

Abstract
While postsynaptic ionotropic and metabotropic glutamate receptors have received the lions share of attention in studies of long-term activity-dependent synaptic plasticity, it is becoming clear that presynaptic metabotropic glutamate receptors play critical roles in both short-term and long-term plasticity of vesicular transmitter release, and that they act both at the level of voltage-dependent calcium channels and directly on proteins of the vesicular release machinery. Activation of G protein-coupled receptors can transiently inhibit vesicular release through the release of Gβγ which binds to both voltage-dependent calcium channels to reduce calcium influx, and directly to the C-terminus region of the SNARE protein SNAP-25. Our recent work has revealed that the binding of Gβγ to SNAP-25 is necessary, but not sufficient, to elicit long-term depression (LTD) of vesicular glutamate release, and that the concomitant release of Gαi and the second messenger nitric oxide are also necessary steps in the presynaptic LTD cascade. Here, we review the current state of knowledge of the molecular steps mediating short-term and long-term plasticity of vesicular release at glutamatergic synapses, and the many gaps that remain to be addressed.
Keywords: Group II metabotropic glutamate receptors, G protein-coupled receptors, Giα, Gβγ, long-term synaptic depression, SNAP-25, SNARE protein, synaptic plasticity, vesicular release
1. Introduction
Neurons communicate with one another via chemical messengers called neurotransmitters. Synaptic transmission, i.e. the process of transmitting electrical impulses from one neuron to the other via a chemical intermediate, is crucial for the normal function of a neural network, and perturbations of these junctions have been correlated with many neurological diseases, such as Alzheimer's, schizophrenia and Parkinson's (Cook and Leuchter, 1996; Huang et al., 2011; Nathan et al., 2011). Synapse strength can be bi-directionally modified in an activity dependent manner, and these changes can be brief or persistent. William James (1890), at a time when the neuron doctrine and the role of synapses in intercellular communication had yet to emerge, postulated that long-lasting alterations in the strength of connections between excitable elements of the brain could be a way in which memories are stored, an early “connectionist” view that is as current over a century later.
Homosynaptic long-term potentiation (LTP) is a persistent, synapse-specific strengthening of synaptic transmission induced by brief bursts of high frequency synaptic activity (100–200 Hz), whereas long-term synaptic depression (LTD) is weakening of synaptic strength, usually triggered by low frequency activation (1–5 Hz) for prolonged periods (10–15 min). These persistent changes in synaptic strength are widely considered to be leading candidates for cellular mechanisms of memory storage (Bliss et al., 2006; Pastalkova et al., 2006; Whitlock et al., 2006). Glutamate serves as the main excitatory neurotransmitter in the central nervous system and uses a myriad of receptor subtypes to activate ionic channels to change membrane potential, and G protein-coupled receptors to initiate downstream signal transduction. The glutamate receptor family can be divided into two types: a) fast, ionotropic glutamate receptors that include 2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl)propanoic acid (AMPA), N-Methyl-D-aspartate (NMDA) and kainate receptors (Madden, 2002) and b) G-protein coupled metabotropic receptors that act via second messenger cascades (mGluR) (Nicoletti et al., 2011; Niswender and Conn, 2010; Pinheiro and Mulle, 2008). The mGluR superfamily can be divided into eight different receptor subtypes on the bases of their sequence homology, pharmacologic properties and activation of downstream signal transduction pathways (Conn and Pin, 1997). Group I metabotropic receptors consist of mGluR1 and mGluR5, which are primarily expressed postsynaptically with a somatodendritic distribution. These receptors are selectively activated by 3,5-dihydroxyphenyl-glycine (DHPG) and coupled to heterotrimeric Gq proteins that stimulate PLC-β to produce the intracellular messengers inositol trisphosphate (IP3) and diacylglycerol (DAG) through phosphatidylinositol hydrolysis. IP3 triggers release of calcium from intracellular endoplasmic reticulum calcium stores (Fagni et al., 2000), while DAG recruits protein kinase C (PKC) to the membrane and activates it (Ferraguti et al., 2008; Houamed et al., 1991; Masu et al., 1991), resulting in enhanced excitability of hippocampal neurons via modulation of Ca2+, K+ and non-selective cation channels, as well as many longer term effects mediated by serine-threonine phosphorylation of numerous target proteins.
Group II mGluRs (mGluR2 and mGluR3), and group III mGluRs (mGluR4, mGluR7 and mGluR8) are coupled to Gi/o proteins that inhibit adenylate cyclase, preventing the formation of cyclic adenosine 3'5'-monophosphate (cAMP). These receptors are expressed both presynaptically and postsynaptically. Presynaptically, their activation can decrease transmitter release by reducing voltage-dependent Ca2+ channel conductance through direct binding of Gβγ to the channel, by interfering directly with the presynaptic release apparatus, or both (Anwyl, 1999; Cartmell and Schoepp, 2000). In the hippocampus, group II mGluRs are believed to be localized primarily in presynaptic terminals, while group III mGluR are located in or near presynaptic active zones (Shigemoto et al., 1997). While these mGluRs are perfectly positioned to regulate synaptic transmission, the role of presynaptic mGluR in persistent plasticity of vesicular transmitter release is still relatively under-explored.
2. Presynaptic component of LTD of synaptic transmission
Homosynaptic LTD is an input-specific, long-lasting reduction in synaptic strength induced by prolonged low-frequency stimulation that has been observed at a variety of glutamatergic synapses in the hippocampus (Bear and Abraham, 1996), neocortex and other brain regions (Collingridge et al., 2010). Homosynaptic, associative LTD can be evoked at a synaptic input that is activated out of phase with a second bursting input that converges on the same neuron (Chattarji et al., 1989; Stanton and Sejnowski, 1989), or temporally mismatching presynaptic and postsynaptic activation to mimic such activity (Debanne et al., 1994; Stanton and Sejnowski, 1989). The N-methyl-D-aspartate subtype of glutamate receptor (NMDAR) has been found to be essential for the induction of some forms of long-term synaptic plasticity because a) it gates the influx of Ca2+ and b) its voltage-dependent Mg2+ channel block allows the NMDAR to detect the level of coincident pre- and postsynaptic activity at individual synapses (Yuste et al., 1999). NMDAR-gated Ca2+ influx leads to the downstream activation of kinases and phosphatases required for inducing LTP and/or LTD in frequency-dependent patterns that elicit differing levels of [Ca2+] increase in dendritic spines. Prolonged stimulation of Schaffer collateral-CA1 synapses at low frequencies (LFS: 1–5 Hz for 10–15 min) elicits a form of LTD (LFS-induced LTD) whose induction is blocked by the NMDAR-selective antagonist D-AP5 (Dudek and Bear, 1992; Mulkey et al., 1993). This stimulation has been suggested to elicit LTD, rather than LTP, because the smaller amplitude and slower rate of postsynaptic [Ca2+] increase selectively activates the high affinity calcium sensitive phophatases calcineurin (PP2B) and PP1, that mediate postsynaptic changes resulting in LTD (Collingridge et al., 2010; Lisman, 1989; Mulkey et al., 1993). This hypothesis is supported by studies showing that inhibition of PP1/PP2A (Mulkey et al., 1993), or of PP2B (Mulkey et al., 1994), blocks the induction of LTD at Schaffer collateral-CA1 synapses. Postsynaptically, calcineurin dephosphorylates and inactivates inhibitor-1 (Mulkey et al., 1994), leading to activation of PP1, which dephosphorylates AMPAR GluR1 subunits at serine-845, thereby both decreasing AMPAR open probability (Lee et al., 1998) and triggering AMPAR internalization (Beattie et al., 2000; Ehlers, 2000). PP1 can also regulate gene expression via the dephosphorylation and the inactivation of the transcriptional factor cAMP response element-binding protein (CREB) (Bito et al., 1996; Deisseroth et al., 1996; Hagiwara et al., 1992), preventing activity-dependent expression of early genes such as c-fos, BDNF and Arc, all found to promote LTP (Barco et al., 2005). Additional mechanisms involved in the postsynaptic induction and expression of a significant component of LTD have been addressed in previous reviews and the reader is referred to them for these considerations (Collingridge et al., 2010; Kessels and Malinow, 2009; Poschel and Stanton, 2007)
Calcineurin can also dephosphorylate presynaptic proteins such as synapsin I (Chi et al., 2003; Jovanovic et al., 2001), whose dephosphorylated state is associated with reduced neurotransmitter release through decreasing the size of the readily-releasable vesicle pool (RRP) (Bykhovskaia, 2011; Hilfiker et al., 1999). Activation of NMDAR (Stanton et al., 2003; Zhang et al., 2006), group I (Zakharenko et al., 2002) and group II (Santschi et al., 2006) mGluR have all been shown to play roles in a presynaptic component of LFS-induced LTD at Schaffer collateral synapses. This presynaptic component of LTD appears to require a retrograde, diffusible intercellular messenger (Bolshakov and Siegelbaum, 1994; Stanton et al., 2003), perhaps arachidonic acid (Bolshakov and Siegelbaum, 1995) and/or nitric oxide (NO) (Stanton et al., 2003; Zhang et al., 2006) that are generated in the postsynaptic neuron. Retrograde messengers such as NO are membrane permeable, rapidly diffusible gaseous messenger that can diffuse from the postsynaptic compartment to presynaptic terminals within a small three-dimensional volume (≤50 μm3; Wood and Garthwaite, 1994). A principal physiological enzyme target of NO is soluble guanylate cyclase (Southam and Garthwaite, 1993) and studies have shown that NO mediated elevation of presynaptic [cGMP] requires release of Ca2+ from ryanodine-sensitive intracellular stores via the second-messenger cyclic ADP ribose, and that this cascade is a key component of the induction of stimulus-evoked LTD at Schaffer collateral–CA1 synapses (Gage et al., 1997; Izumi and Zorumski, 1993; Reyes-Harde et al., 1999).
At dentate granule cell mossy fiber-CA3 synapses, it is generally agreed that, like LTP, LTD induced by electrical stimulation leads to a largely presynaptically expressed form of LTD. A low frequency stimulus train of 1–2 Hz for 10–15 min can induce LTD at mossy fiber synapses (Kobayashi et al., 1996; Nicholls et al., 2006). This form of LTD depends on the presynaptic G-protein coupled receptors (GPCR) mGluR2 and A1 adenosine receptors, which, upon activation, release Gα subunits that inhibit adenylate cyclase (Kobayashi et al., 1996; Nicholls et al., 2006) and reduce activity of the cAMP-PKA pathway (Tzounopoulos et al., 1998).
2.1 Role of G-protein coupled receptors and Gαi2 in presynaptic LTD
Studies have shown that inhibition of PKA can augment the magnitude of LTD induced by a subthreshold low-frequency stimulus train (1Hz/400seconds; (Santschi et al., 1999), and that pairing inhibition of PKA with production of cGMP induces a robust form of chemical LTD (CLTD) with a presynaptic locus of expression requiring activation of cyclic GMP-dependent protein kinase (PKG; Santschi et al., 1999). These studies also found that presynaptic GPCRs such as A1 adenosine receptors and group II mGluRs can supply adenylate cyclase inhibition necessary for induction of LTD (Santschi et al., 1999). Indeed, simultaneous activation of A1 adenosine receptors and elevation of [cGMP] is sufficient to elicit CLTD, and LTD is impaired by selective antagonists for either group II mGluR (EGLU) or A1 receptors (DPCPX) (Santschi et al., 2006), supporting a role for increased PKG and reduced PKA activity in the induction of presynaptic LTD (Broome et al., 1994; Gage et al., 1997; Izumi and Zorumski, 1993; Reyes-Harde et al., 1999; Santschi et al., 1999).
A presynaptic locus of expression of cyclic GMP-dependent LTD is further supported by experiments using the styryl dye FM1-43 to visualize vesicular release rates from Schaffer collateral presynaptic terminals (Pyle et al., 1999; Stanton et al., 2003; Stanton et al., 2005; Tyler et al., 2006; Zakharenko et al., 2002), and by measurements of release of [3H]glutamate from isolated presynaptic hippocampal synaptosomes (Bailey et al., 2003). Transient inhibition of glutamate release is thought to be due to modulation of voltage-dependent calcium channels, particularly N-type presynaptic calcium channels that are downstream targets of GPCRs (Tedford and Zamponi, 2006), but whether the same signal transduction and expression mechanisms are involved in any forms of LTD was unknown. Transgenic mice that express a mutant, constitutively active Gαi2 which inhibits adenylate cyclase, exhibit enhanced stimulus-induced LTD at both mossy fiber-CA3 (MF-CA3; Nicholls et al., 2006) and Schaffer collateral-CA1 (SCH-CA1; Bailey et al., 2008) synapses without significant changes in basal synaptic transmission. Interestingly, pharmacological elevations in [cGMP] which elicit a transient presynaptic depression in wildtype rodent slices (Boulton et al., 1994) evokes a presynaptic LTD in slices from these Gαi2 transgenic animals (Bailey et al., 2008; Nicholls et al., 2006). Furthermore, LFS-induced LTD at both MF-CA3 and SCH-CA1 synapses is also enhanced in slices from these animals (Bailey et al., 2008), implying a decreased threshold for chemically and stimulus-evoked LTD. Overall, these studies suggest that activation of inhibitory Gαi subunits by presynaptic G-protein coupled receptors contribute to inhibition of presynaptic adenylate cyclase/suppression in cyclic AMP production in the presynaptic terminal that is essential for induction of a presynaptic form of LTD. There are multiple presynaptic GPCRs that liberate Gαi, (Santschi et al., 2006) found that antagonists of either A1 adenosine receptors or group II mGluRs (both coupled to Gi/o) impair the induction of stimulus-evoked SCH-CA1 LTD. The adenylate cyclase isoforms AC1 and AC8 are candidates as immediate targets of inhibitory Gαi subunits, because they are sensitive to inhibition by Gi-coupled receptors (Nielsen et al., 1996). Gα-mediated inhibition of presynaptic adenylate cyclase would lower both basal and stimulus-evoked PKA activity, in turn altering phosphorylation states of a wide range of presynaptic PKA substrate proteins. These substrates could include, but are not restricted to, proteins that are essential in synaptic vesicle recycling, such as Rab3A and its effector protein Rim1, both believed to regulate presynaptic activity downstream of PKA (Chevaleyre et al., 2007; Huang et al., 2005; Lonart et al., 1998), or members of the SNARE complex (Nagy et al., 2005).
In the above-referenced study at mossy fiber-CA3 synapses in transgenic mice expressing constitutively active Gαi2 (Nicholls et al., 2006), these investigators observed both a doubling of the magnitude of stimulus-evoked LTD, and an occlusion of the transient suppression of mossy fiber synaptic transmission normally elicited by pharmacological activation of group II mGluRs, suggesting that, while group II mGluR activation elicits only transient depression of presynaptic glutamate release, there must be additional factors, such as NO activation of guanylyl cyclase and release from interterminal Ca2+ stores (Reyes and Stanton, 1996; Gage et al., 1997; Reyes-Harde et al., 1999), required to convert this depression to LTD. Similarly, constitutively active Gαi2 leads to suppression of MF-CA3 and SCH-CA1 LTP, which was rescued by exogenous application of SP-adenosine 3',5'-monophosphorothioate (SP-cAMPS), implying that the cAMP-PKA pathway is important for presynaptic LTP, at least at these synapses (Bailey et al., 2008; Nicholls et al., 2006). Previous studies have shown LTP at MF-CA3 synapses to be expressed presynaptically and depend on cAMP and PKA (Huang et al., 1994; Villacres et al., 1998), consistent with presynaptic group II mGluRs acting via inhibitory Gαi2 to suppress activity in the cAMP-PKA pathway.
2.2 mGluR-mediated LTD in different stages of brain development
Immature synapses (postnatal days 1–2) exhibit a form of mGluR-dependent LTD that is induced postsynaptically via co-activation of group I mGluRs and voltage-dependent calcium channels, and expressed presynaptically at SCH-CA1 synapses in the hippocampus (Bolshakov and Siegelbaum, 1994). Other studies have reported that, in neonatal rats, low-frequency stimulus-evoked LTD is mGluR-dependent, and not affected by NMDAR blockade (Feinmark et al., 2003; Li et al., 2002). mGluR-LTD in neonates is also independent of protein synthesis and does not require changes in the surface expression of AMPARs. This form of LTD is expressed as a long-term decrease in transmitter release, as demonstrated by increases in paired-pulse facilitation (Nosyreva and Huber, 2005), coefficient of variation of EPSC's (Bolshakov and Siegelbaum, 1994) and FM1-43 destaining (Zakharenko et al., 2002). The retrograde messenger in this neonatal mGluR-LTD has been suggested to be arachidonic acid (Bolshakov and Siegelbaum, 1994; Feinmark et al., 2003), and to involve the postsynaptic p38 MAPK-PLA2 pathway in the generation of this intercellular messenger.
In juvenile rats, mGluR-LTD can be induced by low frequency trains of paired-pulse stimulation (paired-pulse interval 50ms, delivered at a frequency of 1–2 Hz) in the presence of an NMDAR antagonist; (Huber et al., 2002; Kemp et al., 2000) or by a 5Hz/3min LFS train (Oliet et al., 1997). This group 1 mGluR-mediated LTD most likely involves postsynaptic activation of the mGluR5 subtype of mGluRs (Huang and Hsu, 2006; Huber et al., 2001; Oliet et al., 1997). Group 1mGluR-LTD that far outlasts washout of drug can be induced chemically by the group I agonist (S)-3,5-dihydroxyphenylglycine (Huber et al., 2001), is independent of both NMDAR and group II mGluR activation (Huang and Hsu, 2006) and occludes paired-pulse stimulus-evoked LTD, suggesting overlapping induction/expression mechanisms. The induction of group 1 mGluR-LTD required a postsynaptic increase in [Ca2+] and activation of PKC (Nicholls et al., 2006; Oliet et al., 1997). Interestingly, expression of DHPG-LTD also requires persistent activation of group I mGluRs, since established DHPG-LTD can be reversed by mGluR antagonists, but is reestablished once these blockers are removed from the extracellular perfusate (Huang and Hsu, 2006). Group 1 mGluR-LTD in juvenile animals requires protein synthesis (Connelly et al., 2011; Huber et al., 2001), and is thought to be mediated by endocytosis of postsynaptic AMPA and NMDA receptors (Nosyreva and Huber, 2005; Snyder et al., 2001). Additional evidence for a postsynaptic site of expression of DHPG-LTD comes from variance-mean analyses of release probability (Pr) at Schaffer collateral presynaptic terminals (Zhang et al., 2006) and studies of FM1-43 destaining kinetics from the readily releasable vesicle pool (Zhang et al., 2006). A presynaptic site of expression has also been reported for group 1 mGluR-LTD (Connelly et al., 2011; Fitzjohn et al., 2001; Manzoni and Bockaert, 1995) accompanied by enhanced paired-pulse facilitation (Fitzjohn et al., 2001; Manzoni and Bockaert, 1995), increased EPSC failures, changes in EPSC coefficient of variation, decreased mEPSC frequency (Fitzjohn et al., 2001) and a lack of postsynaptic change in responses to focal application of glutamate by uncaging (Rammes et al., 2003). Taken together, the above data lead to the conclusion that DHPG-induced LTD is expressed postsynaptically in the young brain, can co-exist with presynaptic alterations in release probability, and can also lead to postsynaptic endocytosis of AMPARs that creates silent synapses appearing to be presynaptic increases in release failures (Liao et al., 1995).
In adult rats, mGluR-LTD is not affected by inhibiting PKC or PKA, (Schnabel et al., 1999, 2001) or depleting intracellular calcium stores (Schnabel et al., 1999). This form of LTD is induced postsynaptically and may require CaMKII-mediated protein synthesis (Mockett et al., 2011) and postsynaptic protein tyrosine phosphatases (PTPs) dependent internalization of surface GluR2-containing AMPARs (Gladding et al., 2009), but also entail persistent presynaptic inhibition of voltage-sensitive Ca2+ and K+ channels (Watabe et al., 2002) that leads to reduced presynaptic Ca2+ influx (Tan et al., 2003). LTD cascades can include crosstalk with other receptors, as has been shown in a study of CA3 pyramidal neuron-specific M1 muscarinic receptor knockout animals, which exhibited deficiencies in mGluR-LTD (induced by bath application of DHPG), but normal stimulus-evoked LTD (Kamsler et al., 2010) This loss of mGluR-LTD was attributed to increased presynaptic release probability as a result of M1 receptor knockout, suggesting a presynaptic modulatory role for M1 receptors in mGluR-LTD (Kamsler et al., 2010).
3. Gβγ modulation of neurotransmission
Activation of GPCRs leads to the obligatory release of Gα and Gβγ subunits in a 1:1 stoichiometric ratio. While plasticity research has focused on downstream pathways activated by Gα subunits (such as Gs, Gq, Gi/o), the roles of Gβγ subunits in short and long-term plasticity of presynaptic function have been far less studied. Receptors such as CB1 endocannabinoid (Matsuda, 1997; Matsuda et al., 1990), 5HT-1 serotonin (Gerachshenko et al., 2005; Photowala et al., 2006), D2 dopamine (Congar et al., 2002), M4 muscarinic (Shirey et al., 2008), and α2-adrenergic receptors, are known to inhibit evoked transmitter release (Stephens and Mochida, 2005). Gβγ modulation of presynaptic vesicular release can occur in two different ways; ion channel regulation and direct regulation of vesicle exocytosis machinery. Ion channels that can alter their properties due to Gβγ binding include voltage-dependent calcium channels and G-protein coupled inward rectifying potassium channels, both of which can alter presynaptic action potential stimulated influx of calcium, thereby affecting probability of vesicular release. There is strong evidence to suggest that high voltage-activated N and P/Q type calcium channels are direct downstream targets for presynaptic Gβγ proteins (Herlitze et al., 1996; Ikeda, 1996; Tedford and Zamponi, 2006; Zhang et al., 2008). Binding of these G-proteins leads to a voltage-dependent inhibition of calcium currents that decreases vesicular release probability (Ikeda, 1996; Jarvis et al., 2000; Zhang et al., 2008). Another target of Gβγ modulation is G- protein activated inward rectifying potassium (GIRK) channels. GIRK channels are regulated by Gi/o coupled GPCRs and present at presynaptic terminals (Ponce et al., 1996) localized to presynaptic active zones (Ladera et al., 2008). Activation of GIRK channels by direct interactions with Gβγ proteins (Sadja et al., 2001) leads to membrane hyperpolarization, thus decreasing local membrane excitability and reducing the amount of local Ca2+ influx into presynaptic terminals, although this has not been demonstrated in a presynaptic terminal.
3.1 Gβγ modulation of vesicular release by direct interaction with SNARE proteins
Evidence suggests that there may be two different actions of Gβγ on the presynaptic release machinery. First, Gβγ can modify spontaneous mEPSP frequency, first demonstrated at the neuromuscular junction (Silinsky, 1984). Secondly, a Ca2+-independent pathway for presynaptic modulation of neurotransmitter release by Gβγ, first identified by studies in the lamprey, where (Blackmer et al., 2001) demonstrated that presynaptic injection of Gβγ into these large terminals mimicked serotonin inhibition of presynaptic transmitter release. To determine the role of Gβγ in serotonin-mediated inhibition, these authors used a potent, selective Gβγ scavenger (ct-GRK2), which completely occluded the ability of serotonin to inhibit chemical neurotransmission, suggesting a crucial role for Gβγ subunits in transient, serotonin-mediated presynaptic inhibition of release via GPCRs. Fluorescent calcium imaging revealed that presynaptic Gβγ injection did not alter evoked Ca2+ entry through voltage-gated calcium channels (Blackmer et al., 2001) and that serotonin application can still inhibit synaptic transmission when paired with Ca2+ release by intraterminal uncaging (Gerachshenko et al., 2005) suggesting that Gβγ is likely to act on binding sites downstream of calcium entry, such as proteins of the presynaptic vesicular release apparatus.
SNAP (soluble NSF attachment protein) receptor (SNARE) proteins seem likely candidates as Gβγ binding targets that modulate transmitter release, since Gβγ can directly bind to syntaxin-1A, SNAP-25 and the ternary SNARE complex (Blackmer et al., 2005; Blackmer et al., 2001; Gerachshenko et al., 2005; Yoon et al., 2007). Indeed, SNAP-25 became a prime candidate Gβγ binding target when it was found that pretreatment of lamprey synapses with type A botulinum toxin (BoNT/A, a protease that selectively cleaves 9 amino acids from the C-terminus of SNAP-25) (Binz et al., 1994; Gerachshenko et al., 2005; Xu et al., 1998), both reduced release probability and prevented the ability of Gβγ to inhibit exocytosis (Gerachshenko et al., 2005). Furthermore, presynaptic infusion with the C-terminal 14-amino-acid fragment of SNAP-25 (ct-SNAP-25) also blocked 5-HT mediated presynaptic inhibition, reinforcing the idea that G-protein binding to the C-terminus of SNAP-25 could serve as a mechanism for inhibition of neurotransmitter release, at least at lamprey synapses (Gerachshenko et al., 2005). This inhibition of vesicle fusion downstream of calcium entry could have been as a result of a) decreased efficiency of the SNARE complex, b) uncoupling of interactions between the putative calcium sensor synatotagmin and the SNARE complex (Blackmer et al., 2005; Gerachshenko et al., 2005; Yoon et al., 2007; Figure 1B) or c) a shift in the preferred mode of vesicle fusion to a kiss-and-run mode of release (Photowala et al., 2006; Schwartz et al., 2007; Gerachshenko et al., 2009), decreasing quantal size and cleft glutamate release concentration. Photowala et al. (2006) used a method of sensing trapped FM1-43 within vesicles by quenching with the hydrophilic fluorophore sulforhodamine101, to conclude that 5-HT led to an enhanced recruitment of a kiss and run mode of release. In addition, it was demonstrated that 5-HT, via Gβγ acting at the SNARE complex, led to transient vesicle fusion of synaptic vesicles that resulted in decreased quantal size (Blackmer et al., 2001; Chen et al., 2005; Photowala et al., 2006).
Figure 1.
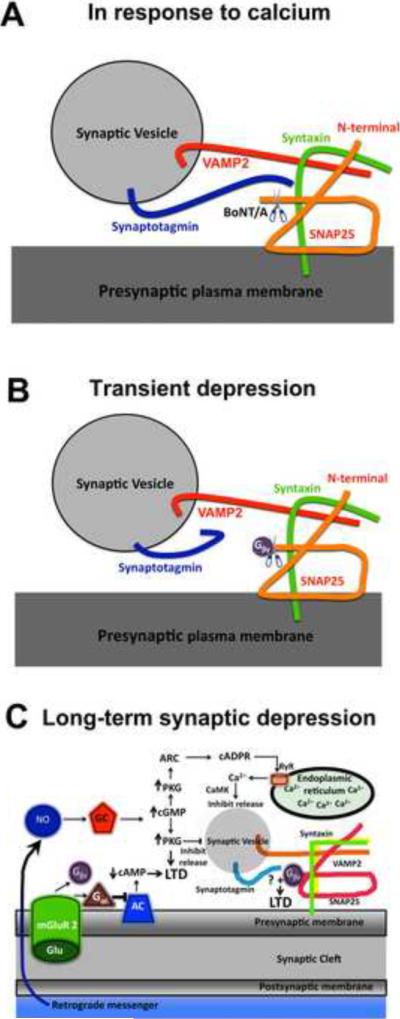
A: In response to presynaptic calcium entry, synaptotagmin, a synaptic vesicle protein that is the putative calcium sensor controlling evoked neurotransmitter release, binds to the C-terminus of SNAP-25 (Gerona et al., 2000; Zhang et al., 2002), one of three proteins in the SNARE complex of vesicle fusion machinery. Cleavage of the last 9 amino acids of the C-terminus of SNAP-25 with type A Botulinum Toxin (BoNT/A) reduces synaptotagmin binding (Gerona et al., 2000; Zhang et al., 2002), and shifts the calcium dependence of release (Gerona et al., 2000; Capogna et al.,. 1997). B: Gβγ binding to the C-terminus of SNAP-25 directly but transiently inhibits release. When G protein-coupled receptors such as group II metabotropic glutamate receptors are activated, Gβγ subunits are liberated and bind to the C-terminus of SNAP-25, where they interfere competitively with calcium-dependent binding of synaptotagmin to inhibit release (Blackmer et al., 2005; Gerachshenko et al., 2005). Cleavage of the last 9 C-terminus amino acids of SNAP-25 with BoNT/A eliminates Gβγ binding (Blackmer et al., 2005; Gerachshenko et al., 2005), mGluR2-mediated presynaptic depression (Zhang et al., 2011), and LTD (Zhang et al., 2011), implicating this site in both transient and long-term presynaptic depression. C: Signal transduction cascades necessary for induction of presynaptic long-term depression in the Schaffer collateral-CA1 presynaptic terminal. Abbreviations: mGluR2 - group II metabotropic glutamate receptor; NO – nitric oxide; GC – soluble guanylyl cyclase; AC – adenylate cyclase; cAMP – cyclic adenosine 3',5' monophosphate; cGMP – cyclic guanosine 3',5' monophosphate; LTD – long-term depression; PKG – cyclic GMP-dependent protein kinase; ARC – ADP-ribosyl cyclase/hydrolase; cADPR – cyclic ADP ribose; RyR – ryanodine receptor; CaMK – Ca2+/calmodulin-dependent protein kinase; VAMP2 – vesicle-associated membrane protein 2; SNAP25 – synaptosomal-associated protein 25.
Interestingly, Gβγ has been shown to interact with SNAP-25 and syntaxin1A (both members of the core SNARE complex), but, more importantly at the fully formed ternary SNARE complex, the interaction sites are near those used by synaptotagmin (Gerona et al., 2000; (Yoon et al., 2007). During the resting state, docked presynaptic vesicles are primed by a switch of plasma membrane t-SNARE associated syntaxin from its closed to its open conformation triggered by dissociation of the munc18-syntaxin complex (Carr and Rizo, 2010; Sudhof and Rothman, 2009). In the open conformation, the syntaxin-1 SNARE motif is unbound from the Habc domain and made available to complex with SNARE motifs in v-SNAREs (synaptobrevin). Once the action potential invades the presynaptic terminal, neurotransmitter release is initiated within 200μs and shows a steep dependence on [Ca2+] influx through high voltage gated Ca2+ channels (Schneggenburger and Neher, 2005; Tsien et al., 1991; Uchitel et al., 1992) with a half maximal activation at as high as 190μM [Ca2+] (Heidelberger et al., 1994; Sheng et al., 1996). Synaptotagmin-1 is believed to be the putative Ca2+ sensor that intiates synchronized vesicle fusion (Brose et al., 1992; Martens and McMahon, 2008; Yoshihara and Littleton, 2002). GPCR activation close to the active zone has the potential to release Gβγ that could compete with synaptotagmin binding, thus, it is possible that at low to moderate presynaptic [Ca2+]i,, calcium-independent Gβγ/SNARE binding wins out, inhibiting exocytosis (Blackmer et al., 2005; Blackmer et al., 2001; Gerachshenko et al., 2005), while high [Ca2+]i produces a steep increase in affinity of synaptotagmin for SNARE proteins, promoting exocytosis. Indeed, even after BoNT/A-mediated cleavage of the C-terminus of SNAP-25, exocytosis is restored by high [Ca2+]i (Gerachshenko et al., 2005), overcoming the loss of C-terminus binding sites by promoting more efficient SNARE fusion. However, the roles of Ca2+ and SNARE protein binding of Gβγ could be united if Gβγ binding to one or more SNARE proteins results in a lower affinity of synaptotagmin for Ca2+ that leads to reduced probability of release.
In mammalian synapses of the central amygdala, norepinephrine inhibits neurotransmitter release via activation of presynaptic α2 adrenoceptors on excitatory parabrachial inputs (Delaney et al., 2007). Presynaptic application of the Gβγ-binding peptide mSIRK (Chen et al., 2005), as well as incubation with BoNT/A, both significantly reduce noradrenergic inhibition at these terminals (Delaney et al., 2007), supporting an essential role for Gβγ in this presynaptic inhibition. In the hypothalamus, dynorphin mediated inhibition of glutamate release that occurs via activation of presynaptic κ-opioid receptors also appears to recruit direct Gβγ-mediated inhibition of exocytosis, since this action is independent of both inhibition of adenylyl cyclase and voltage-dependent calcium channels (Iremonger and Bains, 2009). In a non-neuronal system (INS 832/13 β-cells), norepinephrine also inhibits exocytosis via Gβγ interaction with SNAP-25, indicated by studies showing that intracellular application of antibodies against Gβγ or BoNT/A block this inhibition, while myristoylated βγ-binding/activating peptide mSIRK inhibited exocytosis to a similar degree as norepinephrine (Zhao et al., 2010).
4. Gβγ and the C-terminus of SNAP-25 are required for LTD of transmitter release
The above studies strongly argue for a role for Gβγ binding to the C-terminus region of SNAP-25 in mediating transient inhibition of neurotransmission elicited by a variety of GPCRs (Figure 1A, B), but could a similar mechanism be involved in the induction/expression of long-term presynaptic plasticity, and could it involve group II mGluRs and/or other GPCRs that liberate Gβγ?
To test the hypothesis that Gβγ binding to the C-terminus region of SNAP-25 might play a necessary role in the induction of LTD at Schaffer collateral-CA1 synapses in the mammalian hippocampus, (Zhang et al., 2011) treated hippocampal slices with BoNT/A, which, by itself, produced a ~50% reduction in fEPSPs associated with a presynaptic reduction in release probability (demonstrated by variance-mean analysis). In this study, slices pretreated with BoNT/A showed impaired induction of LTD by low frequency (2 Hz/10min) Schaffer collateral stimulation, while homosynaptic LTP induced by theta burst stimulation and, interestingly, dedepression induced by low frequency stimulation at the same synapse were not affected (Zhang et al., 2011). These findings support the notion that, in addition to GPCR-mediated transient presynaptic inhibition of release, the C-terminus of SNAP-25 is also essential in some way for the induction of LTD.
Zhang et al., (2011) also showed that stimulus-evoked LTD and BoNT/A-mediated presynaptic depression mutually occlude one another, since saturation of SCH-CA1 LTD eliminated any further effect of BoNT/A on synaptic transmission, further suggesting shared mechanisms of induction. Interestingly, the ability of BoNT/A to inhibit vesicular release was restored when, after saturating LTD, theta burst stimulation was applied to repotentiate synaptic strength, a sharp contrast from the lack of effect of BoNT/A pretreatment on de novo LTP (Zhang et al., 2011). However, BoNT/A pretreatment did not occlude cGMP- mediated presynaptic depression (Gage et al., 1997; Izumi and Zorumski, 1993; Reyes-Harde et al., 1999) induced by application of the selective type V phosphodiesterase inhibitor zaprinast, suggesting divergent mechanisms for decrease in synaptic strength (Zhang et al., 2011). Moreover, stimulus-evoked LTD was not impaired by reducing extracellular [Ca2+] as it was by BoNT/A, and elevating extracellular [Ca2+] after BoNT/A did not rescue LTD, indicating that the effects of BoNT/A were not simply due to the reduction in probability of release (Zhang et al., 2011).
To directly test whether the presynaptic role of the C-terminus of SNAP-25 in LTD might involve binding of Gβγ in this region, these authors utilized two molecularly distinct methods used previously to scavenge free Gβγ, to determine whether they could impair the induction of LTD at SCH-CA1 synapses. Schaffer collateral presynaptic terminals were selectively filled by electroporating pyramidal cell soma in field CA3 to fill them with either a Gβγ scavenging peptide, consisting of 14 amino acids of the C-terminus sequence (Ct-SNAP-25 peptide) of SNAP-25 (Gerachshenko et al., 2005), or the Gβγ binding peptide mSIRK (Chen et al., 2005). In both cases, there was a significant reduction in the magnitude of LTD when compared to LTD induced in the presence of scrambled control peptides, confirming the conclusion that Gβγ binding to its targets is crucial for the full expression of LFS-LTD (Zhang et al., 2011). The Ct-SNAP-25 peptide also markedly reduced the presynaptic inhibition produced by group II mGluR agonists, supporting a role for Gβγ in mGluR-dependent transient synaptic depression at mammalian synapses (Zhang et al., 2011) similar to that observed at lamprey synapses (Blackmer et al., 2005; Blackmer et al., 2001; Gerachshenko et al., 2005). Since Gβγ is known to directly bind to and inhibit voltage-gated calcium channels, this study also tested the possibility that such binding could play a role in presynaptic LTD. Calcium imaging revealed that induction of LTD was associated with only a transient decrease in stimulus-evoked Ca2+ influx that, while possibly contributing to short-term plasticity of release, could not underlie LTD, suggesting rather mechanisms downstream of calcium entry (Blackmer et al., 2005; Blackmer et al., 2001; Chen et al., 2005; Delaney et al., 2007; Gerachshenko et al., 2005; Takahashi et al., 2001)
5. Conclusion
Recent work has revealed a critical role for metabotropic glutamate GPCRs in the induction of a presynaptic form of LTD of synaptic transmission at hippocampal synapses, one that appears to recruit the same GPCR-mediated biochemical cascade involved in transient presynaptic depression at both lamprey and mammalian synapses. This cascade requires the simultaneous GPCR-mediated release of Gαi, which inhibits adenylate cyclase, and Gβγ, which binds to the C-terminus of the SNARE protein SNAP-25 to reduce vesicular glutamate release, and this cascade can be activated by a number of GPCRs, including group II mGluRs and A1 adenosine receptors. It is clear however that, while these G-proteins are essential components in the induction of a presynaptic form of LTD, they are not sufficient to elicit long-term changes in transmitter release. Both at lamprey and mammalian synapses, GPCR activation alone, whether by serotonin, glutamate or adenosine, elicits only transient presynaptic depression of transmitter release that recovers upon removal of receptor agonist. Previous studies at the SCH-CA1 synapse (Figure 1C), where both presynaptic LTD and LTP require postsynaptic activation, have shown that it is the production of the second messenger nitric oxide (NO) that appears to be an essential additional intercellular messenger in presynaptic plasticity (Gage et al., 1997; Izumi and Zorumski, 1993). Postsynaptically-generated NO acts to report to the presynaptic terminal that a “Hebbian” coincident activation of presynaptic and postsynaptic compartments has occurred. This NO, through activation of guanylyl cyclase that leads to increased cyclic GMP-dependent protein kinase activation, causes the production of the ryanodine receptor agonist cyclic ADP ribose that releases Ca2+ from intraterminal stores (Reyes-Harde et al., 1999). This Ca2+ appears to activate CamKII that is also an essential component of the LTD cascade (Stanton and Gage, 1996). Evidence now supports direct actions on proteins of the vesicular release apparatus underlying long-term plasticity of release (Figure 1C), rather than persistent alterations at the level of voltage-dependent calcium channels (Zhang et al., 2011).
While a role for GPCRs, including group II mGluRs, in presynaptic activity-dependent plasticity is now becoming evident, the downstream molecular mechanisms are still far from elucidated. One key question is how SNAP-25 is modified molecularly to persistently reduce glutamate release, since persistent binding of Gβγ for many hours or longer is unlikely. Another is whether other SNARE proteins are also targets for persistent plastic modifications. Ca2+-dependent kinases and phosphatases, and PKG, may all play critical roles, suggesting phosphorylation sites on SNAP-25 and other SNARE proteins as prime targets. While occlusion experiments suggest that LTD has something in common with BoNT/A-mediated cleavage of the C-terminus of SNAP-25, the notion of an endogenous BoNT/A-like activity may be too irreversible to be computationally useful. Nevertheless, it is now becoming clear that, in addition to postsynaptic dendritic spines in all their complexity, the presynaptic terminal is also capable of long-term activity-dependent plasticity of transmitter release that offers the potential for far different, complex and frequency-dependent plasticity of synaptic transmission that is likely to play important roles in neural network development, learning and memory and information storage, warranting further investigation of the potential for nootropic therapeutics that target group II mGluRs and other presynaptic GPCRs.
Highlights
Presynaptic mGluRs are key to short and long-term plasticity of transmitter release.
mGluRs transiently inhibit vesicular release via release of Gβγ.
Gβγ binding the SNAP-25 C-terminus is necessary, not sufficient, for LTD of release.
Gαi2, Nitric oxide, cyclic GMP, cyclicADP ribose, CaMKinase are all necessary for LTD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Nicholls RE, Zhang XL, Zhou ZY, Muller W, Kandel ER, Stanton PK. Galpha(i2) inhibition of adenylate cyclase regulates presynaptic activity and unmasks cGMP-dependent long-term depression at Schaffer collateral-CA1 hippocampal synapses. Learn Mem. 2008;15:261–270. doi: 10.1101/lm.810208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Trejos JA, Schanne FA, Stanton PK. Pairing elevation of [cyclic GMP] with inhibition of PKA produces long-term depression of glutamate release from isolated rat hippocampal presynaptic terminals. Eur J Neurosci. 2003;17:903–908. doi: 10.1046/j.1460-9568.2003.02507.x. [DOI] [PubMed] [Google Scholar]
- Barco A, Patterson SL, Alarcon JM, Gromova P, Mata-Roig M, Morozov A, Kandel ER. Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron. 2005;48:123–137. doi: 10.1016/j.neuron.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Bear MF, Abraham WC. Long-term depression in hippocampus. Annu Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Binz T, Blasi J, Yamasaki S, Baumeister A, Link E, Sudhof TC, Jahn R, Niemann H. Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J Biol Chem. 1994;269:1617–1620. [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Bartleson C, Kowalchyk JA, Yoon EJ, Preininger AM, Alford S, Hamm HE, Martin TF. G protein betagamma directly regulates SNARE protein fusion machinery for secretory granule exocytosis. Nat Neurosci. 2005;8:421–425. doi: 10.1038/nn1423. [DOI] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Takahashi M, Martin TF, Alford S, Hamm HE. G protein betagamma subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science. 2001;292:293–297. doi: 10.1126/science.1058803. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL, Laroche S. Neuroscience. ZAP and ZIP, a story to forget. Science. 2006;313:1058–1059. doi: 10.1126/science.1132538. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Hippocampal long-term depression: arachidonic acid as a potential retrograde messenger. Neuropharmacology. 1995;34:1581–1587. doi: 10.1016/0028-3908(95)00127-r. [DOI] [PubMed] [Google Scholar]
- Boulton CL, Irving AJ, Southam E, Potier B, Garthwaite J, Collingridge GL. The nitric oxide--cyclic GMP pathway and synaptic depression in rat hippocampal slices. Eur J Neurosci. 1994;6:1528–1535. doi: 10.1111/j.1460-9568.1994.tb00543.x. [DOI] [PubMed] [Google Scholar]
- Broome MR, Collingridge GL, Irving AJ. Activation of the NO-cGMP signalling pathway depresses hippocampal synaptic transmission through an adenosine receptor-dependent mechanism. Neuropharmacology. 1994;33:1511–1513. doi: 10.1016/0028-3908(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Brose N, Petrenko AG, Sudhof TC, Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- Bykhovskaia M. Synapsin regulation of vesicle organization and functional pools. Semin Cell Dev Biol. 2011;22:387–392. doi: 10.1016/j.semcdb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Carr CM, Rizo J. At the junction of SNARE and SM protein function. Curr Opin Cell Biol. 2010;22:488–495. doi: 10.1016/j.ceb.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75:889–907. doi: 10.1046/j.1471-4159.2000.0750889.x. [DOI] [PubMed] [Google Scholar]
- Chattarji S, Stanton PK, Sejnowski TJ. Commissural synapses, but not mossy fiber synapses, in hippocampal field CA3 exhibit associative long-term potentiation and depression. Brain Res. 1989;495:145–150. doi: 10.1016/0006-8993(89)91228-6. [DOI] [PubMed] [Google Scholar]
- Chen XK, Wang LC, Zhou Y, Cai Q, Prakriya M, Duan KL, Sheng ZH, Lingle C, Zhou Z. Activation of GPCRs modulates quantal size in chromaffin cells through G(betagamma) and PKC. Nat Neurosci. 2005;8:1160–1168. doi: 10.1038/nn1529. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Sudhof TC, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Congar P, Bergevin A, Trudeau LE. D2 receptors inhibit the secretory process downstream from calcium influx in dopaminergic neurons: implication of K+ channels. J Neurophysiol. 2002;87:1046–1056. doi: 10.1152/jn.00459.2001. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Connelly T, Fan Y, Schulz PE. Distinct mechanisms contribute to agonist and synaptically induced metabotropic glutamate receptor long-term depression. Eur J Pharmacol. 2011;667:160–168. doi: 10.1016/j.ejphar.2011.04.063. [DOI] [PubMed] [Google Scholar]
- Cook IA, Leuchter AF. Synaptic dysfunction in Alzheimer's disease: clinical assessment using quantitative EEG. Behav Brain Res. 1996;78:15–23. doi: 10.1016/0166-4328(95)00214-6. [DOI] [PubMed] [Google Scholar]
- Debanne D, Gahwiler BH, Thompson SM. Asynchronous pre- and postsynaptic activity induces associative long-term depression in area CA1 of the rat hippocampus in vitro. Proc Natl Acad Sci U S A. 1994;91:1148–1152. doi: 10.1073/pnas.91.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Crane JW, Sah P. Noradrenaline modulates transmission at a central synapse by a presynaptic mechanism. Neuron. 2007;56:880–892. doi: 10.1016/j.neuron.2007.10.022. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc Natl Acad Sci U S A. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Fagni L, Chavis P, Ango F, Bockaert J. Complex interactions between mGluRs, intracellular Ca2+ stores and ion channels in neurons. Trends Neurosci. 2000;23:80–88. doi: 10.1016/s0166-2236(99)01492-7. [DOI] [PubMed] [Google Scholar]
- Feinmark SJ, Begum R, Tsvetkov E, Goussakov I, Funk CD, Siegelbaum SA, Bolshakov VY. 12-lipoxygenase metabolites of arachidonic acid mediate metabotropic glutamate receptor-dependent long-term depression at hippocampal CA3-CA1 synapses. J Neurosci. 2003;23:11427–11435. doi: 10.1523/JNEUROSCI.23-36-11427.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraguti F, Crepaldi L, Nicoletti F. Metabotropic glutamate 1 receptor: current concepts and perspectives. Pharmacol Rev. 2008;60:536–581. doi: 10.1124/pr.108.000166. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL. A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J Physiol. 2001;537:421–430. doi: 10.1111/j.1469-7793.2001.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage AT, Reyes M, Stanton PK. Nitric-oxide-guanylyl-cyclase-dependent and -independent components of multiple forms of long-term synaptic depression. Hippocampus. 1997;7:286–295. doi: 10.1002/(SICI)1098-1063(1997)7:3<286::AID-HIPO4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Gerachshenko T, Blackmer T, Yoon EJ, Bartleson C, Hamm HE, Alford S. Gbetagamma acts at the C terminus of SNAP-25 to mediate presynaptic inhibition. Nat Neurosci. 2005;8:597–605. doi: 10.1038/nn1439. [DOI] [PubMed] [Google Scholar]
- Gladding CM, Fitzjohn SM, Molnar E. Metabotropic glutamate receptor-mediated long-term depression: molecular mechanisms. Pharmacol Rev. 2009;61:395–412. doi: 10.1124/pr.109.001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara M, Alberts A, Brindle P, Meinkoth J, Feramisco J, Deng T, Karin M, Shenolikar S, Montminy M. Transcriptional attenuation following cAMP induction requires PP-1-mediated dephosphorylation of CREB. Cell. 1992;70:105–113. doi: 10.1016/0092-8674(92)90537-m. [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houamed KM, Kuijper JL, Gilbert TL, Haldeman BA, O'Hara PJ, Mulvihill ER, Almers W, Hagen FS. Cloning, expression, and gene structure of a G protein-coupled glutamate receptor from rat brain. Science. 1991;252:1318–1321. doi: 10.1126/science.1656524. [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Sustained activation of metabotropic glutamate receptor 5 and protein tyrosine phosphatases mediate the expression of (S)-3,5-dihydroxyphenylglycine-induced long-term depression in the hippocampal CA1 region. J Neurochem. 2006;96:179–194. doi: 10.1111/j.1471-4159.2005.03527.x. [DOI] [PubMed] [Google Scholar]
- Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Huang YY, Zakharenko SS, Schoch S, Kaeser PS, Janz R, Sudhof TC, Siegelbaum SA, Kandel ER. Genetic evidence for a protein-kinase-A-mediated presynaptic component in NMDA-receptor-dependent forms of long-term synaptic potentiation. Proc Natl Acad Sci U S A. 2005;102:9365–9370. doi: 10.1073/pnas.0503777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YZ, Rothwell JC, Lu CS, Chuang WL, Chen RS. Abnormal bidirectional plasticity-like effects in Parkinson's disease. Brain. 2011;134:2312–2320. doi: 10.1093/brain/awr158. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis--dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86:321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Iremonger KJ, Bains JS. Retrograde opioid signaling regulates glutamatergic transmission in the hypothalamus. J Neurosci. 2009;29:7349–7358. doi: 10.1523/JNEUROSCI.0381-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF. Nitric oxide and long-term synaptic depression in the rat hippocampus. Neuroreport. 1993;4:1131–1134. [PubMed] [Google Scholar]
- Jarvis SE, Magga JM, Beedle AM, Braun JE, Zamponi GW. G protein modulation of N-type calcium channels is facilitated by physical interactions between syntaxin 1A and Gbetagamma. J Biol Chem. 2000;275:6388–6394. doi: 10.1074/jbc.275.9.6388. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Jr., Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci. 2001;21:7944–7953. doi: 10.1523/JNEUROSCI.21-20-07944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamsler A, McHugh TJ, Gerber D, Huang SY, Tonegawa S. Presynaptic m1 muscarinic receptors are necessary for mGluR long-term depression in the hippocampus. Proc Natl Acad Sci U S A. 2010;107:1618–1623. doi: 10.1073/pnas.0912540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp N, McQueen J, Faulkes S, Bashir ZI. Different forms of LTD in the CA1 region of the hippocampus: role of age and stimulus protocol. Eur J Neurosci. 2000;12:360–366. doi: 10.1046/j.1460-9568.2000.00903.x. [DOI] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Manabe T, Takahashi T. Presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapse. Science. 1996;273:648–650. doi: 10.1126/science.273.5275.648. [DOI] [PubMed] [Google Scholar]
- Ladera C, del Carmen Godino M, Jose Cabanero M, Torres M, Watanabe M, Lujan R, Sanchez-Prieto J. Pre-synaptic GABA receptors inhibit glutamate release through GIRK channels in rat cerebral cortex. J Neurochem. 2008;107:1506–1517. doi: 10.1111/j.1471-4159.2008.05712.x. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Li ST, Kato K, Tomizawa K, Matsushita M, Moriwaki A, Matsui H, Mikoshiba K. Calcineurin plays different roles in group II metabotropic glutamate receptor- and NMDA receptor-dependent long-term depression. J Neurosci. 2002;22:5034–5041. doi: 10.1523/JNEUROSCI.22-12-05034.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci U S A. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Janz R, Johnson KM, Sudhof TC. Mechanism of action of rab3A in mossy fiber LTP. Neuron. 1998;21:1141–1150. doi: 10.1016/s0896-6273(00)80631-5. [DOI] [PubMed] [Google Scholar]
- Madden DR. The structure and function of glutamate receptor ion channels. Nat Rev Neurosci. 2002;3:91–101. doi: 10.1038/nrn725. [DOI] [PubMed] [Google Scholar]
- Manzoni O, Bockaert J. Metabotropic glutamate receptors inhibiting excitatory synapses in the CA1 area of rat hippocampus. Eur J Neurosci. 1995;7:2518–2523. doi: 10.1111/j.1460-9568.1995.tb01051.x. [DOI] [PubMed] [Google Scholar]
- Martens S, McMahon HT. Mechanisms of membrane fusion: disparate players and common principles. Nat Rev Mol Cell Biol. 2008;9:543–556. doi: 10.1038/nrm2417. [DOI] [PubMed] [Google Scholar]
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- Matsuda LA. Molecular aspects of cannabinoid receptors. Crit Rev Neurobiol. 1997;11:143–166. doi: 10.1615/critrevneurobiol.v11.i2-3.30. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Mockett BG, Guevremont D, Wutte M, Hulme SR, Williams JM, Abraham WC. Calcium/calmodulin-dependent protein kinase II mediates group I metabotropic glutamate receptor-dependent protein synthesis and long-term depression in rat hippocampus. J Neurosci. 2011;31:7380–7391. doi: 10.1523/JNEUROSCI.6656-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- Nagy G, Milosevic I, Fasshauer D, Muller EM, de Groot BL, Lang T, Wilson MC, Sorensen JB. Alternative splicing of SNAP-25 regulates secretion through nonconservative substitutions in the SNARE domain. Mol Biol Cell. 2005;16:5675–5685. doi: 10.1091/mbc.E05-07-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan PJ, Cobb SR, Lu B, Bullmore ET, Davies CH. Studying synaptic plasticity in the human brain and opportunities for drug discovery. Curr Opin Pharmacol. 2011;11:540–548. doi: 10.1016/j.coph.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Nicholls RE, Zhang XL, Bailey CP, Conklin BR, Kandel ER, Stanton PK. mGluR2 acts through inhibitory Galpha subunits to regulate transmission and long-term plasticity at hippocampal mossy fiber-CA3 synapses. Proc Natl Acad Sci U S A. 2006;103:6380–6385. doi: 10.1073/pnas.0601267103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD, Wroblewski JT, Pin JP. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology. 2011;60:1017–1041. doi: 10.1016/j.neuropharm.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MD, Chan GC, Poser SW, Storm DR. Differential regulation of type I and type VIII Ca2+-stimulated adenylyl cyclases by Gi-coupled receptors in vivo. J Biol Chem. 1996;271:33308–33316. doi: 10.1074/jbc.271.52.33308. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2005;25:2992–3001. doi: 10.1523/JNEUROSCI.3652-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Photowala H, Blackmer T, Schwartz E, Hamm HE, Alford S. G protein betagamma-subunits activated by serotonin mediate presynaptic inhibition by regulating vesicle fusion properties. Proc Natl Acad Sci U S A. 2006;103:4281–4286. doi: 10.1073/pnas.0600509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro PS, Mulle C. Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat Rev Neurosci. 2008;9:423–436. doi: 10.1038/nrn2379. [DOI] [PubMed] [Google Scholar]
- Ponce A, Bueno E, Kentros C, Vega-Saenz de Miera E, Chow A, Hillman D, Chen S, Zhu L, Wu MB, Wu X, Rudy B, Thornhill WB. G-protein-gated inward rectifier K+ channel proteins (GIRK1) are present in the soma and dendrites as well as in nerve terminals of specific neurons in the brain. J Neurosci. 1996;16:1990–2001. doi: 10.1523/JNEUROSCI.16-06-01990.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poschel B, Stanton PK. Comparison of cellular mechanisms of long-term depression of synaptic strength at perforant path-granule cell and Schaffer collateral-CA1 synapses. Prog Brain Res. 2007;163:473–500. doi: 10.1016/S0079-6123(07)63026-X. [DOI] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Choi S, Tsien RW. Visualization of synaptic activity in hippocampal slices with FM1-43 enabled by fluorescence quenching. Neuron. 1999;24:803–808. doi: 10.1016/s0896-6273(00)81028-4. [DOI] [PubMed] [Google Scholar]
- Rammes G, Palmer M, Eder M, Dodt HU, Zieglgansberger W, Collingridge GL. Activation of mGlu receptors induces LTD without affecting postsynaptic sensitivity of CA1 neurons in rat hippocampal slices. J Physiol. 2003;546:455–460. doi: 10.1113/jphysiol.2002.033514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M, Stanton PK. Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J Neurosci. 1996;16:5951–5960. doi: 10.1523/JNEUROSCI.16-19-05951.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Harde M, Potter BV, Galione A, Stanton PK. Induction of hippocampal LTD requires nitric-oxide-stimulated PKG activity and Ca2+ release from cyclic ADP-ribose-sensitive stores. J Neurophysiol. 1999;82:1569–1576. doi: 10.1152/jn.1999.82.3.1569. [DOI] [PubMed] [Google Scholar]
- Sadja R, Smadja K, Alagem N, Reuveny E. Coupling Gbetagamma-dependent activation to channel opening via pore elements in inwardly rectifying potassium channels. Neuron. 2001;29:669–680. doi: 10.1016/s0896-6273(01)00242-2. [DOI] [PubMed] [Google Scholar]
- Santschi L, Reyes-Harde M, Stanton PK. Chemically induced, activity-independent LTD elicited by simultaneous activation of PKG and inhibition of PKA. J Neurophysiol. 1999;82:1577–1589. doi: 10.1152/jn.1999.82.3.1577. [DOI] [PubMed] [Google Scholar]
- Santschi LA, Zhang XL, Stanton PK. Activation of receptors negatively coupled to adenylate cyclase is required for induction of long-term synaptic depression at Schaffer collateral-CA1 synapses. J Neurobiol. 2006;66:205–219. doi: 10.1002/neu.20213. [DOI] [PubMed] [Google Scholar]
- Schnabel R, Kilpatrick IC, Collingridge GL. An investigation into signal transduction mechanisms involved in DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology. 1999;38:1585–1596. doi: 10.1016/s0028-3908(99)00062-3. [DOI] [PubMed] [Google Scholar]
- Schnabel R, Kilpatrick IC, Collingridge GL. Protein phosphatase inhibitors facilitate DHPG-induced LTD in the CA1 region of the hippocampus. Br J Pharmacol. 2001;132:1095–1101. doi: 10.1038/sj.bjp.0703905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol. 2005;15:266–274. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Sheng ZH, Rettig J, Cook T, Catterall WA. Calcium-dependent interaction of N-type calcium channels with the synaptic core complex. Nature. 1996;379:451–454. doi: 10.1038/379451a0. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey JK, Xiang Z, Orton D, Brady AE, Johnson KA, Williams R, Ayala JE, Rodriguez AL, Wess J, Weaver D, Niswender CM, Conn PJ. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat Chem Biol. 2008;4:42–50. doi: 10.1038/nchembio.2007.55. [DOI] [PubMed] [Google Scholar]
- Silinsky EM. On the mechanism by which adenosine receptor activation inhibits the release of acetylcholine from motor nerve endings. J Physiol. 1984;346:243–256. doi: 10.1113/jphysiol.1984.sp015019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Southam E, Garthwaite J. The nitric oxide-cyclic GMP signalling pathway in rat brain. Neuropharmacology. 1993;32:1267–1277. doi: 10.1016/0028-3908(93)90021-t. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Gage AT. Distinct synaptic loci of Ca2+/calmodulin-dependent protein kinase II necessary for long-term potentiation and depression. J Neurophysiol. 1996;76:2097–2101. doi: 10.1152/jn.1996.76.3.2097. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Sejnowski TJ. Associative long-term depression in the hippocampus induced by hebbian covariance. Nature. 1989;339:215–218. doi: 10.1038/339215a0. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Bailey CP, Kyrozis A, Raginov I, Laube G, Veh RW, Nguyen CQ, Muller W. Long-term depression of presynaptic release from the readily releasable vesicle pool induced by NMDA receptor-dependent retrograde nitric oxide. J Neurosci. 2003;23:5936–5944. doi: 10.1523/JNEUROSCI.23-13-05936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Zhang XL, Muller W. Imaging LTP of presynaptic release of FM1-43 from the rapidly recycling vesicle pool of Schaffer collateral-CA1 synapses in rat hippocampal slices. Eur J Neurosci. 2005;22:2451–2461. doi: 10.1111/j.1460-9568.2005.04437.x. [DOI] [PubMed] [Google Scholar]
- Stephens GJ, Mochida S. G protein {beta}{gamma} subunits mediate presynaptic inhibition of transmitter release from rat superior cervical ganglion neurones in culture. J Physiol. 2005;563:765–776. doi: 10.1113/jphysiol.2004.080192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Freed R, Blackmer T, Alford S. Calcium influx-independent depression of transmitter release by 5-HT at lamprey spinal cord synapses. J Physiol. 2001;532:323–336. doi: 10.1111/j.1469-7793.2001.0323f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Hori N, Carpenter DO. The mechanism of presynaptic long-term depression mediated by group I metabotropic glutamate receptors. Cell Mol Neurobiol. 2003;23:187–203. doi: 10.1023/A:1022949922364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedford HW, Zamponi GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol Rev. 2006;58:837–862. doi: 10.1124/pr.58.4.11. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Ellinor PT, Horne WA. Molecular diversity of voltage-dependent Ca2+ channels. Trends Pharmacol Sci. 1991;12:349–354. doi: 10.1016/0165-6147(91)90595-j. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, Zhang XL, Hartman K, Winterer J, Muller W, Stanton PK, Pozzo-Miller L. BDNF increases release probability and the size of a rapidly recycling vesicle pool within rat hippocampal excitatory synapses. J Physiol. 2006;574:787–803. doi: 10.1113/jphysiol.2006.111310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounopoulos T, Janz R, Sudhof TC, Nicoll RA, Malenka RC. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- Uchitel OD, Protti DA, Sanchez V, Cherksey BD, Sugimori M, Llinas R. P-type voltage-dependent calcium channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proc Natl Acad Sci U S A. 1992;89:3330–3333. doi: 10.1073/pnas.89.8.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villacres EC, Wong ST, Chavkin C, Storm DR. Type I adenylyl cyclase mutant mice have impaired mossy fiber long-term potentiation. J Neurosci. 1998;18:3186–3194. doi: 10.1523/JNEUROSCI.18-09-03186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe AM, Carlisle HJ, O'Dell TJ. Postsynaptic induction and presynaptic expression of group 1 mGluR-dependent LTD in the hippocampal CA1 region. J Neurophysiol. 2002;87:1395–1403. doi: 10.1152/jn.00723.2001. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Wood J, Garthwaite J. Models of the diffusional spread of nitric oxide: implications for neural nitric oxide signalling and its pharmacological properties. Neuropharmacology. 1994;33:1235–1244. doi: 10.1016/0028-3908(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Xu T, Binz T, Niemann H, Neher E. Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat Neurosci. 1998;1:192–200. doi: 10.1038/642. [DOI] [PubMed] [Google Scholar]
- Yoon EJ, Gerachshenko T, Spiegelberg BD, Alford S, Hamm HE. Gbetagamma interferes with Ca2+-dependent binding of synaptotagmin to the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. Mol Pharmacol. 2007;72:1210–1219. doi: 10.1124/mol.107.039446. [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Littleton JT. Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron. 2002;36:897–908. doi: 10.1016/s0896-6273(02)01065-6. [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Cash SS, Denk W. Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J Neurosci. 1999;19:1976–1987. doi: 10.1523/JNEUROSCI.19-06-01976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron. 2002;35:1099–1110. doi: 10.1016/s0896-6273(02)00898-x. [DOI] [PubMed] [Google Scholar]
- Zhang XL, Upreti C, Stanton PK. Gbetagamma and the C terminus of SNAP-25 are necessary for long-term depression of transmitter release. PLoS One. 2011;6:e20500. doi: 10.1371/journal.pone.0020500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XL, Zhou ZY, Winterer J, Muller W, Stanton PK. NMDA-dependent, but not group I metabotropic glutamate receptor-dependent, long-term depression at Schaffer collateral-CA1 synapses is associated with long-term reduction of release from the rapidly recycling presynaptic vesicle pool. J Neurosci. 2006;26:10270–10280. doi: 10.1523/JNEUROSCI.3091-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen YH, Bangaru SD, He L, Abele K, Tanabe S, Kozasa T, Yang J. Origin of the voltage dependence of G-protein regulation of P/Q-type Ca2+ channels. J Neurosci. 2008;28:14176–14188. doi: 10.1523/JNEUROSCI.1350-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Fang Q, Straub SG, Lindau M, Sharp GW. Noradrenaline inhibits exocytosis via the G protein betagamma subunit and refilling of the readily releasable granule pool via the alpha(i1/2) subunit. J Physiol. 2010;588:3485–3498. doi: 10.1113/jphysiol.2010.190090. [DOI] [PMC free article] [PubMed] [Google Scholar]