl termini, two extracellular loops (ECLs), and one intracellular loop. The membrane-spanning regions of the connexins are important structural elements that participate in intraprotomer interactions and contribute to determining channel properties, including gating and permeability. Recently, Maeda et al. (2009) reported the crystal structure of gap junction channels formed by human Cx26 at 3.5 Å resolution. (A more complete and refined structural model generated by all atom molecular dynamics was also recently published [ Kwon et al., 2011].) This structure shows that the walls of the channel pore are formed from the first half of the amino terminus, parts of TM1 and TM2, and the N-terminal part of ECL1.
Although the hexameric nature of the connexons has been known for some time, the information available about the determinants of connexin oligomerization is relatively limited. Several studies implicate the connexin TM domains. Ahmad et al. (2001) have reported that a Cx32 chimera in which TM3 was replaced by a TM domain from CFTR does not oligomerize and is retained in the cytoplasm. Using a chimeric strategy, Maza et al. (2005) have obtained results suggesting that sequences in TM3 and ECL2 regulate Cx43 oligomerization by preventing formation of Cx43 hexamers in the endoplasmic reticulum (ER). We recently published data suggesting that TM3 is critical for the oligomerization of Cx43, while a region in the first half of the protein (likely TM1) is most important for oligomerization of Cx26 (Martinez et al., 2011).
TOXCAT is a dual-reporter system that allows detection of molecular interactions between plasma membrane α-helices (Russ and Engelman, 1999; Lis and Blumenthal, 2006). This system is based on bacterial expression of DNA constructs coding for chimeric proteins that contain a transactivation domain (ToxR), a TM domain of interest, and a C-terminal β-lactamase (Lis and Blumenthal, 2006). If a chimera is correctly oriented and inserted into the bacterial inner membrane, the β-lactamase domain (located within the periplasmic space) confers ampicillin resistance. Interaction between the TM domains confers chloramphenicol resistance by allowing dimerization of the cytoplasmic ToxR domains, leading to transactivation of chloramphenicol acetyltransferase at the ctx promoter. The maximal concentration of chloramphenicol that allows bacterial growth correlates with the strength of TM dimerization (Russ and Engelman, 1999).
Recently TOXCAT has been used to characterize the dimerization motifs of TM domains from both monotopic and polytopic proteins, including the receptor tyrosine-protein kinase ErbB4 (Mendrola et al., 2002), the Helicobacter pylori vacuolating cytotoxin VacA (McClain et al., 2003), the integrin αIIb subunits (Li et al., 2004), glycophorin A (Duong et al., 2007), and the human ATP-binding cassette half-transporter ABCG2 (Polgar et al., 2010). Moreover, Duong and collaborators (2007) found that the changes in the free energy of dimerization produced by point mutations assessed using TOXCAT were similar to previous calculations using sedimentation equilibrium in detergents.
In the current study, we have taken advantage of the TOXCAT system to analyze the homodimerization properties of the TM domains of Cx26. Using this system, we identified TM1 as a critical domain for oligomerization. Because two Cx26 deafness mutations are located in the critical region of TM1, we also used the TOXCAT system to examine the oligomerization abilities of constructs containing these mutant TM1s. We also studied the effects of the mutant TM1s in the context of full-length Cx26 and found impairments of oligomerization and function that parallel the severity of disease they cause.
RESULTS
TOXCAT assay reveals that Cx26-TM1 can homodimerize and that amino acids 37–40 are important for this process
To test for homodimerization of the four Cx26 TM domains utilizing the TOXCAT system (Kashlan et al., 2006; Lis and Blumenthal, 2006), we transformed bacteria with the pML27 plasmid constructs containing the sequences of each of these TM domains. Bacteria transformed with constructs containing TM1 and TM2 grew in ampicillin, whereas those containing TM3 or TM4 did not (Table 1). The results obtained with the TM3 and TM4 constructs imply that either β-lactamase production was insufficient or it was not properly localized within the periplasmic space, possibly due to lack of proper membrane insertion of the TM domain; therefore TM3 and TM4 were not studied further. The construct containing TM1 allowed bacterial growth in chloramphenicol, while that containing TM2 did not (Table 1 and Figure 1). These data suggested that Cx26-TM1 formed homodimers, unlike Cx26-TM2. Therefore, we focused our work on Cx26-TM1. A similarly expressed Cx43-TM1 construct grew in ampicillin, but not in chloramphenicol (Table 1), further supporting the unique importance of TM1 for dimerization of Cx26, but not of Cx43.
TABLE 1:
Homodimerization of expressed connexin TM domains in the bacterial plasma membrane as determined by TOXCAT analysis.
| Growth in | |||
|---|---|---|---|
| Domain | Sequence | Ampicillin | Chloramphenicol |
| Cx26 TM1 | GKIWLTVLFIFRIMILVVAA | + | 120 μg/ml |
| Cx26 TM2 | LWALQLIMVSTPALLVAMHV | + | − |
| Cx26 TM3 | IFFRVIFEAVFMYVFYIMYNGFF | − | |
| Cx26 TM3A | LWWTYTTSIFFRVIFEAVF | − | |
| Cx26 TM4 | VFTVFMISVSGICILLNITEL | − | |
| Cx43 TM1 | TAGGKVWLSVLFIFRILLLGTAV | + | − |
| Cx26 TM1 DelVVAA | GKIWLTVLFIFRIMIL | + | − |
| Cx26 TM1 GTAV | GKIWLTVLFIFRIMILGTAV | + | − |
| Cx26 TM1 V37I | GKIWLTVLFIFRIMILIVAA | + | 80 μg/ml |
| Cx26 TM1 A40G | GKIWLTVLFIFRIMILVVAG | + | − |
Bacteria were transformed with TOXCAT constructs containing the indicated TM domains and tested for growth in 100 μg/ml ampicillin and increasing concentrations of chloramphenicol (20–120 μg/ml). Bacterial growth is indicated as positive (+); lack of bacterial growth is indicated as negative (−). The maximal concentration of chloramphenicol at which there was bacterial growth is indicated.
FIGURE 1:
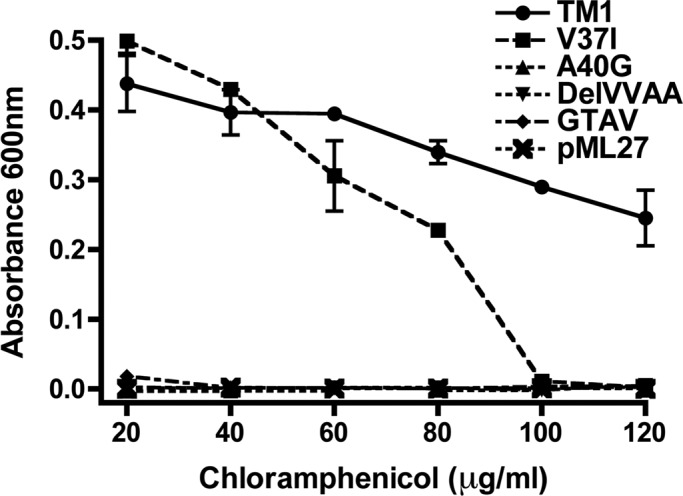
Self-interaction of Cx26TM1 at the bacterial plasma membrane is affected by mutation or deletion of amino acids 37–40. The graph depicts the growth of bacteria (absorbance at 600 nm) transformed with constructs containing the wild-type Cx26TM1 (TM1), deafness-related variants (V37I and A40G), the truncated form (DelVVAA), Cx43-like Cx26TM1chimera (GTAV), or the pML27 plasmid alone that were subjected to TOXCAT analysis after incubation at increasing concentrations of chloramphenicol (20–120 μg/ml) for 10 h.
Because Cx26-TM1 dimerized (in the TOXCAT assay) but Cx43-TM1 did not, we focused on the amino acids that differ between the TM1 domains of these two connexins to identify residues in Cx26-TM1 conferring dimerization ability. The longest continuous stretch of differing amino acids is found at positions 37–40, located at the extracellular end of TM1 (Table 1). Bacteria transformed with pML27 containing a deletion construct of Cx26-TM1 lacking amino acids 37–40 (Cx26TM1DelVVAA) grew in ampicillin but failed to grow in chloramphenicol (Table 1 and Figure 1), indicating that deletion of the VVAA sequence abolished dimerization of Cx26-TM1. We also replaced the VVAA sequence in Cx26-TM1 with the corresponding sequence from Cx43 (GTAV). Bacteria transformed with this chimera grew in ampicillin but not in chloramphenicol (Table 1 and Figure 1), implying that the presence of the GTAV sequence did not restore the dimerization ability.
Two human Cx26 mutations associated with recessively inherited deafness have been identified within this region, V37I and A40G; they produce mild and severe nonsyndromic deafness, respectively (Rabionet et al., 2000; Bason et al., 2002; Martinez et al., 2009). Therefore, we used the TOXCAT assay to examine the consequences of these substitutions on the ability of Cx26-TM1 to dimerize. Bacteria transformed with a pML27-Cx26TM1 construct containing the V37I mutation grew as well as those expressing wild-type Cx26TM1 at chloramphenicol concentrations ≤40 μg/ml, but their growth decreased with higher concentrations of chloramphenicol (Figure 1); no growth was observed at chloramphenicol concentrations ≥100 μg/ml. These results suggest that the V37I missense mutation reduced the strength of TM1 dimerization (Table 1 and Figure 1). In contrast, bacteria transformed with a construct containing the A40G mutation did not grow even in the lowest concentrations of chloramphenicol used, suggesting that this substitution abolished dimerization of TM1 (Table 1 and Figure 1).
The VVAA sequence in TM1 is important for Cx26 oligomerization and gap junction formation
To analyze the role of the VVAA sequence in the context of the full-length Cx26 protein, we replaced this sequence with the corresponding sequence from Cx43, GTAV (Cx26GTAV; Figure 2A). HeLa cells were stably transfected with a construct encoding Cx26GTAV, and the cellular localization of the protein was studied by immunofluorescence. When expressed by itself, Cx26GTAV showed a diffuse or vesicular intracellular distribution, but it was never observed at gap junction plaques (Figure 2B). In contrast, wild-type Cx26 localized mainly at gap junction plaques (Figure 2C).
FIGURE 2:

Cx26GTAV formed gap junction plaques when coexpressed with wild-type Cx26 but not when expressed alone or in combination with wild-type Cx43. (A) Diagram of the Cx26GTAV mutant. In this mutant, amino acids 37–40 from Cx26 were substituted for the corresponding sequence from Cx43, GTAV; a hemagglutinin epitope (HA) was appended to the carboxy terminus of the protein. The location of the GTAV sequence at the extracellular end of the first TM domain (TM1) is indicated. ECL1 and ECL2, first and second extracellular loops; ICL, intracellular loop; NH2 and COOH, amino and carboxyl termini. (B and C) Cellular distributions of Cx26GTAV (B) and wild-type Cx26 (C) in HeLa cells stably transfected with Cx26GTAV (HeLaCx26GTAV) or wild-type Cx26 (HeLaCx26) subjected to immunofluorescence using anti-HA (B) or anti-Cx26 (C) antibodies. (D–F) Immunolocalization of Cx26GTAV (D) and wild-type Cx26 (E) in HeLaCx26 transiently transfected with Cx26GTAV after double immunofluorescence using anti-HA (D) and anti-Cx26 (E) antibodies. The merged image is shown in (F). (G–I) Images of HeLaCx26GTAV transiently transfected with wild-type Cx43EGFP showing immunolocalization of Cx26GTAV (G) using anti-HA antibodies and GFP fluorescence localization of wild-type Cx43-EGFP (H). The merged image is shown in (I). Gap junction plaques are indicated by arrows. Scale bar: 15 μm. (J) Graphs represent the levels of Cx26 in sucrose gradient fractions detected by immunoblotting using anti-Cx26 antibodies. Triton X-100–soluble extracts from HeLaCx26 cells treated with SDS (open circles) or left untreated (closed circles) were subjected to sedimentation velocity through 5–20% sucrose gradients. (K) Graphs represent the levels of Cx26GTAV detected by immunoblotting of sucrose gradient fractions using anti-HA antibodies. Triton X-100–soluble material from HeLaCx26GTAV cells (open squares) or HeLaCx26 cells transiently transfected with Cx26GTAV (closed squares) was subjected to sedimentation velocity through 5–20% sucrose gradients. Levels of connexin are presented in arbitrary units (a.u.) after densitometric analysis of the respective immunoblots. Arrows indicate the percentage of sucrose at which Cx26 monomers and hexamers sediment.
To test whether wild-type connexins could restore normal distribution of Cx26GTAV, we coexpressed Cx26GTAV with wild-type Cx26 or wild-type Cx43. When HeLa cells expressing wild-type Cx26 were transfected with Cx26GTAV, Cx26GTAV localized to gap junction plaques (Figure 2D); however, expression of Cx26GTAV decreased the size and frequency of gap junction plaques (Figure 2, D–F). In contrast, Cx26GTAV did not localize at gap junction plaques when coexpressed with wild-type Cx43–enhanced green fluorescent protein (Cx43-EGFP; Figure 2, G–I). These results suggest that Cx26GTAV can co-oligomerize with Cx26, but not with Cx43 and are consistent with the oligomerization incompatibility between wild-type Cx26 and Cx43 (Gemel et al., 2004).
The change in cellular localization of Cx26GTAV suggested that the GTAV sequence affected oligomerization of Cx26, a phenomenon that could be reversed, at least in part, by coexpression with wild-type Cx26. Because connexin monomers and oligomers can be distinguished by their different sedimentation velocities, we used velocity sedimentation through sucrose gradients to analyze the oligomerization state of Cx26 and Cx26GTAV in cells expressing these proteins by themselves or coexpressing these proteins. When analyzed using a linear gradient of 5–20% sucrose, Cx26 showed a two-peak pattern, with one peak centered at 5–6% sucrose, and a second peak centered at 15–16% sucrose (Figure 2J). Treatment of HeLaCx26 cell homogenates with SDS to disrupt connexin oligomers collapsed immunoreactive Cx26 into a single peak at 5–8% sucrose (Figure 2J). Thus the two peaks observed in the absence of SDS treatment correspond to monomeric and oligomerized Cx26. When material solubilized from HeLaCx26GTAV cells was analyzed by sedimentation velocity, only one peak of Cx26GTAV was detected at 5–6% sucrose, corresponding to monomers (Figure 2K); these results confirmed that this mutant is impaired in its ability to homo-oligomerize. However, when Cx26GTAV was coexpressed with wild-type Cx26 (HeLaCx26/Cx26GTAV), Cx26GTAV was observed in two peaks, one centered at 7–9% sucrose and the other centered at 15–16% sucrose (Figure 2K), the same percentages of sucrose at which Cx26 monomers and oligomers sedimented. The change in sedimentation velocity of Cx26GTAV when coexpressed with wild-type Cx26 in HeLa cells implies that Cx26GTAV co-oligomerized with Cx26.
Missense mutations in the VVAA domain affect the efficiency of oligomerization and gap junction plaque formation
Previous studies have shown that the deafness-associated mutants Cx26V37I and Cx26A40G do not support intercellular communication, but their abilities to form gap junction plaques have not been examined (Bruzzone et al., 2003; Zhang et al., 2005). To test whether these proteins could form gap junction plaques, we expressed them in HeLa cells as green fluorescent protein (GFP) fusion proteins (Cx26V37I-GFP and Cx26A40G-GFP) and analyzed their cellular distributions by fluorescence microscopy. Both Cx26V37I-GFP and Cx26A40G-GFP localized to the plasma membrane in gap junction plaques in a manner similar to wild-type Cx26-GFP (Figure 3, A–C). Gap junction plaques were clearly visible in 77, 81, and 77% of the pairs of contacting cells expressing Cx26-GFP, Cx26V37I-GFP, and Cx26A40G-GFP, respectively. However, the mutants were less efficient at forming gap junction plaques and showed a higher proportion of intracellular immunostaining as compared with Cx26-GFP; the ratios of intracellular to appositional fluorescence intensity for Cx26-GFP, Cx26V37I-GFP, and Cx26A40G-GFP, were 0.15, 0.96, and 2.4, respectively.
FIGURE 3:

The deafness-associated Cx26 mutants, Cx26V37I, and Cx26A40G, oligomerized and formed gap junction plaques and hemichannels in the plasma membrane. (A–C) Photomicrographs show the distribution of fluorescence in HeLa cells transfected with Cx26-GFP (A), Cx26V37I-GFP (B), or Cx26A40G-GFP (C). Arrows point to gap junction plaques. Scale bar: 15 μm. (D) Immunoblot of streptavidin-bound (“B”) and not bound (“NB”) proteins after cell surface biotinylation of HeLa cells transiently transfected with Cx26-GFP, Cx26V37I-GFP, or Cx26A40G-GFP using anti-GFP (top) or anti–β-tubulin (bottom) antibodies. The absence of a β-tubulin band in the bound (biotinylated) fractions confirms the integrity of the plasma membrane during cell surface biotinylation. Graph illustrates the quantification of the immunoblot data. Bars represent the percentage of the total protein that was biotinylated (average + SEM; n = 3). (E and F) Graphs represent the levels of Cx26V37I-GFP (E) and Cx26A40G-GFP (F) in sucrose gradient fractions detected by immunoblotting using anti-Cx26 antibodies. Triton X-100–soluble extracts from HeLaCx26V37I-GFP or HeLaCx26A40G-GFP cells treated with SDS (open symbols) or left untreated (closed symbols) were subjected to sedimentation velocity through 5–20% sucrose gradients.
To quantify the proportion of the protein present at the plasma membrane, we performed cell surface biotinylation experiments. About 50% of the total levels of wild-type Cx26 and of each mutant were labeled with biotin (Figure 3D). These results indicated that a significant fraction of the mutant proteins was at the cell surface and exposed to the extracellular milieu.
When analyzed by sedimentation velocity through linear gradients of 5–20% sucrose, Cx26V37I-GFP (Figure 3E) and Cx26A40G-GFP (Figure 3F) showed two broad peaks at 6–10% sucrose and 13–18% sucrose for Cx26V37I-GFP. Treatment of cell homogenates with SDS collapsed immunoreactive mutant Cx26 into single peaks centered at 5–8% sucrose (Figure 3, E and F), indicating that the two peaks observed in the absence of SDS treatment corresponded to monomeric and oligomerized mutant connexins. While most of the wild-type Cx26 or Cx26V37I-GFP was present in the oligomeric fraction (Figures 2J and 3E), the monomer peak represented a greater proportion of Cx26A40G-GFP (Figure 3F). These results imply that Cx26A40G-GFP may have decreased efficiency of homo-oligomerization. Quantification of oligomer peaks showed that the efficiencies of oligomerization were 83, 68, and 33% for Cx26-GFP, Cx26V37I-GFP, and Cx26A40G-GFP, respectively.
We also studied the consequences of coexpression of Cx26V37I-GFP or Cx26A40G-GFP with Cx26GTAV. In coexpressing cells, Cx26V37I-GFP and Cx26A40G-GFP localized at gap junction plaques (Figure 4, A and D), whereas Cx26GTAV localized intracellularly and was not found at gap junction plaques (Figure 4, B and E). There was little colocalization between Cx26GTAV and either Cx26V37I-GFP or Cx26A40G-GFP (Figure 4, C and F). Sedimentation velocity of solubilized cellular extracts from coexpressing cells revealed no major oligomer peak of Cx26GTAV (Figure 4, G and H). Although oligomer peaks were observed for Cx26V37I-GFP and Cx26A40G-GFP, their relative proportions appeared decreased as compared with extracts from cells expressing these mutants alone (compare Figure 4, G and H, with Figure 3, E and F). These results indicate that Cx26GTAV does not hetero-oligomerize with Cx26V37I-GFP or Cx26A40G-GFP or it does so in a very small proportion that could not be detected under our experimental conditions.
FIGURE 4:

The Cx26 mutants V37I and A40G did not co-oligomerize with Cx26GTAV. (A–F) Photomicrographs show the distribution of GFP fluorescence (A, D) and immunolocalization of Cx26GTAV (B,E) in HeLa cells cotransfected with Cx26V37I-GFP (A) and Cx26GTAV (B) or Cx26A40G-GFP (D) and Cx26GTAV (E). The merged images are shown in panels C and F. (G and H) Graphs represent the levels of Cx26V37I-GFP, Cx26A40G-GFP and Cx26GTAV detected by immunoblotting of 5–20% sucrose gradient fractions of Triton X-100–soluble material from HeLa cells cotransfected with Cx26GTAV and Cx26V37I-GFP (G) or with Cx26GTAV and Cx26A40G-GFP (H). Cx26GTAV was detected using anti-HA antibodies (open squares), and Cx26V37I-GFP and Cx26A40G-GFP were detected using anti-GFP antibodies (closed symbols). Levels of connexin are presented in arbitrary units (a.u.) after densitometric analysis of the respective immunoblots.
If Cx26 oligomerization involves homointeractions between VVAA domains of adjacent protein subunits, we reasoned that cysteine substitutions in the VVAA domain might lead to the formation of disulfide bonds. To test this possibility, we replaced residues Val-37 or Ala-40 with cysteines and analyzed the cellular distribution and oligomerization of these mutants.
Cx26V37C-GFP was detected mainly intracellularly, with some cells containing gap junction plaques (Figure 5A). Analysis by sedimentation velocity revealed three peaks of Cx26V37C-GFP: a broad peak centered at 5–8% sucrose (similar sedimentation velocity to wild-type Cx26 monomers), an intermediate peak centered at 11% sucrose, and a narrow peak centered at 14% sucrose (matching the sedimentation velocity of wild-type Cx26 hexamers). Treatment of cell homogenates with SDS prior to ultracentrifugation collapsed Cx26V37C-GFP into monomeric and intermediate peaks. When cell homogenates were treated with the disulfide-reducing agent dithiothreitol (DTT) and SDS, Cx26V37C was found only in the monomeric fraction (Figure 5C). The disruption of the intermediate peak by DTT suggests that Cys-37 formed disulfide bonds that allowed the formation of stable dimers.
FIGURE 5:

Replacement of Val-37 (but not Ala-40) with cysteine allows disulfide bond formation and stabilization of intermediate oligomers. (A and B) Photomicrographs show the distribution of GFP fluorescence in HeLa cells transfected with Cx26V37C-GFP (A) and Cx26A40C-GFP (B). Nuclei were stained with 4′,6-diamidino-2-phenylindole. The arrow in (A) points to a gap junction plaque. Scale bar: 30 μm. (C and D) Graphs represent the levels of Cx26V37C-GFP (C) and Cx26A40C-GFP (D) in sucrose gradient fractions detected by immunoblotting using anti-Cx26 antibodies. Triton X-100–soluble extracts from HeLaCx26V37C-GFP or HeLaCx26A40C-GFP cells left untreated (closed symbols) or treated only with SDS (open triangles) or with DTT and SDS (gray triangles) were subjected to sedimentation velocity through 5–20% sucrose gradients. Levels of connexin are presented in arbitrary units (a.u.) after densitometric analysis of the respective immunoblots. The arrow in (C) indicates the intermediate oligomer peak (likely dimers).
The cysteine substitution of Ala-40 resulted in a protein that localized intracellularly without any detectable gap junction plaques (Figure 5B). Cx26A40C-GFP was detected by sedimentation velocity mainly as a single peak at 8–9% sucrose; no peak was present at higher sucrose densities, as would have been expected for hexamers. When cell homogenates were treated with SDS, Cx26A40C-GFP was detected in a peak at 5–6% sucrose with a broad shoulder at higher percentages of sucrose (Figure 5D). These data suggest that Cx26A40C did not form hexamers but did form some oligomers containing fewer subunits (subhexameric forms) that were not stabilized by disulfide bonds.
Mutations in the VVAA sequence affect gap junction channel function
To evaluate the effect of mutations in the VVAA sequence on gap junction channel function, we studied electrical coupling between pairs of HeLa cells transfected with wild-type Cx26-GFP, Cx26V37I-GFP, or Cx26A40G-GFP. Large gap junction currents were consistently recorded between cells expressing Cx26-GFP (Figure 6); the junctional conductance (4.4 ± 0.63 nS; n = 6 pairs) was significantly different from that determined in untransfected cells. Treatment of HeLaCx26-GFP cells with 18-β-glycyrrhetinic acid (18-βGA) blocked these currents. Most pairs of cells expressing Cx26V37I-GFP were also electrically coupled, but their average conductance (1.3 ± 0.2 nS; n = 6) was significantly less than that of pairs of cells expressing wild-type Cx26-GFP. Very small junctional currents were detected between pairs of cells expressing mutant Cx26A40G-GFP; the conductance value (0.4 ± 0.15 nS; n = 4) was not significantly different from that determined for pairs of untransfected cells or HeLa cells transfected with Cx26-GFP treated with the gap junction blocker 18-βGA.
FIGURE 6:
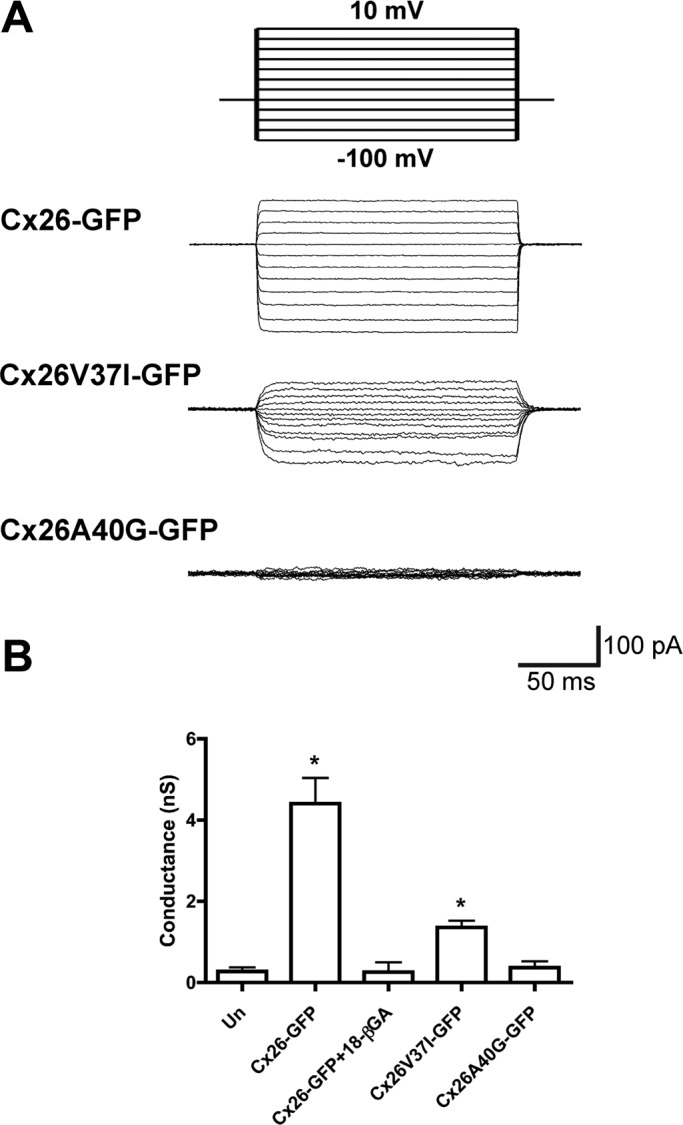
The deafness mutant, Cx26V37I, but not Cx26A40G, forms functional gap junction channels. (A) Representative family of gap junction current traces from pairs of HeLa cells transfected with Cx26-GFP, Cx26V37I-GFP, or Cx26A40G-GFP recorded by double whole-cell patch-clamp. Voltage-clamp steps were applied to voltages between −100 and +10 mV in 10-mV increments from a holding potential of −60 mV (top panel). (B) The bars in the graph depict the average ± SEM of gap junction conductance in pairs of untransfected HeLa cells (Un) or HeLa cells transfected with Cx26-GFP (in the absence or in the presence of 18-βGA), Cx26V37I-GFP, or Cx26A40G-GFP.
We also assessed intercellular transfer of tracer molecules of varying mass and charge (Neurobiotin, ethidium, YO-PRO-1, propidium, and Lucifer yellow) in HeLa cells transfected with wild-type Cx26 and mutants within the VVAA domain. Untransfected HeLa cells did not show transfer of any of the tracers (Table 2), confirming that these cells were communication-deficient. In HeLa cells stably transfected with wild-type Cx26 or Cx26-GFP, transfer was observed after microinjection of all five tracers (Table 2). In contrast, transfer was never observed after microinjection of Neurobiotin or Lucifer yellow into cells expressing Cx26GTAV (Table 2). HeLa cells expressing Cx26V37I-GFP showed almost uniform transfer of Neurobiotin and ethidium, rare transfer of Lucifer yellow, and no transfer of YO-PRO-1 or propidium. Substitution of Val-37 with a cysteine allowed Neurobiotin transfer and Lucifer yellow transfer in 71 and 29% of the microinjected cells, respectively. In cells expressing Cx26A40G-GFP, transfer of Neurobiotin or Lucifer yellow was only very rarely observed; no transfer of the other, larger, positively charged tracers tested was ever detected (Table 2). Similarly, Cx26A40C-GFP did not support either Neurobiotin or Lucifer yellow transfer. Thus, gap junction function was abolished by substitutions of amino acids 37–40 or Ala-40 alone. Substitution of Val-37 resulted in channels with a dramatic reduction in electrical conductance and transfer of larger tracers (Table 2). When either Cx26V37I-GFP or Cx26A40G-GFP was coexpressed with wild-type Cx26, transfer of Neurobiotin or Lucifer yellow was observed after 80–100% of the microinjections (Table 2). These data confirm that neither mutant acted as a dominant negative inhibitor of wild-type function (Bruzzone et al., 2003; Zhang et al., 2005).
TABLE 2:
Effects of mutations in TM1 on gap junction intercellular communication produced by homomeric or heteromeric channels.
| Incidence of coupling | |||||
|---|---|---|---|---|---|
| Expressed construct | Neurobiotin (287 Da/+1) |
Ethidium (314 Da/+1) |
YO-PRO-1 (375 Da/+2) |
Propidium (414 Da/+2) |
Lucifer yellow (430 Da/–2) |
| None | 0 (15) | 0 (5) | 0 (5) | 0 (6) | 0 (15) |
| Cx26 | 100 (30) | 100 | 100 | 100 | 100 (30) |
| Cx26GTAV | 0 (15) | ND | ND | ND | 0 (15) |
| Cx26-GFP | 100 (16) | 100 (9) | 100 (7) | 100 (6) | 100 (16) |
| Cx26V37I-GFP | 93.3 (15) | 100 (7) | 0 (10) | 0 (7) | 13.3 (15) |
| Cx26V37C-GFP | 70.6 (17) | ND | ND | ND | 29.4 (17) |
| Cx26A40G-GFP | 5.3 (19) | 0 (8) | 0 (9) | 0 (6) | 5.3 (19) |
| Cx26A40C-GFP | 0 (13) | ND | ND | ND | 0 (13) |
| Cx26 + Cx26V37I-GFP | 100 (17) | ND | ND | ND | 85 (17) |
| Cx26 + Cx26A40G-GFP | 80 (10) | ND | ND | ND | 80 (10) |
| Cx43 | 100 (54) | ND | ND | ND | 98.1 (54) |
| Cx43 + Cx26V37I-GFP | 85.7 (7) | ND | ND | ND | 85.7 (7) |
| Cx43 + Cx26A40G-GFP | 75 (8) | ND | ND | ND | 75 (8) |
The incidence of coupling is reported as the percent of microinjections in which transfer of the gap junction tracer was observed from the injected cell to its neighbors in untransfected HeLa cells (None, mock-transfected) and in HeLa cells transfected with each of the DNA constructs. The number of microinjected cells is indicated in parentheses. The molecular weights and charge of the gap junction tracers are indicated in parentheses underneath the tracer names. ND, not determined.
Deafness mutants, Cx26V37I-GFP and Cx26A40G-GFP, do not form functional hemichannels
To examine whether amino acid substitutions in the VVAA domain affected the function of Cx26 hemichannels, we studied the uptake of tracers in HeLa cells expressing GFP-tagged wild-type Cx26 or one of the two deafness-associated mutants during incubation in calcium-free media. HeLaCx26-GFP cells took up all three tracers tested: YO-PRO-1, ethidium, and Neurobiotin (Figure 7, A–C). This uptake was substantially reduced by incubation with the hemichannel blockers, La3+ or 18-βGA (Figure 7, A–C). In contrast, HeLa cells expressing Cx26V37I-GFP or Cx26A40G-GFP did not show significant uptake of YO-PRO-1 or ethidium (Figure 7, A and B). Moreover, the uptake of Neurobiotin in HeLaV37I-GFP cells was no greater than that seen in untransfected HeLa cells (Figure 7C). Taken together, these results suggest that neither mutant formed functional hemichannels in calcium-free media.
FIGURE 7:
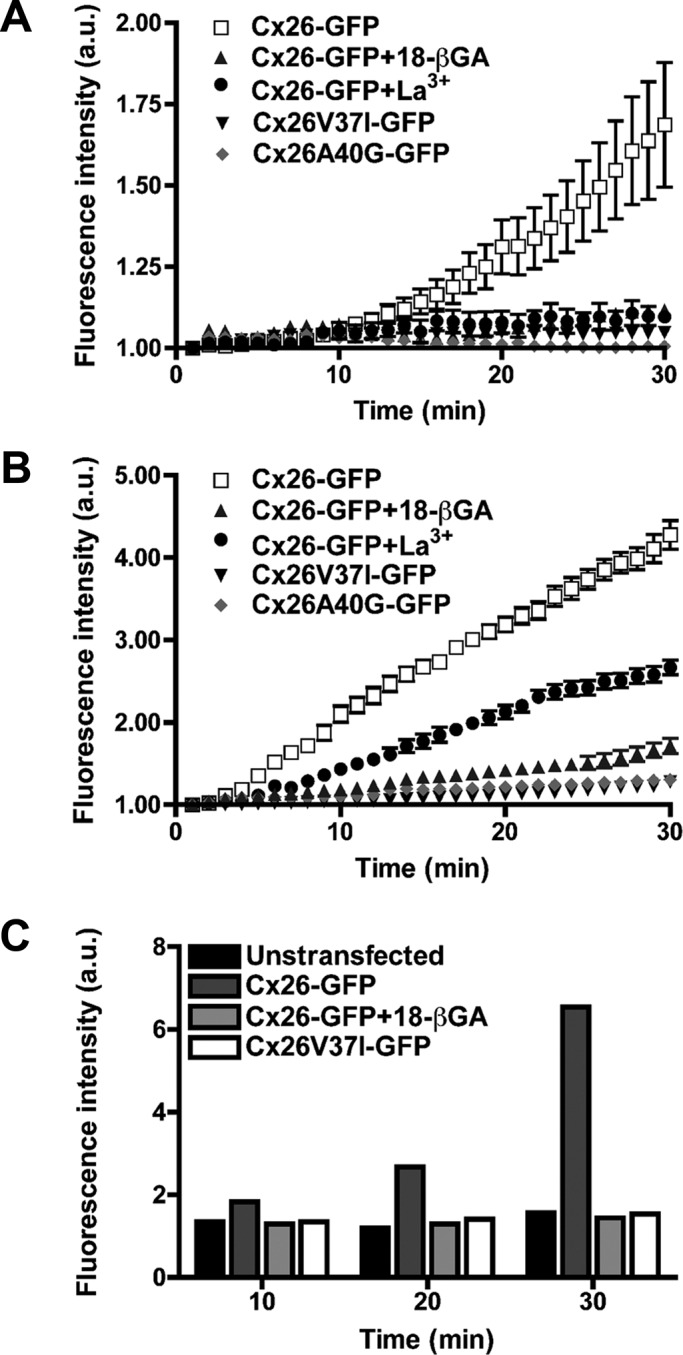
Hemichannels formed by the Cx26 deafness mutants, V37I and A40G, are nonfunctional. (A–C) Graphs show the time course of uptake of YO-PRO-1 (A), ethidium (B), and Neurobiotin (C) in HeLa cells expressing Cx26-GFP (in the absence or in the presence of 18-βGA or La+3), Cx26V37I-GFP, or Cx26A40G-GFP under conditions of free extracellular calcium (to induce the opening of hemichannels).
DISCUSSION
The oligomerization of ion channels is a complex process that involves interaction of multiple protein subunits. Using the TOXCAT analysis to study the homodimerization properties of different connexin TM domains, we found that the TM1 of Cx26 has the strongest propensity to homodimerize. We also determined that the VVAA motif (from Val-37 to Ala-40) is critical for dimerization of Cx26TM1, since expression of the truncated version of Cx26TM1 (Cx26TM1DelVVAA) or the TM1Cx26/Cx43 chimera in which the VVAA motif was replaced by the Cx43 TM1 sequence GTAV (TM1-GTAV) did not allow bacterial growth in chloramphenicol. In addition, two amino acid substitutions in the VVAA motif (corresponding to nonsyndromic deafness-associated mutations) also affected Cx26 TM1 dimerization in the TOXCAT assay: V37I reduced the strength of TM1 dimerization, and A40G impeded it completely. Our data suggest that dimerization (at least for Cx26) may involve interaction between TM1 domains (specifically the VVAA motif). These results extend previous studies suggesting that the TM domains are important for oligomerization of connexins (Ahmad et al., 2001; Lagree et al., 2003; Maza et al., 2005; Martinez et al., 2011) and that formation of dimers and tetramers are intermediate steps in the oligomerization of connexins into gap junction hemichannels (Ahmad et al., 2001).
The data presented here have shown that the sequence of amino acids 37–40 (VVAA) within the TM1 of Cx26 is also a critical determinant of homo-oligomerization into hexameric connexons. Mutations of a single residue within this four–amino acid motif resulted in proteins with reduced oligomerization efficiencies. Cx26V37I and Cx26A40G were accessible to cell surface labeling with biotin and produced gap junction plaques, suggesting they were assembled and transported to the plasma membrane; however, the fractions of the protein detected as hexamers were reduced for both mutants as compared with wild-type Cx26. Moreover, substitution of three residues in this motif (Cx26GTAV) resulted in a protein that did not form gap junction plaques or oligomers. A TM1–TM1 interaction during the oligomerization process is supported by the limited or absent oligomerization of Cx26GTAV when coexpressed with Cx26V37I or Cx26A40G. However, we cannot discard a possible interaction between Cx26GTAV and the mutants, because the amount of Cx26A40G or Cx26V37I in the oligomeric fraction was significantly diminished in cells that coexpress either of these mutants with Cx26GTAV.
The structure of Cx26 channels (Maeda et al., 2009) and physiological studies of the roles of individual amino acids (Kronengold et al., 2003; Tang et al., 2009) suggest that TM1 is the major TM domain contributing to the Cx26 channel pore. In the Cx26 structure, the proximity between TM1 domains of two adjacent protomers is closest near the VVAA region, but there are no apparent interactions between TM1 domains in the mature channel (Figure 8). This implies that Cx26 dimerization through interaction of TM1 domains (as identified by the TOXCAT assays) must be only a transient step in the oligomerization process. This inference is supported by the results obtained with the Val-37 cysteine-substitution mutant of Cx26. Cx26V37C formed incomplete hexamers likely representing formation of a disulfide bond between cysteines at position 37. Thus, it is possible that stabilization of the dimeric intermediate prevented rearrangements required for formation of mature hexamers and gap junction plaques. The wild-type Cx26 structure also shows that Ala-40 participates in intramolecular hydrophobic interactions between amino acid side chains of the TM helices (especially with Ile-74 of TM2), but not in interactions with TM1 amino acids from adjacent subunits (Maeda et al., 2009 and Figure 8). Replacement of Ala-40 with cysteines would not allow disulfide bond formation between Cys-40 from adjacent protomers in the mature channel, due to the orientation of their side chains and the distance between them. Consistent with the structural model of Cx26, we did not observe hexamers or gap junction plaques formed by Cx26A40C. Taken together, these results imply that Cx26 subunits undergo rearrangements between the formation of dimers and mature hemichannels in which the Val-37 (but not the Ala-40) side chains come closer together than in the mature channel.
FIGURE 8:

Structural models of rat Cx26 and the nonsyndromic deafness-associated mutants V37I and A40G. (A) Ribbon representation of the overall structure of the Cx26 hemichannel colored by monomer. The cyan dashed lines demarcate the location of the residues shown in detail in (B–D). (B) View from inside the pore of the Cx26 hemichannel illustrating the main interactions of residues Val-37 and Ala-40 in two protomers shown in ribbon representation (red and green). For clarity, the amino terminus (NTH) of the protomer shown in green is not shown. Relevant residues are shown in Licorice representation colored by element (C = cyan, N = blue, O = red, H = white, S = yellow) and surrounded by a transparent surface. Val-37 is on the same face (but approximately one turn away) of the TM1 α-helix as Met-34 which interacts with Trp-3 from an adjacent protomer. Ala-40 is involved in a TM1–TM2 intraprotomer interaction with Ile-74. (C) Detail of the Cx26A40G model. It is possible that replacement of Ala-40 by glycine might decrease the interaction with Ile-74, perhaps increasing the mobility of TM1 (curved arrow). (D) Detail of the Cx26V37I model. It is possible that the increased size and volume of the isoleucine residue at position 37 (TM1 from the protomer shown in green) might enhance the interaction of this residue with Trp-3 of the adjacent monomer (NTH from the adjacent protomer shown in red), perhaps leading to movement of the amino terminal helix (arrow).
Assembly of other multisubunit membrane channels is a multiple-step process that also involves formation of intermediates containing fewer subunits than the mature form (TRP channels, AMPA receptors, NMDA receptors, nicotinic acetylcholine receptors, etc.; Feng et al., 2008; Nakagawa, 2010; Mayer, 2011; Tsetlin et al., 2011). Oligomerization is determined by nonmembrane domains in many such proteins. However, similar to our findings for Cx26, TM region interactions are important for oligomerization of other proteins, such as the inositol trisphosphate receptor, G protein–coupled receptors (GPCRs), and two-pore channels (TPCs; Galvan et al., 1999; Lee et al., 2003; Fanelli and Felline, 2011; Churamani et al., 2012). Homo-oligomerization of TMs is involved in dimerization of GPCRs. TPCs assemble as dimers through differential interactions between TM regions. Although structural rearrangements have been reported during assembly of many of these channels/receptors, we do not know of a previous identification of subunit dimerization through regions that do not interact in the mature channel.
Cx26GTAV formed oligomers and gap junction plaques when it was coexpressed with wild-type Cx26, but it did not oligomerize when coexpressed with Cx26V37I or Cx26A40G. These results imply that at least one wild-type subunit is required for formation of Cx26 hemichannels. Similarly, the A40G mutation only partially affected oligomerization into hexamers as seen using sedimentation velocity. Both groups of studies imply that the VVAA motif is not the only determinant of Cx26 oligomerization. Moreover, the partial ability of Cx26A40G to oligomerize into hexamers but the absence of dimerization of the TM1s containing this mutant in the TOXCAT assay suggest that oligomerization into a hexamer is a less stringent process than dimerization.
Previous studies have shown that Cx26 and Cx43 do not hetero-oligomerize (Gemel et al., 2004) and that Cx43 oligomerization compatibility is determined by its TM3 (Martinez et al., 2011). Consistently, we found that Cx26GTAV did not form oligomers or gap junction plaques when it was coexpressed with wild-type Cx43. This implies that the VVAA motif does not determine hetero-oligomerization.
All Cx26 mutants within the VVAA region showed impaired intercellular channel function in our experiments. HeLa cells expressing Cx26V37I-GFP contained functional gap junction channels (albeit with lower total junctional conductances than cells expressing wild-type Cx26); they were permeable to the smaller tracers, Neurobiotin and ethidium, but not to the larger ones, YO-PRO-1, propidium, and Lucifer yellow. Similarly, Cx26V37C channels transferred Neurobiotin more frequently than Lucifer yellow. Cells expressing Cx26A40G-GFP did not show significant electrical coupling and showed only rare transfer of Lucifer yellow or Neurobiotin; Cx26A40C channels did not allow transfer of either of these tracers. Interestingly, patients with the A40G mutation have more severe deafness than those with the V37I mutation; thus the extent of impairment of intercellular coupling due to these mutations correlates with disease severity. In previous studies, no intercellular coupling was observed when Cx26V37I or Cx26A40G was expressed in Xenopus oocytes and HEK293 cells (Bruzzone et al., 2003; Zhang et al., 2005). Thus it is possible that HeLa cells are more permissive for the weakly functional mutant Cx26V37I.
Wild-type Cx26 formed functional hemichannels that allowed uptake of multiple different tracers when cells were incubated in low calcium concentrations. In contrast, neither Cx26V37I nor Cx26A40G allowed the uptake of any of the tracers tested. The discrepancy in function observed between intercellular channels and hemichannels formed by Cx26V37I suggests that docking of these hemichannels leads to conformational changes that result in a permeable aqueous pore.
MATERIALS AND METHODS
Generation of DNA constructs
pML-27-CxTM constructs.
DNA sequences encoding the TM domains were subcloned between the NheI and BamHI restriction sites of pML-27 vector, a gift of K. M. Blumenthal (Kashlan et al., 2006; Lis and Blumenthal, 2006). The DNA fragments encoding the TM domains were generated either by annealing of synthetic oligonucleotides (containing the overlapping and complementary NheI and BamHI restriction enzyme sequences) or by PCR amplification using sense and antisense primers, LA Taq DNA polymerase (TaKaRa, Otsu, Japan), and the coding regions of rat Cx26 (accession: NM_001004099) or rat Cx43 (accession: NM_012567) as templates (Beyer et al., 1987; Zhang and Nicholson, 1989). The sequences of the synthetic oligonucleotides are listed in Supplemental Table S1. Equimolar ratios of sense and antisense oligonucleotides were incubated at 95°C for 5 min in T4 DNA ligase buffer and then incubated for 1 h at room temperature. Then the annealed DNA was ligated into pML-27 linearized with NheI and BamHI. The PCR-amplified TM DNAs were subcloned into pGEM-T Easy (Promega, Madison, WI), digested with NheI and BamHI, and then subcloned into pML-27. In-frame insertion of the TM DNAs was confirmed by sequencing. Transformed bacteria were selected in 50 μg/ml spectinomycin.
Generation of Cx26GTAV.
The Cx26GTAV mutant was generated by PCR using Phusion High-Fidelity DNA polymerase (New England Biolabs, Ipswich, MA) and wild-type rat Cx26 DNA with a hemagglutinin (HA) tag appended to its C-terminus in pcDNA3.1/Hygro(+) (Invitrogen, Carlsbad, CA) as a template, according to the strategy used previously by Minogue et al. (2005). Primers facing opposite directions and spanning the DNA region encoding GTAV (Table S1) were designed to amplify the sequence of the full construct (including the vector sequence). The plasmid was regenerated by religation of the PCR product.
Generation of GFP-tagged Cx26 mutant constructs in pcDNA3.1.
The Cx26 GFP fusion protein (Cx26-GFP) was generated by subcloning the coding region of rat Cx26 into pcDNA3.1/CT-GFP-TOPO (Invitrogen) according to the manufacturer's instructions. Mutations were introduced into Cx26 by site-directed mutagenesis using the QuickChange kit (Stratagene, Agilent, Santa Clara, CA) and employing Cx26-GFP as the template and the sense and antisense primers listed in Table S1. The coding regions of all constructs were fully sequenced to ensure that PCR amplification did not introduce unwanted mutations.
TOXCAT analysis
Bacteria (strain JM109; Promega) were transformed with each of the pML-27-CxTM constructs and plated on LB-agar containing 50 μg/ml spectinomycin. Then single bacterial colonies were grown overnight in LB media containing 100 μg/ml ampicillin and 50 μg/ml spectinomycin. (Only colonies that grew in this concentration of ampicillin were used for the assay.) Aliquots of these bacterial cultures were inoculated into different tubes each containing 5 ml of LB plus 100 μg/ml ampicillin, 50 μg/ml spectinomycin, and increasing concentrations of chloramphenicol (20–120 μg/ml) to obtain an initial A600 of 0.025 (as a measure of bacterial density) in each tube. The bacterial cultures were then grown at 37°C with shaking at 250 rpm for 8–10 h, and the bacterial density was assessed by measuring the A600. These experiments were performed five times each time in triplicate.
Cell culture and transfection
Parental HeLa cells were grown as previously described (Martínez et al., 2002, 2011). When cultures reached 60–70% confluence, HeLa cells were transiently transfected with the DNA constructs using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. HeLa cells stably transfected with Cx43-EGFP or wild-type Cx26 were cultured as previously described (Contreras et al., 2003; Martinez et al., 2011).
Immunofluorescence
Single- and double-labeling immunofluorescence microscopy was performed essentially as described previously (Martinez et al., 2002, 2011). Cells grown on glass coverslips were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS; pH 7.4) for 30 min and permeabilized with 1% Triton X-100 in PBS prior to incubation with antibodies. A rabbit polyclonal antibody directed against the HA tag (Invitrogen) to detect Cx26GTAV and mouse monoclonal antibody to detect Cx26 (Invitrogen) were used in this study. Cy3-conjugated goat anti–mouse immunoglobulin G (IgG) and Cy2-conjugated goat anti–rabbit IgG antibodies were obtained from Jackson ImmunoResearch (West Grove, PA). Cells were examined using a Nikon TE-2000U inverted microscope equipped for epifluorescence and digital microscopy (Nikon ACT-2U, Tokyo, Japan). Images were acquired using a Nikon DS-2WBc fast-cooled monochromatic digital camera. Composite figures and overlays were generated using Adobe Photoshop CS4 (San Jose, CA).
Cell surface biotinylation
HeLa cells were grown on 60-mm culture dishes. When the cells reached 70% confluence, they were subjected to cell surface biotinylation according to a previously published method (Cooper and Lampe, 2002). Briefly, cells were rinsed twice with ice-cold PBS and labeled twice with EZ-Link NHS-LC-Biotin (0.5 mg/ml; Pierce, Rockford, IL) in PBS on ice for 10 min. Then cells were rinsed five times with cold PBS containing 15 mM glycine on ice. After lysis in 0.5 ml PBS supplemented with 0.5% SDS, 0.25% Triton X-100, and protease and phosphatase inhibitors, biotinylated proteins were affinity-purified by incubation with ImmunoPure Immobilized Avidin (Pierce) for 30 min at 4°C. After three washes in PBS supplemented with 0.25% Triton X-100 plus protease and phosphatase inhibitors followed by a wash in PBS, bound proteins were eluted in 1X Laemmli sample buffer containing 5% β-mercaptoethanol and boiled for 3 min. Nonbiotinylated proteins (intracellular proteins) and cell surface biotinylated proteins (plasma membrane proteins exposed to the extracellular milieu bound to immobilized avidin) were analyzed by immunoblotting using anti-GFP and anti–β-tubulin antibodies.
Immunoblotting
Cultures of parental or stably transfected HeLa cells at 70% confluence were rinsed with PBS and harvested in ice-cold 2 mM phenylmethylsulfonyl fluoride (PMSF) in PBS. The cell suspensions were centrifuged, the supernatants were discarded, and the cell pellets were flash-frozen in liquid nitrogen and stored at −80°C until further analysis. On the day immunoblotting was performed, cell pellets were resuspended and lysed by sonication in water containing 200 μg/ml soybean trypsin inhibitor, 1 mg/ml benzamidine, 1 mg/ml ε-aminocaproic acid, 2 mM PMSF, 20 μM Na4P2O7, and 100 μM NaF. Western blot analysis was performed essentially as previously described (Martinez et al., 2002). Proteins (aliquots of whole homogenates containing 100 μg total protein, 50–100 μl aliquots of biotinylated cell surface proteins and nonbiotinylated proteins, or aliquots from sucrose gradient fractions) were resolved on 8 or 10% polyacrylamide gels containing SDS (SDS–PAGE). Proteins were electrotransferred onto Immobilon-P membranes (Millipore, Bedford, MA) at 300 mA for 1.5 h. Membranes were incubated in 5% nonfat milk and 0.1% Tween-20 in Tris-buffered saline (pH 7.4; TBST), overnight at 4°C. Then they were incubated with the respective primary antibodies (rabbit anti-HA [Invitrogen], rabbit anti-GFP [Invitrogen], or mouse anti–β-tubulin [Promega]) diluted in 5% nonfat milk in TBST for 3 h at room temperature. Membranes were rinsed in TBST several times and then incubated for 30 min at room temperature with horseradish peroxidase–conjugated goat anti–mouse or anti–rabbit IgG antibodies (Jackson ImmunoResearch). After six rinses in TBST, antibody binding was detected by enhanced chemiluminescence (Amersham, Arlington Heights, IL) using the imaging system Epichemi3 Darkroom (UVP BioImaging Systems, Upland, CA).
Sedimentation velocity through sucrose gradients
Transfected HeLa cells from two 100-mm culture dishes at 70% confluence were harvested in ice-cold PBS containing 2 mM PMSF. The cell suspensions were centrifuged, and the supernatants were discarded. Cell pellets were lysed in 200 μl of incubation buffer (0.14 M NaCl, 5.3 mM KCl, 0.35 mM Na2HPO4, 0.35 mM KH2PO4, 0.8 mM MgSO4, 2.7 mM CaCl2, 20 mM HEPES, pH 7.5) containing protease and phosphatase inhibitors (as described in Immunoblotting) by repeated passages through a needle (20 passages through a 20-gauge needle followed by 10 passages through a 27-gauge needle). Cell homogenates were incubated on ice for 30 min in 1% Triton X-100 and mixed every 5 min by inversion. Homogenates were centrifuged at 100,000 × g for 30 min at 4ºC. The resulting Triton X-100–soluble fractions (supernatants) were subjected to sedimentation velocity through 5–20% (wt/vol) linear sucrose gradients containing 0.1% Triton as previously described (Berthoud et al., 2001). After centrifugation for 22 h at 100,000 × g, 250-μl fractions were collected and analyzed by immunoblotting. For determination of the percentage of sucrose at which connexin monomers sediment, the Triton X-100–soluble fractions were treated with SDS to disaggregate the oligomers before the fractions were subjected to sedimentation velocity through sucrose gradients. To test for the formation of disulfide bonds, we treated the Triton X-100–soluble fractions with DTT in addition to SDS before subjecting them to sedimentation velocity through sucrose gradients. The intensities of the bands were quantified by densitometry, and the values were normalized with respect to the maximal signal.
Measurement of intercellular coupling
Cells were grown on coverslips and allowed to reach ∼80% confluence. Then intercellular coupling was assessed using gap junction tracers (Martínez et al., 2002, 2011). One cell from a cell cluster was impaled with a micropipette filled with a solution of 4% Lucifer yellow (Sigma-Aldrich) and 4% Neurobiotin (Vector Laboratories, Burlingame, CA) or ethidium bromide (5 mg/ml; Sigma-Aldrich), YO-PRO-1 (0.6 mg/ml; Invitrogen) or propidium iodide (5 mg/ml; Invitrogen) in water, except for YO-PRO-1 that was diluted in dimethyl sulfoxide (0.01%). The tracer-containing solution was microinjected into the cell using an InjectMan-FemtoJet system (Eppendorf, Hamburg, Germany) and 0.2- to 0.3-s pulses of 1–2 psi for 1–2 min. Cells containing fluorescent tracers were visualized by microscopy under ultraviolet illumination. For visualization of Neurobiotin, cells were fixed with 4% paraformaldehyde in PBS for 30 min, permeabilized with methanol/acetone (1:1) for 2 min at room temperature, and incubated with streptavidin-Cy3 conjugate (Sigma-Aldrich). The results are reported as the incidence of coupling, that is, the percentage of the microinjections that resulted in diffusion of the tracer to two or more adjacent cells.
Electrophysiology
HeLa cells in coverslips were placed on an experimental chamber containing Ham's F-12 media as the extracellular solution. Cells were observed using an inverted BX51WI Olympus microscope. Micropipettes were pulled from microcapillary borosilicate glass using a micropipette puller (Sutter Instrument). The resistance of the resulting micropipettes was 10–15 megaOhms. The micropipettes were filled with a solution containing 10 mM KCl, 10 mM HEPES, 2 mM ethylene glycol tetraacetic acid, 125 mM potassium gluconate, 2 mM sodium adenosine 5′-triphosphate. Electrophysiological recordings in pairs of HeLa cells were made by double whole-cell patch-clamp using two patch-clamp amplifiers (HEKA EPC7 Plus, Lambrecht, Germany; and Warner PC-501 A, Hamden, CT). Cells were clamped at −60 mV and a series of 200-ms voltage steps from −100 to +10 mV in 10-mV steps were applied to one cell of the pair. Data were acquired with IGOR 6.0 software using an analogue–digital converter card (BNC-2090; National Instruments, Austin, TX). Total gap junction conductance (gj) was calculated by dividing the transjunctional current in the neighboring nonstimulated cell (Ij) by the difference in voltage between the neighboring cells (gj = Ij/Vj).
Assessment of hemichannel function
Hemichannel function was assessed by time-lapse imaging of the uptake of tracers of different size and charge (Neurobiotin: 287 Da, +1; ethidium: 314 Da, +1; YO-PRO-1: 375 Da, +2; propidium: 414 Da, +2) as previously described (Contreras et al., 2002). Specifically, cells plated on glass coverslips were rinsed twice with calcium-free Hanks’ balanced salt solution (HBSS; Invitrogen). Cells were then incubated in HBSS containing 5 μM ethidium bromide, 20 μM YO-PRO-1, 50 μM propidium iodide or 100 μM Neurobiotin. Fluorescence images of different cells (defined as regions of interest) during uptake of the nucleic acid stains were obtained using a 40× objective in a Nikon TE-2000U inverted microscope and captured with a Nikon DS-2WBc fast-cooled monochromatic digital camera (8-bit) every 30 s (exposure time = 30 ms, gain = 0.5). Detection of Neurobiotin was performed as described above in cells that were incubated for 10, 20, or 30 min in calcium-free medium. In some experiments, cells were preincubated with the connexin hemichannel blocker 18-βGA (50 μM) or La3+ (100 μM LaCl3; Sigma-Aldrich). Image analysis and quantification of fluorescence intensity were performed using ImageJ (http://rsbweb.nih.gov/ij).
Structural modeling of rat Cx26, Cx26V37I, and Cx26A40G
Three-dimensional models of wild-type rat Cx26, Cx26V37I, and Cx26A40G hemichannels were constructed by comparative modeling with MODELLER version 9.10 (Eswar et al., 2006), using the crystal structure of human Cx26 (PDB-ID: 2zw3) as a template (Maeda et al., 2009). One hundred different models were generated, and the best model determined according to MODELLER´s internal PDF score was selected for further analyses. Structure validation was performed using the tools available at the SAVES Web server (http://nihserver.mbi.ucla.edu/SAVES).
Supplementary Material
Acknowledgments
This work was partially supported by a grant from Fondo Nacional de Desarrollo Científico y Tecnológico-FONDECYT (1090573) to A.D.M., an ANILLO grant ACT-71 to A.D.M. and J.C.S., and grants from the National Institutes of Health (HL59199 and EY08368) to E.C.B. The “Centro Interdisciplinario de Neurociencia de Valparaíso” is a Chilean Science Millennium Institute.
Abbreviations used:
- 18-βGA
18-β-glycyrrhetinic acid
- a.u.
arbitrary units
- Cx
connexin
- DTT
dithiothreitol
- ECL
extracellular loop
- EGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- GPCR
G protein–coupled receptor
- HA
hemagglutinin
- HBSS
Hanks’ balanced salt solution
- IgG
immunoglobulin G
- PBS
phosphate-buffered saline
- PMSF
phenylmethylsulfonyl fluoride
- TBST
0.1% Tween-20 in Tris-buffered saline
- TM
transmembrane
- ToxR
transactivation domain
- TPC
two-pore channel
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-12-1058) on July 11, 2012.
REFERENCES
- Ahmad S, Martin PE, Evans WH. Assembly of gap junction channels: mechanism, effects of calmodulin antagonists and identification of connexin oligomerization determinants. Eur J Biochem. 2001;268:4544–4552. doi: 10.1046/j.1432-1327.2001.02380.x. [DOI] [PubMed] [Google Scholar]
- Bason L, Dudley T, Lewis K, Shah U, Potsic W, Ferraris A, Fortina P, Rappaport E, Krantz ID. Homozygosity for the V37I Connexin 26 mutation in three unrelated children with sensorineural hearing loss. Clin Genet. 2002;61:459–464. doi: 10.1034/j.1399-0004.2002.610611.x. [DOI] [PubMed] [Google Scholar]
- Berthoud VM, Montegna EA, Atal N, Aithal NH, Brink PR, Beyer EC. Heteromeric connexons formed by the lens connexins, connexin43 and connexin56. Eur J Cell Biol. 2001;80:11–19. doi: 10.1078/0171-9335-00132. [DOI] [PubMed] [Google Scholar]
- Beyer EC, Paul DL, Goodenough DA. Connexin43: a protein from rat heart homologous to a gap junction protein from liver. J Cell Biol. 1987;105:2621–2629. doi: 10.1083/jcb.105.6.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, Veronesi V, Gomes D, Bicego M, Duval N, Marlin S, Petit C, D'Andrea P, White TW. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003;533:79–88. doi: 10.1016/s0014-5793(02)03755-9. [DOI] [PubMed] [Google Scholar]
- Churamani D, Hooper R, Brailoiu E, Patel S. Domain assembly of NAADP-gated two-pore channels. Biochem J. 2012;441:317–323. doi: 10.1042/BJ20111617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras JE, Saez JC, Bukauskas FF, Bennett MV. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc Natl Acad Sci USA. 2003;100:11388–11393. doi: 10.1073/pnas.1434298100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras JE, Sánchez HA, Eugenin EA, Speidel D, Theis M, Willecke K, Bukauskas FF, Bennett MV, Sáez JC. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci USA. 2002;99:495–500. doi: 10.1073/pnas.012589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CD, Lampe PD. Casein kinase 1 regulates connexin-43 gap junction assembly. J Biol Chem. 2002;277:44962–44968. doi: 10.1074/jbc.M209427200. [DOI] [PubMed] [Google Scholar]
- Duong MT, Jaszewski TM, Fleming KG, MacKenzie KR. Changes in apparent free energy of helix-helix dimerization in a biological membrane due to point mutations. J Mol Biol. 2007;371:422–434. doi: 10.1016/j.jmb.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics. 2006;5 doi: 10.1002/0471250953.bi0506s15. Unit 5.6.1–5.6.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanelli F, Felline A. Dimerization and ligand binding affect the structure network of A2A adenosine receptor. Biochim Biophys Acta. 2011;1808:1256–1266. doi: 10.1016/j.bbamem.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Feng S, Okenka GM, Bai CX, Streets AJ, Newby LJ, DeChant BT, Tsiokas L, Obara T, Ong ACM. Identification and functional characterization of an N-terminal oligomerization domain for polycystin-2. J Biol Chem. 2008;283:28471–28479. doi: 10.1074/jbc.M803834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan DL, Borrego-Diaz E, Perez PJ, Mignery GA. Subunit oligomerization, and topology of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1999;274:29483–29492. doi: 10.1074/jbc.274.41.29483. [DOI] [PubMed] [Google Scholar]
- Gemel J, Valiunas V, Brink PR, Beyer EC. Connexin43 and connexin26 form gap junctions, but not heteromeric channels in co-expressing cells. J Cell Sci. 2004;117:2469–2480. doi: 10.1242/jcs.01084. [DOI] [PubMed] [Google Scholar]
- Harris AL. Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys. 2001;34:325–472. doi: 10.1017/s0033583501003705. [DOI] [PubMed] [Google Scholar]
- Kashlan OB, Maarouf AB, Kussius C, Denshaw RM, Blumenthal KM, Kleyman TR. Distinct structural elements in the first membrane-spanning segment of the epithelial sodium channel. J Biol Chem. 2006;281:30455–30462. doi: 10.1074/jbc.M604615200. [DOI] [PubMed] [Google Scholar]
- Kronengold J, Trexler EB, Bukauskas FF, Bargiello TA, Verselis VK. Single-channel SCAM identifies pore-lining residues in the first extracellular loop and first transmembrane domains of Cx46 hemichannels. J Gen Physiol. 2003;122:389–405. doi: 10.1085/jgp.200308861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon T, Harris AL, Rossi A, Bargiello TA. Molecular dynamics simulations of the Cx26 hemichannel: evaluation of structural models with Brownian dynamics. J Gen Physiol. 2011;138:475–493. doi: 10.1085/jgp.201110679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagree V, Brunschwig K, Lopez P, Gilula NB, Richard G, Falk MM. Specific amino-acid residues in the N-terminus and TM3 implicated in channel function and oligomerization compatibility of connexin43. J Cell Sci. 2003;116:3189–3201. doi: 10.1242/jcs.00604. [DOI] [PubMed] [Google Scholar]
- Lee SP, O'Dowd BF, George SR. Homo- and hetero-oligomerization of G protein-coupled receptors. Life Sci. 2003;74:173–180. doi: 10.1016/j.lfs.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Li R, Gorelik R, Nanda V, Law PB, Lear JD, DeGrado WF, Bennett JS. Dimerization of the transmembrane domain of integrin αIIb subunit in cell membranes. J Biol Chem. 2004;279:26666–26673. doi: 10.1074/jbc.M314168200. [DOI] [PubMed] [Google Scholar]
- Lis M, Blumenthal K. A modified, dual reporter TOXCAT system for monitoring homodimerization of transmembrane segments of proteins. Biochem Biophys Res Commun. 2006;339:321–324. doi: 10.1016/j.bbrc.2005.11.022. [DOI] [PubMed] [Google Scholar]
- Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- Martínez AD, Acuña R, Figueroa V, Maripillán J, Nicholson B. Gap-junction channels dysfunction in deafness and hearing loss. Antioxid Redox Signal. 2009;11:309–322. doi: 10.1089/ars.2008.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez AD, Hayrapetyan V, Moreno AP, Beyer EC. Connexin43 and connexin45 form heteromeric gap junction channels in which individual components determine permeability and regulation. Circ Res. 2002;90:1100–1107. doi: 10.1161/01.res.0000019580.64013.31. [DOI] [PubMed] [Google Scholar]
- Martínez AD, Maripillán JM, Acuña R, Minogue PJ, Berthoud VM, Beyer EC. Different domains are critical for oligomerization compatibility of different connexins. Biochem J. 2011;436:35–43. doi: 10.1042/BJ20110008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML. Structure and mechanism of glutamate receptor ion channel assembly, activation and modulation. Curr Opin Neurobiol. 2011;21:283–290. doi: 10.1016/j.conb.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maza J, Das Sarma J, Koval M. Defining a minimal motif required to prevent connexin oligomerization in the endoplasmic reticulum. J Biol Chem. 2005;280:21115–21121. doi: 10.1074/jbc.M412612200. [DOI] [PubMed] [Google Scholar]
- McClain MS, Iwamoto H, Cao P, Vinion-Dubiel AD, Li Y, Szabo G, Shao Z, Cover TL. Essential role of a GXXXG motif for membrane channel formation by Helicobacter pylori vacuolating toxin. J Biol Chem. 2003;278:12101–12108. doi: 10.1074/jbc.M212595200. [DOI] [PubMed] [Google Scholar]
- Mendrola JM, Berger MB, King MC, Lemmon MA. The single transmembrane domains of ErbB receptors self-associate in cell membranes. J Biol Chem. 2002;277:4704–4712. doi: 10.1074/jbc.M108681200. [DOI] [PubMed] [Google Scholar]
- Minogue PJ, Liu X, Ebihara L, Beyer EC, Berthoud VM. An aberrant sequence in a connexin46 mutant underlies congenital cataracts. J Biol Chem. 2005;280:40788–40795. doi: 10.1074/jbc.M504765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T. The biochemistry, ultrastructure, and subunit assembly mechanism of AMPA receptors. Mol Neurobiol. 2010;42:161–184. doi: 10.1007/s12035-010-8149-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgar O, Ierano C, Tamaki A, Stanley B, Ward Y, Xia D, Tarasova N, Robey RW, Bates SE. Mutational analysis of threonine 402 adjacent to the GXXXG dimerization motif in transmembrane segment 1 of ABCG2. Biochemistry. 2010;49:2235–2245. doi: 10.1021/bi902085q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabionet R, Zelante L, López-Bigas N, D'Agruma L, Melchionda S, Restagno G, Arbonés ML, Gasparini P, Estivill X. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum Genet. 2000;106:40–44. doi: 10.1007/s004390051007. [DOI] [PubMed] [Google Scholar]
- Russ WP, Engelman DM. TOXCAT: a measure of transmembrane helix association in a biological membrane. Proc Natl Acad Sci USA. 1999;96:863–868. doi: 10.1073/pnas.96.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáez JC, Berthoud VM, Branˇes MC, Martínez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- Tang Q, Dowd TL, Verselis VK, Bargiello TA. Conformational changes in a pore-forming region underlie voltage-dependent “loop gating” of an unapposed connexin hemichannel. J Gen Physiol. 2009;133:555–570. doi: 10.1085/jgp.200910207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsetlin V, Kuzmin D, Kasheverov I. Assembly of nicotinic and other Cys-loop receptors. J Neurochem. 2011;116:734–741. doi: 10.1111/j.1471-4159.2010.07060.x. [DOI] [PubMed] [Google Scholar]
- Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Güldenagel M, Deutsch U, Söhl G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem. 2002;383:725–737. doi: 10.1515/BC.2002.076. [DOI] [PubMed] [Google Scholar]
- Zhang JT, Nicholson BJ. Sequence and tissue distribution of a second protein of hepatic gap junctions, Cx26, as deduced from its cDNA. J Cell Biol. 1989;109:3391–3401. doi: 10.1083/jcb.109.6.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tang W, Ahmad S, Sipp JA, Chen P, Lin X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc Natl Acad Sci USA. 2005;102:15201–15206. doi: 10.1073/pnas.0501859102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.