

Abstract
Substituted ureas with a carboxylic acid ester as a secondary pharmacophore are potent soluble epoxide hydrolase (sEH) inhibitors. Although the ester substituent imparts better physical properties, such compounds are quickly metabolized to the corresponding less potent acids. Toward producing biologically active ester compounds, a series of esters were prepared and evaluated for potency on the human enzyme, stability in human liver microsomes, and physical properties. Modifications around the ester function enhanced in vitro metabolic stability of the ester inhibitors up to 32-fold without a decrease in inhibition potency. Further, several compounds had improved physical properties.
Keywords: sEH, sEH inhibitors, substituted urea-ester derivatives, metabolic stability
Epoxyeicosatrienoic acids (EETs), which are metabolites of arachidonic acid by cytochrome P450 epoxygenases, have important roles in the regulation of hypertension,1–6 inflammation,7–11 and other cardiovascular related diseases.12–13 Recently a role in modulating pain was also reported.14 However, metabolism of EETs (1) to their corresponding hydrated products (2) by soluble epoxide hydrolase (sEH) generally reduces these biological activities (Figure 1).1 Both in vitro and in vivo studies have demonstrated that the anti-hypertensive and cardio protective effects mediated by the EETs are reversely dependent on the extent of sEH hydrolysis of the EETs.2–4,6–8,13,15 Thus, maintaining the in vivo concentration of EETs through sEH inhibition is a promising therapeutic pathway to treat cardiovascular and other diseases. Urea, amide, and carbamate compounds substituted with hydrophobic groups are very potent and stable inhibitors of sEH with significant biological activities in both in vitro and in vivo models.3–4,16 However, poor physical properties of the early compounds, such as low solubilities and high melting points, likely result in limited in vivo availability.17 The addition of a polar functional group on specific positions of one of the urea-substitution is effective in increasing solubility in either water or organic solvents, and also in improving in vivo availability while maintaining the inhibition potency on the target enzyme.18–21 Among a variety of polar functional groups explored previously, ester functionalities located on the fifth atom from the urea function were found to be among the most suitable polar groups for making potent inhibitors with improved physical properties.18 However, as expected, the ester functionality is rapidly hydrolyzed both in vitro and in vivo producing the corresponding short chain acid metabolite that has dramatically reduced inhibition potency on the human sEH.20 Although rapid enzymatic hydrolysis of the ester compound is not favorable for high in vivo efficacy, it offers some advantage. First, alcohol moieties of ester groups can be rapidly altered to explore binding properties of the right hand side of the inhibitors such as those shown in Scheme 1. Second, the introduction of a secondary pharmacophore roughly 7–8 Å from the urea or amide carbonyl of the primary pharmacophore often improves solubility while retaining high potency.18,21–23 Finally, to date the urea inhibitors of the sEH appear highly selective with low toxicity. The epoxy-lipids stabilized by these inhibitors have numerous biological activities. Although the activities of the epoxy-lipids are so far found to be beneficial, it may be important to direct the activities to individual tissues. Administering short acting compounds may be also important for the treatment of pulmonary inflammation by inhalation. Thus for these latter two approaches it is important to develop esters of varying metabolic stability with increasing stability for classical systemic administration and decreasing stability for short term or local effects.24 In the present study, we explored structural modifications of urea-ester inhibitors toward altering the enzymatic stability of the ester function in an in vitro metabolic system while maintaining sEH inhibition potency and appropriate physical properties.
Figure 1.

Metabolism of biologically active EETs (1; e.g. 14,15-EET) to the less active DHETs (2; e.g. 14,15-DHET) by sEH. The sEH converts a variety of lipid epoxides to their corresponding diols. The sEHIs act by blocking this enzyme, increasing the in vivo concentrations of EETs which have direct actions on hypertension, inflammation, and pain. EET, epoxyeicosatrienoic acid; DHET, dihydroxyeicosatrienoic acid; sEHI, soluble epoxide hydrolase inhibitor.
Scheme 1.

Syntheses of 4-(3-adamantan-1-yl-ureido)-butyric acid or -cyclohexanecarboxylic acid derivatives: (a) EDCI, DMAP, a corresponding alcohol (2-hexanol for compound 5, 3-heptanol for compounds 6 and 12, and diethylene glycol monoethyl ether for compounds 8 and 13) in MC or 1-bromopentane for compounds 4 and 11 in DMF, rt, (b) pentanal, TMSCN, ZnI, chloroform, rt / 3N HCl (c) coupling of compound 3 with the product of reaction (b) by using EDCE/DMAP in MC, rt, (d) 2-ethoxyethanol, NaH, DMF, rt.
Compound 3, which was prepared as previously reported,18,21 was coupled with a corresponding alcohol by using 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide (EDCI) in the presence of 4-dimethylaminopyridine (DMAP) in dichloromethane to afford compounds 5, 6, and 8 or was alkylated with 1-bromopentane in the presence of K2CO3 in DMF to afford compound 4 in a range of 64–95% yield (Scheme 1 (A)). For the synthesis of cyanoalkyl ester (7), 1-pentanal was first reacted with trimethylsilyl cyanide in the presence of zinc iodide in chloroform at 0°C, followed by hydrolysis with an aqueous solution of 3N HCl (80%). The resulting cyanohydrin intermediate was then coupled with compound 3 by using EDCI and DMAP in dichloromethane to provide 7 in 70% yield. In addition, ethoxyethanol with a phenyl group, which was prepared by the reaction of 2-ethoxyethanol with 2-phenyloxirane, was coupled with compound 3 in the above same method as that used for the preparation of compound 7 to afford compound 9 in 48% yield. Scheme 1 (B) shows syntheses of derivatives with a cyclohexyl linker (10–13) between the urea and ester pharmacophores.21 Compound 10, which was prepared from the coupling of 1-adamantyl isocyanate with 3-aminocyclohexanoic acid, was alkylated or coupled with a corresponding bromide or alcohol in the same reaction condition as that used for the preparation of compounds 4–9 in Scheme 1 (A) to give target compounds 11–13 in 65–95% yield.21
Five derivatives (5–9) were prepared to first explore the modification effect of the alcohol moiety of the ester function of compound 4 on inhibition potency. As shown in Table 1, the incorporation of a methyl group (5) on the alpha carbon of the alcohol moiety showed a 2.5-fold improvement in inhibition potency compared to compound 4. An ethyl group (6) substituted on the alpha carbon of compound 4 exhibited a similar enhancement in potency, indicating that an alkyl substituent on the alpha position is effective for improving potency. On the other hand, the introduction of a cyano group (7) induced a 3-fold decrease in inhibition potency in comparison with that of methyl (5) or ethyl (6) groups. This suggests that hydrophobic alkanes are better than a polar group to be incorporated on the alpha carbon of the alcohol moiety of the ester function for enhanced inhibition potency. In a previous report, we demonstrated that diethylene glycol groups attached to hydrophobic urea inhibitors are useful for improving water solubility and bioavailability without a loss in inhibition potency.21 Two derivatives with a diethylene glycol group (8 and 9) were prepared to investigate whether the polar ethylene glycol group affects inhibition potency of urea-ester compounds. As illustrated by compound 8, attachment of a diethylene glycol resulted in a 7–17-fold reduction in inhibition when compared to those of non-substituted (4) and alkyl substituted compounds (5 and 6). However, interestingly, a big improvement in inhibition was obtained by incorporating a phenyl group (9) on the alpha carbon of the alcohol moiety of compound 8, which was as potent as that of the alkyl derivatives (5 and 6). This further indicates that a hydrophobic substituent on the alpha carbon of the ester function is highly effective to lead to potent urea-butyrate compounds as inhibitors of the human sEH. As described, the corresponding acid compound (3) of butyrate derivatives in Table 1 was poor as an inhibitor of the human enzyme. In addition, when a corresponding cyclohexylcarboxylic acid was synthesized as seen in compound 10 in Table 2, further reduced inhibition was exhibited. This indicates that a short chain acid attached to the urea function like urea-cyclohexylcarboxylic acid is a poor inhibitor and substitution of the acid could also result in potent compounds. When cyclohexylcarboxylic acid esters were prepared as seen in Table 2, as expected, a dramatic improvement in inhibition on the target human enzyme was observed in compound substituted with a pentyl group (11). The incorporation of an ethyl group (12) on the alpha carbon of the alcohol moiety of the pentyl ester 11 provided a highly potent inhibitor, as well. A diethyleneglycol group with a phenyl also made a potent inhibitor (13), although it was less potent than compounds 11 or 12. The results in Table 2 imply that alkylation of the acid function of urea-cyclohexylcarboxylic acid (10) is effective for producing highly potent inhibitors like butyrate derivatives in Table 1.
Table 1.
Inhibition of human sEH of 4-(3-adamantan-1-yl-ureido)-butyric acid derivatives.
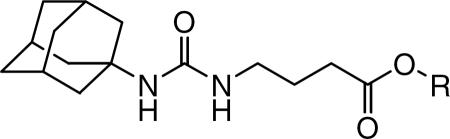
| Compound | R | Human sEH IC50 (nM)a |
|---|---|---|
| 3 | H | 684 |
| 4 |
|
10 |
| 5 |
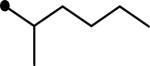
|
4 |
| 6 |
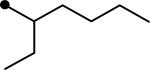
|
4 |
| 7 |
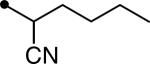
|
12 |
| 8 |
|
67 |
| 9 |
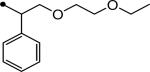
|
4 |
Human sEH (0.96 nM) was incubated with inhibitors for 5 min in 25 mM Bis-Tris/HCl buffer (200 μL; pH 7.0) at 30°C before fluorescent substrate introduction ([S] = 5 μM). Results are averages of three separate measurements. The fluorescent assay as performed here has a standard error between 10 and 20%, suggesting that differences of 2-fold or greater are significant.
Table 2.
Inhibition of human sEH of 3-(3-adamantan-1-yl-ureido)-cyclohexylcarboxylic acid derivatives.
| Compound | Structure | Human sEH IC50 (nM)a |
|---|---|---|
| 10 |
|
1160 |
| 11 |
|
2 |
| 12 |
|
3 |
| 13 |
|
9 |
Human sEH (0.96 nM) was incubated with inhibitors for 5 min in 25 mM Bis-Tris/HCl buffer (200 μL; pH 7.0) at 30°C before fluorescent substrate introduction ([S] = 5 μM). Results are averages of three separate measurements. The fluorescent assay as performed here has a standard error between 10 and 20%, suggesting that differences of 2-fold or greater are significant.
In general, liphophilicity of compounds causes limited solubility in water, which probably affects in vivo efficacy of compounds.17,21,25 In addition, the stability of the crystals of compounds, indicated by their high melting points, leads to a general lack of solubility even in organic solvents. These poor physical properties result in undesirable pharmacokinetic properties and difficulty in compound formulation in either an aqueous or oil base. We examined physical properties of the above potent derivatives in Tables 1 and 2. As seen in Table 3, butyric acid (3) and the corresponding ester derivatives (4 and 6) had very low solubility in water, while dramatic improvement in solubility was observed in diethylene glycol compounds (8 and 9). Similar increase in solubility was obtained in cyclohexylcarboxylic acid ester with a diethylene glycol group (13). The diethylene glycol derivatives (8, 9, and 13) also possessed much lower melting points in comparison with the acids (3 and 10) or other ester compounds (4, 6, and 12). This result indicates that hydrophilic alcohol moiety of the ester function of compounds 9 and 13 is highly beneficial for improving physical properties while producing potent inhibition. In order to see in vitro metabolism of the urea-ester compounds we used human liver microsomes, which contain various kinds of enzymes such as esterases, amidases, and cytochrome P450s playing a primary role in drug metabolism. When compounds 3 and 10 were tested for microsomal incubation, >95% of the parent acids were determined. This implies that the adamantylurea-acid structure of compounds 3 and 10 are stable under the incubation conditions and the human liver microsomal incubation is useful to investigate relative in vitro metabolic stability of urea-ester compounds. As expected, pentyl ester of butyric acid (4) was all metabolized to the corresponding butyric acid (3) in the incubation. On the other hand, a little increased stability was shown in compound 6 with an ethyl branch on the alpha carbon of the ester function. Further, diethylene glycol compounds (8 and 9) had >2.5-fold improved metabolic stability compared to compound 6, suggesting that a polar alcohol moiety of the ester function is more effective for improving the ester stability over a hydrophobic alkyl group. Interestingly, cyclohexylcarboxylic acid ester derivatives (12 and 13) exhibited much higher metabolic stability than the corresponding butyric acid esters (6 and 9). Comparing both compounds with an ethyl branch on the alpha carbon (6 and 12) a 2.5-fold increase in stability was observed in the cyclohexane derivative (12). Approximately a 2-fold improved stability was also shown in compound 13 compared to that of the corresponding ethyleneglycol butyric acid ester (9), indicating that sterically rigid cyclohexane linker present between the urea primary pharmacophore and the ester secondary pharmacophore is highly effective for increasing the ester stability. The results in Table 3 showed that solubility and stability of ureaester compounds are varied up to 390-fold and 32-fold, respectively, through incorporation of a substituent and/or modification in the linker structure.
Table 3.
Physical properties and in vitro metabolic stability of 4-(3-adamantan-1-yl-ureido)-butyric acid or -cyclohexylcarboxylic acid derivatives.
| Compound | Solubility in watera (μg/mL) | MP (°C) | Stabilityb (% remaining) |
|---|---|---|---|
| 3 | 7 | 165 | 97 |
| 4 | 2 | 114 | <1 |
| 6 | 3 | 72 | 4 |
| 8 | 878 | oil | 10 |
| 9 | 89 | oil | 14 |
| 10 | NDc | >250 | 95 |
| 12 | 1 | 124 | 10 |
| 13 | 41 | oil | 32 |
An excess of the test compound was added to a vial containing sodium phosphate buffer, 0.1 M pH 7.4 (1 mL), and a suspension of the mixture was equilibrated during 1 hr of sonication and 24 hrs of shaking, followed by centrifugation. The water supernatant was analyzed by LC-MS/MS. A regression curve for each compound was obtained from five standard stock solutions by using LC-MS/MS. Then, the absolute amount of each compound was calculated.19
Human liver microsomal protein (0.125 mg) was incubated with a solution of test compound under a NADPH generating system. Incubation mixture was shaken at 37°C for 60 min. The absolute quantity of parent compounds remaining after the incubation was analyzed by LC-MS/MS and was converted to a percentage. The corresponding acid metabolites (3 and 10) of ester compounds were analyzed in all determinations (data not shown). The results given are averages of triplicate independent analyses.
Not-determined because the crystal of compound 10 was not dissolved in water which seems to be due to the high melting point (> 250°C).
In conclusion, we synthesized a series of urea-ester compounds as inhibitors for the human soluble epoxide hydrolase (sEH) to produce compounds of varying metabolic stability by improving the biodegradable ester function while possessing potent inhibition and improved physical properties. The SAR study showed that the incorporation of an alkyl group on the alpha carbon of the alcohol moiety of the ester increased inhibition potency on the sEH, while a polar group in the alcohol moiety resulted in a big decrease in potency. However, interestingly, a substituted polar group with a hydrophobic function made potent inhibitors (9 and 13). Moreover, the potent polar compounds (9 and 13) exhibited approximately a 20–45-fold improved water solubility and low melting points when compared to those of alkyl substituted inhibitors (4 or 6). Because we found that adamantylureas with a short chain acid (3 and 10 in Table 3) are not metabolized in incubation with human liver microsomal enzymes, this incubation was used to investigate relative ester bond stability of urea-ester compounds. Among the modified urea-ester derivatives, the highest improvement (32-fold) in stability was obtained from a cyclohexyl analogue with a polar substituent (13). Approximately a 2-fold lower stability was observed when the cyclohexyl linker of compound 13 was replaced by an alkyl chain linker (9). In compounds with a cyclohexyl linker (12) or polar group (8), a 3-fold decreased stability was shown compared to that of 13. A dramatic decrease in stability was exhibited in butyrate derivatives with an alkyl substitution (4 and 6). The overall structural modifications altered the relative metabolism of the ester inhibitors up to 32-fold without a decrease in inhibition potency. Further, several compounds had improved physical properties. These findings will facilitate development of biologically active urea-ester compounds as potent inhibitors of the sEH. The ester compounds described in this study could be particularly applicable for administration directly to inflamed tissue by inhalation for chronic obstructive pulmonary disorder (COPD) or asthma. Alternatively they could be given by slow release formulations to address inflammatory bowel diseases.26 sEH inhibitors have been shown to be active on a variety of cardiovascular, inflammatory, and pain related disorders in rodents.27,28
Supplementary Material
Acknowledgments
This work was supported in part by NIEHS Grant R01 ES02710, NIEHS Center for Environmental Health Sciences P30 ES05707, NIH/NIEHS Superfund Basic Research Program P42 ES04699, NIH/NHLBI R01 HL59699-06A1, and UCDMC Translational Technology Research Grant. BDH is a George and Judy Marcus Senior Fellow of the American Asthma Foundation.
Abbreviations
- sEH
soluble epoxide hydrolase
- EET
epoxyeicosatrienoic acid
- EDCI
1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide
- DMAP
4-dimethylaminopyridine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data Syntheses and detailed analytical data for compounds 5–13, IC50 assay conditions, in vitro metabolic stability in human liver microsomes, and water solubility are available.
References
- (1).Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation: molecular and functional properties on the arachidonate monooxygenase. J. Lipid Res. 2000;41:163. [PubMed] [Google Scholar]
- (2).Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ. Res. 2000;87:992. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- (3).Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- (4).Zhao X, Yamamoto T, Newman JW, Kim I-H, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J. Am. Soc. Nephrol. 2004;15:1244. [PubMed] [Google Scholar]
- (5).Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45:759. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- (6).Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2006;46:975. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Smith KR, Pinkerton KE, Watanabe T, Pedersen TL, Ma SJ, Hammock BD. Attenuation of tobacco smoke-induced lung inflammation by treatment with a soluble epoxide hydrolase inhibitor. Proc. Natl. Acad. Sci. USA. 2005;102:2186. doi: 10.1073/pnas.0409591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc. Natl. Acad. Sci. USA. 2005;102:9772. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Campbell WB. New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends Pharmacol. Sci. 2000;21:125. doi: 10.1016/s0165-6147(00)01472-3. [DOI] [PubMed] [Google Scholar]
- (11).Yang B, Graham L, Dikalov S, Mason RP, Falck JR, Liao JK, Zeldin DC. Overexpression of cytochrome P450 CYP2J2 protects against hypoxia-reoxygenation injury in cultured bovine aortic endothelial cells. Mol. Pharmacol. 2001;60:310. doi: 10.1124/mol.60.2.310. [DOI] [PubMed] [Google Scholar]
- (12).Node K, Ruan X, Dai J, Yang S, Graham L, Zeldin DC, Liao JK. Activation of Gs mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J. Biol. Chem. 2001;276:15983. doi: 10.1074/jbc.M100439200. [DOI] [PubMed] [Google Scholar]
- (13).Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai H-J, Kim IH, Tuteja D, Mateo RKP, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc. Natl. Acad. Sci. USA. 2006;103:18733. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Inceoglu B, Wagner K, Schebb NH, Morisseau C, Jinks SL, Ulu A, Hegedus C, Rose T, Brosnan R, Hammock BD. Analgesia mediated by soluble epoxide hydrolase inhibitors is dependent on cAMP. Proc. Natl. Acad. Sci. USA. 2011;108:5093. doi: 10.1073/pnas.1101073108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Moghaddam MF, Grant DF, Cheek JM, Greene JF, Williamson KC, Hammock BD. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat. Med. 1997;3:562. doi: 10.1038/nm0597-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Newman JW, Denton DL, Morisseau C, Koger CS, Wheelock CE, Hinton DE, Hammock BD. Evaluation of fish models of soluble epoxide hydrolase inhibition. Environ. Health Perspect. 2001;109:61. doi: 10.1289/ehp.0110961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD. Structural refinement of inhibitors of urea based soluble epoxide hydrolase. Biochem. Pharmacol. 2002;63:1599. doi: 10.1016/s0006-2952(02)00952-8. [DOI] [PubMed] [Google Scholar]
- (18).Kim IH, Morisseau C, Watanabe T, Hammock BD. Design, synthesis, and biological activity of 1,3-disubstituted ureas as potent inhibitors of the soluble epoxide hydrolase of increased water solubility. J. Med. Chem. 2004;47:2110. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- (19).Kim IH, Heirtzler FR, Morisseau C, Nishi K, Tsai HJ, Hammock BD. Optimization of amide-based inhibitors of soluble epoxide hydrolase with improved water solubility. J. Med. Chem. 2005;48:3621. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kim IH, Nishi K, Tsai H-J, Bradford T, Koda Y, Watanabe T, Morisseau C, Blanchfield J, Toth I, Hammock BD. Design of bioavailable derivatives of 12-(3-adamantan-1-yl-ureido)dodecanoic acid, a potent inhibitor of the soluble epoxide hydrolase. Bioorg. Med. Chem. 2007;15:312. doi: 10.1016/j.bmc.2006.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Kim IH, Tsai H-J, Nishi K, Kasagami T, Morisseau C, Hammock BD. 1,3-Disubstituted ureas functionalized with ether groups are potent inhibitors of the soluble epoxide hydrolase with improved pharmacokinetic properties. J. Med. Chem. 2007;50:5217. doi: 10.1021/jm070705c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kasagami T, Kim IH, Tsai H-J, Nishi K, Hammock BD, Morisseau C. Salicylate-urea-based soluble epoxide hydrolase inhibitors with high metabolic and chemical stabilities. Bioorg. Med. Chem. Lett. 2009;19:1784. doi: 10.1016/j.bmcl.2009.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kim IH, Park YK, Hammock BD, Nishi K. Structure-activity relationships of cycloalkylamide derivatives as inhibitors of the soluble epoxide hydrolase. J. Med. Chem. 2011;54:1752. doi: 10.1021/jm101431v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).(a) Anandan S-K, Gless RD. Exploration of secondary and tertiary pharmacophores in unsymmetrical N,N'-diarylurea inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett. 2010;20:2740. doi: 10.1016/j.bmcl.2010.03.074. [DOI] [PubMed] [Google Scholar]; (b) Shen HC, Ding F-X, Wang S, Xu S, Chen H-S, Tong X, Tong V, Mitra K, Kumar S, Zhang X, Chen Y, Zhou G, Pai L-Y, Alonso-Galicia M, Chen X, Zhang B, Tata JR, Berger JP, Colletti S. Discovery of spirocyclic secondary amine-derived tertiaryureas as highly potent, selective and bioavailable soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett. 2009;19:3398. doi: 10.1016/j.bmcl.2009.05.036. [DOI] [PubMed] [Google Scholar]
- (24).Richard BS. The organic chemistry of drug design and drug action, Second Edition. Elsevier Academic Press. 2004:471–473. [Google Scholar]
- (25).Hwang SH, Tsai H-J, Liu J-Y, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J. Med. Chem. 2007;50:3825. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Pillarisetti S, Khanna I. Targeting soluble epoxide hydrolase for inflammation and pain - an overview of pharmacology and the inhibitors. Inflamm. Allergy Drug Targets. 2012;11:143. doi: 10.2174/187152812800392823. [DOI] [PubMed] [Google Scholar]
- (27).Gross GJ, Gauthier KM, Moore J, Falck JR, Hammock BD, Campbell WB, Nithipatikom K. Effects of the selective EET antagonist, 14,15-EEZE, on cardioprotection produced by exogenous or endogenous EETs in the canine heart. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H2838. doi: 10.1152/ajpheart.00186.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shen H, Hammock BD. Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J. Med. Chem. 2012;55:1789. doi: 10.1021/jm201468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.